

Abstract
Hepatocellular carcinoma (HCC) has high mortality and no adequate treatment. Endocannabinoids interact with hepatic CB1 receptors (CB1R) to promote hepatocyte proliferation in liver regeneration via inducing cell-cycle proteins involved in mitotic progression, including Forkhead Box M1 (FOXM1). Because FOXM1 is highly expressed in HCC and contributes to its genesis and progression, we analyzed the involvement of the endocannabinoid/CB1R system in murine and human HCC. Postnatal diethylnitrosamine (DEN) treatment induced HCC within 8 months in wild-type mice, but fewer and smaller tumors in CB1R−/− mice or in wild-type mice treated with the peripheral CB1R antagonist JD5037, as monitored in vivo by serial magnetic resonance imaging. Genome-wide transcriptome analysis revealed CB1R-dependent, tumor-induced upregulation of the hepatic expression of CB1R, its endogenous ligand anandamide, and a number of tumor promoting genes, including the GRB2 interactome as well as FOXM1 and its downstream target the tryptophan-catalyzing enzyme indoleamine 2,3-dioxygenase (IDO2). Increased IDO2 activity and consequent induction of immunosuppressive Treg cells in tumor tissue promote immune tolerance.
Conclusion
The endocannabinoid/CB1R system is upregulated in chemically induced HCC resulting in the induction of various tumor promoting genes, including IDO2, and attenuation of these changes by blockade or genetic ablation of CB1R suppresses the growth of HCC and highlights the therapeutic potential of peripheral CB1R blockade.
Liver cancer is the third most prevalent cancer with 695,000 deaths worldwide (1). The American Cancer Society’s estimates for 2013 are about 30,640 new cases of liver cancer in the United States and about 21,670 deaths from these cancers. (2). The 5-year survival rate of individuals with liver cancer is only 8.9% despite aggressive conventional therapy, marking this malignancy the second most lethal after pancreatic cancer (3). Hepatocellular carcinoma (HCC) is the most common type of liver cancer and it accounts for 4 out of 5 liver tumors. The most common underlying factor is cirrhosis caused by viral hepatitis, chronic alcoholism or obesity (4, 5), although HCC can also develop in the absence of cirrhosis. Most treatments are inadequate and there is also a lack of biomarkers for early diagnosis of HCC.
Diethylnitrosamine (DEN)-induced liver cancer in mice is the most widely used animal model of HCC, with a gene expression profile similar to that in a subset of human HCC with low survival (6). The near 100% effectiveness of DEN treatment to induce HCC with a reproducible time-course has made this model uniquely suited to analyze mechanisms of HCC.
Endocannabinoids are lipid mediators that interact with the same G protein-coupled receptors – CB1R and CB2R – that mediate the effects of the psychoactive ingredient of marijuana, Δ9-tetrahydrocannabinol. CB1R expression is very high in the brain and much lower yet functionally relevant in peripheral tissues including the liver, whereas CB2R expression is restricted primarily to immune and hematopoietic cells (7). Activation of hepatic CB1R stimulates de novo lipogenesis and induces hepatic insulin resistance (7, 8), and their expression is induced by chronic alcohol intake, high-fat diets and liver fibrosis (9–11), conditions known to predispose to cirrhosis and HCC. Hyperactivation of the biosynthesis of the endocannnabinoid anandamide and induction of CB1R following partial hepatectomy in mice contribute to the early stages of liver regeneration via inducing key cell cycle proteins involved in mitotic progression, such as FOXM1 (12). FOXM1 is also upregulated in HCC and contributes to tumor progression and various aspects of tumorogenesis (13), and CB1R is induced in HCC (14). Such observations suggest a functional link between the endocannabinoid/CB1R system and FOXM1 in the development and progression of HCC.
We explored the possible such a functional link by monitoring the development and growth of DEN-induced HCC in wild-type mice treated with vehicle or a peripheral CB1R antagonist as well as in CB1R−/− mice. Furthermore, we identified tumor-induced, CB1R-mediated changes in hepatic gene expression by analyzing the transcriptome of normal and cancerous liver tissue from individuals with HCC and from wild-type and CB1R−/− mice. The results highlight the role of CB1R in inducing multiple tumor promoting pathways, including the Gi/o/CREBP/FOXM1/Bin1/IDO2 (indoleamine 2,3-dioxygenase 2) pathway that promotes tumor immune tolerance.
Materials and Methods
Animals
Procedures were approved by the Institutional Animal Care and Use Committee and performed in accordance with the NIH Guide for the Care and Use of Laboratory Animals. C57BL/6J mice were from Jackson Laboratories. CB1R−/− and CB1R+/+ littermates were obtained by breeding heterozygotes that had been backcrossed to a C57BL/6J background (8). Mice were housed under 12 hour light-dark cycle, fed standard rodent chow and water ad libitum. Two week-old male CB1R+/+ and CB1R−/− mice were injected intraperitoneally with 25 mg/kg of diethylnitrosamine (DEN, Sigma) (15) and were subjected to MRI at 8 months to verify the presence and measure the size of the tumor. DEN-treated CB1R+/+ mice were then started on the peripheral CB1R antagonist JD5037 (3mg/kg/day by oral gavage) or vehicle for 8 weeks followed by a repeat MRI scan and then sacrificed for obtaining HCC and adjacent non-tumor tissue for in vitro analyses.
Human HCC Biopsy Samples
Paraffin embedded blocks of human HCC and normal liver samples were purchased from Capitol Biosciences, Liver Tissue Cell Distribution System, UMN and Matrix Biological, LLC (Washington, DC). Twelve sets of non-tumor and corresponding tumor samples were obtained, with the donors de-identified. All patients were free from hepatitis A, B or C infection.
In vivo Monitoring of HCC by MRI
Mice were anesthetized with 5% isoflurane in 30% oxygen/70% nitrogen. The isoflurane was then reduced to 1–2% and anesthesia was maintained via a nose cone. Core body temperature was monitored rectally by a fiberoptic probe (FISO™) and maintained by blowing heated air into the magnet bore (Bair Hugger™). Breath rate was kept at 35–45 bpm by adjusting the isoflurane level. MRI imaging was performed on a 7T Bruker Bioscan with a 9 cm shielded gradient system. Following an initial locator scan, a block of 16 consecutive, coronal planes covering the mouse liver were scanned using a RARE sequence with the following parameters: slice thickness 1mm, 6×4cm FOV, 256×256 matrix, RARE factor 4, 4 averages, and TR/TE 2000/10.3. Scanning was gated to the breathing cycle so that data were only acquired in between breaths. The captured images were analyzed and quantified using NIH ImageJ software. For each animal, all 16 coronal planes were subjected to morphometric analysis and the plane showing the highest relative area occupied by HCC tissue, expressed in pixels, was used for statistical analyses. Mean values and standard errors were calculated from measurements obtained from 15–30 mice per treatment group.
RESULTS
CB1R Modulate the Incidence and Progression of DEN-induced HCC in Mice
The presence and size of HCC in DEN-treated mice was assessed at 8 months of age by MRI. As illustrated in Fig. 1A/B, the cross-sectional area of tumor tissue was significantly higher in wild-type than in CB1R−/− liver. CB1R+/+ mice were then divided into two groups, one group receiving vehicle and the other 3 mg/kg/day of JD5037 (16) for 8 weeks. Two months later, the size of tumors was significantly smaller in JD5037-treated than in vehicle-treated mice (Fig. 1C/D). Thus, genetic ablation or pharmacological blockade of CB1R attenuates HCC progression.
Figure 1. Genetic or Pharmacological Ablation of CB1R Attenuates HCC Progression.

A. Representative MRI scans in DEN-treated, 8 months-old CB1R+/+ and CB1R−/− mice. B. Total tumor area in CB1R−/− and CB1R+/+ mice was assessed and statistically analyzed as described in Methods. * P<0.05 n=25–30 mice/group. C. Representative MRI scans from DEN-treated CB1R+/+ mice treated with JD5037, 3 mg/kg/day, or vehicle from 8 to10 months. D. Quantifications of the data in C was done as described for panel B. * P<0.05 vehicle vs JD5037 treatment, n=15–20 mice/group.
Activation of the Endocannabinoid/CB1R System in Murine and Human HCC
CB1R were readily detectable by immunohistochemistry in HCC from CB1R+/+ mice, but were barely detectable in the adjacent normal liver tissue (Figure 2A). The specificity of CB1R staining was verified by its elimination by blocking peptide or in livers from CB1R−/− mice. CB1R mRNA was similarly increased in HCC versus adjacent non-tumor tissue from vehicle-treated but not from JD5037-treated mice (Figure 2B). CB1R immunoblots revealed a similar pattern of change at the protein level (Fig. 2C). Additionally, tumors from CB1R+/+ mice contained significantly more anandamide (AEA) compared to non-tumor tissue, and AEA levels were attenuated in HCC samples from JD5037-treated mice. There was no difference in AEA content in non-tumor vs HCC tissue from CB1R−/− mice (Fig. 2D). Increased AEA in tumors is likely due to increased synthesis, as indicated by the increased expression of the AEA biosynthetic enzyme N-arachidonoyl phosphatidylethanolamide-specific phospholipase D (NAPE-PLD) in both mouse and human HCC tissue (Fig. S1).
Figure 2. The CB1R/Endocannabinoid System is Activated in Murine HCC.
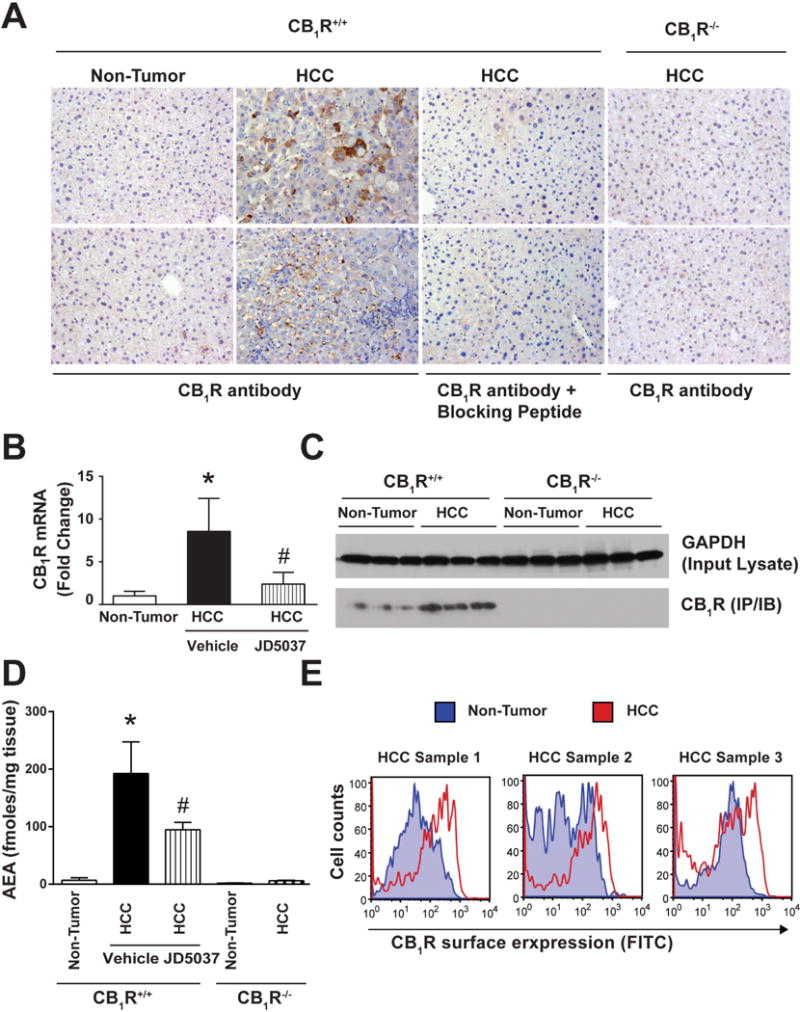
A. Immuno-histochemical analyses of CB1R in non-tumor and tumor area of mice liver. Blocking peptides and tumors from CB1R−/− mice were used as negative control. Induction of CB1R demonstrated by RT-PCR (B) and Western blotting (C). D. Induction of the AEA in tumor vs non-tumor tissue. *P<0.05; n=6–10 mice/group. E. Flow-cytometric measurements of CB1R surface expression in suspensions of single cells isolated from tumor and non-tumor region of CB1R+/+ mice.
CB1R activation contributed to increased cell proliferation in HCC versus non-tumor tissue, as deduced from a marked increase in Ki67 staining and its attenuation by CB1R but not by CB2R blockade (Fig. S2A), and from similar changes in in vivo BrdU uptake, quantified ex vivo by ELISA (Fig. S2B). No difference in AEA levels in DEN-treated vs control mice was detected in heart, kidney, brain and lung, except for a small increase in the spleen (Fig. S3). Tissue levels of the endocannabinoid 2-AG remained unchanged in DEN-treated vs control mice in all tissues including the liver (data not shown).
Human HCC had increased levels of immunoreactive CB1R relative to tumor-free areas (Figure 3A), which was confirmed by Western blots (Fig. 3B) and paralleled by increased CNR1 gene expression (Fig. 3C). Similar to mice, AEA content was higher in human HCC versus tumor-free tissue (Fig. 3D). The diagnosis of HCC in these samples was verified in H&E counterstained sections highlighting nuclear atypia, involving enlarged nuclei with irregular nuclear membranes and prominent nucleids as well as aberrant architecture (absence of sinusoids and portal areas), as illustrated in Fig. S4.
Figure 3. Induction of CB1R and AEA in Human HCC.

A. Immunohistochemical analyses of CB1R in non-tumor and tumor area of human liver samples. Blocking peptides were used as negative control, scale bar: 100 μm. CB1R protein detected by Western blot (B) and CNR1 expression quantified by RT-PCR (C). D. Increased AEA content in HCC vs. normal human liver tissue. *P<0.05 vs non-tumor tissue. n=8–12 subjects/group.
Transcriptome Profile of Human and Murine HCC
Gene expression profiles of tumor vs non-tumor tissue from four human HCC samples were obtained by RNAseq. The uniformly high percentage (86–94%) of matched reads documented the high quality of the sequencing data (Table S1). Induced genes were further screened for CB1R-dependent regulation by analyzing RNA sequencing data from pooled non-tumor and tumor liver tissue from CB1R+/+ and CB1R−/− mice and JD5037-treated CB1R+/+ mice, as detailed in Methods. CB1R expression itself was markedly induced in both human and mouse tumors, and the increase in mouse tumors was abrogated in animals treated with JD5037 (Fig. S5).
CB1R-dependent Induction of FOXM1 in Human and Murine HCC
Among the genes overexpressed in HCC in a CB1R-dependent manner, we further analyzed the regulation of FOXM1, a transcription factor induced in HCC (13). Immunofluorescent staining of FOXM1 protein in human HCC samples indicates induction and nuclear localization (Fig. 4A), which was further validated by real-time PCR and Western blotting (Fig. 4B and C). FoxM1 was also overexpressed in tumor samples from CB1R+/+ but not CB1R−/− mice (Fig. 4E and F). Nuclear localization of FoxM1 was observed in CB1R+/+ tumor tissue (Fig. 4D).
Figure 4. CB1R-dependent Up-regulation of FOXM1 in Murine and Human HCC.

A–C: Human HCC. A: Immunofluorescence analyses of FoxM1 in liver biopsy samples. Nuclear Stain (green) and anti-Foxm1 (red) were visualized with Sytox Green and Texas Red-conjugated secondary Abs, respectively. Tumor-induction of FOXM1 by RT-PCR (B) and immunoblot analyses (C). *P<0.05 tumor vs non-tumor; n=8–12/group. D–F: Murine HCC. D: Immunofluorescence analyses of FOXM1 in tumor vs. non-tumor areas of CB1R+/+ vs CB1R−/− liver. Nuclear Stain (red) and anti-Foxm1 (green) were visualized with PI and FITC-conjugated secondary Abs. Similar changes observed by RT-PCR (E) and immunoblot analyses (F). *P<0.05, tumor vs non-tumor; n=8–12/group.
The mechanism of CB1R-dependent FOXM1 expression was further analyzed in cultured Hepg2 liver tumor cells (Fig. 5A). FOXM1 was robustly induced by 1–5 μM AEA, which was abrogated by pertussis toxin, indicating the involvement of Gi/o-coupled signaling via cAMP. Because of the presence of a canonical cAMP response element (CRE) in the FOXM1 promoter (Fig. 5B), we tested the role of cAMP in the regulation of FOXM1 transcription using FOXM1 promoter/luciferase constructs. AEA at 1 μM markedly stimulated luciferase activity, blocked in the presence of 100 nM JD5037 and abrogated using a CRE deletion construct (Fig. 5B).
Figure 5. Mechanism of CB1R-Induced FOXM1 Expression.

A: FOXM1 protein in HepG2 cells is increased by anandamide (AEA) in the absence but not presence of pertussis toxin. Cells were incubated with 1–5 μM anandamide for 1 h either in the absence or presence of 100 ng/mL PTX. Cell extracts were analyzed by Western immunoblotting. B: FOXM1 promoter activity is increased by AEA in a CRE-dependent manner. Promoter activity was quantified using a luciferase reporter system. The site of deletion of a canonical CRE site is schematically illustrated.
A number of cell cycle-related genes, many of them downstream targets of FoxM1, were also induced in tumors in a CB1R-dependent manner, most prominently the cyclin-dependent kinase inhibitor 1a and 3 (Cdkn1a, Cdkn3), as well as factors promoting angiogenesis, such as vascular endothelial growth factor C (VegfC) and platelet-derived growth factor B (Pdgfb) (Table S2). As a functional correlate of these latter, angiogenesis was increased in CB1R+/+ HCC vs non-tumor tissue, as reflected by an increase in isolectin+ vascular endothelial cells (Fig. S6).
Additional genes induced in a CB1R-dependent manner included genes linked to tumor promotion, but also some with tumor inhibitory activity, as listed in Table S2. There were also genes down-regulated in HCC by a CB1R-dependent manner, and these also included both tumor promoters and tumor suppressors (Table S2).
CB1R-dependent Induction of the Tryptophan Catabolic Enzyme IDO2 in HCC
Two isoforms of the enzyme indoleamine 2,3-dioxygenase, IDO1 and IDO2, as well as tryptophan 2,3-dioxygenase (TDO) catalyze the breakdown of the essential amino-acid tryptophan, which leads to immune suppression, a well-documented pathway of immune tolerance in cancer (17). Based on its transcriptome profile, IDO2 was one of the most robustly induced genes in human HCC, whereas IDO1 and TDO expression remained unchanged. IDO2 induction in human HCC was confirmed by immunohistochemistry, immunoblots and real-time PCR (Fig. 6A–C). Ido2 protein and mRNA were also increased in HCC compared to tumor-free tissue from CB1R+/+ but not CB1R−/− mice (Fig. 6D,F,G). Hepatocyte-specific expression of Ido2 is indicated by its co-localization with albumin (Fig. 6E). HCC also increased Ido2 enzyme activity in wild-type but not in CB1R−/− mice (Fig. 7A). In primary cultured mouse isolated hepatocytes, 10 μM anandamide caused a 4-fold increase in Ido2 mRNA, blocked by 100 nM JD5037 and absent in hepatocytes from CB1R−/− mice, indicating CB1R involvement (Fig. 7B).
Figure 6. IDO2 is Up-regulated in Human and Murine HCC.
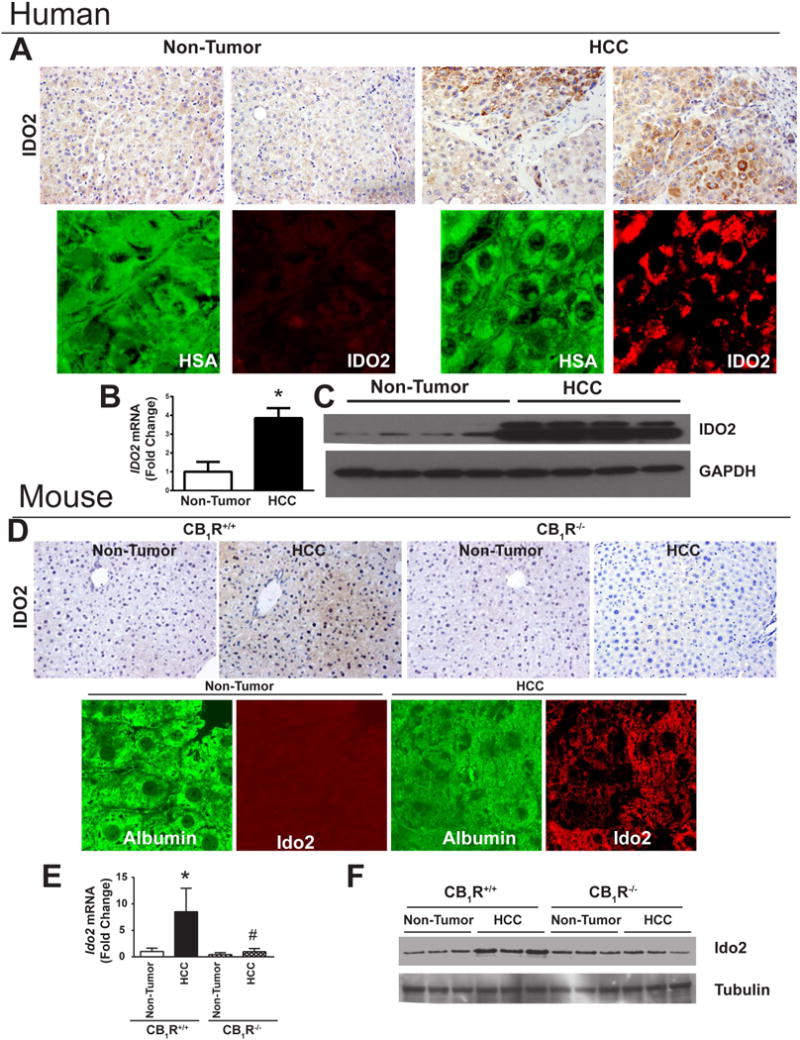
A–C: Human HCC. A: Immuno-histochemical analyses of IDO2 protein in tumor vs non-tumor liver tissue (top), with hepatocyte-specific expression of IDO2 indicated by colocalization with hepatocyte-specific antigen (HAS –green, IDO2 red). B: RT-PCR and C: immunoblot analyses of IDO2; *P<0.05 tumor vs non-tumor; n=8–12/group. D–F: Tumor and non-tumor tissue from CB1R+/+ and CB1R−/− mice were analyzed as described for human liver, *P<0.05 tumor vs non-tumor; n=8–12 mice/group.
Figure 7. CB1R- and FOXM1-dependent Induction of IDO2 Increases Treg Cells in HCC Tissue.

A. CB1R-dependent induction of Ido2 expression by AEA in mouse isolated hepatocytes. Ido2 mRNA was quantified by RT-PCR; *P<0.05 relative to vehicle; n=6/group B. IDO activity in tumor and non-tumor tissue. C. Chromatin immunoprecipitation (CHIP) indicates direct interaction of FOXM1 with IDO2 promoter in CB1R+/+ tumor samples. D. Representative flow-cytometry scatter plot indicates CB1R-mediated increase in CD4+/CD25+/Foxop3+ Treg cells in mouse liver 8 weeks following DEN-treatment. *P<0.05 vs vehicle; n=6/group.
The tumor suppressor amphyphysin 2 (Bin1) negatively regulates IDO2 expression (18). Bin1 was markedly down-regulated in both human and mouse HCC relative to tumor-free tissue (Fig. S7).
FOXM1 Regulates IDO2 which Promotes Tumor Growth by Altering Microenvironment
To test whether the CB1R-dependent induction of Ido2 is via FoxM1, we analyzed the interaction of FoxM1 with the Ido2 promoter in HCC and tumor-free tissue from CB1R+/+ and CB1R−/− mice. CHIP analyses using FoxM1 antibody followed by PCR of the IDO2 promoter region demonstrated a clear, CB1R-dependent regulation of Ido2 expression by FoxM1 in HCC (Fig. 7C). Cancer promotion by IDO is thought to be due to immunosuppression secondary to the breakdown of tryptophan in the tumor microenvironment, which renders effector T cells inactive and dendritic cells immunosuppressive. Indeed, immunosuppressive Treg cells identified by the surface markers CD4, CD25 and FoxP3 were upregulated as early as 8 weeks following DEN treatment in the liver of wild-type but not CB1R−/− mice (Fig. 7D).
Increased Ido2 activity contributes to tumor growth in mice
To test the contribution of increased Ido2 activity to tumor growth, 8-month old wild-type mice with DEN-induced HCC were treated for 6 weeks with the Ido2 inhibitor 1-methyl-L-tryptophan provided in the drinking water at 500 mg/kg/day. Vehicle-treated mice displayed marked progression of tumor growth, which was completely blunted in 1-methyl-L-tryptophan treated mice, as verified by repeat MRI scan at the end of treatment (Fig. S8).
CB1R-GRB2 Interaction in HCC
The GRB2 interactome is implicated in cancer development (19), and GRB2 is also associated with the progression of HCC (20). Transcriptome analyses of human HCC indicated up-regulation of the expression of GRB2 and its downstream target RAS, which was confirmed by Western blotting (Fig. S9A). A similar up-regulation was evident in HCC samples from CB1R+/+ but not from CB1R−/− liver (Fig. S9B). Furthermore, GRB2 interacted directly with CB1R, as indicated by their co-immunoprecipation (Fig. S9C). In addition, flow cytometric cell cycle analyses of mouse HCC cells with CB1R surface expression demonstrated a direct correlation between the level of CB1R expression and the number cells in S-phase (Fig. S9D). These data suggest that surface expression and activation of CB1R is a marker of dividing tumor cells.
DISCUSSION
Previous studies have documented tumor inhibitory effects of plant-derived cannabinoids and their analogs, which could be abrogated by CB1R blockade (21), although the alternative involvement of CB2R (22–24), TRPV1 receptors (25), or receptor-independent effects has also been reported (26). These tumor inhibitory effects occur invariably at micromolar concentrations, well above the CB1 binding constants of the compounds, and thus may be due to down-regulation rather than activation of CB1R. In contrast, endocannabinoids acting at nanomolar concentrations close to their Kd values for CB1R promote the proliferation of a variety of tumor cells, which suggests their involvement in promoting tumor growth (27), also supported by documented tumor inhibitory effects of CB1R antagonists, although off-target action of the antagonists could not be excluded (28). Here we have documented dramatic reductions in the size and number of chemically induced HCC in mice either by genetic ablation or pharmacological blockade of CB1R. This strongly supports the tumor-promoting role of endocannabinoids acting via hepatic CB1R, which are significantly overexpressed in both mouse and human HCC. These findings also agree with earlier observations that activation of hepatic CB1R by newly synthesized anandamide promotes hepatocyte proliferation in the early stages of liver regeneration (12). Reversal of the tumor-induced overexpression of both CB1R and anandamide by CB1R blockade is also in agreement with earlier findings in the steatotic (11, 29), diabetic (30) or regenerating liver (12), and is compatible with autoinduction of both the receptor and the synthesis of its endogenous ligand, a common feature of tumor promoting growth factors (31).
The strategy of analyzing the transcriptome of tumor and adjacent tumor-free liver tissue from both wild-type and CB1R−/− mice and from wild-type mice treated with a CB1R antagonist helped identifying tumor-related genes regulated by CB1R. The putative link between HCC and the genes thus identified was further validated using human HCC specimens. Among the genes up-regulated in HCC in a CB1R-dependent manner, both Cnr1 (the gene encoding CB1R itself) and FoxM1 are also upregulated in the regenerating liver (12). The hepatic level of anandamide was also markedly increased in the tumor, which support an increase in CB1R signaling.
The transcription factor FOXM1 is exclusively expressed in proliferating cells, where it activates the transcription of cell cycle proteins involved in G1/S and G2/M transition and is essential for M phase progression (13). FOXM1 is essential for the development of HCC (32), and silencing FOXM1 expression inhibits HCC growth (33). Here we show that tumor promotion by FOXM1 may also involve altering the tumor microenvironment through increased expression of IDO2. Direct regulation of IDO2 expression by FOXM1 is indicated by the binding of FOXM1 to the promoter region of IDO2, likely resulting in transcriptional activation. Increased IDO2 expression may additionally result from down-regulation of Bin1, a known transcriptional inhibitor of IDO2 (18), in both human and mouse HCC. In turn, CB1R up-regulates FOXM1 gene transcription via a Gi/o/cAMP/CREBP pathway as indicated by the anandamide-induced increase in FOXM1 expression in HepG2 cells and its abrogation by PTX or by deletion of a canonical CRE site from the FOXM1 promoter (Fig. 5).
The growth of HCC is facilitated by the diminished anti-tumor immune response of natural killer (NK) cells, which has been linked to increased expression and activity of Treg cells (34). Tryptophan depletion and/or the accumulation of tryptophan metabolites such as kynurenine is a well-established mechanism whereby tumors suppress the anti-tumor activity of NK cells by boosting the formation and activity of Treg cells (35). The observed increase in Treg cells in liver tumor tissue is compatible with tryptophan depletion and could contribute to suppression of an antitumor immune response. Of the three enzymes involved in tryptophan catabolism (IDO, IDO2 and TDO) only IDO2 was induced in HCC, which supports its key role in suppressing the anti-tumor immune response. Tumor promotion by IDO2 also involves increased angiogenesis (36), indicated by the CB1R-dependent increase in the expression of the pro-angiogenic genes Pdgfb and Vegfc and the parallel increase in vascular endothelial cells in HCC compared to non-tumor tissue. The CB1R-mediated induction of IDO2 contributes to the progression of DEN-induced HCC, as indicated by the inhibition of tumor growth by chronic in vivo treatment with the IDO inhibitor 1-methyl-L-tryptophan.
The role of proinflammatory immune cells and cytokines in HCC progression has been well documented (37, 38). Recently, a unique set of CD14+CTLA4+ dendritic cells isolated from peripheral blood of HCC patients was found to suppress CD4+ T cell proliferation in vitro via increased expression of interleukin (IL)-10 and IDO (39). Here we show that hepatocytes themselves express IDO2 that is increased in HCC or in normal hepatocytes via CB1R activation, and could thus contribute to the immune suppressive microenvironment in HCC.
In addition to the CB1R/Bin1/FOXM1/IDO2 interactome targeting immune suppressive and pro-angiogenic mechanisms, CB1R are involved in inducing a number of other tumor-related genes in HCC. Of these, the GRB2 interactome plays a critical role in regulating receptor tyrosine kinases, cell motility, and EGF-regulated pathways involved in cancer progression, invasion and metastasis (19, 40). As documented here, CB1R interacts directly with GRB2 in both HCC and tumor-free samples, and its downstream effector RAS, another key cell cycle regulator, is also increased in HCC in a CB1R-dependent manner. Interestingly, genes up-regulated in HCC included not only tumor promoter but also tumor inhibitory genes, and the reverse was also true regarding down-regulated genes, which included some tumor promoters (Table S2). While these latter changes may represent compensatory mechanisms, they may also account for the often contradictory data in the literature on tumor promoting or anti-tumor effects of cannabinoids.
In summary, the present findings reveal a tumor promoting function of the endocannabinoid/CB1R system in HCC via inducing a number of tumor promoting pathways, including immune suppression in the tumor microenvironment through activation of the FOXM1/Bin1/IDO2 interactome. These findings raise the possibility that peripheral CB1R blockade may have a role in the treatment of HCC.
Supplementary Material
Acknowledgments
We thank Dr. Avi Z. Rosenberg for tumor pathology assessment of liver sections and Dr. Raouf Kechrid for help with in vivo drug administrations.
Financial support: This work was supported by funds from the intramural research program of NIAAA.
Abbreviations
- HCC
hepatocellular carcinoma
- CB1R
Cannabinoid receptor 1
- DEN
diethylnitrosamine
- FOXM1
Forkhead Box M1
- IDO2
indoleamine 2,3-dioxygenase 2
- AEA
arachidonoyl ethanolamide or anandamide
- CREBP
cyclic AMP response element binding protein
Footnotes
Potential conflict of interest: nothing to report.
SUPPLEMENTAL INFORMATION
Additional Supporting Information may be found in the online version of this article at the publisher’s website
References
- 1.WHO. 2008 http://www.who.int/mediacentre/factsheets/fs297/en/
- 2.Spangenberg HC, Thimme R, Blum HE. Targeted therapy for hepatocellular carcinoma. Nat Rev Gastroenterol Hepatol. 2009;6:423–432. doi: 10.1038/nrgastro.2009.86. [DOI] [PubMed] [Google Scholar]
- 3.Hertl M, Cosimi AB. Liver transplantation for malignancy. Oncologist. 2005;10:269–281. doi: 10.1634/theoncologist.10-4-269. [DOI] [PubMed] [Google Scholar]
- 4.Farazi PA, DePinho RA. Hepatocellular carcinoma pathogenesis: from genes to environment. Nat Rev Cancer. 2006;6:674–687. doi: 10.1038/nrc1934. [DOI] [PubMed] [Google Scholar]
- 5.Morgan TR, Mandayam S, Jamal MM. Alcohol and hepatocellular carcinoma. Gastroenterology. 2004;127:S87–96. doi: 10.1053/j.gastro.2004.09.020. [DOI] [PubMed] [Google Scholar]
- 6.Fausto N, Campbell JS. Mouse models of hepatocellular carcinoma. Semin Liver Dis. 2010;30:87–98. doi: 10.1055/s-0030-1247135. [DOI] [PubMed] [Google Scholar]
- 7.Pacher P, Batkai S, Kunos G. The endocannabinoid system as an emerging target of pharmacotherapy. Pharmacol Rev. 2006;58:389–462. doi: 10.1124/pr.58.3.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu J, Zhou L, Xiong K, Godlewski G, Mukhopadhyay B, Tam J, Yin S, et al. Hepatic cannabinoid receptor-1 mediates diet-induced insulin resistance via inhibition of insulin signaling and clearance in mice. Gastroenterology. 2012;142:1218–1228. doi: 10.1053/j.gastro.2012.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jeong WI, Osei-Hyiaman D, Park O, Liu J, Batkai S, Mukhopadhyay P, Horiguchi N, et al. Paracrine activation of hepatic CB1 receptors by stellate cell-derived endocannabinoids mediates alcoholic fatty liver. Cell Metab. 2008;7:227–235. doi: 10.1016/j.cmet.2007.12.007. [DOI] [PubMed] [Google Scholar]
- 10.Teixeira-Clerc F, Julien B, Grenard P, Tran Van Nhieu J, Deveaux V, Li L, Serriere-Lanneau V, et al. CB1 cannabinoid receptor antagonism: a new strategy for the treatment of liver fibrosis. Nat Med. 2006;12:671–676. doi: 10.1038/nm1421. [DOI] [PubMed] [Google Scholar]
- 11.Jourdan T, Djaouti L, Demizieux L, Gresti J, Verges B, Degrace P. CB1 antagonism exerts specific molecular effects on visceral and subcutaneous fat and reverses liver steatosis in diet-induced obese mice. Diabetes. 2010;59:926–934. doi: 10.2337/db09-1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mukhopadhyay B, Cinar R, Yin S, Liu J, Tam J, Godlewski G, Harvey-White J, et al. Hyperactivation of anandamide synthesis and regulation of cell-cycle progression via cannabinoid type 1 (CB1) receptors in the regenerating liver. Proc Natl Acad Sci U S A. 2011;108:6323–6328. doi: 10.1073/pnas.1017689108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Alvarez-Fernandez M, Medema RH. Novel functions of FoxM1: from molecular mechanisms to cancer therapy. Front Oncol. 2013;3:30. doi: 10.3389/fonc.2013.00030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xu X, Liu Y, Huang S, Liu G, Xie C, Zhou J, Fan W, et al. Overexpression of cannabinoid receptors CB1 and CB2 correlates with improved prognosis of patients with hepatocellular carcinoma. Cancer Genet Cytogenet. 2006;171:31–38. doi: 10.1016/j.cancergencyto.2006.06.014. [DOI] [PubMed] [Google Scholar]
- 15.Naugler WE, Sakurai T, Kim S, Maeda S, Kim K, Elsharkawy AM, Karin M. Gender disparity in liver cancer due to sex differences in MyD88-dependent IL-6 production. Science. 2007;317:121–124. doi: 10.1126/science.1140485. [DOI] [PubMed] [Google Scholar]
- 16.Tam J, Cinar R, Liu J, Godlewski G, Wesley D, Jourdan T, Szanda G, et al. Peripheral cannabinoid-1 receptor inverse agonism reduces obesity by reversing leptin resistance. Cell Metab. 2012;16:167–179. doi: 10.1016/j.cmet.2012.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Prendergast GC. Immune escape as a fundamental trait of cancer: focus on IDO. Oncogene. 2008;27:3889–3900. doi: 10.1038/onc.2008.35. [DOI] [PubMed] [Google Scholar]
- 18.Muller AJ, DuHadaway JB, Donover PS, Sutanto-Ward E, Prendergast GC. Inhibition of indoleamine 2,3-dioxygenase, an immunoregulatory target of the cancer suppression gene Bin1, potentiates cancer chemotherapy. Nat Med. 2005;11:312–319. doi: 10.1038/nm1196. [DOI] [PubMed] [Google Scholar]
- 19.Zheng Y, Zhang C, Croucher DR, Soliman MA, St-Denis N, Pasculescu A, Taylor L, et al. Temporal regulation of EGF signalling networks by the scaffold protein Shc1. Nature. 2013;499:166–171. doi: 10.1038/nature12308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Carloni V, Mazzocca A, Pantaleo P, Cordella C, Laffi G, Gentilini P. The integrin, alpha6beta1, is necessary for the matrix-dependent activation of FAK and MAP kinase and the migration of human hepatocarcinoma cells. Hepatology. 2001;34:42–49. doi: 10.1053/jhep.2001.25224. [DOI] [PubMed] [Google Scholar]
- 21.Guzman M. Cannabinoids: potential anticancer agents. Nat Rev Cancer. 2003;3:745–755. doi: 10.1038/nrc1188. [DOI] [PubMed] [Google Scholar]
- 22.Sanchez C, Galve-Roperh I, Canova C, Brachet P, Guzman M. Delta9-tetrahydrocannabinol induces apoptosis in C6 glioma cells. FEBS Lett. 1998;436:6–10. doi: 10.1016/s0014-5793(98)01085-0. [DOI] [PubMed] [Google Scholar]
- 23.Galve-Roperh I, Sanchez C, Cortes ML, Gomez del Pulgar T, Izquierdo M, Guzman M. Anti-tumoral action of cannabinoids: involvement of sustained ceramide accumulation and extracellular signal-regulated kinase activation. Nat Med. 2000;6:313–319. doi: 10.1038/73171. [DOI] [PubMed] [Google Scholar]
- 24.Vara D, Salazar M, Olea-Herrero N, Guzman M, Velasco G, Diaz-Laviada I. Anti-tumoral action of cannabinoids on hepatocellular carcinoma: role of AMPK-dependent activation of autophagy. Cell Death Differ. 2011;18:1099–1111. doi: 10.1038/cdd.2011.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Maccarrone M, Lorenzon T, Bari M, Melino G, Finazzi-Agro A. Anandamide induces apoptosis in human cells via vanilloid receptors. Evidence for a protective role of cannabinoid receptors. J Biol Chem. 2000;275:31938–31945. doi: 10.1074/jbc.M005722200. [DOI] [PubMed] [Google Scholar]
- 26.Flygare J, Sander B. The endocannabinoid system in cancer-potential therapeutic target? Semin Cancer Biol. 2008;18:176–189. doi: 10.1016/j.semcancer.2007.12.008. [DOI] [PubMed] [Google Scholar]
- 27.Hart S, Fischer OM, Ullrich A. Cannabinoids induce cancer cell proliferation via tumor necrosis factor alpha-converting enzyme (TACE/ADAM17)-mediated transactivation of the epidermal growth factor receptor. Cancer Res. 2004;64:1943–1950. doi: 10.1158/0008-5472.can-03-3720. [DOI] [PubMed] [Google Scholar]
- 28.Pisanti S, Picardi P, D’Alessandro A, Laezza C, Bifulco M. The endocannabinoid signaling system in cancer. Trends Pharmacol Sci. 2013;34:273–282. doi: 10.1016/j.tips.2013.03.003. [DOI] [PubMed] [Google Scholar]
- 29.Mukhopadhyay B, Liu J, Osei-Hyiaman D, Godlewski G, Mukhopadhyay P, Wang L, Jeong WI, et al. Transcriptional regulation of cannabinoid receptor-1 expression in the liver by retinoic acid acting via retinoic acid receptor-gamma. J Biol Chem. 2010;285:19002–19011. doi: 10.1074/jbc.M109.068460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jourdan T, Godlewski G, Cinar R, Bertola A, Szanda G, Liu J, Tam J, et al. Activation of the Nlrp3 inflammasome in infiltrating macrophages by endocannabinoids mediates beta cell loss in type 2 diabetes. Nat Med. 2013;19:1132–1140. doi: 10.1038/nm.3265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim SJ, Angel P, Lafyatis R, Hattori K, Kim KY, Sporn MB, Karin M, et al. Autoinduction of transforming growth factor beta 1 is mediated by the AP-1 complex. Mol Cell Biol. 1990;10:1492–1497. doi: 10.1128/mcb.10.4.1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kalinichenko VV, Major ML, Wang X, Petrovic V, Kuechle J, Yoder HM, Dennewitz MB, et al. Foxm1b transcription factor is essential for development of hepatocellular carcinomas and is negatively regulated by the p19ARF tumor suppressor. Genes Dev. 2004;18:830–850. doi: 10.1101/gad.1200704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen T, Xiong J, Yang C, Shan L, Tan G, Yu L, Tan Y. Silencing of FOXM1 transcription factor expression by adenovirus-mediated RNA interference inhibits human hepatocellular carcinoma growth. Cancer Gene Ther. 2014;21:133–138. doi: 10.1038/cgt.2014.8. [DOI] [PubMed] [Google Scholar]
- 34.Cai L, Zhang Z, Zhou L, Wang H, Fu J, Zhang S, Shi M, et al. Functional impairment in circulating and intrahepatic NK cells and relative mechanism in hepatocellular carcinoma patients. Clin Immunol. 2008;129:428–437. doi: 10.1016/j.clim.2008.08.012. [DOI] [PubMed] [Google Scholar]
- 35.Prendergast GC, Smith C, Thomas S, Mandik-Nayak L, Laury-Kleintop L, Metz R, Muller AJ. Indoleamine 2,3-dioxygenase pathways of pathogenic inflammation and immune escape in cancer. Cancer Immunol Immunother. 2014 doi: 10.1007/s00262-014-1549-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nonaka H, Saga Y, Fujiwara H, Akimoto H, Yamada A, Kagawa S, Takei Y, et al. Indoleamine 2,3-dioxygenase promotes peritoneal dissemination of ovarian cancer through inhibition of natural killercell function and angiogenesis promotion. Int J Oncol. 2011;38:113–120. [PubMed] [Google Scholar]
- 37.Gao Q, Zhao YJ, Wang XY, Qiu SJ, Shi YH, Sun J, Yi Y, et al. CXCR6 upregulation contributes to a proinflammatory tumor microenvironment that drives metastasis and poor patient outcomes in hepatocellular carcinoma. Cancer Res. 2012;72:3546–3556. doi: 10.1158/0008-5472.CAN-11-4032. [DOI] [PubMed] [Google Scholar]
- 38.Leonardi GC, Candido S, Cervello M, Nicolosi D, Raiti F, Travali S, Spandidos DA, et al. The tumor microenvironment in hepatocellular carcinoma (review) Int J Oncol. 2012;40:1733–1747. doi: 10.3892/ijo.2012.1408. [DOI] [PubMed] [Google Scholar]
- 39.Han Y, Chen Z, Yang Y, Jiang Z, Gu Y, Liu Y, Lin C, et al. Human CD14+ CTLA-4+ regulatory dendritic cells suppress T-cell response by cytotoxic T-lymphocyte antigen-4-dependent IL-10 and indoleamine-2,3-dioxygenase production in hepatocellular carcinoma. Hepatology. 2014;59:567–579. doi: 10.1002/hep.26694. [DOI] [PubMed] [Google Scholar]
- 40.Lin CC, Melo FA, Ghosh R, Suen KM, Stagg LJ, Kirkpatrick J, Arold ST, et al. Inhibition of basal FGF receptor signaling by dimeric Grb2. Cell. 2012;149:1514–1524. doi: 10.1016/j.cell.2012.04.033. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.