

Abstract
Many recent advances in genomics and the expectations of personalized medicine are made possible thanks to power of high throughput sequencing (HTS) in sequencing large collections of human genomes. There are tens of different sequencing technologies currently available, and each HTS platform have different strengths and biases. This diversity both makes it possible to use different technologies to correct for shortcomings; but also requires to develop different algorithms for each platform due to the differences in data types and error models. The first problem to tackle in analyzing HTS data for resequencing applications is the read mapping stage, where many tools have been developed for the most popular HTS methods, but publicly available and open source aligners are still lacking for the Complete Genomics (CG) platform. Unfortunately, Burrows-Wheeler based methods are not practical for CG data due to the gapped nature of the reads generated by this method. Here we provide a sensitive read mapper (sirFAST) for the CG technology based on the seed-and-extend paradigm that can quickly map CG reads to a reference genome. We evaluate the performance and accuracy of sirFAST using both simulated and publicly available real data sets, showing high precision and recall rates.
Keywords: Complete Genomics, read mapping, gapped reads, high throughput sequencing
1. Introduction
High throughput sequencing (HTS) technologies continue to evolve in a fascinating speed since their first use for paired-end sequencing in [1], and whole genome shotgun sequencing (WGS) [2]. Many sequencing technologies were developed, including the Roche/454 [3], Illumina [4], AB SOLiD [5], and Pacific Biosciences [6], among others [7]. Many tools for read mapping [8], variant calling [9, 10], and genome assembly [11] have been developed for most of these platforms.
The availability of many different algorithms and their open source implementations helps researchers take more advantage of the data generated by the HTS technologies. However, there are no publicly available algorithms devised to map and analyze data generated by the Complete Genomics [12] (CG) platform. This is mainly due to the fact that the Complete Genomics company offers only sequencing service, and provides its own analysis results to their customers through the use of proprietary software. The only set of algorithms devised to analyze CG data are described by a group of scientists from the company [13], but the implementations of these algorithms remain proprietary and unreleased. Without doubt, the company’s own analysis pipeline is fine tuned for data with different characteristics. Still, research projects that incorporate new algorithm development with the analysis of many genomes, such as the 1000 Genomes Project [14], may benefit from the availability of open-source tools and algorithms to further improve the analysis results. Having an open source tool to analyze CG data can enable other researchers to develop new findings that are otherwise not possible to discover.
In this paper, we present a new read mapper, sirFAST, designed for the data generated by the CG platform. Complete Genomics reads present characteristics different from all other HTS technologies, where a read is composed of several subreads (read sections) that may either overlap, or have gaps between each other. We designed sirFAST to efficiently align such reads to the reference assembly, at the presence of the flexible “expected” gaps/overlaps within each read. Due to the fact that the Burrows-Wheeler transform [15] based methods do not scale well with indels, we implemented sirFAST as a hash based seed-and-extend algorithm. Similar to its Illumina and SOLiD based counterparts [16, 17, 18, 19], sirFAST supports both SAM [20] and DIVET [21] file formats. The implementation of sirFAST is publicly available at http://sirfast.sf.net
Observation and Problem
Unique characteristics of Complete Genomics reads
The Complete Genomics (CG) platform is based on DNA nanoball technology. Similar to the reads generated by other DNA sequencing technologies, CG provides paired-end reads where each read is 29–35 base-pairs (bps), and the fragment size (distance between the two ends) can be between 400 to 1500 bps.
In contrast with the other DNA sequencing technologies that generate reads with consecutive bases (typically 100–8000 bps depending on the platform, as shown Figure 1(a)), a CG read shows a gapped read structure that consists of groups of consecutive bases denoted as read sections. There may be either gaps or overlaps between each read sections, denoted as expected gaps. Overlaps between read sections are represented as negative gaps. As shown in Figure 1(b), each read includes four read sections of lengths 5, 10, 10 and 10 bases (35 bps length in total) respectively. Between these read sections, there are expected gaps with sizes that follow a normal distribution with specific mean values (−2, 1 and 6 respectively). As a result, the typical composition of a CG read consists of the following: i) first read section (5 bps), ii) expected gap ([−3, −1] bps), iii) second section (10 bps), iv) expected gap ([0, 2] bps), v) third section (10 bps), vi) expected gap ([5, 7] bps), and vii) fourth section (10 bps). More recent CG data sets now support slightly different read structure, where 29bp reads are divided into three read sections of size 10bp, 9bp, and 10bp, respectively2. Note that, this difference in the read structure requires only small changes in the implementation of our algorithm, and support for such reads are implemented through a parameter in sirFAST. In the remainder of the paper, we explain our mechanisms based on the assumption that the reads are in the former structure. This is mainly due to the lack of availability of publicly released data generated in this structure. However, we provide simulation-based test results in Table 2.
Figure 1.
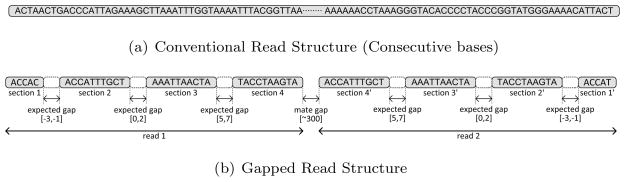
Conventional Read Structure vs. Gapped Read Structure. Note that the read section structures of paired-end reads generated by the Complete Genomics platform are symmetric to each other. Read structure of more recent CG data is changed slightly, however, it is straightforward to provide support for the newer data type using the same algorithms presented in this manuscript.
Table 2.
Mapping sensitivity, precision, and recall values for sirFAST using simulated data sets with different error thresholds. We mapped 500,000 read pairs, and compared their “best” mapping locations with their true locations as simulated from the reference genome. Note that all numbers given above are for number of read pairs.
| Format | Data set | TP | FP | FN | Sensitivity | Precision (PPV) | Recall |
|---|---|---|---|---|---|---|---|
|
| |||||||
| 1 | set 1 (e=0) | 481,379 | 17,528 | 1,093 | 96.28% | 96.49% | 99.77% |
| set 2 (e=1) | 481,438 | 17,602 | 960 | 96.29% | 96.47% | 99.80% | |
| set 3 (e=2) | 480,966 | 18,137 | 897 | 96.19% | 96.37% | 99.81% | |
|
| |||||||
| 2 | set 1 (e=0) | 489,158 | 10,842 | 0 | 97.83% | 97.83% | 100.00% |
| set 2 (e=1) | 488,142 | 11,775 | 62 | 97.62% | 97.64% | 99.98% | |
| set 3 (e=2) | 487,793 | 12,099 | 74 | 97.55% | 97.57% | 99.98% | |
The challenge of mapping CG reads
Two paradigms for mapping reads to the reference genome are 1) seed-and-extend, and 2) Burrows-Wheeler Transformation with FM-indexing (BWT-FM) methods [8]. Unfortunately, all existing mappers are designed for mapping consecutive reads to the reference genome, thereby facing challenges or requiring much longer time to map the reads in a gapped read structure (as in CG). Specifically, mapping CG reads with BWT-FM is a harder problem since the expected gaps in CG reads show themselves as indels in an alignment, which increases the run time of BWT-FM search exponentially. None of the currently available BWT-FM based aligners perform gapped alignment per read; instead, they anchor one end with a gap-free alignment, and apply gapped alignment for its mate in regions of close proximity. It is therefore impractical to use BWT-FM based approach for CG reads as both ends contain expected gaps. Although BWT-FM methods outperform seed-and-extend methods in aligning consecutive basepair reads with very low sequence errors, seed-and-extend techniques scale better with high error rate, especially when the errors are indels. We therefore designed our mapper to use the seed-and-extend technique similar to several others available in the literature [8].
Figure 2 shows the typical operation of a seed-and-extend mapper. The mapper first selects a set of short subsequences of a read as seeds, and quickly queries their locations in the genome using a hash table or a similar data structure. The mapper then tries to extend these initial seed locations by calculating the alignment of the full read against pieces of reference genome at these seed locations. This extension step is commonly called verification where the similarity score is calculated between the read and the reference. Each insertion, deletion and substitution has a positive score while each matching base has a zero score. A smaller score denotes higher similarity between the read and the reference. The alignment algorithms used in this step are dynamic programming algorithms which tackles insertions and deletions. The complexity of the verification step per seed location is quadratic, which can be reduced to O(kn) if k is an upper bound for allowed indels in the alignment. In the remainder of the paper, we refer to the alignment algorithms as bp-granularity mapping algorithms.
Figure 2.

Mapping consecutive reads using conventional hash-table based mapper
2. Methods
We now explain how we map the reads generated by the CG platform to the reference genome using the seed-and-extend methodology (Figure 3).
Figure 3.

Mapping gapped read structure using conventional hash-table based mapper
Selecting Seeds. The performance and accuracy of seed-and-extend mappers depend on how the seeds are selected in the first stage. The seeds should be selected in a way that guarantees: 1) very high sensitivity to make sure real mapping locations are captured, 2) high specificity to avoid unnecessary alignments, 3) fast seed placement, preferably in O(1) time, and 4) low memory usage. The two main types of seeds are 1) consecutive seeds as used in BLAST [22], and 2) spaced seeds, first defined in PatternHunter [23]. Spaced seeds carefully select bases from a larger consecutive region of the read to form seeds with gaps in them in order to reduce seed locations. Spaced seeds are considered to be more powerful in terms of higher sensitivity and specificity than consecutive seeds as they provide fewer locations to verify. However, spaced seeds are not suitable for CG reads due to the lack of large consecutive regions in the read (the lengths of expected gaps are flexible and the read sections are only 10-base-long). We, therefore, use consecutive seeds of length 10, and implement our seed index using a traditional hash table. We also develop the concept of “combined seeds” to effectively reduce seed locations, since the seed length of 10bp will yield too many initial locations due to the repetitive nature of the human genomes.
Verification. The seeds serve as a first-step quick filter to find potential locations for the reads, which then have to be verified with an alignment algorithm for the full-length read. This can be done using the Levenshtein [24] or Smith-Waterman [25] algorithms or their variants. Although these originally O(n2) algorithms can be accelerated to O(kn) using Ukkonen’s method [26], the high number of expected gaps (up to 12bp) limit the run time improvements. We, therefore take a slightly different approach, and use the Hamming distance in our verification while keeping track of the expected gaps in the alignment.
Problem
Verifying CG reads is more computationally expensive than verifying conventional reads of consecutive bases. This is because CG reads have gaps of flexible lengths between read sections. Given a seed location of one of the read sections, we cannot deduce the exact locations of other read sections from the same read. Rather, we know that they belong to a few short ranges. For each read section other than the seed, there exist multiple possible mappings of it within a short range. Therefore, for each seed location, we need to verify potential mappings of read sections under all combinations of expected gaps in order to guarantee full sensitivity. The similarity score of a CG read to a reference, is calculated as the sum of the minimum similarity scores of all read sections. This is computationally expensive because for each seed location it requires multiple verifications of many potential mappings of all read sections.
For example, in Figure 3, if we use the second read section (first 10-bp read section) as the seed, then there are three possible mappings respectively for the first and the third read section with different expected gaps between the second read section. For each possible mapping of the third read section, there exist three more possible mappings of the forth read section with different expected gaps from the third one. As a result, there exists 3 × 3 × 3 = 27 possible combinations of expected gaps between all four read sections. In this example, the minimum similarity score of the read is obtained with a gap of −2 between the first and the second read sections, a gap of 2 between the second and the third read sections and a gap of 6 between the third and the forth read sections (circled in boxes with “error=0”).
Our goal is to build a mechanism that reduces this large search space of possible combinations of expected gaps in the CG read to improve mapping performance while maintaining the mapping sensitivity.
Combined seeds
As shown in the previous section, CG reads create a large search space of possible combinations of expected gaps between read sections. This search space grows exponentially with the number of expected gaps in the read. To help eliminate this burden, we propose the idea of combined seeds, which drastically reduces the search space of CG reads. The key idea is to merge any two of the 10bp read sections together as a single combined seed including the gap between them. With this mechanism, the number of expected gaps in a gapped CG read is reduced from three to two (For example, if we examine Figure 4, we see that there are originally three expected gaps in the read. By combining section 2 and 3, the gap between these two seeds would be eliminated). This reduces the search space of the CG read significantly. This combined seed mechanism is based on two key observations: 1) the distance of mapping locations of two read sections in the reference genome should be within the size of the expected gap and 2) the hash table contains information about the relative distances of any two seeds. Therefore, by obtaining the locations of two read sections from the hash table and comparing them to each other, we can easily determine whether the distance between two read sections are within the range of the expected gap. (We will show an example of this below.)
Figure 4.

Mechanism to merge the seeds for generating the combined seed
Although we can combine all three read sections of a CG read together, we choose only two of them at a time in order to maintain high sensitivity (i.e., if three sections are combined, we cannot find all the errors). We require at least two 10-bp read sections matches that follow the aforementioned rule to invoke the verification step. Note that, for a 35bp read, allowing up to 5% substitutions (a typically used cutoff) will mean a maximum Hamming distance of 2. Hence, by the pigeonhole principle, unless the two allowed substitutions are in two different 10bp read sections, this seeding step will still provide 100% sensitivity.
We now explain the detailed operation of our combined seed technique. The location list of the combined seed is the subset of the location lists of two seeds that can be combined. Figure 4 shows the mechanism to combine seeds. First, we determine the number of seeds within a read that can be potentially merged into a combined seed (two seeds in Figure 4). Next, we select two 10bp read sections as seeds (section 2 and 3 in Figure 4). We then query the hash table to load the location lists of the two seeds, and calculate the location list of the combined seed. In this merging step, we use the information in the CG protocol [13] regarding values of the expected gap sizes between read sections. In Figure 4, we show how to merge read sections 2 and 3, where the expected gap size is in the range [0,2], and the seed length is 10bp. Therefore, the map locations of these two seeds in the reference genome should be separated by 10 to 12 basepairs (10 + [0,2]: length of seed + expected gap). Finally, we calculate the effective length of the combined seed as the summation of the lengths of two seeds and the size of the gap in between. Note that a 2-tuple combined seed where the individual seed length is 10bp, and expected gap size is in the range [0,2] is similar to creating three spaced seeds [23] with weight 20 and lengths 20, 21, and 22. Such spaced seeds are in the form 110110, 1100110, and 11000110 (1|seed| for seeds, 0|gap| for gaps between seeds). Similarly, combined seeds of sections 3 and 4 can be denoted as a combination of 11005110, 11006110, and 11007110. Finally, we would need to create 9 different forms of spaced seeds to represent the combined seeds of sections 2 and 4.3 Due to these issues with spaced seeds, we instead develop and use the combined seeds approach.
There are two main advantages that combined seeds offer in mapping CG reads. They help reduce the potential locations, and also reduce the overhead introduced by the flexible-sized expected gaps. The number of potential mapping locations of the combined seed is drastically reduced when compared to a single seed, because the location list of a combined seed is the intersection of the potential mapping locations of two different seeds. Therefore, mapping using the combined seed decreases the mapping time. Additionally, the expected gaps between the combined sections are automatically detected and removed from consideration in the verification step.
Multiple combinations of merging seeds
Given a seed length of 10bp, we can potentially use three seeds in a CG read that are separated by expected gaps. Therefore, to satisfy the high sensitivity constraint, we can merge three different pairs of read sections to generate combined seeds.
Mapping CG reads using combined seeds and Hamming distance calculation
The entire mapping procedure is shown in Figure 3, with the exception of using the combined seeds. To describe this briefly, we map a CG read by doing the following steps: 1) finding and merging seed locations to generate combined seeds and list of their locations, 2) verifying the map location of the full-length read through Hamming distance calculation [27]. In the verification step, we apply the Hamming distance algorithm to different read sections using a sliding window technique to ensure that we cover all possible combinations of possible expected gaps. The Hamming distance test is not performed on the read sections that are merged into a combined seed for that location, since the hash table and the combined seed procedure guarantees perfect matches for these sections.
3. Results
We used both simulated and real data sets to assess the performance of our methods. We first generated three simulated sets of reads using the human reference genome assembly GRCh37 (hg19), by selecting one subsequence of length 5bp, and three with 10bp, in order, and separated with the expected gaps as previously described [12, 13] (Figure 1(b)). We also repeated this procedure with three 10bp subsequences followed with a 5bp subsequence to construct the second end of matepairs. The first set of reads we generated assumed no sequencing errors compared to the reference genome, and the second and third sets included randomly inserted single base mutations at 1% and 2% probability, respectively. For the real data set experiment, we selected one entire slide of reads from the genome of NA12878 that was sequenced by the Complete Genomics corporation, and released publicly 4. Note that, since there are no other available tools to map CG data, and the official CG mapper is unreleased, we cannot provide performance comparisons against other read mappers. However, in the case of real data set we compared sirFAST with the available mapping result which is released on the CG website.
3.1. Mapping reads independently
We first tested sirFAST by mapping all reads independently, without using the matepair information (Table 1) with three different Hamming distance cut offs of 0 to 2bp. Our aim here was to demonstrate the mapping speed and comprehensiveness. sirFAST was able to map all simulated reads in both first and second sets when allowing same number of errors to the generated errors (e=0, e=1, e=2 for set 1, set 2, set 3, respectively), showing full mappability.
Table 1.
The performance of sirFAST with three simulated data sets. Each data set included 1 million reads. Note that 1% of the simulated reads in set 1 have one error respect to the reference, thus the best performance is to map 99% of the reads when the error tolerance of sirFAST is set to zero. In the second set, 2% of the reads have one error, therefore the best possible mappability ratio is 98% when the error tolerance is set to zero.
| Error | Data set | Time (min.) | Map Locations | Reads Mapped (%) |
|---|---|---|---|---|
|
| ||||
| e=0 | Simulated set 1 | 185 | 169,840,573 | 100 |
| Simulated set 2 | 187 | 166,584,186 | 99 | |
| Simulated set 3 | 189 | 164,985,213 | 98 | |
|
| ||||
| e=1 | Simulated set 1 | 220 | 870,605,211 | 100 |
| Simulated set 2 | 220 | 860,656,526 | 100 | |
| Simulated set 3 | 222 | 852,321,616 | 99.4 | |
|
| ||||
| e=2 | Simulated set 1 | 265 | 1,816,245,093 | 100 |
| Simulated set 2 | 265 | 1,802,365,053 | 100 | |
| Simulated set 3 | 262 | 1,783,516,970 | 100 | |
In Table 1, we also report the total number of mapped locations to demonstrate the comprehensiveness and potential use of sirFAST for duplication detection [16]. Note that since we treated each read independently in this test, we did not calculate mapping accuracy, which is reported in the next section.
3.2. Paired-end read mapping and precision/recall tests
As a second test to demonstrate the high precision5 and recall rates6 of sirFAST mapping, we mapped our simulated and real data sets in paired-end mode. The size distribution of the inferred fragment lengths followed a Gaussian-like distribution as expected in both real (Figure 5(a)) and simulated (Figure 5(b)) data sets.
Figure 5.

The histogram of insert size obtained from mapping the reads in paired-end and single-end mode. The x-axis is the insert size and the y-axis is the number of paired-end mapping (frequency) having that insert size.
We used the simulated read set mapping result also to estimate the precision and recall rates of sirFAST. Since sirFAST can track all possible mapping locations for each read, and all pairwise combinations of paired-end reads, theoretically, the precision and recall rates can be 100%. In fact, our single-end mapping test as explained in Section 3.1 demonstrates the perfect recall. However, although it is powerful to keep track all possible locations for structural variation discovery (SV) [16, 21], it is not practical to do so for other purposes, such as SV discovery. We, therefore, implemented a strategy to select a single “best” mapping, and only report the multiple map locations for read pairs that are candidates for structural variation. We consider the paired-end read mapping location as “best” if:
the orientation of the end reads are as expected by the library specifications (i.e., no inversion or tandem duplication candidates)
the inferred fragment size is closest to the library mean compared to other mapping alternatives of the same read pair
the total number of mismatches in the mappings of both end reads is minimal among all mapping mapping alternatives of the same read pair
We consider a read pair to be concordant (w.r.t. library specifications) if its orientation is as expected and the inferred fragment size is within the user specified minimum and maximum cutoff values (typically μ ± 4 × σ). All other read pairs are denoted as discordant. We also select a “best” mapping location for discordant pairs, but for such pairs we also allow them to be in inverted orientation. In addition, all possible map locations for the discordant pairs are reported in a separate file.
Using the best mapping locations as assigned by sirFAST, we calculated the precision and recall rates (Table 2). We mapped 500,000 simulated pairs of reads to the human reference genome, and compared the sirFAST-assigned best locations with the actual locations where the reads were selected from. We repeated the experiment on three simulated data sets, demonstrating high precision (>96%) and recall (>99%). The very low false negative rate (<4%) can be explained by the read pairs that, incorrectly, were mapped to transchromosomal locations, that are not reported in the output file in the current version of sirFAST.
3.3. Mapping a real dataset and comparison against the CG mapper
We show comparison between the mapping results generated by sirFAST and the CG mapper using a real data set is in Table 3. We first downloaded 5,360,139,478 reads and their mapping output provided by the company from the Complete Genomics public data archive (URL provided above). We remapped the reads using sirFAST with a maximum edit distance of 2 to the reference human genome assembly (GRCh37) with the paired-end mapping options. Then, we removed PCR duplicates and obtained a total of 2,558,246,235 (47.73%) reads that mapped to GRCh37. Using the CG mapping files, and applied the same parameters (filter out those mappings with e > 2; and PCR duplicate removal). We observed that 2,450,884,557 (45.7%) of the reads were mapped by the official CG mapper. However, we also note that the CG mapper can also map reads with more errors, and the raw mapping ratio for CG was 50.79%. We note that the mapping ratio comparisons might be slightly different if other genomes are used for benchmarking. However, since the dataset we used is at extremely high depth (62.5X), we expect to observe only negligible differences in benchmarking other data sets. We also would like to point out that, the NA12878 genome was sequenced many times with many other HTS methods, which also lends itself for downstream analysis comparisons.
Table 3.
The comparison of sirFAST and CG mappers on the real data set (n=5,360,139,478 reads). We considered the mapping positions with at most 2 errors for both sirFAST and the CG mapper.
| sirFAST | CG mapper | |||
|---|---|---|---|---|
| Data set | Mapped | Mapping % | Mapped | Mapping % |
| NA12878 | 2,558,246,235 | 47.73 | 2,450,884,557 | 45.7 |
4. Conclusion
In this manuscript, we described the first publicly-available read mapper, sirFAST, designed to align reads generated by the Complete Genomics platform to the reference genome. The CG reads have very different properties when compared with data from other platforms. The reads are composed of several read sections where there may be overlaps or gaps in between. Our algorithms search for all possible combinations of expected gaps, while maintaining high sensitivity and mapping speed. Performance tests on both simulated and real data revealed high precision and recall rates in our mapping results with sirFAST.
To develop sirFAST, we made a number of design choices, such as using combined seeds and calculating Hamming distance over the seeds instead of implementing the Smith-Waterman [25] algorithm, and using hash tables for seed lookups instead of using suffix arrays. We note that different choices may affect the performance slightly, and in practice, some designs may increase or decrease performance. However, theoretically, full dynamic programming requires O(n2) run time, where our combined seed approach takes O(kl) where k is the total gap size in the read, and l is the seed size (l = 10 in our dataset). 7 Similarly, we use hash tables for seed queries, which can be performed in O(1) time, where suffix index requires binary search that takes O(log n) time.
Because of the relatively low cost of generating CG data, and because the CG data generation is provided as a service only, many researchers are now using the Complete Genomics service to analyze their genomes of interest. However, the lack of alternative analysis tools, from read mappers to SNP and SV callers, limit the discovery efforts to running a single set of proprietary algorithms developed by the Complete Genomics company. In fact, it is well known that different algorithms have different power in detection of genomic variants, especially structural variations [10, 28]. We hope and believe the open source implementation of our algorithms (sirFAST), which are introduced in this paper, will not only enable researchers and practitioners to more effectively use the Complete Genomics data sets but also pave the way to new research that can greatly improve the variation detection mechanisms for the Complete Genomics platform, thereby increasing the scientific community’s ability to find important genomic variants.
Acknowledgments
This study is supported by an NIH grant (HG006004) to C.A. and O.M. and a Marie Curie Career Integration Grant (PCIG-2011-303772) to C.A. under the Seventh Framework Programme. C.A. also acknowledges support from The Science Academy of Turkey, under the BAGEP program.
Footnotes
Note that the negative gaps (overlaps) can be represented in a similar fashion with shorter seeds. For example if the expected gap distribution is within [−2,0] between two 10bp read sections, then corresponding spaced seeds would be 118, 119, and 120.
where T P indicates the number of reads that map to the same position that was simulated and F P indicates the number of reads that map to a different position. However, the reads have the right insert size and orientation.
where F N indicates the number of reads that map to a different position than that was simulated and the insert size or the read orientation is not as expected.
This run time estimation is calculated for the current read length and seed length, which may change if the read format drastically changes.
Competing Interests
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Onur Mutlu, Email: onur@cmu.edu.
Can Alkan, Email: calkan@cs.bilkent.edu.tr.
References
- 1.Korbel JO, Urban AE, Affourtit JP, et al. Paired-end mapping reveals extensive structural variation in the human genome. Science. 2007;318(5849):420–426. doi: 10.1126/science.1149504. URL http://dx.doi.org/10.1126/science.1149504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wheeler DA, Srinivasan M, Egholm M, et al. The complete genome of an individual by massively parallel DNA sequencing. Nature. 2008;452(7189):872–876. doi: 10.1038/nature06884. URL http://dx.doi.org/10.1038/nature06884. [DOI] [PubMed] [Google Scholar]
- 3.Margulies M, Egholm M, Altman WE, et al. Genome sequencing in microfabricated high-density picolitre reactors. Nature. 2005;437(7057):376–380. doi: 10.1038/nature03959. URL http://dx.doi.org/10.1038/nature03959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bentley DR, Balasubramanian S, Swerdlow HP, et al. Accurate whole human genome sequencing using reversible terminator chemistry. Nature. 2008;456(7218):53–59. doi: 10.1038/nature07517. URL http://dx.doi.org/10.1038/nature07517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McKernan KJ, Peckham HE, Costa GL, et al. Sequence and structural variation in a human genome uncovered by short-read, massively parallel ligation sequencing using two-base encoding. Genome Res. 2009;19(9):1527–1541. doi: 10.1101/gr.091868.109. URL http://dx.doi.org/10.1101/gr.091868.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Eid J, Fehr A, Gray J, et al. Real-time DNA sequencing from single polymerase molecules. Science. 2009;323(5910):133–138. doi: 10.1126/science.1162986. URL http://dx.doi.org/10.1126/science.1162986. [DOI] [PubMed] [Google Scholar]
- 7.Metzker ML. Sequencing technologies - the next generation. Nat Rev Genet. 2010;11(1):31–46. doi: 10.1038/nrg2626. URL http://dx.doi.org/10.1038/nrg2626. [DOI] [PubMed] [Google Scholar]
- 8.Fonseca NA, Rung J, Brazma A, Marioni JC. Tools for mapping high-throughput sequencing data. Bioinformatics. 2012;28(24):3169–3177. doi: 10.1093/bioinformatics/bts605. URL http://dx.doi.org/10.1093/bioinformatics/bts605. [DOI] [PubMed] [Google Scholar]
- 9.Nielsen R, Paul JS, Albrechtsen A, Song YS. Genotype and snp calling from next-generation sequencing data. Nat Rev Genet. 2011;12(6):443–451. doi: 10.1038/nrg2986. URL http://dx.doi.org/10.1038/nrg2986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Alkan C, Coe BP, Eichler EE. Genome structural variation discovery and genotyping. Nat Rev Genet. 2011;12(5):363–376. doi: 10.1038/nrg2958. URL http://dx.doi.org/10.1038/nrg2958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bradnam KR, Fass JN, Alexandrov A, et al. Assemblathon 2: evaluating de novo methods of genome assembly in three vertebrate species. Gigascience. 2013;2(1):10. doi: 10.1186/2047-217X-2-10. URL http://dx.doi.org/10.1186/2047-217X-2-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Drmanac R, Sparks AB, Callow MJ, et al. Human genome sequencing using unchained base reads on self-assembling dna nanoarrays. Science. 2010;327(5961):78–81. doi: 10.1126/science.1181498. URL http://dx.doi.org/10.1126/science.1181498. [DOI] [PubMed] [Google Scholar]
- 13.Carnevali P, Baccash J, Halpern AL, et al. Computational techniques for human genome resequencing using mated gapped reads. J Comput Biol. 2012;19(3):279–292. doi: 10.1089/cmb.2011.0201. URL http://dx.doi.org/10.1089/cmb.2011.0201. [DOI] [PubMed] [Google Scholar]
- 14.1000 Genomes Project Consortium, An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491(7422):56–65. doi: 10.1038/nature11632. URL http://dx.doi.org/10.1038/nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Burrows M, Wheeler DJ. Tech rep. Digital Equipment Corporation; 1994. A block sorting lossless data compression algorithm. [Google Scholar]
- 16.Alkan C, Kidd JM, Marques-Bonet T, et al. Personalized copy number and segmental duplication maps using next-generation sequencing. Nat Genet. 2009;41(10):1061–1067. doi: 10.1038/ng.437. URL http://dx.doi.org/10.1038/ng.437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hach F, Hormozdiari F, Alkan C, et al. mrsFAST: a cache-oblivious algorithm for short-read mapping. Nat Methods. 2010;7(8):576–577. doi: 10.1038/nmeth0810-576. URL http://dx.doi.org/10.1038/nmeth0810-576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hormozdiari F, Hach F, Sahinalp SC, Eichler EE, Alkan C. Sensitive and fast mapping of di-base encoded reads. Bioinformatics. 2011;27(14):1915–1921. doi: 10.1093/bioinformatics/btr303. URL http://dx.doi.org/10.1093/bioinformatics/btr303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xin H, Lee D, Hormozdiari F, et al. Accelerating read mapping with fasthash. BMC Genomics. 2013;14(Suppl 1):S13. doi: 10.1186/1471-2164-14-S1-S13. URL http://www.biomedcentral.com/1471-2164/14/S1/S13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li H, Handsaker B, Wysoker A, et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25(16):2078–2079. doi: 10.1093/bioinformatics/btp352. URL http://samtools.sourceforge.net. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hormozdiari F, Alkan C, Eichler EE, Sahinal SC. Combinatorial algorithms for structural variation detection in high-throughput sequenced genomes. Genome Res. 2009;19(7):1270–1278. doi: 10.1101/gr.088633.108. URL http://dx.doi.org/10.1101/gr.088633.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215(3):403–410. doi: 10.1016/S0022-2836(05)80360-2. URL http://dx.doi.org/10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 23.Ma B, Tromp J, Li M. PatternHunter: faster and more sensitive homology search. Bioinformatics. 2002;18(3):440–445. doi: 10.1093/bioinformatics/18.3.440. [DOI] [PubMed] [Google Scholar]
- 24.Levenshtein V. Binary codes capable of correcting deletions, insertions and reversals. Soviet Physics Doklady. 1966;10(8):707–710. [Google Scholar]
- 25.Smith TF, Waterman MS. Identification of common molecular subsequences. J Mol Biol. 1981;147(1):195–197. doi: 10.1016/0022-2836(81)90087-5. [DOI] [PubMed] [Google Scholar]
- 26.Ukkonen E. On approximate string matching. Proceedings of the International FCT-Conference on Fundamentals of Computation Theory; 1983. pp. 487–495. [Google Scholar]
- 27.Hamming RW. Error detecting and error correcting codes. Bell System Technical Journal. 1950;26(2):147–160. [Google Scholar]
- 28.Mills RE, Walter K, Stewart C, et al. Mapping copy number variation by population-scale genome sequencing. Nature. 2011;470(7332):59–65. doi: 10.1038/nature09708. URL http://dx.doi.org/10.1038/nature09708. [DOI] [PMC free article] [PubMed] [Google Scholar]