

Abstract
The MYC transcription factor plays a crucial role in the regulation of cell cycle progression, apoptosis, angiogenesis, and cellular transformation. Due to its oncogenic activities and overexpression in a majority of human cancers, it is an interesting target for novel drug therapies. MYC binding to the E-box (5′-CACGTGT-3′) sequence at gene promoters contributes to more than 4000 MYC-dependent transcripts. Owing to its importance in MYC regulation, we designed a novel sequence-specific DNA-binding pyrrole–imidazole (PI) polyamide, Myc-5, that recognizes the E-box consensus sequence. Bioinformatics analysis revealed that the Myc-5 binding sequence appeared in 5′- MYC binding E-box sequences at the eIF4G1, CCND1, and CDK4 gene promoters. Furthermore, ChIP coupled with detection by quantitative PCR indicated that Myc-5 has the ability to inhibit MYC binding at the target gene promoters and thus cause downregulation at the mRNA level and protein expression of its target genes in human Burkitt's lymphoma model cell line, P493.6, carrying an inducible MYC repression system and the K562 (human chronic myelogenous leukemia) cell line. Single i.v. injection of Myc-5 at 7.5 mg/kg dose caused significant tumor growth inhibition in a MYC-dependent tumor xenograft model without evidence of toxicity. We report here a compelling rationale for the identification of a PI polyamide that inhibits a part of E-box-mediated MYC downstream gene expression and is a model for showing that phenotype-associated MYC downstream gene targets consequently inhibit MYC-dependent tumor growth.
Keywords: Cell cycle, E-Box, MYC, pyrrole-imidazole polyamide, transcription therapy
The transcription factor c-MYC possesses an exclusive and extensive set of biological actions that underlie its role as a salient oncogene and therefore could be the key to anticancer drug development. The c-MYC proto-oncogene belongs to the family of MYC genes that includes B-MYC, L-MYC, N-MYC, and S-MYC.1 Among them, c-MYC (here after referred as MYC) is found in almost all proliferating cells and expression of N- and L-MYC is more constrained to specific cell types.2 MYC is a basic helix–loop–helix leucine zipper transcription factor that binds DNA in a sequence-specific manner3 and activates the transcription of genes whose products are involved in crucial aspects of cancer biology such as cell proliferation, cell growth, apoptosis, and differentiation.4 The biological activities of MYC depends on its ability to heterodimerize with its protein partner, MAX, to bind the enhancer box (E-box) sequence and stimulate transcription of a number of its downstream genes.4,5 The MAX:MAX homodimer or MAD:MAX heterodimer is supposed to antagonize the functions of MYC by binding to the same core E-box sequences.6
MYC targets approximately 10–15% of all cellular promoters in human cells, which is higher than any other typical transcription factor.7 Target genes of MYC identified in mammalian cells include genes involved in cell cycle, ribosomal biogenesis, protein synthesis, and mitochondrial function.4,5,8 Among the direct target genes of MYC, those with E-box binding sites include ODC, ECA39, eIF4E, CDC25, CAD, CDK4, eIF4G1, and CCND1.9,10 MYC has been known to play a crucial role in malignant transformation.11 Several attempts have been made using small molecule inhibitors to inhibit MYC binding at gene promoters.11–15 Pyrrole–imidazole (PI) polyamides are a class of sequence-specific DNA-binding small molecules that have been shown to be effective inhibitors of transcription factors by disrupting essential protein–DNA interactions.16–20 Our group and others have designed PI polyamides to specifically target critical regulatory proteins, including MMP-9, transforming growth factor-β1, vascular endothelial growth factor, hypoxia-inducible factor 1-α, androgen receptor, epidermal growth factor receptor, prostate-specific antigen, and lectin-type oxidized LDL receptor-1.16,21–24 Recently, we have demonstrated that E-Box recognizing PI polyamide, Myc-6 was found to significantly suppress malignant phenotypes of human osteosarcoma MG63 cells both in vitro and in vivo, showing the potential of PI polyamides in cancer therapy.25
In the present study, we developed an E-box-binding PI polyamide, Myc-5 (target sequence 5′-WCWCGWGW-3′, where W = A or T) to inhibit MYC target genes. Myc-5 showed inhibition of MYC-driven cell growth by downregulation of a subgroup of MYC downstream genes, including eIF4GI and CCND1, at an early stage of transcription. In animal tumor model studies, Myc-5 inhibits tumor growth by inhibiting cell proliferation and inducing apoptosis in tumor tissue. Collectively, our results establish a transcriptional regulation at an early stage of MYC regulatory proteins by using PI polyamides and provide a novel antitumor agent targeting MYC function.
Materials and Methods
Cell culture
The human Burkitt's lymphoma model cell line, P493.6, carrying Tet-repressible c-MYC system and chronic myeloid leukemia cell line K562, were used in this study. To suppress MYC expression, P493.6 cells were treated with 0.1 μg/mL tetracycline for 3 days before treatment with Myc-5. The P493.6 cell line was kindly provided by C. Grandori (Fred Hutchinson Cancer Research Center, Seattle, WA, USA) and K562 cells were obtained from the Riken cell bank (Ibaraki, Japan).
Synthesis of PI polyamides targeting the E-box consensus sequence
Myc-5 was designed to target the E-box consensus sequence and mismatch PI polyamide was designed to target by exchanging the CG dinucleotide with GC in the center of the E-box (Fig.1a,c). Fluorescein isothiocyanate-conjugated Myc-5 was also synthesized for nuclear localization experiments (Fig.1b). All of the PI polyamides were synthesized according to previously established methods.21
Fig 1.
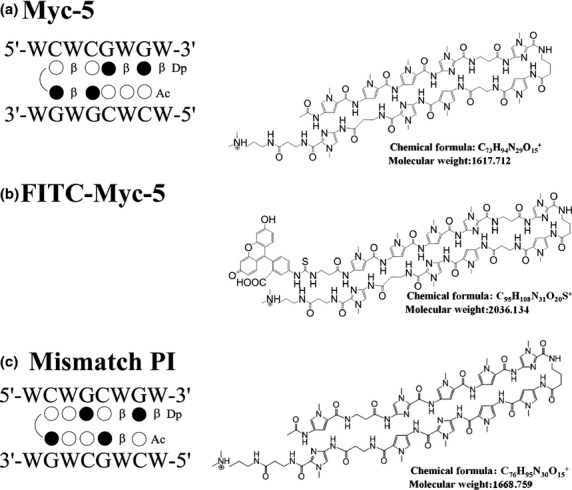
Designed pyrrole–imidazole (PI) polyamide structure and binding site. Structures and binding sites for synthesized PI polyamides Myc-5 (a), FITC-labeled Myc-5 (b), and mismatch PI polyamide (c). Base sequence specificity depends on side-by-side pairing of pyrrole and imidazole amino acids in the minor groove of DNA. Black and white circles represent imidazole and pyrrole rings, respectively; curved lines represent hairpin junctions. β, β-alanine; Dp, diaminomethylene propylamide; W, A or T.
Electrophoretic mobility shift assay and surface plasmon resonance
FITC-labeled matching hairpin oligonucleotide (46 base pair) corresponding to eIF4G1 gene promoters having E-box consensus binding sites and mismatch promoter were synthesized (Table S1) for EMSA. Results were visualized by luminescent image analyzer LAS3000 (Fujifilm, Tokyo, Japan). The kinetic measurements of the polyamides’ binding curves to the biotin-labeled double-stranded DNA (having the E-box consensus sequence) and data processing were carried out on a Biacore 2000 system as described previously.26
Quantitative real-time PCR
Total RNA was extracted and digested with DNase I using the RNeasy kit according to the manufacturer's protocol (Qiagen, Valencia, CA, USA). The primer sequences are listed in Table S2. Relative gene expression was determined by normalizing the gene expression of each target gene to GAPDH.
Conventional ChIP followed by real-time PCR
The status of polyamide binding at the target promoter region (Table S3) was detected using a ChIP assay kit (Upstate Biotechnology, Upstate Biotechnology, Inc., Lake Placid, NY, USA) following the manufacturer's protocol. ChIP DNA was further analyzed by quantitative PCR using primers encompassing the regions of interest on the eIF4G1, CCND1, and CDK4 promoters.
Tumorigenicity studies in SCID mice
Seven-week-old SCID mice were housed under specific pathogen-free conditions. Experiments were approved by the committee for laboratory animal welfare and ethics of Nihon University School of Medicine (Tokyo, Japan). The effect of Myc-5 on the xenograft model was examined as follows. P493.6 cells (1 × 107) were inoculated s.c. into the flank of mice and they were divided into three treatment groups: control group (PBS i.v.; n = 8); a Myc-5 treated group (7.5 mg/kg i.v., single dose; n = 8); and a doxycycline treated group (0.01% doxycycline in regular drinking water; n = 5). Doxycycline induces repression of P493.6 xenograft tumor growth in a MYC-dependent manner. The treatment was started on day 7 after cell inoculation and mice were killed after 30 days of treatment.
In vivo nuclear localization
For in vivo nuclear localization analysis by fluorescence microscopy, tumor-bearing mice were injected with FITC-labeled Myc-5 (0.15 mg) into the lateral tail vein of the animals. Tumor tissues, along with adjacent normal tissues, were collected 5 days after the injection for analysis using propidium iodide as a nuclear dye to identify nucleated cells.
Statistical analysis
Results are shown as mean ± SD. Each experiment was carried out independently three times. The level of significance (**P < 0.05 and ***P < 0.001) was determined using Student's t-test.
Additional supplementary materials and methods detailing cell viability assay, detection of nuclear localization by confocal microscopy, histopathology, Western blot analysis, microarray analyses, and references can be found in Documents S1 and S2.
Results
Identification of MYC target genes that harbor Myc-5 consensus sequence
E-boxes are present in many genes, but not all are known to be direct transcriptional targets of MYC. Based on compilations by Fernandez et al.,27 we found that 105 genes harbor the Myc-5 binding site (WCWCGWGW) in their E-box consensus sequence (Table S4). These genes can be classified into a wide range of functional classes. Here we focused on genes related to translation and proliferation. Among the ten top MYC-regulated genes we chose two proliferation genes (CDK4 and CCND1) and a translation gene (eIF4G1). The selected genes have been well characterized by previous researchers and are also well known direct targets of MYC.10,9,28
DNA-binding affinity and specificity of designed Myc-5 PI polyamide
The EMSA and surface plasmon resonance (SPR) assay are used to determine the binding affinity and specificity of PI polyamide to its target DNA. The EMSA results demonstrated that a clear mobility band shift (Fig.2a,b) was detected when Myc-5 was incubated with eIF4G1 gene promoter oligonucleotide, whereas no shift was detected for the mismatch PI polyamide (Fig.2a, lane 4) or eIF4G1 mismatch gene promoter (Fig.2a, lane 5) in which the core recognition sequence, CACGTG, was replaced by CAGCTG. Increasing concentrations of Myc-5, but not of the mismatch PI polyamide, bound to the eIF4G1 promoter oligo (Fig.2b, lanes 2–5), suggesting that Myc-5 can specifically bind to sequences of their target gene-promoters. To further confirm binding of the E-box to the target gene promoter, we used a biosensor-SPR assay to explore the interaction of Myc-5 with biotinylated hairpin duplexes having WCWCGWGW sequences (Fig.2c). The kinetic profile of Myc-5 revealed a relatively fast “on-rate” and slow “off-rate” in binding with a KD of 4.81 ± 2.79 × 10−8 M. The mismatch PI polyamide showed slow “on-rate” and fast “off-rate” with KD was calculated at 5.1 ± 0.54 × 10−5. In summary, these results confirm that the presence of the E-box in the DNA sequence has a pronounced influence on the binding of Myc-5 and yields a 654-times higher affinity than the mismatch PI polyamide (Fig.2d,e, Table S5).
Fig 2.

Myc-5 binding at the target gene promoters. (a) EMSA of eIF4G1 gene match and mismatch promoter and with Myc-5 and mismatch pyrrole–imidazole (PI) polyamide. (b) EMSA of eIF4G1 gene promoter with Myc-5 and mismatch PI polyamide. FITC-labeled hairpin oligonucleotide was incubated at 37°C for 60 min in Myc-5 or mismatch PI polyamide. (c) Typical surface plasmon resonance sensograms for the interaction between PI polyamides and the hairpin duplex with 5′-biotin labeled and immobilized E-box (CACGTG) sequences. (d, e) Remarkable differences in binding kinetics were observed: fast on/off kinetics for Myc-5 (d), and slower kinetics for the mismatch PI polyamide (e).
Myc-5 inhibited cell proliferation and localized into nucleus in P493.6 and K562 cell lines
P493.6 and K562 cells were incubated with different concentrations (1–10 μM) of Myc-5 and mismatch PI polyamide and viability was determined at 24, 48, and 72 h after treatment, respectively. As shown in Figure S1, cell viability was significantly reduced (P < 0.001 vs control) in both cell lines treated with Myc-5 in a time- and concentration-dependent manner. Nuclear localization of Myc-5 was determined by FITC-conjugated Myc-5 using laser confocal fluorescence microscopy. Green fluorescence indicates the presence of Myc-5 and red fluorescence depicts the cell nuclei, indicating that Myc-5 localizes into nuclei within 2 h (Fig. S2a,c,d). In contrast, cells incubated with FITC solution (control) at the same concentration did not localize into nuclei (Fig. S2b) in either cell line.
Myc-5 attenuates MYC binding at the gene promoter, causing downregulation of MYC target genes
Myc-5 inhibited target gene expression at protein and mRNA levels (Fig.3a,b). Cells treated with Myc-5 at 10 μM concentration for 72 h caused statistically significant suppression of eIF4G1 mRNA compared with control or mismatch PI polyamide treated cells in both systems. The CCND1 mRNA was unaffected in all treated and untreated groups of P493.6 cells; however, its expression was significantly (P < 0.001) downregulated in the K562 cell line compared to control and mismatch PI polyamide treated groups. The mRNA expression correlated well with protein expression in the Myc-5 administration using both cell systems (Fig.3c,d). To investigate whether target gene expression is directly regulated by Myc-5, we used ChIP assays of the E-box and exonic region (Figs4a,b,S3a). The exonic region was taken as an arbitrary negative control region during the analysis. The ChIP analysis revealed that, compared to control, the Myc-5 and tetracycline groups significantly inhibited binding of MYC to the E-box region in P493.6 cells (Figs4c,S3b) at the eIF4G1 and CDK4 gene promoters. In contrast, the E-box and control regions (exon) of CCND1 gene promoter showed restrained enrichment in all treated groups (Fig.4d). Similarly, MYC was specifically enriched near the E-box site at the eIF4G1, CCND1, and CDK4 gene promoters but not in the Myc-5 treated group in K562 cells (Figs4e,f,S3c). Moreover, our microarray analysis results showed (Fig. S4a,b) that differentially expressed genes in K562 cells (29 upregulated and 21 downregulated) and P943.6 cells (20 genes upregulated and 20 downregulated) were identified by unpaired two-class significance analysis of microarray with 0% false discovery rate. Among the top 50 modulated genes, five were carrying the Myc-5 consensus sequence in their promoter, as identified from Table S4.
Fig 3.

Myc-5 downregulates mRNA expression and protein expression of target genes. (a, b) Expression of target genes was detected by quantitative real-time PCR after 72 h of treatment with control, Myc-5 (1, 5, and 10 μM concentration), or mismatch PI polyamide (MM) at 10 μM concentration and normalized with GAPDH in P493.6 (a) and K562 (b) cells. Data are shown as mean values with error bars representing ±SD. Statistical significance was calculated by Student's t-test. **P < 0.05, ***P < 0.001 when compared to control. (c, d) P493.6 and K562 cells were treated with Myc-5 (1, 5, and 10 μM) or mismatch PI polyamide (10 μM; MM) for 72 h. β-actin was used as the loading control. The relative band intensities in P493.6 and K562 cells were determined by dividing the intensity of the band by β-actin followed by normalization to the control. Tet, tetracycline.
Fig 4.

In vivo binding of Myc-5 to the E-box at its target gene promoter. (a, b) Schematic depiction of the Myc-5 target gene promoter with MYC binding site (underline) indicated. (c–f) ChIP assay of Myc-5 target genes in the P493.6 (c, d) and K562 (e, f) cell systems. Labeled regions (E-box and exon) of each gene were quantitatively amplified by real-time PCR. Data are representative of three independent experiments. Tet, tetracycline.
Myc-5 retards growth in animal tumor models
To investigate whether the in vitro efficacy of Myc-5 can also be recapitulated in vivo, therapeutic animal studies were carried out using P493.6 s.c. xenografts. Mice were inoculated s.c. with P493.6 (high MYC expressing) cells. One week after inoculating the cells, when the tumor volumes reached approximately 100 mm3, mice were split into three groups and treated with either saline, doxycycline, or 7.5 mg/kg Myc-5 injected i.v. into the lateral tail vein of animals at day 7 (single dose; Fig.5a). Myc-5 was formulated based on previous xenograft studies with PI polyamide.20,29,30 Growth curve data indicated that Myc-5 (7.5 mg/kg) and doxycycline treated groups had a significantly smaller tumor volume during the growth phase (P < 0.001 vs control; Fig.5b) by the end of the study. Representative images of each group of mice are shown in Fig.5b (inset). All mice with Myc-5 treatment continued to gain weight at an equal rate throughout the treatment period ( Fig.5c). The average tumor weight results further confirmed inhibition of tumor growth as Myc-5 and doxycycline treated groups were found to be significantly lower (P < 0.001 vs control; Fig.5d) at the termination of the study.
Fig 5.
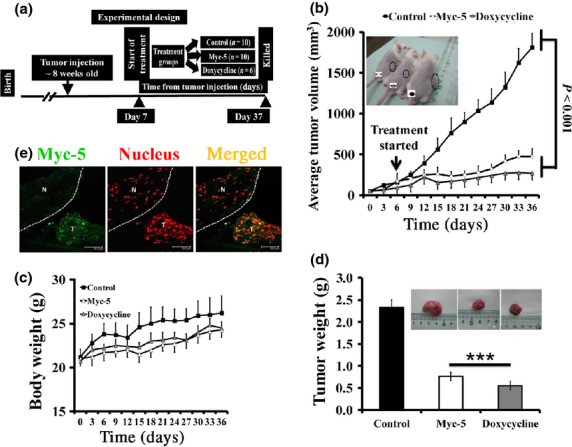
Myc-5 blocks the growth of P493.6 xenografts. (a) Schematic diagram of the xenograft model illustrating timing of tumor implantation and treatment. Eight-week-old SCID mice were s.c. injected with P493.6 cells. (b) Tumor growth chart showing the effect of treatment in vivo. Myc-5 (7.5 mg/kg) and doxycycline significantly slowed tumor growth (P < 0.001) at the termination point in comparison to the control group. Representative picture shows each group of mice (inset). (c) Mean body weight for each treatment group plotted as function of day after post-injection. (e) Comparisons of excised tumor weights for three different treatment groups at the end of study. Data in (b, d, e) are shown as the mean ± SD. Statistical significance was calculated by Student's t-test. ***P < 0.001. (e) FITC-labeled Myc-5 localizes to nucleus of P493.6 xenograft leaving normal tissue unaffected (separated by white line). N: Normal tissue, T: tumor tissue.
Myc-5 localizes into tumor and causes decreased cell proliferation and induced apoptosis in P493.6 tumor xenografts
To evaluate in vivo nuclear localization, single i.v. injection of FITC-conjugated Myc-5 was given to P493.6 cell-derived xenografts. Twenty-four hours after injection, animals were killed and tumor tissues were obtained. The tumor-derived tissues were found to display strong and characteristic nuclear staining (Fig.5e). In contrast, adjacent normal tissues were found to be devoid of nuclear fluorescence (Fig.5e). Myc-5 was found throughout the tumor, indicating its capacity to enter the tumor through the vascular system. To assess the activity of Myc-5, tumors were harvested from all treatment groups and examined by histopathology. Microscopic analysis of H&E staining showed that Myc-5 and doxycycline treated tumors showed areas with necrosis, cellular debris, and swollen cells with cytoplasmic vacuoles as compared with the vehicle-treated control (Fig.6a; left). In order to investigate the mechanism underlying Myc-5-mediated tumor growth inhibition in P493.6 xenografts, immunohistochemical analyses were carried out for BrdU uptake, and TUNEL reaction assay. BrdU-positive nuclei were detected in a small number of cells in Myc-5 treated tumor as compared to control group (Fig6a; middle). In contrast, a large number of cells stained positively for TUNEL in Myc-5 or doxycycline treated group (Fig.6a; right) compared to the control group. Quantitative data were consistent with the expression pattern of BrdU and TUNEL staining assays (Fig.6b). Quantification of BrdU-positive cells in the control group were significantly (P < 0.001) reduced (approximately 83% and 76% reductions in Myc-5 and doxycycline groups, respectively) compared to treated groups. The TUNEL analysis showed that the number of apoptotic cells was significantly higher (P < 0.001; approximately 38% and 30% higher in Myc-5 and doxycycline groups, respectively) in treated groups. Overall, we found that Myc-5 is well tolerated, inhibits tumor growth, and induces apoptosis in P493.6 xenograft mouse models.
Fig 6.
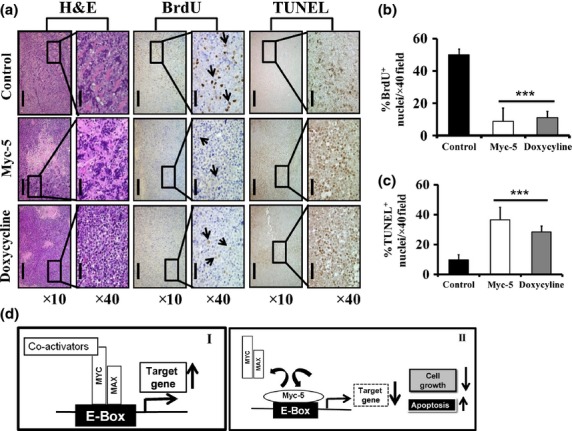
Histopathology of xenografts in nude mice and illustration of potential mechanism by Myc-5. (a) Tissue sample were analyzed qualitatively for morphological changes. Magnification, ×10 (scale bar = 200 μm); magnification, ×40 (scale bar = 50 μm). (b, c) Quantitative data of immunohistochemical analysis of BrdU and TUNEL positive staining in each group. Data in (b, c) are shown as the mean ± SD of three tumor samples from an individual mouse in each group. Statistical significance was calculated by Student's t-test. ***P < 0.001. (d) Schematic diagram of the mechanism by which pyrrole–imidazole polyamide inhibits MYC/MAX interaction to the E-box. (I) MYC:MAX dimer binds to E-box and activates MYC target gene expression. (II) Myc-5 occupied the E-box by binding, thereby inhibiting the MYC/MAX interaction to the E-box, causing further suppression of target gene expression.
Discussion
In this study, we synthesized two β-alanine-linked polyamides of different lengths: Myc-5 (AcPyPyPyImβImγPyβPyImβImβDp), where Py is pyrrole, Im is imidazole, Ac is acetyl, β is β-alanine, Dp is dimethylaminopropylamine, and γ is γ-turn to target the 8-bp site of 5′-WCWCGWGW-3′; and mismatch polyamide (AcPyβImPyPyImγPyPyImPyβImβDp) to target the 8-bp site 5′-WCWGCWGW-3′, flipping dinucleotide CG to GC at the central portion (Fig.1a,b). A search for Myc-5-binding sites from the published database27 revealed that the Myc-5 consensus sequence was flanking the E-box of genes involved in apoptosis, cell cycle, nucleolar function, ribosomal proteins, and translation initiation factors. Among them, we focused on the eIF4G1, CCND1, and CDK4 genes because they carry a Myc-5 consensus sequence including a MYC binding site (E-box) in the promoter region (Fig. S3d) and their transcription is modulated by MYC.9,10,28 Using EMSA and Biacore analyses, our results showed that Myc-5 binds specifically and with high affinity (654 times; Table S5) to the eIF4G1, CCND1, and CDK4 gene promoters at the E-box region (Fig.2). The effective concentration of Myc-5 in the EMSA was higher than the concentration for biological effect, which is consistent with results obtained for other compounds;31 possibly, PI polyamide accumulates in cells to effectively reach the intracellular and intranuclear levels.32 The higher binding affinity of Myc-5 in comparison to mismatch PI polyamide might be explained as the targeted PI polyamide has more aliphatic β-alanine unit, giving it more flexibility and optimizing the positioning of the imidazole amino acids on binding to its targeted sequence.33 In vivo binding of Myc-5 to their target gene-promoter was confirmed by ChIP assay. Results of ChIP indicated that MYC transcription factor bound E-box in the control group whereas, in treated groups, Myc-5 inhibited MYC binding on its target gene promoters. However, in the P493.6 cell line, the CCND1 gene-promoter showed only a background signal that was obtained at the E-box region as well as in the control (exon) regions (Fig.4d). These results were consistent with previous reports of the absence of this regulator in the B cell line.34
Myc-5 was used at the concentration of 10 μM based on previous studies using PI polyamides.20,35,19 Myc-5 significantly reduced mRNA and protein expression of MYC target genes at this given concentration. The correlation between MYC binding and mRNA expression of Myc-5 was established by real-time mRNA expression analysis and Western blot analysis. The eIF4G1 gene has a downregulated expression and might be a direct target of MYC in both systems, which was also favored by EMSA and ChIP data. CCND1 mRNA expression was downregulated in the K562 cell line with Myc-5 treatment in a dose-dependent manner, which is known to exert positive growth effects and to be regulated by MYC.36 The mRNA expression of CDK4, a positive growth controller of MYC,28 was unaffected by Myc-5 treatment as the Myc-5 binding site (E-box#2) has no role in MYC-dependent gene regulation.28 We analyzed the CDK4 promoter region for putative MYC binding sites. Our sequence analysis revealed the presence of four E-box sequences, which could be recognized by MYC (Fig. S2). Among them, only E-box#2 is a putative binding site for Myc-5 as it contains the Myc-5 consensus sequence. As shown in Figure S2, Myc-5 bound to the E-box#2 of the CDK4 promoter. The CDK4 gene is established as a direct target of MYC identified by serial analysis of gene expression with essential E-boxes in their promoters.28 The CDK4 promoter contains four highly conserved E-box elements where E-box#3 and #4 were the most important for CDK4 promoter activity.28 The Myc-5 effect on m-RNA expression of the MYC gene and further its transcriptional target genes were analyzed by real-time PCR in K562 and P493.6 cell lines. In K562 cells, MYC mRNA expression is unaffected by Myc-5 treatment and a similar trend was observed in its target gene CDK4 mRNA expression. However MYC mRNA expression in Myc-5 treated group is significantly increased as compared to control group in P493.6 cells. P493.6 B lymphoma cells overexpress MYC, which indicates that P493.6 cell lines may have some mutations on its promoter or regulator regions. The MYC promoter or regulatory regions do not have any Myc-5 binding consensus sequence. This may be due to some specific mutation in these regions that might have generated a novel Myc-5 binding site at the MYC promoter of P493.6 cells. This novel Myc-5 binding site in the promoter could be acting as a transcriptional activator of the MYC gene and helping it to further overexpress. Furthermore, Myc-5 has very high affinity for its consensus sequence and may act as a non-competitive inhibitor of MYC protein. Therefore, overexpression of MYC protein in P493.6 cells does not have an effect on MYC downstream genes with active Myc-5 binding sites, such as eIF4G1.
Taken together, our data are compatible with a mechanism that involves the recruitment of Myc-5 at E-box sites within the promoter of the MYC target gene, thereby inhibiting MYC binding at particular sites and inhibiting target gene function instead of blocking MYC. Myc-5 downregulates its target gene transcription, supporting the notion that polyamides bind to DNA with affinity and sequence-specificity comparable to DNA-binding proteins and gene expression can be regulated by competitive displacement of transcription factor from DNA target sequences,37 where Myc-5 consensus sequences are not contained.
Myc-5 exerts its therapeutic role in tumor maintenance through selective effects on the translation of specific downstream genes. In our in vivo study we used 7.5 mg/kg on the basis of prior pharmacokinetic profiles and previous analytical studies.20,38 We identified strong in vivo nuclear localization of Myc-5 and also its inhibitory effect on tumor growth in a P493.6 mouse model. One possible reason for this activity can be explained by previous reports showing that changes in the levels or activity of eIF4F mediate the translational regulation of specific genes involved (as in the P493.6 cell line, eIF4G1 is completely suppressed) in survival and apoptosis.39 In nasopharyngeal carcinoma, knockdown of eIF4G1 expression markedly inhibited cell-cycle progression, proliferation, and suppressed in vivo xenograft tumor growth,40,41 which might be applicable to our system as well.
In conclusion, the results reported here identify a novel PI polyamide, Myc-5, as a lead compound targeting against the E-box and thereby modulating expression of some genes. Our approach can be an efficient tool to identify the sequence-specific compound that targets the E-box and specifically modulates MYC downstream genes and interferes with their pathways. Myc-5 competes with the MYC:MAX heterodimer and inhibits transregulation of the target gene promoter (Fig.6d) at the E-box. The extension of this approach could result in the identification of more potent inhibitory PI polyamides in multiple pathways and reveal a new E-box-regulated target or combination of targets for a distinct MYC function, which may have formidable therapeutic opportunities for the future.
Acknowledgments
We thank Mr. Shigeki Nakai, Ms. Asako Oguni, Mr. Motoaki Kataba, Ms. Yuki Yamada, and Ms.Yukari Obana, for their technical support and Ms. Paula Jones, Dr. Manisha Tripathi, and Dr. Sandrine Billet for critical reading. This work was supported by the Academic Frontier Project for 2006 Project for Private Universities, a matching fund subsidy and Grant-in-Aid for Scientific Research (B) (#23300344 and #26290060) from the Ministry of Education, Culture, Sports, Science and Technology of Japan (to H. Nagase), the Setsuro Fujii Memorial, and the Osaka Foundation for the Promotion of Fundamental Medical Research, Osaka (to H. Nagase). Support was also received from the Nihon University Strategic Projects for Academic Research and R01 ES012249 from the National Institutes of Health/National Institute of Environmental Health Science, North Carolina, USA. (to H. Nagase).
Disclosure Statement
The authors have no conflict of interest.
Supporting Information
Additional supporting information may be found in the online version of this article:
Doc. S1. Supplementary materials and methods.
Doc. S2. Reference list.
Fig. S1. Antiproliferative effect of Myc-5.
Fig. S2. Myc-5 localizes into nucleus.
Fig. S3. In vivo binding of Myc-5 at CDK4 gene promoter.
Fig. S4. Heat map represents the functional annotated transcripts that are identified by unpaired two-class significance analysis of microarray analysis with a false discovery rate of 0% from cells treated with DMSO (control), mismatch polyamide, and Myc-5.
Table S1. FITC-labeled oligomer sequences designed for EMSA
Table S2. Primers used in quantitative RT-PCR
Table S3. Primers used for ChIP real-time PCR
Table S4. Promoter-associated E-box genes
Table S5. Kinetic constant for Myc-5 pyrrole–imidazole polyamide and mismatch binding
References
- Adhikary S, Eilers M. Transcriptional regulation and transformation by Myc proteins. Nat Rev Mol Cell Biol. 2005;6:635–45. doi: 10.1038/nrm1703. [DOI] [PubMed] [Google Scholar]
- Luscher B. Function and regulation of the transcription factors of the Myc/Max/Mad network. Gene. 2001;277:1–14. doi: 10.1016/s0378-1119(01)00697-7. [DOI] [PubMed] [Google Scholar]
- Blackwood EM, Eisenman RN. Max: a helix-loop-helix zipper protein that forms a sequence-specific DNA-binding complex with Myc. Science. 1991;251:1211–7. doi: 10.1126/science.2006410. [DOI] [PubMed] [Google Scholar]
- Dang CV, O'Donnell KA, Zeller KI, et al. The c-Myc target gene network. Semin Cancer Biol. 2006;16:253–64. doi: 10.1016/j.semcancer.2006.07.014. [DOI] [PubMed] [Google Scholar]
- Liu YC, Li F, Handler J, et al. Global regulation of nucleotide biosynthetic genes by c-Myc. PLoS One. 2008;3:e2722. doi: 10.1371/journal.pone.0002722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayer DE, Eisenman RN. A switch from Myc: Max to Mad: Max heterocomplexes accompanies monocyte/macrophage differentiation. Genes Dev. 1993;7:2110–9. doi: 10.1101/gad.7.11.2110. [DOI] [PubMed] [Google Scholar]
- Guccione E, Martinato F, Finocchiaro G, et al. Myc-binding-site recognition in the human genome is determined by chromatin context. Nat Cell Biol. 2006;8:764–70. doi: 10.1038/ncb1434. [DOI] [PubMed] [Google Scholar]
- Zeller KI, Jegga AG, Aronow BJ, et al. An integrated database of genes responsive to the Myc oncogenic transcription factor: identification of direct genomic targets. Genome Biol. 2003;4:R69. doi: 10.1186/gb-2003-4-10-r69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huerta M, Munoz R, Tapia R, et al. Cyclin D1 is transcriptionally down-regulated by ZO-2 via an E box and the transcription factor c-Myc. Mol Biol Cell. 2007;18:4826–36. doi: 10.1091/mbc.E07-02-0109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CJ, Cencic R, Mills JR, et al. c-Myc and eIF4F are components of a feedforward loop that links transcription and translation. Cancer Res. 2008;68:5326–34. doi: 10.1158/0008-5472.CAN-07-5876. [DOI] [PubMed] [Google Scholar]
- Albihn A, Johnsen JI, Henriksson MA. MYC in oncogenesis and as a target for cancer therapies. Adv Cancer Res. 2010;107:163–224. doi: 10.1016/S0065-230X(10)07006-5. [DOI] [PubMed] [Google Scholar]
- Wang H, Chauhan J, Hu A, et al. Disruption of Myc-Max heterodimerization with improved cell-penetrating analogs of the small molecule 10074-G5. Oncotarget. 2013;4:936–47. doi: 10.18632/oncotarget.1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Hammoudeh DI, Follis AV, et al. Improved low molecular weight Myc-Max inhibitors. Mol Cancer Ther. 2007;6:2399–408. doi: 10.1158/1535-7163.MCT-07-0005. [DOI] [PubMed] [Google Scholar]
- Yin X, Giap C, Lazo JS, et al. Low molecular weight inhibitors of Myc-Max interaction and function. Oncogene. 2003;22:6151–9. doi: 10.1038/sj.onc.1206641. [DOI] [PubMed] [Google Scholar]
- Delmore JE, Issa GC, Lemieux ME, et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell. 2011;146:904–17. doi: 10.1016/j.cell.2011.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dervan PB, Doss RM, Marques MA. Programmable DNA binding oligomers for control of transcription. Curr Med Chem Anticancer Agents. 2005;5:373–87. doi: 10.2174/1568011054222346. [DOI] [PubMed] [Google Scholar]
- Dickinson LA, Burnett R, Melander C, et al. Arresting cancer proliferation by small-molecule gene regulation. Chem Biol. 2004;11:1583–94. doi: 10.1016/j.chembiol.2004.09.004. [DOI] [PubMed] [Google Scholar]
- Nickols NG, Szablowski JO, Hargrove AE, et al. Activity of a py-im polyamide targeted to the estrogen response element. Mol Cancer Ther. 2013;12:675–84. doi: 10.1158/1535-7163.MCT-12-1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang F, Nickols NG, Li BC, et al. Antitumor activity of a pyrrole-imidazole polyamide. Proc Natl Acad Sci U S A. 2013;110:1863–8. doi: 10.1073/pnas.1222035110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raskatov JA, Nickols NG, Hargrove AE, et al. Gene expression changes in a tumor xenograft by a pyrrole-imidazole polyamide. Proc Natl Acad Sci U S A. 2012;109:16041–5. doi: 10.1073/pnas.1214267109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Nagase H, Watanabe T, et al. Inhibition of MMP-9 transcription and suppression of tumor metastasis by pyrrole-imidazole polyamide. Cancer Sci. 2010;101:759–66. doi: 10.1111/j.1349-7006.2009.01435.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickols NG, Jacobs CS, Farkas ME, et al. Modulating hypoxia-inducible transcription by disrupting the HIF-1-DNA interface. ACS Chem Biol. 2007;2:561–71. doi: 10.1021/cb700110z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda H, Fukuda N, Ueno T, et al. Transcriptional inhibition of progressive renal disease by gene silencing pyrrole-imidazole polyamide targeting of the transforming growth factor-beta1 promoter. Kidney Int. 2011;79:46–56. doi: 10.1038/ki.2010.330. [DOI] [PubMed] [Google Scholar]
- Kang JS, Meier JL, Dervan PB. Design of sequence-specific DNA binding molecules for DNA methyltransferase inhibition. J Am Chem Soc. 2014;136:3687–94. doi: 10.1021/ja500211z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniguchi M, Fujiwara K, Nakai Y, et al. Inhibition of malignant phenotypes of human osteosarcoma cells by a gene silencer, a pyrrole-imidazole polyamide, which targets an E-box motif. FEBS Open Bio. 2014;4:328–34. doi: 10.1016/j.fob.2014.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M, Matsuda H, Wang L, et al. Pretranscriptional regulation of Tgf-beta1 by PI polyamide prevents scarring and accelerates wound healing of the cornea after exposure to alkali. Mol Ther. 2010;18:519–27. doi: 10.1038/mt.2009.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez PC, Frank SR, Wang L, et al. Genomic targets of the human c-Myc protein. Genes Dev. 2003;17:1115–29. doi: 10.1101/gad.1067003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermeking H, Rago C, Schuhmacher M, et al. Identification of CDK4 as a target of c-MYC. Proc Natl Acad Sci U S A. 2000;97:2229–34. doi: 10.1073/pnas.050586197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou CJ, Farkas ME, Tsai SM, et al. Small molecules targeting histone H4 as potential therapeutics for chronic myelogenous leukemia. Mol Cancer Ther. 2008;7:769–78. doi: 10.1158/1535-7163.MCT-08-0130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou CJ, O'Hare T, Lefebvre S, et al. Growth arrest of BCR-ABL positive cells with a sequence-specific polyamide-chlorambucil conjugate. PLoS One. 2008;3:e3593. doi: 10.1371/journal.pone.0003593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg T, Cohen SB, Desharnais J, et al. Small-molecule antagonists of Myc/Max dimerization inhibit Myc-induced transformation of chicken embryo fibroblasts. Proc Natl Acad Sci U S A. 2002;99:3830–5. doi: 10.1073/pnas.062036999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao EH, Fukuda N, Ueno T, et al. A pyrrole-imidazole polyamide targeting transforming growth factor-beta1 inhibits restenosis and preserves endothelialization in the injured artery. Cardiovasc Res. 2009;81:797–804. doi: 10.1093/cvr/cvn355. [DOI] [PubMed] [Google Scholar]
- Farkas ME, Li BC, Dose C, et al. DNA sequence selectivity of hairpin polyamide turn units. Bioorg Med Chem Lett. 2009;19:3919–23. doi: 10.1016/j.bmcl.2009.03.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arvanitakis L, Yaseen N, Sharma S. Latent membrane protein-1 induces cyclin D2 expression, pRb hyperphosphorylation, and loss of TGF-beta 1-mediated growth inhibition in EBV-positive B cells. J Immunol. 1995;155:1047–56. [PubMed] [Google Scholar]
- Wang YD, Dziegielewski J, Wurtz NR, et al. DNA crosslinking and biological activity of a hairpin polyamide-chlorambucil conjugate. Nucleic Acids Res. 2003;31:1208–15. doi: 10.1093/nar/gkg215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mateyak MK, Obaya AJ, Sedivy JM. c-Myc regulates cyclin D-Cdk4 and -Cdk6 activity but affects cell cycle progression at multiple independent points. Mol Cell Biol. 1999;19:4672–83. doi: 10.1128/mcb.19.7.4672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickols NG, Dervan PB. Suppression of androgen receptor-mediated gene expression by a sequence-specific DNA-binding polyamide. Proc Natl Acad Sci U S A. 2007;104:10418–23. doi: 10.1073/pnas.0704217104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Synold TW, Xi B, Wu J, et al. Single-dose pharmacokinetic and toxicity analysis of pyrrole-imidazole polyamides in mice. Cancer Chemother Pharmacol. 2012;70:617–25. doi: 10.1007/s00280-012-1954-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Perlman DM, Peterson MS, et al. Translation initiation factor 4E blocks endoplasmic reticulum-mediated apoptosis. J Biol Chem. 2004;279:21312–7. doi: 10.1074/jbc.M312467200. [DOI] [PubMed] [Google Scholar]
- Tu L, Liu Z, He X, et al. Over-expression of eukaryotic translation initiation factor 4 gamma 1 correlates with tumor progression and poor prognosis in nasopharyngeal carcinoma. Mol Cancer. 2010;9:78. doi: 10.1186/1476-4598-9-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CJ, Nasr Z, Premsrirut PK, et al. Targeting synthetic lethal interactions between Myc and the eIF4F complex impedes tumorigenesis. Cell Rep. 2012;1:325–33. doi: 10.1016/j.celrep.2012.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Doc. S1. Supplementary materials and methods.
Doc. S2. Reference list.
Fig. S1. Antiproliferative effect of Myc-5.
Fig. S2. Myc-5 localizes into nucleus.
Fig. S3. In vivo binding of Myc-5 at CDK4 gene promoter.
Fig. S4. Heat map represents the functional annotated transcripts that are identified by unpaired two-class significance analysis of microarray analysis with a false discovery rate of 0% from cells treated with DMSO (control), mismatch polyamide, and Myc-5.
Table S1. FITC-labeled oligomer sequences designed for EMSA
Table S2. Primers used in quantitative RT-PCR
Table S3. Primers used for ChIP real-time PCR
Table S4. Promoter-associated E-box genes
Table S5. Kinetic constant for Myc-5 pyrrole–imidazole polyamide and mismatch binding