

Abstract
Background
Endothelial growth factors including angiopoietin-2 (Ang-2), its soluble receptor Tie-2 (sTie-2) and hepatocyte growth factor (HGF) play important roles in angiogenesis, vascular remodeling, local tumor growth and metastatic potential of various cancers. Circulating levels of these biomarkers have a heritable component (between 13% and 56%), but the underlying genetic variation influencing these biomarker levels is largely unknown.
Methods and Results
We performed a genome-wide association study for circulating Ang-2, sTie-2, and HGF in 3571 Framingham Heart Study (FHS) participants and assessed replication of the top hits for Ang-2 and sTie-2 in 3184 participants of the Study of Health in Pomerania (SHIP). In multivariable-adjusted models, sTie-2 and HGF concentrations were associated with single nucleotide polymorphisms (SNPs) in the genes encoding the respective biomarkers (top p=2.40×10−65 [rs2273720] and 3.64×10−19 [rs5745687], respectively). Likewise, rs2442517 in the MCPH1 gene (in which the Ang-2 gene is embedded) was associated with Ang-2 levels (p=5.05×10−8 in FHS and 8.39×10−5 in SHIP). Furthermore, SNPs in the AB0 gene were associated with sTie-2 (top SNP rs8176693 with p=1.84×10−33 in FHS; p=2.53×10−30 in SHIP) and Ang-2 (rs8176746 with p=2.07×10−8 in FHS; p=0.001 in SHIP) levels on a genome-wide significant level. The top genetic loci explained between 1.7% (Ang-2) and 11.2% (sTie-2) of the inter-individual variation in biomarker levels.
Conclusions
Genetic variation contributes to the inter-individual variation in growth factor levels and explains a modest proportion of circulating HGF, Ang-2, and Tie-2. This may potentially contribute to the familial susceptibility to cancer, a premise that warrants further studies.
Keywords: Genome Wide Association Study, genetics, endothelial growth factors, HGF, Ang-2, Tie-2
Introduction
Endothelial growth factors play an important role in human physiology. Due to their pivotal effects on angiogenesis and vascular remodeling,1 endothelial growth factors have been implicated in early vascular development,2 the local spread/growth and metastatic potential of various cancers3–4 and the etiology of chronic conditions including cardiovascular and metabolic diseases.5
Previous evidence suggests that circulating levels of these endothelial growth factors are heritable. Specifically, 56%, 38%, and 27% of the inter-individual variation in sTie-2, HGF, and Ang-2 concentrations, respectively, were due to additive genetic factors.6–7 However, the precise genetic underpinnings of circulating Ang-2, Tie-2, and HGF levels are largely unknown. A previous family-based analysis identified significant linkage of sTie-2 levels to a relatively broad locus on chromosome 9, including the Tie-2 gene (logarithm of the odds [LOD] score: 8.31).7 In the present analyses, we report the results of a genome-wide association study (GWAS) for serum levels of Ang-2, sTie-2, and HGF (discovery sample, n=3571; independent replication sample for Ang-2 and sTie-2, n=3184). Given the moderate to substantial heritability estimates,6–7 we hypothesized that circulating concentrations of all three biomarkers will be associated with common genetic variants.
Methods
Discovery sample (Framingham Heart Study, FHS)
The initial genome-wide analyses were performed in participants of the Third Generation cohort of the Framingham Heart Study (FHS).8 At the baseline examination cycle (2002–2005), participants were comprehensively phenotyped, including assessment of standard cardiovascular disease (CVD) risk factors, a physical examination and a standardized interview by a FHS physician. The sample sizes available for genome-wide analyses in the discovery sample varied slightly between biomarkers depending on the availability of covariates: HGF, n=3571; Ang-2, n=3574; sTie-2, n=3574. Written informed consent (including consent to genetic analyses) was provided by all participants and the Institutional Review Board at the Boston University Medical Centre approved the study protocol.
Replication sample (Study of Health in Pomerania, SHIP)
Replication of single nucleotide polymorphisms (SNPs) that reached genome-wide significance (p<5×10−8) for association with Ang-2 or sTie-2 levels in the discovery sample (FHS sample) was assessed in participants of the second examination cycle (2002–2006) of the Study of Health in Pomerania (SHIP-1). SHIP is a community-based sample in Northeast Germany. The study design has been described in detail elsewhere.9 In essence, SHIP represents a random cluster sample, representative of the population of West Pomerania, located in Northeast Germany. The response rate of the SHIP-1 examination cycle was 83.6%.9 The SHIP study was approved by the Ethics Committee of the University of Greifswald. Prior genetic analyses had shown that the SHIP sample is genetically very homogeneous and provided no evidence for a genetic drift.10–11 Furthermore, the design effects (due to cluster sampling) on the p-values caused by deviation from simple random sampling were negligible. Therefore, the cluster sampling was not taken into account in our genetic analyses.
Biomarker measurement
In the FHS sample, blood was drawn in the early morning (usually between 7 and 9 AM) after an overnight fast, immediately centrifuged and stored at −80°C until biomarkers were assayed. Ang-2, sTie-2 and HGF were measured using commercial assays (R&D Inc.), as described previously.6–7 The average inter-assay coefficients of variation for all 3 biomarkers were low: 5.7% for Ang-2; 3.2% for Tie-2 and 1.6% for HGF.6–7 In SHIP, Ang-2 and Tie-2 were measured likewise using commercially available R&D assays. The minimum detectable concentration was 1.20 pg/mL and 0.001 ng/mL for Ang-2 and for Tie-2, respectively. The inter-assay coefficients of variation in SHIP were 10.6% and 13.3% for Ang-2 and 5.6% and 6.7% for Tie-2, for low and high biomarker concentrations, respectively.12
Genotyping, Imputation, and controlling for population stratification
FHS participants were genotyped with the Affymetrix Human Mapping 500K Array Set and the 50K Human Gene Focused Panel. The mean call rate was 98%.13 SNPs with a call rate <95% or deviation from Hardy-Weinberg equilibrium (P <10−6) were excluded. As additional quality control measures in FHS and SHIP, the reported sex in the database had to agree with the called sex in the genetic dataset, identity by descent estimations were performed to identify and exclude duplicated individuals, and individuals with a high genome-wide heterozygosity rate (beyond 5 standard deviations of the mean) were removed prior to analyses. 14–15 Genotypes were imputed to the ~2.5 million SNP of the HapMap CEU panel (release 22; build 36; http://hapmap.ncbi.nlm.nih.gov/) using the Markov Chain Haplotyping (MACH) algorithm.16 Quantile-quantile (Q-Q) plots and λ estimates were obtained to assess whether the observed distribution of p-values fits the p-value distribution expected under the null hypothesis of no association, except at the extreme tail. Principal components analyses were performed using EIGENSTRAT17 to account for potential population substructures within the FHS dataset, but no admixture was observed in the FHS cohort.
SHIP participants were genotyped with the Affymetrix Human SNP Array 6.0 using the Birdseed2 clustering algorithm.11 Pair-wise linkage disequilibrium between SNPs was assessed using the SNAP tool.18 Details of the exact location of SNPs within or between genes were assessed using the UCSC genome browser (http://genome.ucsc.edu/).
Statistical analyses
Due to their skewed distributions, all biomarkers were natural logarithmically-transformed prior to performing genetic analyses. Plots of untransformed and transformed concentrations of HGF, Ang-2 and Tie-2 in the FHS sample (Panel A) and in the SHIP sample (Panel B) are provided in Supplementary Figure 1–3. Assuming an additive genetic model with 1 degree of freedom, we related each of the 2.5 million SNPs in the FHS sample to each circulating biomarker (HGF, Ang-2, sTie-2), using a linear mixed effects model that accounts for the familial relatedness (R package “GWAF”).19 Specifically, we accounted for familial correlations with a linear mixed effects model that uses relationship coefficient matrix as within pedigree correlation matrix. The relationship coefficient (which is twice the kinship coefficient) was calculated based on available pedigree information and structure. Multivariable analyses were adjusted for age, sex and known clinical correlates of each biomarker, as identified in prior analyses in the FHS dataset:6–7 These additional covariates were as follows; for Ang-2: smoking, total cholesterol, diastolic and systolic blood pressure, antihypertensive treatment, diabetes; for Tie-2: diabetes, body mass index, estimated glomerular filtration rate, alcohol consumption, triglycerides; for HGF: diastolic blood pressure, antihypertensive treatment, high-density lipoprotein cholesterol, triglycerides, smoking, body mass index, diabetes. A p-value below 5 × 10−8 was considered genome-wide significant. A top SNP (rs2442517) of the Ang-2 GWAS with a p-value of 5.05 × 10−8 was also listed as the p-value was very close to the genome-wide significance threshold. For each biomarker, we graphically plotted each SNP’s p-value vs. its physical position (Manhattan plot).
The top SNP of each locus associated with Ang-2 and Tie-2 levels in the FHS sample in a genome-wide significant fashion was tested for replication in the SHIP sample, likewise using a linear regression model assuming an additive genetic model and adjusting for the same set of covariates as described above. We did not assess replication of the top SNPs related to HGF concentrations because HGF levels were not available in SHIP.
Pathway analyses
Pathway analyses were conducted for Ang-2 and sTie-2 combined and separately for HGF using the program Ingenuity Pathway Analysis software package (Ingenuity Systems, Redwood, CA). A SNP score was assigned to each genetic variant. This SNP score was equivalent to the lower p-value of the two traits (Ang-2 and Tie-2, respectively) examined. For the pathway analysis for HGF, the SNP score was equivalent to the p-value for association with HGF. Based on the human reference genome (NCBI Build 36, 2006), the location of all SNPs relative to RefSeq genes were obtained from UCSC Genome Browser (http://genome.ucsc.edu/). A gene score was defined as the most significant variant that was located within 110kb upstream and 40kb downstream of the gene's most extreme transcript boundaries. If a SNP could be mapped to multiple genes, all of these genes received the same gene score (the p-value of this SNP) if the respective SNP had the lowest p-value in the region. Of the ~23,000 genes evaluated, 234 genes and 294 genes reached a score less than 1.0 × 10−4 for HGF and for Ang-2/ sTie-2, respectively. These genes were then used for pathway analyses. Enrichment of each of the canonical pathways for HGF- and Ang-2/ sTie-2-related SNPs, respectively, was assessed using Fisher's exact test.
Statistical analyses were performed in R.19 The authors had full access to the data and take responsibility for its integrity. MHC performed the statistical analyses for the FHS sample, AT performed the statistical analyses in the SHIP sample. The pathway analyses were performed by HL. All authors have read and agree to the manuscript as written.
Results
Clinical and biochemical characteristics of the discovery and the replication sample are provided in Table 1. In the discovery sample, the genomic inflation factor did not indicate systematic inflation for each of the biomarkers (between 1.01 and 1.02; Supplementary Figure 4). The top genome-wide significant (p≤5×10−8) hits for Ang-2, sTie-2 and HGF were listed in Table 2. Figure 1 displays the p-values for approximately 2.5 million genetic variants associated with HGF, Ang-2 and soluble Tie-2, respectively.
Table 1.
Clinical and biochemical characteristics of the discovery and replication samples
| Discovery sample (FHS 3rd Generation) | Replication sample (SHIP-1)* | |||
|---|---|---|---|---|
| Variables | Women (n=1904) | Men (n=1670) | Women (n=1654) | Men (n=1546) |
| Clinical features | ||||
| Age, yrs. | 40±9 | 40±9 | 53±15 | 55±15 |
| Systolic blood pressure, mm Hg | 113±14 | 121±12 | 128±19 | 137±19 |
| Diastolic blood pressure, mm Hg | 73±9 | 78±9 | 79±10 | 83±11 |
| Body mass index, kg/m2 | 26±6 | 28±5 | 27±6 | 28±4 |
| Waist circumference, inches | 35±6 | 39±5 | 34±2 | 39±5 |
| Smoking, % | 15 | 16 | 19 | 25 |
| Diabetes, % | 2 | 3 | 9 | 12 |
| Biochemical features | ||||
| Total cholesterol, mg/dL | 185±34 | 193±37 | 217±45 | 211±45 |
| HDL cholesterol, mg/dL | 61±16 | 47±12 | 51±17 | 39±13 |
| Triglycerides, mg/dL | 97±63 | 134±106 | 138±119 | 186±176 |
| Alcohol consumption† | 5.8±7.9 | 13.8±18.1 | 4.4±6.5 | 14.6±21.1 |
| eGFR, ml/min/1.73m2 | 99±19 | 100±17 | 111±28 | 86±22 |
| Endothelial growth factors | ||||
| HGF, Pg/mL | 825 (697, 974) | 814 (702, 967) | Not determined | Not determined |
| Angiopoietin-2, ng/mL | 2.02 (1.51,2.71) | 1.70 (1.33,2.23) | 1.37 (1.06, 1.84) | 1.30 (1.02, 1.75) |
| Soluble Tie-2, ng/mL | 14.3 (12.0,17.3) | 15.0 (12.7,18.2) | 15.0 (12.3, 17.9) | 16.1 (12.9, 19.6) |
FHS indicates Framingham Heart Study; HDL, high-density lipoprotein; HGF, hepatocyte growth factor; eGFR, estimated glomerular filtration rate; SHIP, Study of Health in Pomerania. Data are mean±standard deviation; except for HGF, Angiopoietin-2, and soluble Tie-2 where median (quartile 1, quartile 3) are provided.
Genetic data were missing in 16 participants.
In ounces per month in FHS sample, in g/day in the SHIP-1 sample.
Table 2.
Genome-wide significant loci for circulating concentrations of hepatocyte growth factor (HGF), soluble Tie-2 (sTie-2) and Angiopoietin-2 (Ang-2); The most significant single nucleotide polymorphisms for each locus and biomarker are shown.
| Discovery sample, FHS Third Generation cohort |
Replication sample, SHIP-1 | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Biomarker | rsID | Chr | N | Coded/ Non-Coded Allele |
MAF | Beta (SE) | p-value | Closest Gene |
N | MAF | Beta (SE) | p-value |
| HGF | ||||||||||||
| rs5745687 | 7 | 3571 | T/C | 0.077 | −0.099 (0.011) |
3.64×10−19 | HGF | |||||
| Ang-2 | ||||||||||||
| rs8176746 | 9 | 3574 | T/G | 0.077 | 0.111 (0.020) |
2.07×10−8 | ABO | 3188 | 0.096 | 0.061 (0.019) |
0.001 | |
| rs2442517 | 8 | 3574 | G/A | 0.474 | 0.059 (0.011) |
5.05×10−8 | MCPH1 | 3188 | 0.461 | 0.043 (0.011) |
8.39×10−5 | |
| sTie-2 | ||||||||||||
| rs2273720 | 9 | 3574 | C/A | 0.056 | 0.265 (0.016) | 2.40×10−65 | Tie-2 | 3184 | 0.056 | 0.237 (0.018) | 2.70×10−38 | |
| rs8176693 | 9 | 3574 | T/C | 0.077 | 0.155 (0.013) | 1.84×10−33 | ABO | 3184 | 0.096 | 0.157 (0.014) | 2.53×10−30 | |
Chr. indicates chromosome; FHS, Framingham Heart Study; HGF, hepatocyte growth factor; Ang-2, Angiopoietin-2; MAF, minor allele frequency; SE, standard error; SHIP, Study of Health in Pomerania; SNP, single nucleotide polymorphism.
Figure 1.

Manhattan plot displaying the association of approximately 2.5 million SNPs with circulating concentrations of hepatocyte growth factor (HGF), angiopoietin-2 (Ang-2) and soluble Tie-2 (sTie-2) in the Framingham Heart Study sample. Each dot represents a single nucleotide polymorphism. On the x-axis, the chromosomal location of the genetic variant is shown (with the results being plotted from the p terminal end). On the y-axis, the −log10 (p-value) is shown. The grey horizontal line indicates the threshold of genome-wide significance (5 × 10−8).
Genetic loci associated with circulating HGF concentrations
In the FHS sample, 29 SNPs within or close to the HGF gene on chromosome 7 were associated with circulating HGF levels on a genome-wide significant level (p<5×10−8), most of these SNPs were in moderate linkage disequilibrium with the top SNP (rs5745687; Table 2 and Figure 2). Overall, 24 of these 29 SNPs were genotyped in the FHS sample, and 5 SNPs were imputed (including rs5745687). A regional plot is shown in Figure 2. The top SNP in the HGF gene explained 2.12% of the variation in circulating HGF levels. Mean HGF levels stratified by rs5745687 genotypes are provided in Supplementary Figure 5.
Figure 2.
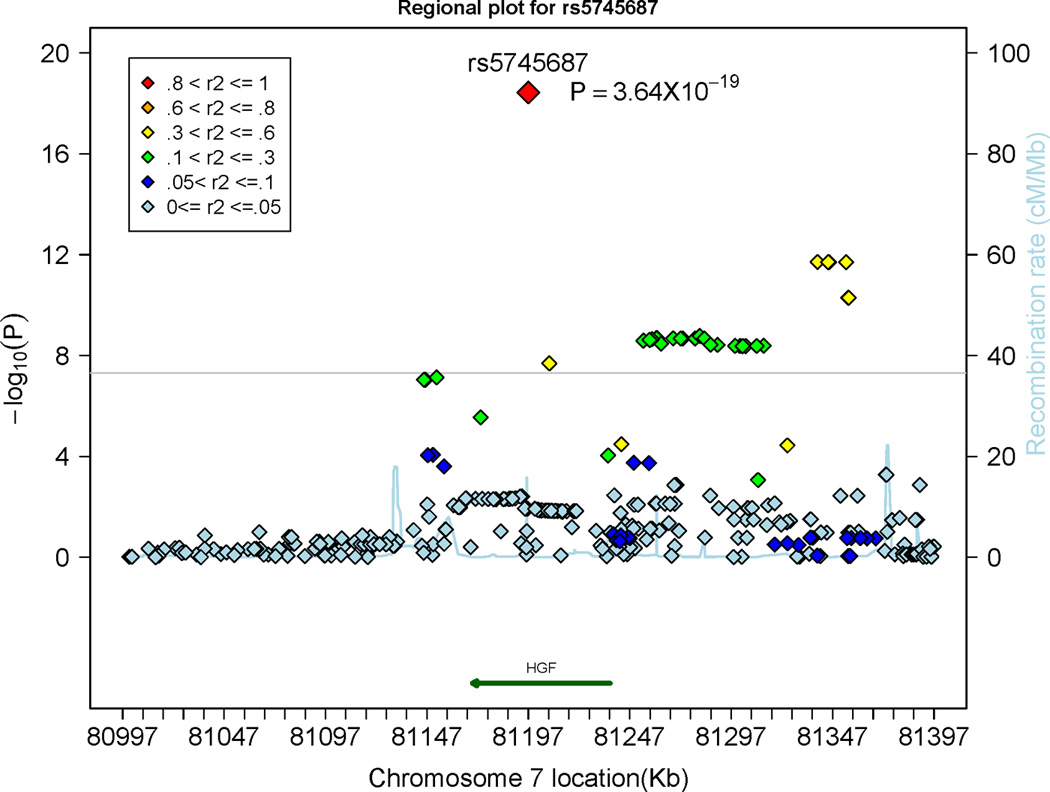
Regional plot displaying the association of single nucleotide polymorphisms (SNPs) at the hepatocyte growth factor (HGF) gene locus with circulating HGF levels. The colors represent the linkage disequilibrium (expressed as r2) between neighboring SNPs. The grey horizontal line indicates the threshold of genome-wide significance (5 × 10−8).
Genetic loci associated with circulating Ang-2 and sTie-2 concentrations
A total of 5 SNPs in the ABO gene on chromosome 9 were associated with circulating Ang-2 concentrations on a genome-wide significance level (p<5×10−8); 4 of these SNPs were genotyped in the FHS sample. The top SNP was imputed in the FHS and SHIP samples; rs2442517 in the MCPH1 gene on chromosome 8 was very close to the genome-wide significance level (p=5.05×10−8). This SNP was genotyped in the FHS sample and imputed in the SHIP sample (Table 2). Regional plots of the MCPH1 locus and the ABO locus are shown in Panel A and Panel B of Figure 3, respectively. The top SNPs of both loci (rs8176746 and rs2442517) combined explained 1.7% and 0.7% of the inter-individual variation in circulating Ang-2 levels in the discovery and in the replication sample, respectively. Ang-2 levels stratified by rs2442517 genotypes (Panel A) and stratified by rs8176746 (Panel B) in the FHS and the SHIP samples are provided in Supplementary Figure 6.
Figure 3.
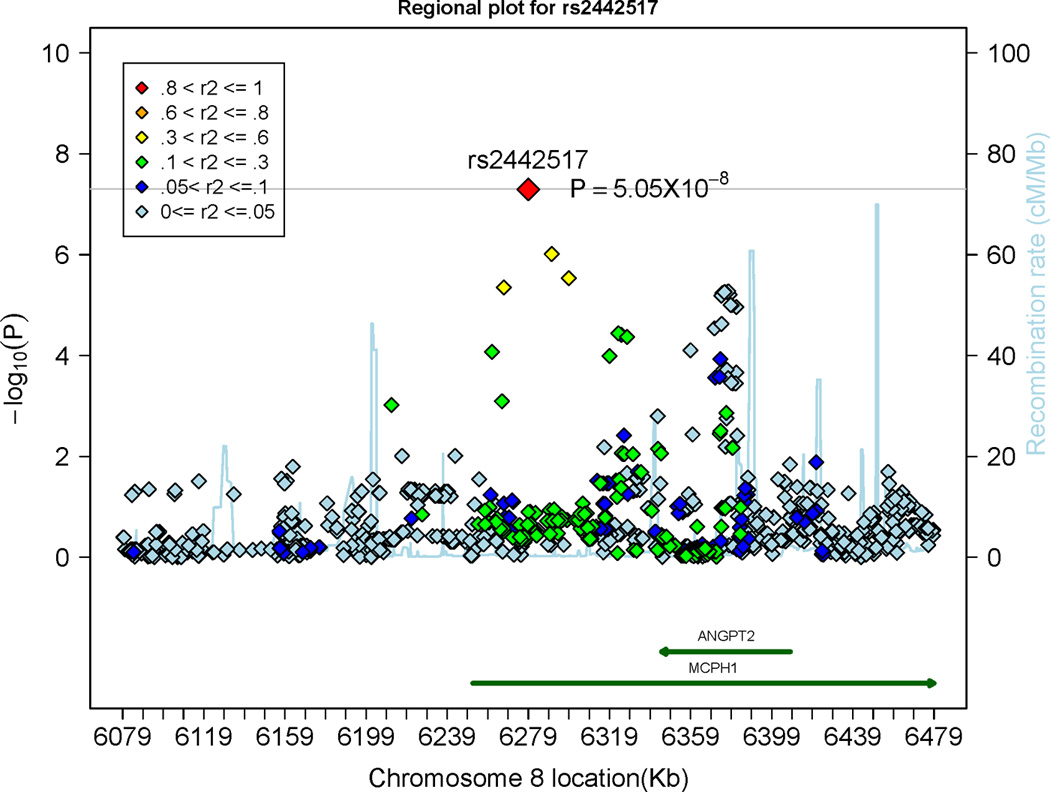
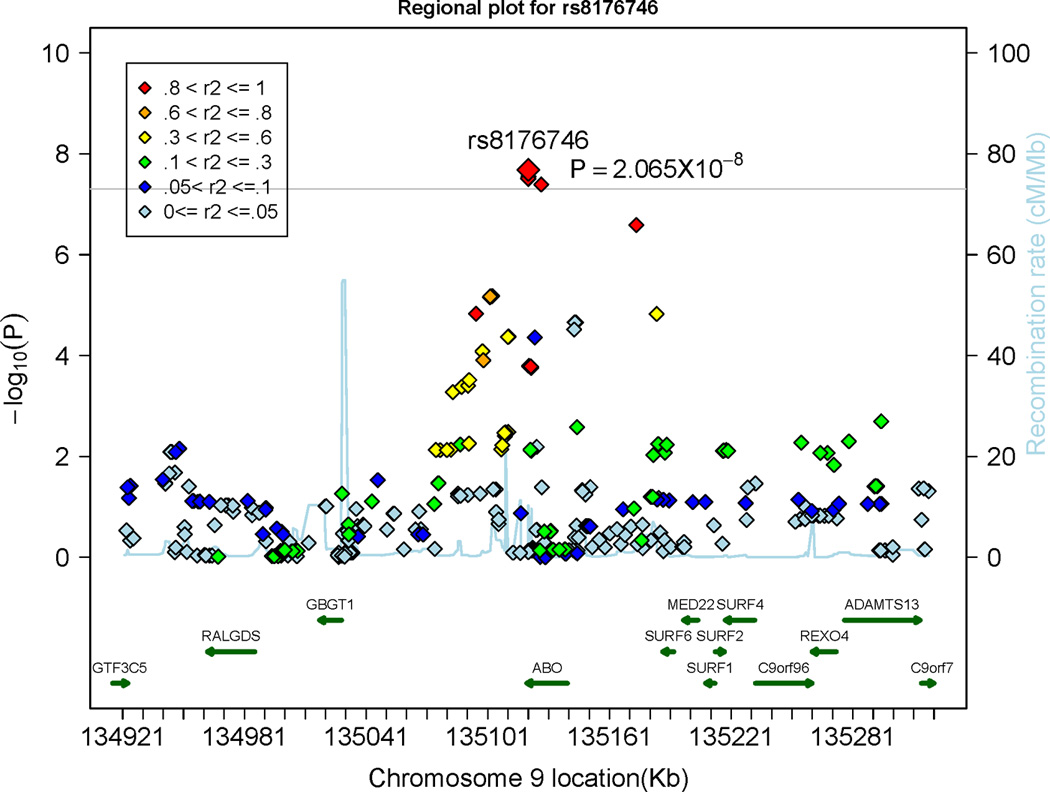
Regional plots displaying the association of single nucleotide polymorphism (SNPs) at the microcephalin 1 (MCPH1) gene locus (Panel A) and at the ABO locus (Panel B), associated with circulating Angiopoietin-2 levels. The colors represent the linkage disequilibrium (expressed as r2) between neighboring SNPs. ANGPT2 indicates Angiopoietin-2. The grey horizontal line indicates the threshold of genome-wide significance (5 × 10−8).
Overall, 17 SNPs within or close to the Tie-2 gene and 56 SNPs within or close to the AB0 gene were associated with sTie-2 levels on a genome-wide significant level (p<5×10−8; Table 2). Regional plots of the Tie-2 and the ABO loci are displayed in Panel A and Panel B of Figure 4, respectively. The most significant SNPs from each significant locus (rs8176693 and rs2273720) combined explained 11.2% and 8.9% of the inter-individual variation in circulating Tie-2 levels in the discovery sample and in the replication sample, respectively. SNP rs2273720 was genotyped in the FHS and SHIP samples, SNP rs8176693 was genotyped in FHS and imputed in the SHIP sample. Tie-2 levels stratified by rs2273720 genotypes (Panel A) and stratified by rs8176693 genotypes (Panel B) in the FHS and the SHIP samples are provided in Supplementary Figure 7. The top SNPs in the ABO gene that were associated with sTie-2 (rs8176693) and Ang-2 (rs8176746) were in perfect linkage disequilibrium (r2=1), underscoring that Ang-2 and Tie-2 levels were associated with the same genetic locus.
Figure 4.
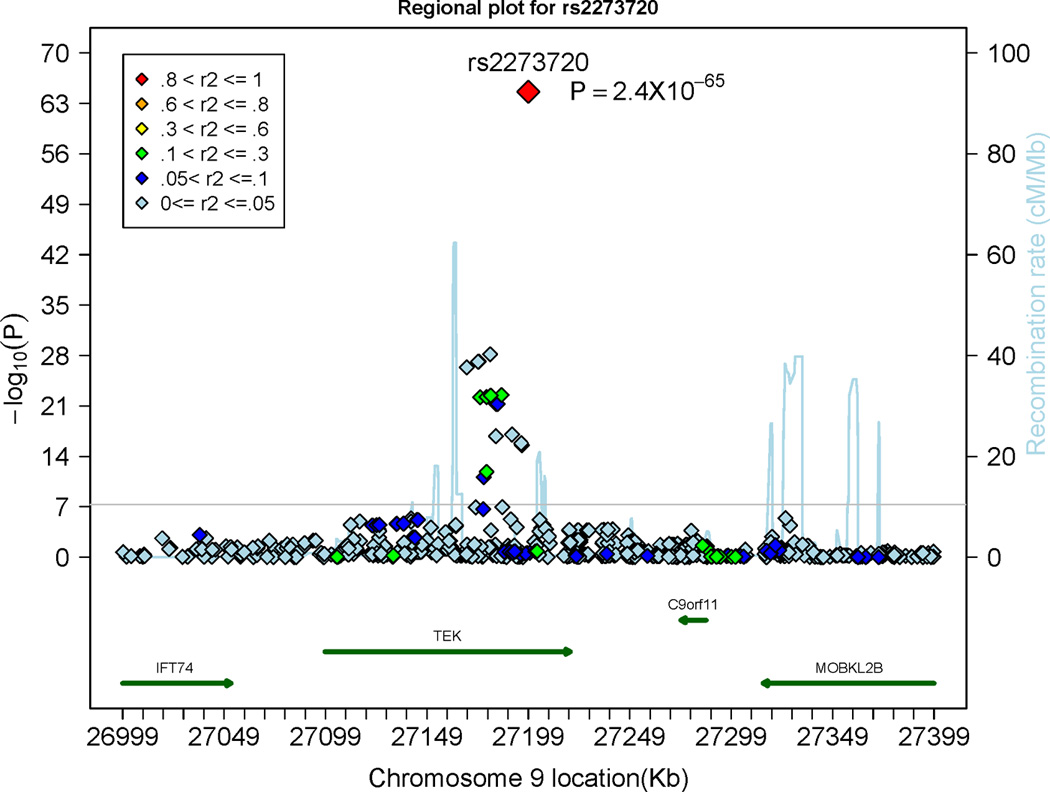
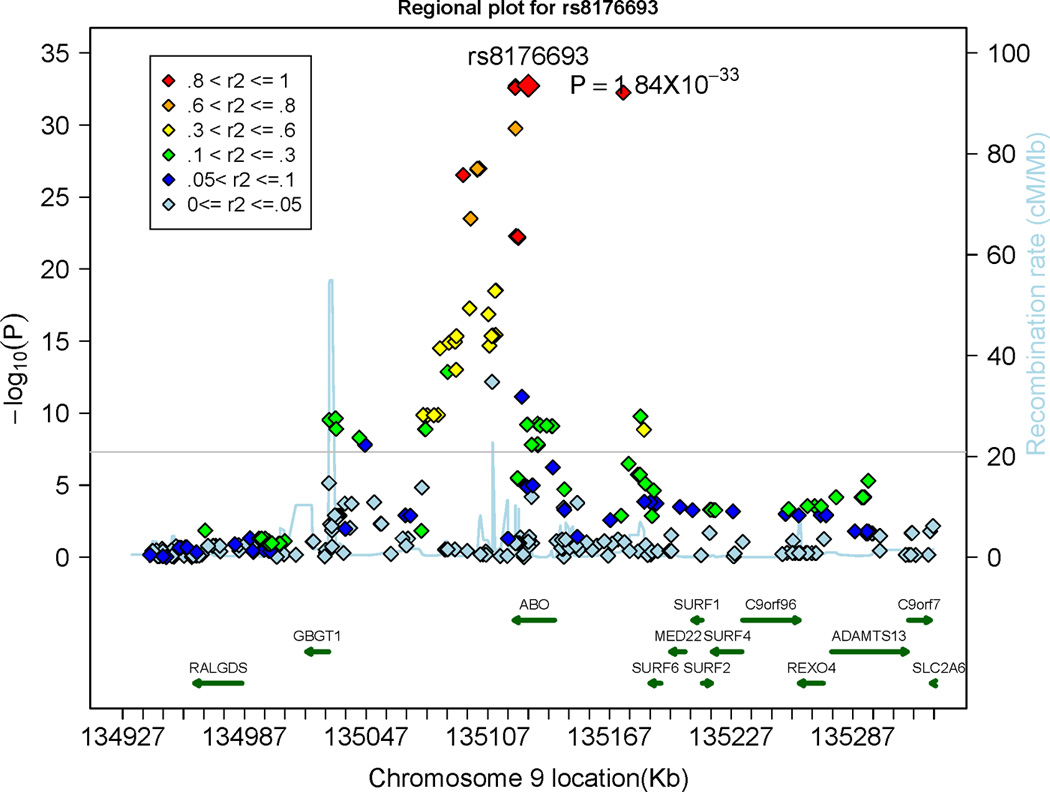
Regional plots displaying the association of single nucleotide polymorphisms (SNPs) at the Tie-2 (denoted as TEK) gene locus (Panel A) and the ABO locus (Panel B) with circulating Tie-2 levels. The colors represent the linkage disequilibrium (expressed as r2) between neighboring SNPs. The grey horizontal line indicates the threshold of genome-wide significance (5 × 10−8).
Pathway analyses
For Ang-2 and sTie-2, no significantly enriched canonical pathway (P<1×10−3) could be identified.
By contrast, several pathways that were overrepresented with HGF-related genes could be observed (Supplementary Table 1), including the mTOR (mammalian Target of Rapamycin) pathway and the Amyotrophic Lateral Sclerosis Signaling pathway.
Discussion
Principal observations
We conducted a genome-wide association study for 3 endothelial growth factors (Ang-2, soluble Tie-2, and HGF) in the Framingham Heart study and assessed replication of the top SNPs associated with Ang-2 and sTie-2 in the SHIP study, a population-based sample from northeastern Germany. Our main findings were four-fold. First, all three biomarker levels were associated with common genetic variants within or very close to the genes encoding the individual biomarkers. Second, Ang-2 and sTie-2 levels were also associated with genetic variants at the ABO locus. Third, the top genetic loci explained a modest (≈2%; HGF, Ang-2) to moderate (11%; sTie-2) proportion of the inter-individual variation in biomarker levels. Fourth, we identified several pathways that were overrepresented with HGF-related genes, including the mTOR (mammalian Target of Rapamycin) and the Amyotrophic Lateral Sclerosis Signaling pathway.
Genetic loci associated with circulating HGF concentrations
Previous family-based analyses reported heritability estimates of 38%6 and 48%,20 respectively, for HGF levels. In small clinical samples, few genetic variants in the HGF gene have been tested for association with HGF levels revealing inconsistent results. In 78 control participants from the Cardiovascular Health Study, SNP rs3735520, which is located in the promoter region of the HGF gene, was statistically significantly associated with HFG levels, whereas SNP rs17501108 (likewise located in the promoter) was not.21 However, SNP rs3735520 was not associated with HGF levels in 133 patients with systemic sclerosis,22 although experimental data indicated that this variant was related to transcriptional activity.22 In our dataset, both SNP rs3735520 (p=0.12) and SNP rs17501108 (p=0.39) were not associated with HGF concentrations. Thus far, there has been no genome-wide analysis regarding the genetic determinants of HGF levels. In our genome-wide approach, only SNPs in the HGF gene reached genome-wide significance for this biomarker. The top SNP (rs5745687) is located in exon 823 and explained about 2.12% of the inter-individual variation in HGF levels. According to the pfam database (http://pfam.xfam.org/protein/P14210),24 SNP rs5745687 leads to the replacement of a negatively charged glutamic acid by a positively charged lysine at position 304. This missense mutation is located just before the third kringle domain of the HGF protein. Such kringle domains are important for the interaction with other proteins.24–25The frequency of the minor allele of rs5745687 is between 0% and 10% in different ethnic subgroups of the 1000 Genomes project26 (Supplementary Table 2). We did not assess replication of the top SNPs related to HGF concentrations, because HGF levels were not available in SHIP.
Genetic loci associated with circulating Tie-2 and Ang-2 concentrations
The statistically strongest association signal in our analyses was observed for SNPs in the Tie-2 gene with circulating Tie-2 concentrations. The top SNP of the Tie-2 locus (rs2273720; located in intron 16) explained about 7% of the variability in sTie-2 levels in the FHS sample and about 5% in the SHIP replication sample. These findings are in good agreement with a recent linkage analysis (displaying strong linkage of sTie-2 levels to a locus on chromosome 9, including the Tie-2 gene)7 and suggest that sTie-2 is moderately influenced by genetic variation in the Tie-2 gene. Furthermore, the ABO locus was associated with sTie-2 on a genome-wide scale (please see below for expanded discussion regarding the ABO locus).
Circulating levels of Ang-2 were associated with rs2442517, an intronic variant in the microcephalin 1 (MCPH1) gene. Interestingly, the Ang-2 gene is nested within the MCPH1 gene (please see regional plot in Figure 3, Panel A) and the most likely explanation for this association signal is that Ang-2 levels are in part determined by genetic variation in the Ang-2 gene. However, the correlation between SNP rs2442517 and variants in the Ang-2 gene is only modest (please see linkage disequilibrium structure in Figure 3, Panel A), and genetic variants in the Ang-2 gene itself did not reach genome-wide significance for Ang-2 levels. Our data add to smaller genetic-epidemiological studies relating SNPs within the Ang-2 gene to circulating Ang-2 concentrations. SNP rs3739391 (located in the 5’UTR; very weakly correlated with rs2442517; r2=0.04) was associated with Ang-2 levels in 360 stroke patients, after adjusting for established vascular risk factors. However, in the same sample, 2 other genetic variants (rs2515507 [located in the promoter] and rs3739390 [5’UTR]) displayed no association with Ang-2 levels.27 In our dataset, none of rs3739391 (p=0.10), rs2515507 (p=0.35) and rs3739390 (p=0.17) displayed statistically significant evidence for association with Ang-2 levels. In another study, SNP rs1868554 (also in very weak LD with rs2442517; r2=0.005), was not associated with plasma Ang-2 levels, but was related to plasma variation in isoforms of Ang-2.28 Interestingly, this variant displayed some evidence of association with Ang-2 levels in our dataset (p=6.5 ×10−6), although the p-value does not meet the threshold for genome-wide significance.
In addition to the MCPH1 locus, also the ABO locus was significantly associated with Ang-2 levels in our dataset, as detailed in the next paragraph.
Association of the ABO locus with circulating Ang-2 and Tie-2 concentrations
Several SNPs in the AB0 gene were associated with sTie-2 levels and Ang-2 levels and the top SNPs of the AB0 locus associated with sTie-2 (rs8176693) and Ang-2 (rs8176746) were perfectly correlated (r2=1). Thus, Ang-2 and sTie-2 add to a growing number of biomarkers that are associated with the ABO locus, including von Willebrand factor,29–30 soluble ICAM-1,31–32 E-selectin33 and P-Selectin,31 tumor necrosis factors alpha34 and Factor VIII29. This suggests that there might be common mechanisms by which the AB0 gene product modulates circulating biomarker concentrations. The genetic architecture of the ABO locus determines the substrate specificity and activity of a glycosyltransferase, which transfers specific carbohydrates to the H antigen and thereby determines the ABO blood group (OMIM: 110300).33, 35–36
There are several potential explanations for the observed association between the ABO locus and circulating Ang-2 and sTie-2 levels. Given that the Tie-2 receptor contains four potential N-glycosylation sites,37 the ABO gene product might modify the Tie-2 receptor and thereby affect its binding activity, its cleavage from the endothelial cells or its renal clearance. These mechanisms have been suggested to explain the association of the ABO locus with other biomarkers including ICAM-1,31, 38 P-selectin,31, 38 E-selectin,33 and von Willebrand factor. 31, 38
In a similar fashion, it is possible that glycosylation of the Ang-2 protein affects its binding ability to the endothelial or soluble Tie-2 receptor and thereby influences circulating Ang-2 serum levels. Such glycosylation (by the ABO gene product) might also modify the Ang-2 clearance rate from the blood stream.(31)
In addition to the association with circulating biomarkers mentioned above, variation in the ABO gene has been associated with different clinical disease traits, including gastric39 and pancreatic cancer,36 duodenal ulcer,40 as well as myocardial infarction41 and stroke,42 and hematological traits.43 Many of these disease conditions depend directly or indirectly on angiogenesis and vascular remodeling – processes that are in part governed by endothelial growth factors. Thus, the association of these biomarkers with the ABO locus might be a mediating mechanism for the observed associations between genetic variation in the ABO gene and clinical disease events, a premise that is speculative.
Across the three biomarkers (HGF, Ang-2, sTie-2) analyzed, the genome-wide significantly associated genetic variants explained a modest proportion (between 2% and 11%) of the inter-individual variation in biomarker levels. This is comparable to other biomarker-related genome-wide analyses, including those for Factor VII, von Willebrand Factor, and ICAM-1, where the identified genome-wide significant loci explained 7.7%, 12.8%, and 8.4%, respectively, of the inter-individual biomarker variation.29, 32 Additional genetic approaches are warranted to explain parts of the ‘missing heritability’,44 including analyses of rare and low-frequency variants or structural genetic variation44 and analyses of all SNPs in the genome.45 Given the reported associations of the analyzed biomarkers with cardiovascular risk factors6–7 and mortality,12 it is of interest, whether these biomarkers and the identified genetic variants (correlated with biomarker levels) improve the prediction of clinically overt cardiovascular events, including myocardial infarction, heart failure and stroke.
Pathway analyses
Pathway analyses identified the mTOR (mammalian Target of Rapamycin) signaling pathway as the most significantly enriched canonical pathway for HGF. Our observations are corroborated by experimental studies, indicating that the mTOR pathway is indeed relevant for modulating HGF expression.46–47 Rapamycin (as inhibitor) and Leucin (as stimulator) exert their influence on HGF expression and secretion through the mTOR pathway.46–47 Furthermore, we revealed a statistically significant overrepresentation of HGF-related genes in the amyotrophic lateral sclerosis signaling pathway. In this context, experimental data support that HGF might improve survival of motor neurons and the clinical course of amyotrophic lateral sclerosis.48–50
Strengths and Limitations
Both study samples (FHS and SHIP) are large and provide a thorough and comprehensive characterization of their study participants. To the best of our knowledge, this is the first genome-wide analysis for Ang-2, Tie-2 and HGF. Some limitations merit consideration. Our samples consisted of young to middle-aged Europeans and European Americans. The applicability of our observations to other ethnicities or age groups is unclear. Furthermore, each biomarker was measured only once in each individual, possibly leading to some non-differential misclassification that would bias our results towards the null hypothesis of no association between genetic variants and biomarker levels. We did not evaluate the impact of rare genetic variants (as could be identified by whole genome sequencing) on biomarker levels.
Conclusion
We identified common genetic variants associated with circulating HGF, Ang-2 and sTie-2 levels. All three biomarkers were related to SNPs located in or close to the genes encoding the respective biomarkers. Furthermore, genetic variants in the ABO gene were associated with Ang-2 and Tie-2 levels. The association findings for Ang-2 and Tie-2 were successfully replicated in an independent sample (SHIP). The genome-wide significantly associated genetic variants explained a modest proportion (between 2% and 11%) of the inter-individual variation in biomarker levels. Furthermore, pathway analyses identified the mTOR and other pathways as being over-represented with HGF-related genes. Given the pivotal role of these biomarkers in angiogenesis, vascular remodeling, local cancer growth and metastatic potential as well as their potential impact on cardiovascular and metabolic disease, our findings suggest that genetically determined endothelial growth factor levels might in part contribute to the known familial susceptibility of conditions like cancer or cardiovascular disease, a premise that warrants further investigation.
Supplementary Material
Acknowledgments
Funding Sources: FHS: This work was supported by the National Heart, Lung, and Blood Institute’s Framingham Heart Study (Contract No. N01-HC-25195; PI, RSV) and its contract with Affymetrix, Inc for genotyping services (Contract No. N02-HL-6-4278), and by grants from the National Heart, Lung, and Blood Institute 2K24HL04334, RO1HL080124, RO1HL077477, and R01HL093328 (all to Dr Vasan). Study of Health in Pomerania (SHIP): SHIP is part of the Community Medicine Research Network, funded by the Federal State of Mecklenburg-West Pomerania and the University Medicine Greifswald. Genome-wide data have been supported by a joint grant from Siemens Healthcare, Erlangen, Germany, and by the Federal State of Mecklenburg- West Pomerania. Furthermore, this work is part of the research project Greifswald Approach to Individualized Medicine (GANI_MED). The GANI_MED consortium is funded by the German Federal Ministry of Education and Research and as well as by the Ministry of Cultural Affairs of the German Federal State of Mecklenburg – West Pomerania (03IS2061A).
Footnotes
Conflict of Interest Disclosures: None
References
- 1.Eklund L, Olsen BR. Tie receptors and their angiopoietin ligands are context-dependent regulators of vascular remodeling. Exp Cell Res. 2006;312:630–641. doi: 10.1016/j.yexcr.2005.09.002. [DOI] [PubMed] [Google Scholar]
- 2.Maisonpierre PC, Suri C, Jones PF, Bartunkova S, Wiegand SJ, Radziejewski C, et al. Angiopoietin-2, a natural antagonist for tie2 that disrupts in vivo angiogenesis. Science. 1997;277:55–60. doi: 10.1126/science.277.5322.55. [DOI] [PubMed] [Google Scholar]
- 3.Shim WS, Ho IA, Wong PE. Angiopoietin: A tie(d) balance in tumor angiogenesis. Mol Cancer Res. 2007;5:655–665. doi: 10.1158/1541-7786.MCR-07-0072. [DOI] [PubMed] [Google Scholar]
- 4.You WK, McDonald DM. The hepatocyte growth factor/c-met signaling pathway as a therapeutic target to inhibit angiogenesis. BMB Rep. 2008;41:833–839. doi: 10.5483/bmbrep.2008.41.12.833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yla-Herttuala S, Rissanen TT, Vajanto I, Hartikainen J. Vascular endothelial growth factors: Biology and current status of clinical applications in cardiovascular medicine. J Am Coll Cardiol. 2007;49:1015–1026. doi: 10.1016/j.jacc.2006.09.053. [DOI] [PubMed] [Google Scholar]
- 6.Lieb W, Safa R, Benjamin EJ, Xanthakis V, Yin X, Sullivan LM, et al. Vascular endothelial growth factor, its soluble receptor, and hepatocyte growth factor: Clinical and genetic correlates and association with vascular function. Eur Heart J. 2009;30:1121–1127. doi: 10.1093/eurheartj/ehp007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lieb W, Zachariah JP, Xanthakis V, Safa R, Chen MH, Sullivan LM, et al. Clinical and genetic correlates of circulating angiopoietin-2 and soluble tie-2 in the community. Circ Cardiovasc Genet. 2010;3:300–306. doi: 10.1161/CIRCGENETICS.109.914556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Splansky GL, Corey D, Yang Q, Atwood LD, Cupples LA, Benjamin EJ, et al. The third generation cohort of the national heart, lung, and blood institute's framingham heart study: Design, recruitment, and initial examination. Am J Epidemiol. 2007;165:1328–1335. doi: 10.1093/aje/kwm021. [DOI] [PubMed] [Google Scholar]
- 9.Volzke H, Alte D, Schmidt CO, Radke D, Lorbeer R, Friedrich N, et al. Cohort profile: The study of health in pomerania. Int J Epidemiol. 2011;40:294–307. doi: 10.1093/ije/dyp394. [DOI] [PubMed] [Google Scholar]
- 10.Stein JL, Medland SE, Vasquez AA, Hibar DP, Senstad RE, Winkler AM, et al. Identification of common variants associated with human hippocampal and intracranial volumes. Nat Genet. 2012;44:552–561. doi: 10.1038/ng.2250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Suhre K, Wallaschofski H, Raffler J, Friedrich N, Haring R, Michael K, et al. A genome-wide association study of metabolic traits in human urine. Nat Genet. 2011;43:565–569. doi: 10.1038/ng.837. [DOI] [PubMed] [Google Scholar]
- 12.Lorbeer R, Baumeister SE, Dorr M, Nauck M, Grotevendt A, Volzke H, et al. Circulating angiopoietin-2, its soluble receptor tie-2, and mortality in the general population. Eur J Heart Fail. 2013;15:1327–1334. doi: 10.1093/eurjhf/hft117. [DOI] [PubMed] [Google Scholar]
- 13.Vasan RS, Glazer NL, Felix JF, Lieb W, Wild PS, Felix SB, et al. Genetic variants associated with cardiac structure and function: A meta-analysis and replication of genome-wide association data. JAMA. 2009;302:168–178. doi: 10.1001/jama.2009.978-a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Psaty BM, O'Donnell CJ, Gudnason V, Lunetta KL, Folsom AR, Rotter JI, et al. Cohorts for heart and aging research in genomic epidemiology (charge) consortium: Design of prospective meta-analyses of genome-wide association studies from 5 cohorts. Circ Cardiovasc Genet. 2009;2:73–80. doi: 10.1161/CIRCGENETICS.108.829747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Johnson AD, Yanek LR, Chen MH, Faraday N, Larson MG, Tofler G, et al. Genome-wide meta-analyses identifies seven loci associated with platelet aggregation in response to agonists. Nat Genet. 2010;42:608–613. doi: 10.1038/ng.604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li Y, Willer CJ, Ding J, Scheet P, Abecasis GR. Mach: Using sequence and genotype data to estimate haplotypes and unobserved genotypes. Genet Epidemiol. 2010;34:816–834. doi: 10.1002/gepi.20533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet. 2006;38:904–909. doi: 10.1038/ng1847. [DOI] [PubMed] [Google Scholar]
- 18.Johnson AD, Handsaker RE, Pulit SL, Nizzari MM, O'Donnell CJ, de Bakker PI. Snap: A web-based tool for identification and annotation of proxy snps using hapmap. Bioinformatics. 2008;24:2938–2939. doi: 10.1093/bioinformatics/btn564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen MH, Yang Q. Gwaf: An r package for genome-wide association analyses with family data. Bioinformatics. 2010;26:580–581. doi: 10.1093/bioinformatics/btp710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vistoropsky Y, Trofimov S, Malkin I, Kobyliansky E, Livshits G. Genetic and environmental determinants of hepatocyte growth factor levels and their association with obesity and blood pressure. Ann Hum Biol. 2008;35:93–103. doi: 10.1080/03014460701822003. [DOI] [PubMed] [Google Scholar]
- 21.Burdon KP, Macgregor S, Bykhovskaya Y, Javadiyan S, Li X, Laurie KJ, et al. Association of polymorphisms in the hepatocyte growth factor gene promoter with keratoconus. Invest Ophthalmol Vis Sci. 2011;52:8514–8519. doi: 10.1167/iovs.11-8261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hoshino K, Satoh T, Kawaguchi Y, Kuwana M. Association of hepatocyte growth factor promoter polymorphism with severity of interstitial lung disease in japanese patients with systemic sclerosis. Arthritis Rheum. 2011;63:2465–2472. doi: 10.1002/art.30415. [DOI] [PubMed] [Google Scholar]
- 23.Campbell DB, Li C, Sutcliffe JS, Persico AM, Levitt P. Genetic evidence implicating multiple genes in the met receptor tyrosine kinase pathway in autism spectrum disorder. Autism Res. 2008;1:159–168. doi: 10.1002/aur.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Finn RD, Bateman A, Clements J, Coggill P, Eberhardt RY, Eddy SR, et al. Pfam: The protein families database. Nucleic Acids Res. 2014;42:D222–D230. doi: 10.1093/nar/gkt1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Patthy L, Trexler M, Vali Z, Banyai L, Varadi A. Kringles: Modules specialized for protein binding. Homology of the gelatin-binding region of fibronectin with the kringle structures of proteases. FEBS Lett. 1984;171:131–136. doi: 10.1016/0014-5793(84)80473-1. [DOI] [PubMed] [Google Scholar]
- 26.Abecasis GR, Altshuler D, Auton A, Brooks LD, Durbin RM, Gibbs RA, et al. A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen J, Yu H, Sun K, Song W, Bai Y, Yang T, et al. Promoter variant of angiopoietin-2 and plasma angiopoietin-2 are associated with risk of stroke recurrence in lacunar infarct patients. Biochem Biophys Res Commun. 2010;398:212–216. doi: 10.1016/j.bbrc.2010.06.062. [DOI] [PubMed] [Google Scholar]
- 28.Meyer NJ, Li M, Feng R, Bradfield J, Gallop R, Bellamy S, et al. Angpt2 genetic variant is associated with trauma-associated acute lung injury and altered plasma angiopoietin-2 isoform ratio. Am J Respir Crit Care Med. 2011;183:1344–1353. doi: 10.1164/rccm.201005-0701OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Smith NL, Chen MH, Dehghan A, Strachan DP, Basu S, Soranzo N, et al. Novel associations of multiple genetic loci with plasma levels of factor vii, factor viii, and von willebrand factor: The charge (cohorts for heart and aging research in genome epidemiology) consortium. Circulation. 2010;121:1382–1392. doi: 10.1161/CIRCULATIONAHA.109.869156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Souto JC, Almasy L, Soria JM, Buil A, Stone W, Lathrop M, et al. Genome-wide linkage analysis of von willebrand factor plasma levels: Results from the gait project. Thromb Haemost. 2003;89:468–474. [PubMed] [Google Scholar]
- 31.Barbalic M, Dupuis J, Dehghan A, Bis JC, Hoogeveen RC, Schnabel RB, et al. Large-scale genomic studies reveal central role of abo in sp-selectin and sicam-1 levels. Hum Mol Genet. 2010;19:1863–1872. doi: 10.1093/hmg/ddq061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pare G, Chasman DI, Kellogg M, Zee RY, Rifai N, Badola S, et al. Novel association of abo histo-blood group antigen with soluble icam-1: Results of a genome-wide association study of 6,578 women. PLoS Genet. 2008;4:e1000118. doi: 10.1371/journal.pgen.1000118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Paterson AD, Lopes-Virella MF, Waggott D, Boright AP, Hosseini SM, Carter RE, et al. Genome-wide association identifies the abo blood group as a major locus associated with serum levels of soluble e-selectin. Arterioscler Thromb Vasc Biol. 2009;29:1958–1967. doi: 10.1161/ATVBAHA.109.192971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Melzer D, Perry JR, Hernandez D, Corsi AM, Stevens K, Rafferty I, et al. A genome-wide association study identifies protein quantitative trait loci (pqtls) PLoS Genet. 2008;4:e1000072. doi: 10.1371/journal.pgen.1000072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wolpin BM, Kraft P, Xu M, Steplowski E, Olsson ML, Arslan AA, et al. Variant abo blood group alleles, secretor status, and risk of pancreatic cancer: Results from the pancreatic cancer cohort consortium. Cancer Epidemiol Biomarkers Prev. 2010;19:3140–3149. doi: 10.1158/1055-9965.EPI-10-0751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Amundadottir L, Kraft P, Stolzenberg-Solomon RZ, Fuchs CS, Petersen GM, Arslan AA, et al. Genome-wide association study identifies variants in the abo locus associated with susceptibility to pancreatic cancer. Nat Genet. 2009;41:986–990. doi: 10.1038/ng.429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Macdonald PR, Progias P, Ciani B, Patel S, Mayer U, Steinmetz MO, et al. Structure of the extracellular domain of tie receptor tyrosine kinases and localization of the angiopoietin-binding epitope. J Biol Chem. 2006;281:28408–28414. doi: 10.1074/jbc.M605219200. [DOI] [PubMed] [Google Scholar]
- 38.Kiechl S, Pare G, Barbalic M, Qi L, Dupuis J, Dehghan A, et al. Association of variation at the abo locus with circulating levels of sicam-1, sp-selectin and se-selectin: A meta-analysis. Circ Cardiovasc Genet. 2011 doi: 10.1161/CIRCGENETICS.111.960682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Aird I, Bentall HH, Roberts JA. A relationship between cancer of stomach and the abo blood groups. Br Med J. 1953;1:799–801. doi: 10.1136/bmj.1.4814.799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tanikawa C, Urabe Y, Matsuo K, Kubo M, Takahashi A, Ito H, et al. A genome-wide association study identifies two susceptibility loci for duodenal ulcer in the japanese population. Nat Genet. 2012;44:430–434. S431–S432. doi: 10.1038/ng.1109. [DOI] [PubMed] [Google Scholar]
- 41.Reilly MP, Li M, He J, Ferguson JF, Stylianou IM, Mehta NN, et al. Identification of adamts7 as a novel locus for coronary atherosclerosis and association of abo with myocardial infarction in the presence of coronary atherosclerosis: Two genome-wide association studies. Lancet. 2011;377:383–392. doi: 10.1016/S0140-6736(10)61996-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Williams FM, Carter AM, Hysi PG, Surdulescu G, Hodgkiss D, Soranzo N, et al. Ischemic stroke is associated with the abo locus: The euroclot study. Ann Neurol. 2013;73:16–31. doi: 10.1002/ana.23838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hong KW, Moon S, Kim YJ, Kim YK, Kim DJ, Kim CS, et al. Association between the abo locus and hematological traits in korean. BMC Genet. 2012;13:78. doi: 10.1186/1471-2156-13-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Manolio TA, Collins FS, Cox NJ, Goldstein DB, Hindorff LA, Hunter DJ, et al. Finding the missing heritability of complex diseases. Nature. 2009;461:747–753. doi: 10.1038/nature08494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yang J, Lee SH, Goddard ME, Visscher PM. Gcta: A tool for genome-wide complex trait analysis. Am J Hum Genet. 2011;88:76–82. doi: 10.1016/j.ajhg.2010.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tomiya T, Yamaoka M, Inoue Y, Nishikawa T, Yanase M, Ikeda H, et al. Effect of rapamycin on hepatocyte function and proliferation induced by growth factors. Chemotherapy. 2007;53:59–69. doi: 10.1159/000098420. [DOI] [PubMed] [Google Scholar]
- 47.Tomiya T, Nishikawa T, Inoue Y, Ohtomo N, Ikeda H, Tejima K, et al. Leucine stimulates hgf production by hepatic stellate cells through mtor pathway. Biochem Biophys Res Commun. 2007;358:176–180. doi: 10.1016/j.bbrc.2007.04.093. [DOI] [PubMed] [Google Scholar]
- 48.Nomura M, Oketa Y, Yasui K, Ishikawa H, Ono S. Expression of hepatocyte growth factor in the skin of amyotrophic lateral sclerosis. Acta Neurol Scand. 2012;125:389–397. doi: 10.1111/j.1600-0404.2011.01579.x. [DOI] [PubMed] [Google Scholar]
- 49.Kadoyama K, Funakoshi H, Ohya W, Nakamura T. Hepatocyte growth factor (hgf) attenuates gliosis and motoneuronal degeneration in the brainstem motor nuclei of a transgenic mouse model of als. Neurosci Res. 2007;59:446–456. doi: 10.1016/j.neures.2007.08.017. [DOI] [PubMed] [Google Scholar]
- 50.Sun W, Funakoshi H, Nakamura T. Overexpression of hgf retards disease progression and prolongs life span in a transgenic mouse model of als. J Neurosci. 2002;22:6537–6548. doi: 10.1523/JNEUROSCI.22-15-06537.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.