

Abstract
Background
Phosphorylative desensitization of G-protein coupled receptors (GPCR) contributes significantly to post-myocardial infarction (MI) remodeling and heart failure (HF). Here, we determined whether adiponectin receptors 1 and 2 (AdipoR1/AdipoR2, the 7-transmembrane domain-containing receptors mediating adiponectin functions) are phosphorylatively modified and functionally impaired post-MI.
Methods and Results
Post-MI HF was induced by coronary artery occlusion. Receptor phosphorylation, kinase expression, and APN function were determined via in vivo, ex vivo, and in vitro models. AdipoR1/AdipoR2 are not phosphorylated in the normal heart. However, AdipoR1 was significantly phosphorylated post-MI, peaking at 7 days and remaining significantly phosphorylated thereafter. The extent of post-MI AdipoR1 phosphorylation positively correlated with the expression level of GPCR kinase 2 (GRK2), the predominant GRK isoform upregulated in the failing heart. Cardiac-specific GRK2 knockout virtually abolished post-MI AdipoR1 phosphorylation, whereas virus-mediated GRK2 overexpression significantly phosphorylated AdipoR1 and blocked APN metabolic-regulatory/anti-inflammatory signaling. Mass spectrometry identified Ser7, Thr24, and Thr53 (residues located in the n-terminal intracellular AdipoR1 region) as the GRK2 phosphorylation sites. Ex vivo experiments demonstrated AMPK activation and anti-TNFα effect of APN were significantly inhibited in cardiomyocytes isolated from non-ischemic area 7 days post-MI. In vivo experiments demonstrated that acute APN administration-induced cardiac GLUT4 translocation and eNOS phosphorylation was blunted 7 days post-MI. Continuous APN administration beginning 7 days post-MI failed to protect the heart from adverse remodeling and HF progression. Finally, cardiac-specific GRK2 knockdown restored APN cardioprotective effect.
Conclusions
AdipoR1 is phosphorylatively modified and desensitized by GRK2 in failing cardiomyocytes, contributing to post-MI remodeling and HF progression.
Keywords: Adipokine, Heart Failure, Phosphorylation, Receptor
Cardiac disease remains a leading cause of mortality worldwide. Although improved reperfusion strategies have led to declined death rates after acute myocardial infarction (AMI), both incidence and prevalence of post-MI heart failure (HF) have continually increased in recent years1. Despite pharmacological advances (e.g. β-blockers, ACE inhibitors), post-MI HF mortality remains very high, with 5-year rates being 30–70%. Defining molecular mechanisms leading to the transition from adaptive to maladaptive remodeling in the post-MI heart, and identifying novel therapeutic strategies capable of blocking/reversing this transition, are therefore in great need.
The capacity of cardiomyocytes to utilize fatty acids and glucose as substrates for energy metabolism is disrupted in chronic HF, resulting in an “energy crisis” significantly contributing to HF progression2. Approaches targeting more efficient substrate use and preserving cardiac metabolism are increasingly recognized as effective therapeutic strategies against HF3. Adiponectin (APN) is a novel adipokine regarded as “adipocyte-derived insulin”, and is an essential molecule maintaining insulin responsiveness4. It increases glucose and free fatty acid utilization, stimulates mitochondrial biogenesis, and inhibits inflammatory response4. Reduced APN levels are correlated with increased AMI risk5, as well as worse cardiac functional recovery after MI with reperfusion6, 7. However, the role of APN in HF is controversial. Despite clear experimental evidence demonstrating APN deficiency significantly exacerbates HF progression8–10, several clinical observations demonstrate hyperadiponectinemia is associated with poor cardiac function and increased mortality in these patient populations11–13. In the skeletal muscle and livers of HF patients, APN biological responses are blunted, suggesting hyperadiponectinemia in severe HF patients is a compensatory protective mechanism countering “APN resistance” development14–16. However, molecular mechanisms responsible for impaired APN signaling remain unclear. Moreover, whether the metabolic regulatory and protective effect of APN is impaired in failing cardiomyocytes themselves, thus directly contributing to HF progression remains unknown.
The adiponectin receptors (AdipoR1 and AdipoR2) belong to a new family of membrane receptors (PAQR) predicted to contain seven transmembrane domains, similar to G protein coupled receptors (GPCRs), but are structurally and topologically distinct17. APN binds the C-terminal extracellular domain of AdipoR, whereas the N-terminal cytoplasmic domain interacts with multiple intracellular molecules and transfers APN signaling. It is well recognized that GPCRs, such as the β-adrenergic receptors (β-AR), are phosphorylated in the failing heart, leading to receptor desensitization, contributing to HF progression18. However, whether AdipoRs are phosphorylated in the failing heart, impairing APN signaling and cardiometabolism, have not been previously investigated.
Therefore, the aims of the current study were 1) to determine whether adiponectin receptors are subjected to phosphorylative modification, and if so, 2) to identify the responsible molecular mechanisms and resultant functional significance.
Materials and Methods
All experiments were performed in adherence to the National Institutes of Health Guidelines on the Use of Laboratory Animals, and were approved by the Thomas Jefferson University Committee on Animal Care. All authors had full access to, and take full responsibility for data integrity. All authors have read and agreed to the manuscript as written.
In vivo Experimental Protocol
Adult male C57/BL6 mice or cardiomyocytes-specific GRK2 knockout mice19 were anesthetized with 2% isoflurane. MI was induced via left anterior descending (LAD) coronary artery ligation as previously described20. Sham-operated control mice (Sham) underwent the same surgical procedures except the suture placed under the LAD was not tied. Animals were re-anesthetized at various time periods after MI. Hearts were removed for immunohistological, biochemical, and Western blot assays as we previously reported20.
To determine the effect of APN treatment upon cardiac function and remodeling, animals were monitored for up to 8 weeks post-MI. At the end of the 8 week observation period, surviving animals were reanesthetized with isoflurane. Two-dimensional echocardiographic views of the mid-ventricular short axis were obtained at the level of the papillary muscle tips below the mitral valve (Vevo 2100, VisualSonic, Toronto, Canada). LV ejection fraction (LVEF) was calculated as previously described20.
To specifically inhibit cardiac GRK2 expression, GRK2 or scramble siRNA (Integrated DNA Technologies, Coralville, IW) was directly injected into the left ventricular wall as we previously reported21. siRNAs were diluted in 5% glucose and mixed with in vivo-jet PEI (Genesee Scientific, San Diego, CA). The heart was exposed via left thoracotomy at the fifth intercostal space. 20 μl GRK2-specific siRNAs (dose 0.8 μg/μl) or scramble control was delivered via three separate intramyocardial injections (by 32G needle), temporarily blanching the left ventricular free wall. 48 hours after siRNA injection, MI was induced as described above. One week after MI, mice were randomized to receive vehicle or APN treatment (0.25 μg/g/day intraperitoneal infusion via mini-osmotic pump, ALZET® DURECT Corporation, Cupertino, CA)20. 7 days after continuous APN infusion, cardiac function was determined by echocardiography, and GLUT4 membrane translocation was determined by Western blot.
Co-Immunoprecipitation
1, 3, 7, or 14-days post-MI, cardiectomy was performed. Cardiac tissue in remote, non-ischemic regions was isolated and homogenized. Supernatant was immunoprecipitated with antibody against AdipoR1 (Bioss, Inc., Woburn, MA) or AdipoR2 (Adipogen Corp., San Diego, CA). Samples were separated on SDS-PAGE gels, transferred to PVDF membranes, and incubated with primary antibodies (anti-PThr or anti-PSer, Cell Signaling, Danvers, MA), followed by horseradish peroxidase-conjugated secondary antibody. Blots were developed via Supersignal Chemiluminescence detection kit (Pierce, Rockford, Ill). Bands were visualized with a Kodak Image Station 4000-Pro (Rochester, NY). The blot densities were analyzed with Kodak 1D software (version 3.6).
Immunoblotting
At the end of the observation period specified in the results, hearts were removed and homogenized. Samples were separated on SDS-PAGE gels, transferred to nitrocellulose membranes, and Western blotted with antibody against AdipoR1, AdipoR2, T-cadherin, GRK2, GRK3, GRK5, AMPK, pAMPK, eNOS, peNOS, GLUT4, Gβ, gp91phox, iNOS, and GAPDH (Cell Signaling). Nitrocellulose membranes were then incubated with HRP-conjugated second antibody (1:2000, Cell Signaling) for 1 hour. Blots were developed and visualized as described above.
Histological Analysis
8 weeks post-MI, surviving animals were anesthetized. Cardiectomy was performed. Hearts were fixed with 4% paraformaldehyde, embedded in paraffin, transversely sectioned (6 μm thick), and mounted upon glass slides. Five sections per heart were stained with Masson trichrome, antibody against PECAM1 (Cell Signaling), or terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling kit (TUNEL, Roche Co., U.S.A.). Slides were examined by an Olympus IX51 fluorescent microscope, and 5 images per slide were captured by a Q-Imaging camera controlled by IP Lab 4.0 software. Cardiac collagen deposition (in remote non-infarcted regions), capillary density (in the infarct border zone), and cardiomyocyte apoptosis (in remote non-infarcted regions) were determined as previously reported20. Results from all slides obtained from the same heart were averaged, and counted as n=1.
Adenoviral Infection
H9C2 cardiac myoblast cells were infected with adenoviral vectors containing cDNAs for GRK2 using a multiplicity of infection of 1000 viral particles per cell (20 infectious units per well). Efficiency of adenoviral gene transfer was monitored 48 hours later by Western blot. Cells infected with adenovirus containing empty vectors served as control. Infected cells were treated with either vehicle or APN (2 μg/ml22) for 60 minutes. Effect of GRK2 overexpression upon APN-induced AMPK phosphorylation and GLUT4 membrane translocation were determined. To determine the effect of GRK2 overexpression upon APN anti-oxidative and anti-nitrative action, control or GRK2-overexpressing cells were treated with rhTNFα (10 ng/ml23) in the presence and absence of APN (2 μg/ml). Cells were collected 8 hours after TNFα treatment; oxidative/nitrative stress were determined by gp91phox and iNOS expression.
Adult Cardiomyocytes
1, 3, or 7 days after MI, hearts were removed and perfused via a Langendorff system for ~3 minutes with a Ca2+-free bicarbonate-based buffer. Enzymatic digestion was initiated by adding collagenase type B/D to the perfusion solution. When the hearts became swollen and hard after digestion (~3 minutes), 50 μM Ca2+ was added to the enzyme solution. Approximately 7 minutes later, the left ventricle was removed, and the ischemic region was dissected away and discarded. Cardiomyocytes from the non-ischemic left ventricular wall were isolated as we previously reported22, and plated at 0.5–1 × 104 cells/cm2 in mouse laminin pre-coated culture dishes. This technique purifies isolated cardiomyocytes >95%. The cellular survival rate is >80% at 12 hours, and exceeds 65% after 24 hours of isolation. After 1 hour of culture in a 5% CO2 incubator at 37°C, cardiomyocytes were treated with either vehicle or APN (2 μg/ml22) for 60 minutes. APN-induced AMPK phosphorylation was determined. To determine the effect of MI upon APN anti-oxidative and anti-nitrative action in cardiomyocytes from non-ischemic remote regions, cells were treated with rhTNFα (10 ng/ml23) in the presence and absence of APN (2 μg/ml). Cells were collected 8 hours after TNFα treatment and oxidative/nitrative stress were determined by gp91phox and iNOS expression, superoxide production (lucigenin-enhanced chemiluminescence), and nitrotyrosine content (ELISA)22.
In vitro kinase assay using purified GST-AdipoR1
A GST-AdipoR1 fusion protein consisting of human AdipoR1 (full length or N-terminal cytosolic domain: amino acid residues 1–136) served as the substrate for reconstitution kinase assays. Phosphorylation reactions were conducted in a 50μl reaction volume in kinase buffer containing 25mM Tris (pH7.5), 10mM MgCl2, 2mM DTT, 5mM β-glycerophosphate, and 0.1mM Na3VO4 and 0.2mM ATP. GRK2-stimulated phosphorylation was initiated by adding 250ng purified GRK2 (Invitrogen) and 100ng of purified Gβγ complex (Calbiochem), to ensure maximal GRK2 activation. Reactions commenced for 40 minutes at 30°C, and were terminated by adding 6x sample loading dye (10μl). Samples were heated for 5 minutes at 95°C, mixed, and centrifuged to pellet agarose beads. 30μl of each reaction mixture was loaded onto a 4–20% Tris-Glycine gel. After electrophoresis, all proteins were transferred to nitrocellulose for Western blot.
Statistical analysis
Data were analyzed with GraphPad Prism-6 statistic software (La Jolla, CA). For cardiac function, cell death, superoxide and peroxynitrite formation, one way ANOVA was conducted across all investigated groups first. Post hoc pairwise tests for certain group pairs with assessment of statistical significance were performed after Bonferroni correction of the overall significance level. Values are presented as the mean±SEM of n independent experiments. Homoscedasticity was determined by Barlett’s test, and sample distribution was determined by the D’Agostino-Pearson omnibus normality test. For Western blot densities (n<10/group), data were analyzed by the Kruskal-Wallis test followed by the Dunn post hoc test. Values are summarized as median. P values less than or equal to 0.05 were considered statistically significant.
Results
AdipoR1 is Significantly Phosphorylated in the Failing Heart
Recent clinical studies suggest reduced AdipoR1 expression in skeletal muscle might be responsible for APN resistance in HF patients14. To determine whether APN receptor expression is downregulated in the failing heart (thus impairing APN cardiac signaling during HF), AdipoR1, AdipoR2, and T-cadherin expression levels were determined in cardiomyocytes isolated from control and MI mice. As illustrated in Figure 1A, AdipoR1/AdipoR2 and T-cadherin expressions were unchanged until 4 weeks post-MI, suggesting that altered receptor expression in cardiomyocytes might be responsible for impaired APN signaling in late HF stages, but not during early HF development.
Figure 1.
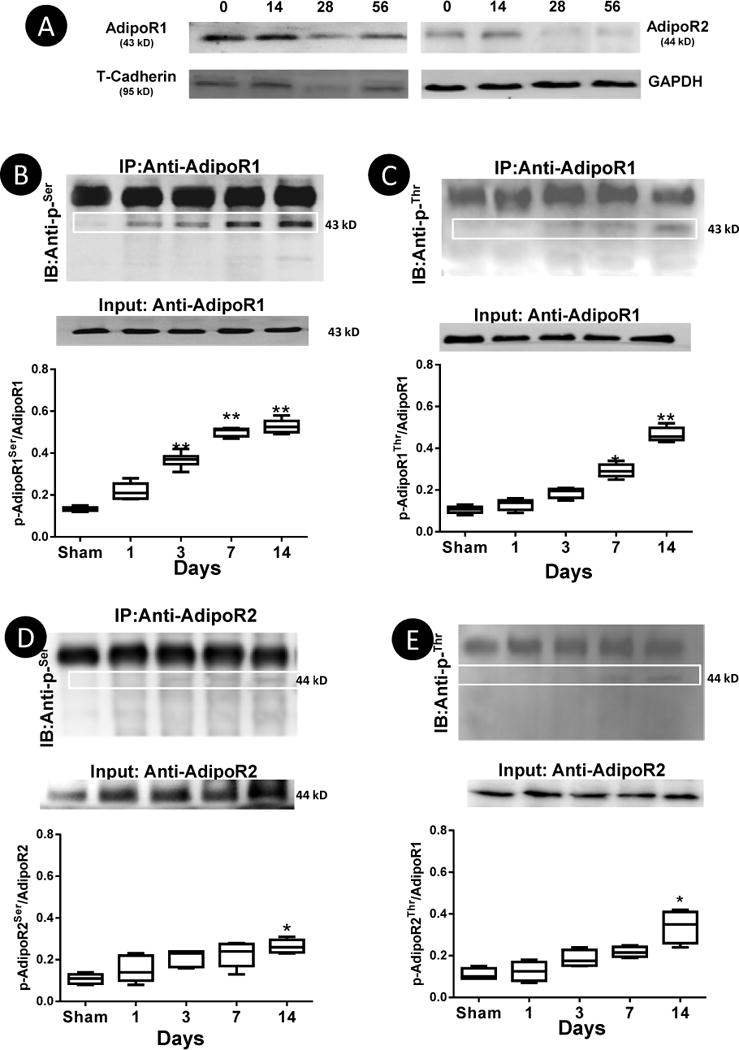
AdipoR1/AdipoR2 and T-cadherin expression are unchanged 7 and 14 days post-MI but significantly reduced 28 days and thereafter (A); AdipoR1 phosphorylation was detected with Anti-pSer (B) and Anti-pThr (C) during HF (days post-MI). AdipoR2 phosphorylation was detected post-MI but to significantly lesser extent than AdipoR1 (D/E). N=5–7/group. *P<0.05, **P<0.01 vs. Sham.
It is well recognized that various GPCRs are rapidly phosphorylated at intracellular C-terminal region during HF development18. Bioinformatical analysis revealed multiple phosphorylation sites in the AdipoR1 and AdipoR2 N-terminal intracellular region. To determine whether AdipoR1 and AdipoR2 might be phosphorylated prior to the reduction of their expression level post-MI, cardiomyocytes from non-ischemic regions were isolated 1, 3, 7, and 14 days post-MI. As illustrated in Figure 1, no receptor phosphorylation at either serine or threonine residue was detected in cardiac tissue from control animals. However, AdipoR1-pSer was detectable as early as 1 day post-MI, becoming statistically significant 3 days post-MI, peaking 7 days post-MI, and remaining elevated thereafter (Figure 1B). AdipoR1 phosphorylation at the threonine residue was also detected post-MI (Figure 1C). However, significant phosphorylation at this site occurred at a later time point (7 days post-MI). AdipoR2 phosphorylation at both serine and threonine sites were observed post-MI, but to markedly lesser extent than AdipoR1 at all-time points observed (Figure 1D/E). This is likely due to lower AdipoR2 expression level in cardiomyocytes, as well as fewer phosphorylation sites predicted in AdipoR2 receptor. Moreover, APN’s activation of AMPK and phosphorylation of eNOS were virtually abolished in cardiomyocytes isolated from AdipoR1 knockout mice, but largely preserved in cardiomyocytes isolated from AdipoR2 knockout mice (Supplemental Figure 1). Based upon the results that 1) AdipoR1 phosphorylation at the serine residue occurred earlier and persisted throughout during the entire observation period, 2) AdipoR1 phosphorylation is much stronger than AdipoR2, and 3) AdipoR1 knockout cardiomyocytes exhibited stronger APN signaling blockade, the current study focused upon AdipoR1 phosphorylation at the serine residue (AdipoR1-pSer).
GRK2 is Largely Responsible for AdipoR1 Phosphorylation
Among 6 potential phosphorylation sites located in the N-terminal intracellular region of AdipoR1, 5 were predicted as GRK sites. The expression levels of GRK2, GRK3, and GRK5 were determined because these are the most biologically important in the adult heart. Significant GRK2 expression was observed 1 day post-MI, peaking (3-fold increase) 3 days post-MI (A time point when significant AdipoR1 phosphorylation was detected, Figure 2A) and remains significantly elevated 7 days post-MI. GRK3 and GRK5 were modestly increased 3 days post-MI, with no significant difference observed 7 days post-MI (Figure 2B/C). Significant positive correlation between GRK2 expression levels and AdipoR-pSer levels were observed in cardiac tissue samples collected 1, 3 and 7 days post-MI (Figure 2D). These results suggest GRK2 is likely responsible for post-MI AdipoR1 phosphorylation.
Figure 2.
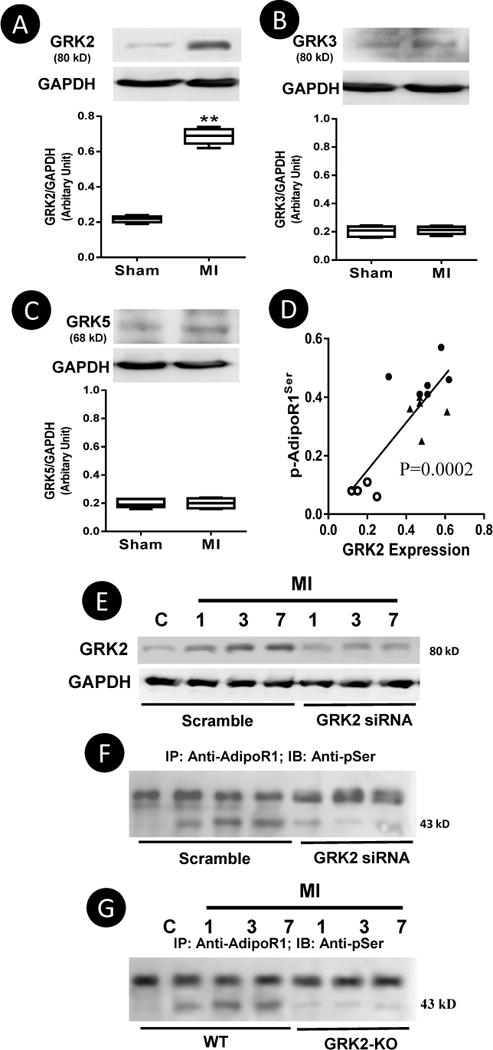
Expression of GRK2, GRK3, and GRK5 in cardiomyocytes isolated from non-ischemic area 7 days post-MI (A–C). Spearman’s rank correlation identified a significant positive correlation between GRK2 expression level and AdipoR1 phosphorylation 1, 3 and 7 days post-MI (D). N=5/group. **P<0.01 vs. Sham. Intramyocardial injection of siRNA effectively inhibited GRK2 overexpression Post-MI (E) and blocked post-MI AdipoR1 phosphorylation (F). Cardiomyocyte-specific GRK2 deletion blocked AdipoR1 phosphorylation (G). Representative blots from 5 repeated experiments.
To obtain direct evidence supporting GRK2 is responsible for post-MI AdipoR1 phosphorylation, cardiac GRK2 expression was genetically inhibited either by intramyocardial siRNA injection (48 hours prior to MI) or cardiomyocyte specific GRK2 deletion19. As illustrated in Figure 2E, GRK2 expression remains significantly inhibited 7 days post-MI in GRK2 siRNA group. More importantly, siRNA mediated GRK2-knockdown (Figure 2F) or cardiomyocyte-specific GRK2 knockout (Figure 2G) markedly inhibited post-MI AdipoR1 phosphorylation.
GRK2 Overexpression Caused AdipoR1 Phosphorylation and Blocked APN Biological Function
To determine whether GRK2-induced AdipoR1 phosphorylation may inhibit APN biological function, several in vitro experiments were performed. H9C2 cardiac myoblast cells were transfected with control or GRK2 expression vectors, and the consequent effect upon AdipoR1 phosphorylation and APN-mediated metabolic-regulation and anti-inflammatory functions were determined. As illustrated in Figure 3A, GRK2 overexpression caused significant AdipoR1 phosphorylation. More importantly, APN metabolic-regulatory signaling (AMPK phosphorylation and GLUT4 translocation, Figure 2B/C) and anti-inflammatory signaling (TNFα-induced NADPH oxidase and iNOS expression, Figure 3D/E) were significantly blunted in cells overexpressing GRK2.
Figure 3.
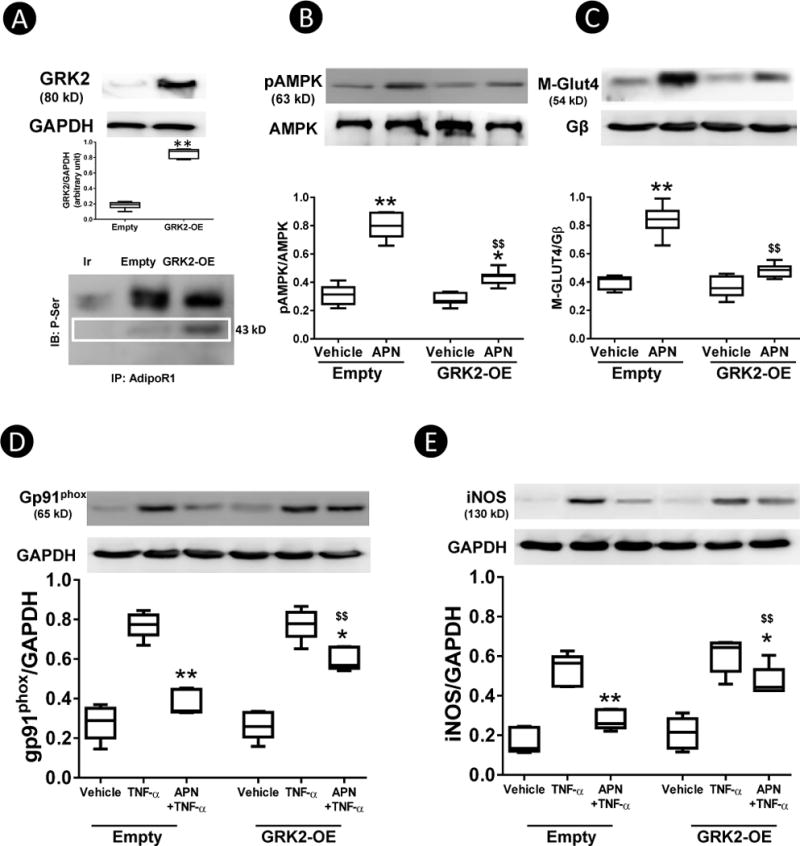
H9C2 cells were transfected with either empty or GRK2 expressing vector. 48 hours after transfection, GRK2 expression and AdipoR1 phosphorylation (A), APN-induced AMPK phosphorylation (B), membrane GLUT4 translocation (C), APN inhibition of TNFα-induced gp91phox (D), and iNOS expression (E) were determined. N=12–14 wells from at least 3 different experiments. *P<0.05, **P<0.01 vs. respective vehicle (B,C,), *P<0.05, **P<0.01 vs. TNFα alone (D, E); $$P<0.01 vs. Empty with same treatment.
AdipoR1 is Directly Phosphorylated by GRK2
To determine whether AdipoR1 is a direct substrate for GRK2, an in vitro kinase assay was performed as we previously reported24. In the absence of GRK2, very low levels of AdipoR1 phosphorylation were detected. However, adding purified GRK2 markedly increased AdipoR1 phosphorylation (Figure 4A). Bioinformatical analysis predicted 6 phosphorylation sites in the N-terminal domain. To further identify the specific residue(s) phosphorylated by GRK2, GST-tagged N-terminal domain of human AdipoR11–136 was incubated with purified GRK2. Mass spectrophotometric analysis was performed. As illustrated in Figure 4C/D/E, serine7, threonine24, and threonine53 were phosphorylated when purified GRK2 was added. To obtain more evidence these residues are responsible for AdipoR1 phosphorylation by GRK2, GST-tagged Ser/Thr-mutated AdipoR1 (AdipoR11–136S7AT24,53A) were generated, utilizing previously reported methods25. As illustrated in Figure 4B, AdipoR11–136S7AT24,53A is not phosphorylated by GRK2. These results provided evidence that AdipoR1 is a direct substrate for GRK2, and Ser7, Thr24 and Thr53 (located in the N-terminal AdipoR1 domain) are sensitive to GRK2 phosphorylation.
Figure 4.
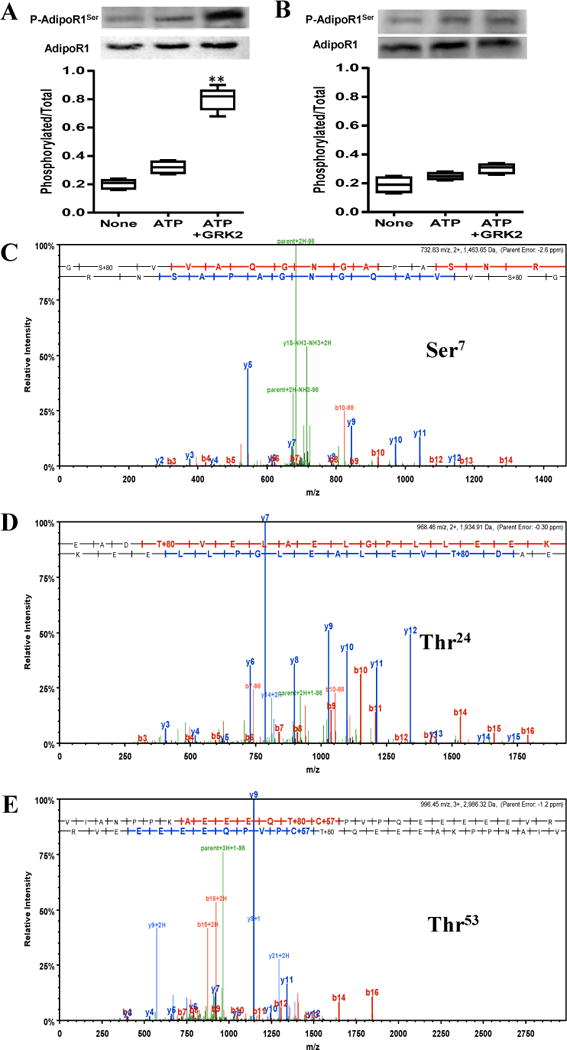
GST-tagged WT AdipoR11–136 (A) or AdipoR11–136S7AT24,53A (B) was incubated in the presence/absence of purified GRK2. Resultant phosphorylation was detected by Western blot. MS analysis revealed Ser7(C), Thr24(D), and Thr53(E) are phosphorylated when incubated with GRK2. N=5–7/group. **P<0.01 vs. ATP group.
Cardioprotective Response to APN is Impaired during the Early Developmental Phase of Post-MI HF
The results presented above demonstrate cardiac AdipoR1 is maximally phosphorylated 7–14 days post-MI and GRK2 overexpression-induced AdipoR1 phosphorylation blocks APN biological function in cultured cells. To determine whether cardiac AdipoR1 phosphorylation in the failing heart may impair APN cardiac biological function, several ex vivo and in vivo experiments were performed. First, left ventricular cardiomyocytes were isolated from normal or MI mice 1, 3, and 7 days post-MI (remote non-ischemic area) and treated with either globular domain APN (gAPN, 2 μg/mL) or full length APN (fAPN, 10 μg/ml). Compared to normal cardiomyocytes, APN-induced AMPK phosphorylation was attenuated in cardiomyocytes from MI mice as early as 1 day post-MI, becoming statistically significant 3 days post-MI, and reaching levels only 50–55% of control 7 days post-MI (Figure 5). Importantly, APN-stimulated AMPK activation (Figure 5G) and GLUT4 membrane translocation (Figure 5I) were preserved in cardiomyocytes isolated from cardiac-specific GRK2 knockout mice subjected to 7 days of MI. Because cardiomyocyte response to both gAPN and fAPN was impaired in the failing heart, subsequent experiments were all performed with gAPN because of its potency. Second, cardiomyocytes isolated from control and MI hearts (at 7 days) were treated with TNFα in the presence and absence of gAPN. The effect of gAPN upon TNFα-induced inflammatory response was determined 8 hours after treatment. As summarized in Figure 6A–D, the anti-inflammatory action of gAPN was significantly attenuated in cardiomyocytes isolated from the failing heart. gAPN markedly inhibited TNFα-induced superoxide production and nitrotyrosine formation in normal cardiomyocytes. However, the suppressive effect of gAPN upon TNFα-induced superoxide production and nitrotyrosine formation was significantly decreased (−34.2% vs. −78.3%, −32.8% vs. −60.7% respectively, P<0.01) in cardiomyocytes from the failing heart (Figure 6A–D). It is noteworthy TNFα significantly upregulated GRK2 expression in cardiomyocytes isolated from non-ischemic control animals (Figure 6A), and further enhanced GRK2 expression in cardiomyocytes isolated from ischemic animals (Figure 6B). Co-treatment with APN significantly inhibited TNFα-induced GRK2 expression in control cardiomyocytes (Figure 6A) but not in cardiomyocytes from ischemic animals (Figure 6B). These results provide insight into the regulatory roles pro- and anti-inflammatory adipokines play concerning GRK2 expression. Adiponectin’s anti-inflammatory role counters TNFα’s pro-inflammatory deleterious function in normal cells, a balance that is disrupted in the ischemic heart due to GRK2-induced AdipoR1 phosphorylation. Third, we determined GLUT-4 translocation and eNOS phosphorylation in response to acute gAPN administration (2 μg/g, intraperitoneal, IP) in control (sham MI) or MI mice (7 days post-MI) in vivo. In control mice, gAPN caused significant cardiac GLUT4 translocation and eNOS phosphorylation. These biological responses were markedly blunted 7 days post-MI (Figure 6E/F). Finally, to determine the long-term cardioprotective effect of chronic gAPN administration, animals were subjected to implantation of abdominal osmotic pumps (capable of gAPN delivery). In opposition to numerous previous studies demonstrating supplementation of gAPN either before or shortly after MI (at a similar dose employed in the current study) significantly protects the heart from MI injury5–7, 26–28, gAPN administration beginning 7 days post-MI failed to protect the heart. Specifically, no significant difference in cardiac function (Figure 7A), cardiac fibrosis (Figure 7B), capillary formation (Figure 7C), or cardiomyocyte apoptosis (Figure 7D) was observed between the vehicle and gAPN treated groups. No difference was observed in infarct size between the vehicle and gAPN treated groups (data not shown).
Figure 5.
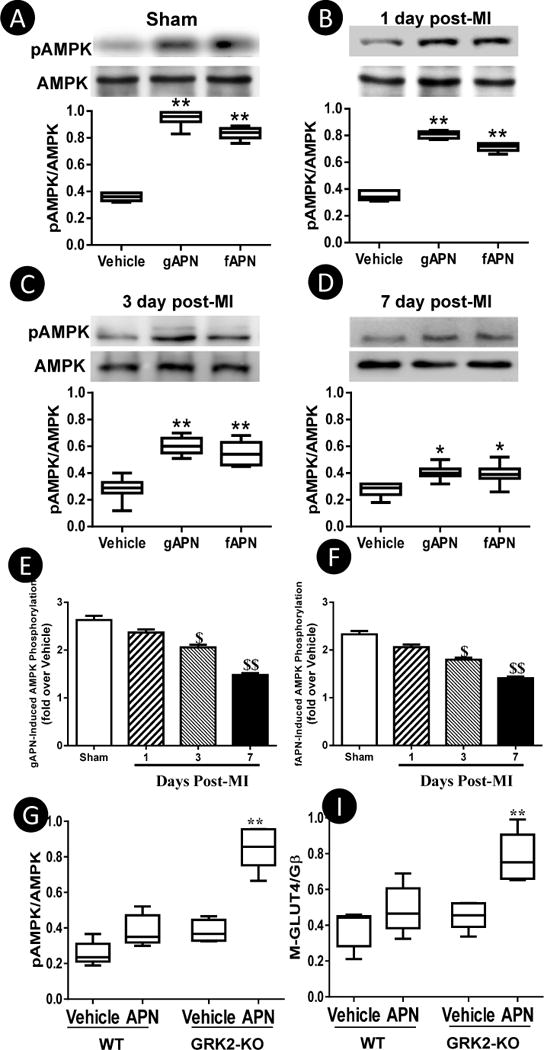
A–F: Cardiomyocytes from remote non-infarct regions were isolated 1, 3, and 7 days post-MI. Cells were treated with gAPN or fAPN for 30 minutes. pAMPK/AMPK were determined. G–I: Cardiomyocytes from remote non-infarct regions of WT or cardiomyocyte-specific GRK2 deletion mice were isolated 7 days post-MI, and treated with APN in vitro. APN-induced AMPK phosphorylation and GLUT4 membrane translocation was determined. N=5 mice/group. *P<0.05, **P<0.01 vs. vehicle, $P<0.05, $$P<0.01 vs. sham.
Figure 6.
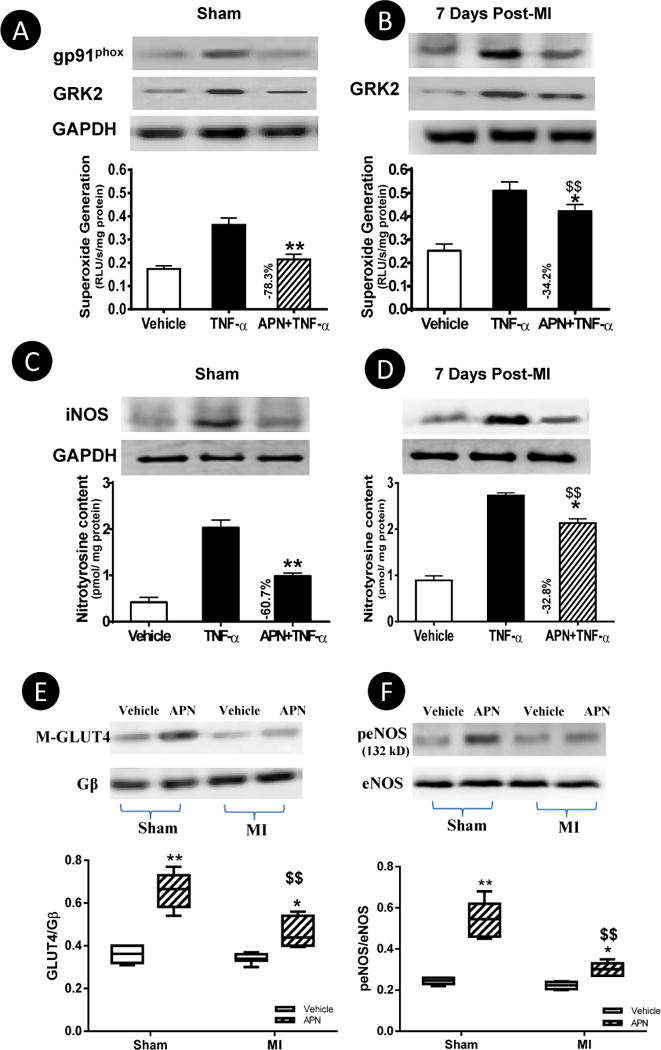
Cardiomyocytes from remote non-infarct regions were isolated 7 days post-MI. Cells were treated with TNFα in the presence or absence of gAPN for 8 hours. gp91phox/iNOS expression (A–D), superoxide production (A/B) and peroxynitrite formation (C/D) were determined. N=5 mice/group. *P<0.05, **P<0.01 vs. TNFα alone, $$P<0.01 vs. sham. E/F: Acute administration of gAPN (intraperitoneal bolus) caused significant GLUT4 membrane translocation (E) and eNOS phosphorylation (F) in control hearts, but these responses were significantly decreased 7 days post-MI. N=6–8 animals/group. *P<0.05, **P<0.01 vs. vehicle; $P<0.05, $$P<0.01 vs. sham with the same treatment.
Figure 7.
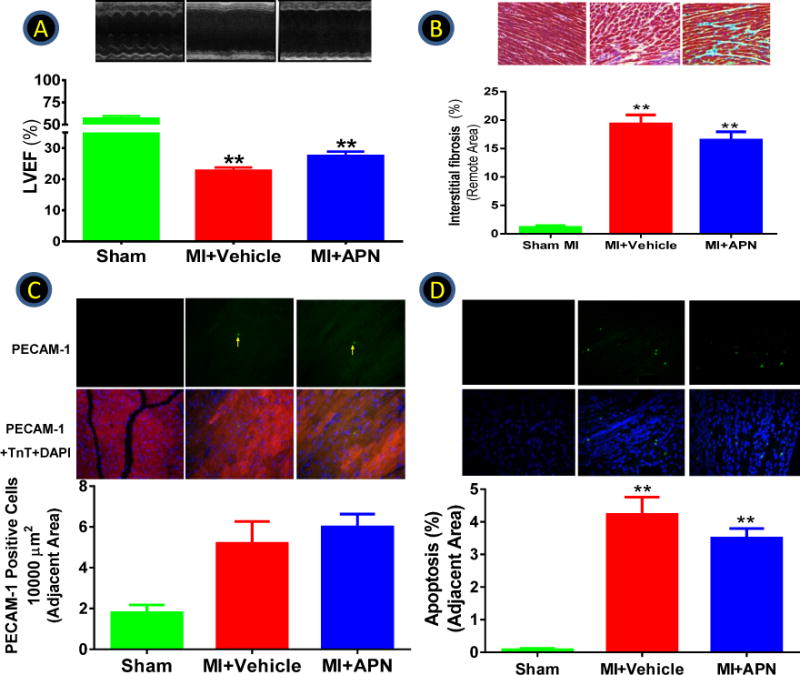
Animals were subjected to MI via LAD occlusion. 7 days post-MI, mice were randomized to receive vehicle or gAPN treatment for the remaining observation period. Effect of gAPN treatment upon left ventricular ejection fraction (A), interstitial fibrosis (B), capillary density (C), and apoptosis (D) were determined 8 weeks post-MI. N=10–21 animals/group. **P<0.01 vs. sham.
GRK2 Knockdown Restored gAPN Cardioprotection Post-MI
In a final attempt to obtain more evidence supporting a causative role for GRK2-induced AdipoR1 phosphorylation and impaired APN cardioprotective action, cardiac GRK2 expression was inhibited by direct intramyocardial injection of GRK2 siRNA. Mice were subjected to MI 2 days after siRNA injection and treated with vehicle or gAPN via osmotic pump beginning 7 days post-MI. The cardioprotective effect of gAPN was determined 1 week after APN treatment. In mice injected with scramble siRNA, continuous APN administration only slightly increased GLUT4 translocation (1.29-fold, Figure 8A left), AMPK phosphorylation (8B, left), and LVEF (1.27-fold, Figure 8C). However, genetic inhibition of GRK2 expression by siRNA restored the effect of gAPN upon GLUT4 translocation (Figure 8A, right), AMPK phosphorylation (8B, right), and almost fully restored the cardioprotective effect of gAPN, even when administered 1 week after MI (Figure 8D). It should be noted the improvement of cardiac function by GRK2 siRNA (without APN treatment) is modest, different from results previously observed in GRK2 knockout mice19. This is likely due to variation in GRK2 inhibition extent. In the current study utilizing GRK2 siRNA, approximately 70% inhibition was achieved (Figure 2E). However, in cardiomyocyte-specific GRK2 knockout mice, cardiac GRK2 is completely deleted. It is unquestionable that complete knockout remains the best approach for investigating the role a particular molecule may play, and establishing a cause-effect relationship. However, the results from partial GRK2 inhibition by siRNA may be more clinically relevant, because it is unlikely complete inhibition of GRK2 expression and/or activity can be achieved by clinical genetic/pharmacological intervention. As such, our current study suggests that although complete GRK inhibition is practically difficult, a combination treatment (e.g. a GRK2 inhibitor and APN) may achieve the best cardioprotective effect.
Figure 8.
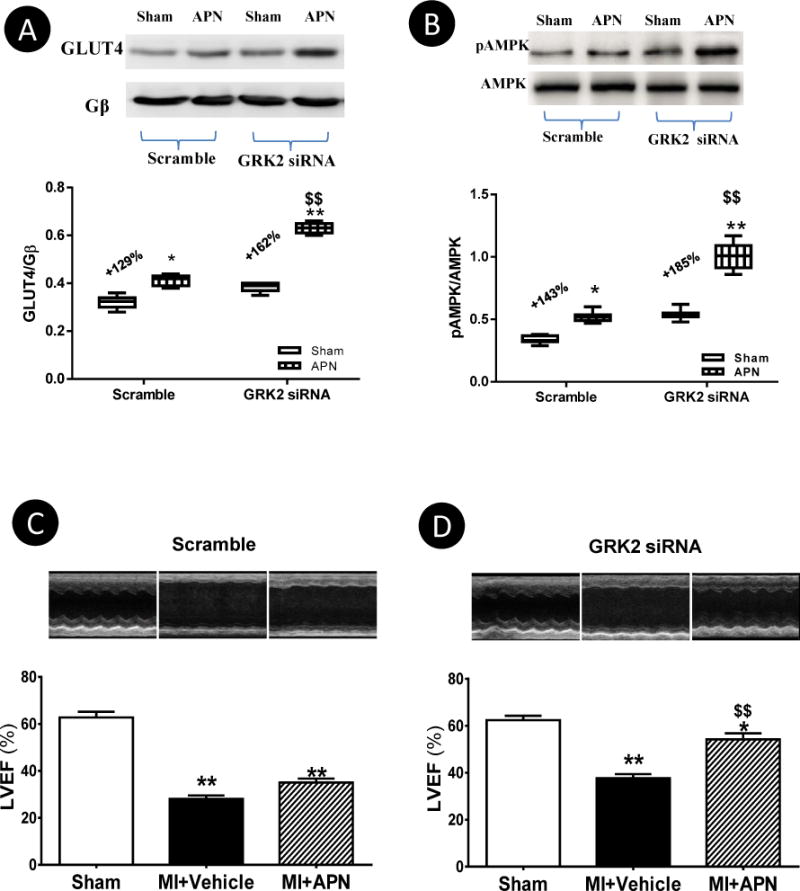
Scramble or GRK2 siRNA was administered via intramyocardial injection 2 days before MI. 7 days post-MI, animals were randomized to receive vehicle or gAPN (abdominal osmotic pump) for another 7 days. The GLUT4 translocation (A), AMPK phosphorylation (B), and cardiac function (LVEF) (C,D) were determined. N=11–14 animals/group. *P<0.05, **P<0.01 vs. sham; $P<0.05, $$P<0.01 vs. scramble group with the same treatment.
Discussion
Despite improvements in HF treatment, the disease prognosis remains poor. The precise mechanisms underlying worsening HF even in patients receiving optimal therapy are unclear. Evidence suggests imbalanced, attenuated, or abnormally activated cardiometabolic pathways in the failing heart contribute to refractoriness to pharmacologic interventions and HF progression2, 3. Insulin resistance is recognized as a major risk factor for adverse cardiac remodeling29. Clinically, insulin responsiveness is inversely correlated with advanced NYHA heart failure classification30, whereas overexpressing a human GLUT4 transgene recovered cardiac dysfunction in mice with insulin resistance31. Therefore, approaches targeting more efficient substrate use and preservation of cardiac metabolism are attractive therapeutic strategies.
Due to increased incidence of obesity and diabetes (and their association with HF), there is great interest in elucidating the underlying molecular mechanisms linking these pathologies. Many adipose-derived cytokines (adipokines) (e.g. TNF-α and resistin) are pro-inflammatory and are causally associated with adverse remodeling in HF32. In contrast, APN is a novel adipokine whose cardioprotective actions in acute MI are well-defined both clinically and experimentally5–7, 26–28. However, the role of APN in chronic HF is controversial. Experimental studies clearly demonstrate APN deficiency significantly exacerbates HF progression8–10. Paradoxically, clinical observations have reported APN increases in relation to worsening HF11–13. Several recent experimental as well as clinical studies demonstrate APN biological responses are significantly impaired in the skeletal muscle and livers of HF individuals, suggesting hyperadiponectinemia in advanced HF patients is a compensatory protective mechanism due to “APN resistance” development14–16. We currently demonstrate for the first time that significant APN resistance develops in the heart itself 7 to 14 days post-MI, a critical transitional period when adaptive to pathological remodeling occurs. The loss of APN-mediated protection during this time period is unlikely due to universal refractoriness to therapy, as cardiac remodeling can be reversed with appropriate treatments even after HF development19. More importantly, preventing AdipoR1 phosphorylation by genetic inhibition of GRK2 restored APN cardioprotection administered 1 week post-MI (Figure 8). Although systemic metabolic disorder caused by previously reported skeletal muscle and liver APN resistance certainly contributes to HF progression via indirect mechanisms, impaired APN signaling in cardiomyocytes themselves directly impacts adverse cardiac remodeling and HF progression.
APN shares biological functions with insulin, including glucose uptake, lipid oxidation, and cardiovascular protection4. APN is not merely an insulin sensitizer, but more importantly, is required for maintaining basal insulin tone responsiveness33. APN deficiency or APN gene variation are major risk factors for insulin resistance and metabolic disorders34. PPARγ agonists improve insulin sensitivity by up-regulating APN expression35. A very recent study further demonstrates APN resistance precedes insulin resistance in high-fat-fed rats36, suggesting APN resistance may contribute to insulin resistance development. Beyond its role in insulin signaling, APN possesses potent anti-oxidative/anti-nitrative stress, anti-inflammatory, and anti-apoptotic properties4. Since insulin resistance, oxidative/nitrative stress, exacerbated inflammatory response, and apoptosis are all well-recognized risk factors for adverse cardiac remodeling post-MI, impaired cardiac APN signaling during early HF development may contribute to metabolic imbalance, oxidative/nitrative stress, inflammation, and apoptosis, promoting adverse remodeling and HF progression.
Impaired APN signaling may occur in multiple levels. Clinical studies demonstrate reduced skeletal muscle AdipoR1 expression levels might be responsible for skeletal muscle APN resistance in HF patients14. However, a recent study reveals, despite significantly impaired skeletal muscle response to APN in high-fat diet-induced diabetic mice, expression levels of molecules involved in APN signaling remains normal37. Our current study demonstrates different molecular mechanisms are involved in cardiac APN resistance at different stages during HF development. The expression levels of AdipoR1 and AdipoR2 are significantly reduced 4–8 weeks post-MI. However, significant AdipoR1 phosphorylation develops much earlier than receptor expression alteration. Importantly, similar to functional impairment of β-ARs by phosphorylation, AdipoR1 phosphorylation at serine residues results in receptor desensitization, blocking APN’s anti-oxidative, anti-nitrative, anti-inflammatory, and cardioprotective function. To our best knowledge, this is the first AdipoR1 post-translational modification identified to date significantly impairing receptor function.
AdipoR1 desensitization after phosphorylation likely involves complicated molecular mechanisms, and must be fully addressed in a separate study. At least 3 possibilities exist that may explain APN signaling inhibition following AdipoR1 phosphorylation. Firstly, AdipoR1 is internalized in ligand-independent fashion38. AdipoR1 phosphorylation may increase its internalization, thereby blocking its function, similar to βAR. Secondly, multiple AdipoR1 intracellular binding proteins, including APPL1, nCDase, Cav3, RACK1 (activated protein kinase C1), and CK2β (protein kinase CK2 β subunit) have been identified23, 25, 39. These interactions are respectively required in APN-mediated metabolic-regulation, anti-inflammation, and cellular protection23, 25, 39. In contrast, binding of APPL2 by AdipoR1 inhibits APN signaling40. AdipoR1 phosphorylation may alter these interactions, thus blocking APN function. Thirdly, APN exerts anti-oxidative function in a cAMP-initiated, PKA-dependent fashion41. Moreover, other PAQR family members (PAQR 5, 7, and 8) interact with G proteins42, 43. These results suggest G protein might be involved with AdipoR1 signaling. Similar to β-AR, phosphorylation of AdipoR1 may alter its affinity, inducing β-arrestin recruitment44, thus blocking AdipoR1 function. These attractive mechanisms will be determined in our future studies.
Another significant finding of the current study is that GRK2, the crucial kinase responsible for post-MI β-AR phosphorylation and desensitization19, is also responsible for post-MI AdipoR1 phosphorylative inhibition. Neurohormonal activation during HF activates GRK2 upregulation, which classically phosphorylates activated GPCRs (such as the cardiac βARs), resulting in reduced signaling. In human HF, increased GRK2 is associated with decreased cardiac function and poor prognosis45. Increasing evidence suggests the biological regulatory functions of GRKs (particularly GRK2) are not limited to GPCRs24, 46. Specifically, increased GRK2 activity after βAR stimulation causes insulin resistance and inhibits insulin-dependent glucose extraction47. We recently demonstrate GRK2 inhibits insulin signaling by phosphorylative modification of insulin substrate-1, but not the insulin receptor itself24. Our current study demonstrates AdipoR1 is a novel substrate for GRK2, indicating that elevated GRK2 activity post-MI not only negatively affects cardiac contractile function by β-ARs phosphorylative desensitization, but also causes abnormal cardiometabolism by phosphorylative inhibition of insulin and APN, two critical hormones necessary for cardiometabolism and cell survival.
In summary, our study demonstrates for the first time that cardiometabolic-regulatory, anti-inflammatory, and cardioprotective functions of APN are significantly impaired by GRK2 mediated AdipoR1 phosphorylative desensitization during a critical period of post-MI HF development. These results add significantly to the dynamic role of APN in HF development, suggesting that inhibiting GRK2-mediated AdipoR1 phosphorylation and restoration of APN signaling might be novel therapeutic targets against post-MI remodeling and HF.
Supplementary Material
Acknowledgments
We are greatly appreciative of Mr. Nadan Wang in the Center for Translational Medicine, Thomas Jefferson University, for his expertise in evaluation of cardiac function by echocardiography.
Funding Sources: This research was supported by grants NIH HL-096686 and HL-123404, American Diabetes Association 1-15-BS-122 (XLM), and American Diabetes Association 1-14-BS-228 (YJW).
Footnotes
Journal Subject Codes: [130] Animal models of human disease, [11] Other heart failure, [107] Biochemistry and metabolism, [93] Receptor pharmacology
Disclosures: None.
References
- 1.Yancy CW, Jessup M, Bozkurt B, Butler J, Casey DE, Jr, Drazner MH, Fonarow GC, Geraci SA, Horwich T, Januzzi JL, Johnson MR, Kasper EK, Levy WC, Masoudi FA, McBride PE, McMurray JJ, Mitchell JE, Peterson PN, Riegel B, Sam F, Stevenson LW, Tang WH, Tsai EJ, Wilkoff BL. 2013 ACCF/AHA guideline for the management of heart failure: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol. 2013;62:e147–e239. doi: 10.1016/j.jacc.2013.05.019. [DOI] [PubMed] [Google Scholar]
- 2.Neubauer S. The failing heart–an engine out of fuel. N Engl J Med. 2007;356:1140–1151. doi: 10.1056/NEJMra063052. [DOI] [PubMed] [Google Scholar]
- 3.Ashrafian H, Frenneaux MP, Opie LH. Metabolic mechanisms in heart failure. Circulation. 2007;116:434–448. doi: 10.1161/CIRCULATIONAHA.107.702795. [DOI] [PubMed] [Google Scholar]
- 4.Goldstein BJ, Scalia RG, Ma XL. Protective vascular and myocardial effects of adiponectin. Nat Clin Pract Cardiovasc Med. 2009;6:27–35. doi: 10.1038/ncpcardio1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pischon T, Girman CJ, Hotamisligil GS, Rifai N, Hu FB, Rimm EB. Plasma adiponectin levels and risk of myocardial infarction in men. JAMA. 2004;291:1730–1737. doi: 10.1001/jama.291.14.1730. [DOI] [PubMed] [Google Scholar]
- 6.Shibata R, Numaguchi Y, Matsushita K, Sone T, Kubota R, Ohashi T, Ishii M, Kihara S, Walsh K, Ouchi N, Murohara T. Usefulness of adiponectin to predict myocardial salvage following successful reperfusion in patients with acute myocardial infarction. Am J Cardiol. 2008;101:1712–1715. doi: 10.1016/j.amjcard.2008.02.057. [DOI] [PubMed] [Google Scholar]
- 7.Shibata R, Sato K, Pimentel DR, Takemura Y, Kihara S, Ohashi K, Funahashi T, Ouchi N, Walsh K. Adiponectin protects against myocardial ischemia-reperfusion injury through AMPK- and COX-2-dependent mechanisms. Nat Med. 2005;11:1096–1103. doi: 10.1038/nm1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liao Y, Takashima S, Maeda N, Ouchi N, Komamura K, Shimomura I, Hori M, Matsuzawa Y, Funahashi T, Kitakaze M. Exacerbation of heart failure in adiponectin-deficient mice due to impaired regulation of AMPK and glucose metabolism. Cardiovasc Res. 2005;67:705–713. doi: 10.1016/j.cardiores.2005.04.018. [DOI] [PubMed] [Google Scholar]
- 9.Shimano M, Ouchi N, Shibata R, Ohashi K, Pimentel DR, Murohara T, Walsh K. Adiponectin deficiency exacerbates cardiac dysfunction following pressure overload through disruption of an AMPK-dependent angiogenic response. J Mol Cell Cardiol. 2010;49:210–220. doi: 10.1016/j.yjmcc.2010.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shibata R, Izumiya Y, Sato K, Papanicolaou K, Kihara S, Colucci WS, Sam F, Ouchi N, Walsh K. Adiponectin protects against the development of systolic dysfunction following myocardial infarction. J Mol Cell Cardiol. 2007;42:1065–1074. doi: 10.1016/j.yjmcc.2007.03.808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.George J, Patal S, Wexler D, Sharabi Y, Peleg E, Kamari Y, Grossman E, Sheps D, Keren G, Roth A. Circulating adiponectin concentrations in patients with congestive heart failure. Heart. 2006;92:1420–1424. doi: 10.1136/hrt.2005.083345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Laoutaris ID, Vasiliadis IK, Dritsas A, Mavrogeni S, Kallistratos MS, Manginas A, Chaidaroglou A, Degiannis D, Panagiotakos DB, Cokkinos DV. High plasma adiponectin is related to low functional capacity in patients with chronic heart failure. Int J Cardiol. 2010;144:230–231. doi: 10.1016/j.ijcard.2008.12.126. [DOI] [PubMed] [Google Scholar]
- 13.Nakamura T, Funayama H, Kubo N, Yasu T, Kawakami M, Saito M, Momomura S, Ishikawa SE. Association of hyperadiponectinemia with severity of ventricular dysfunction in congestive heart failure. Circ J. 2006;70:1557–1562. doi: 10.1253/circj.70.1557. [DOI] [PubMed] [Google Scholar]
- 14.Van Berendoncks AM, Garnier A, Beckers P, Hoymans VY, Possemiers N, Fortin D, Martinet W, Van Hoof V, Vrints CJ, Ventura-Clapier Re, Conraads VM. Functional Adiponectin Resistance at the Level of the Skeletal Muscle in Mild to Moderate Chronic Heart Failure / CLINICAL PERSPECTIVE. Circ Heart Fail. 2010;3:185–194. doi: 10.1161/CIRCHEARTFAILURE.109.885525. [DOI] [PubMed] [Google Scholar]
- 15.Van Berendoncks AM, Garnier A, Beckers P, Hoymans VY, Possemiers N, Fortin D, Van Hoof V, Dewilde S, Vrints CJ, Ventura-Clapier Re, Conraads VM. Exercise training reverses adiponectin resistance in skeletal muscle of patients with chronic heart failure. Heart. 2011;97:1403–1409. doi: 10.1136/hrt.2011.226373. [DOI] [PubMed] [Google Scholar]
- 16.Khan RS, Kato TS, Chokshi A, Chew M, Yu S, Wu C, Singh P, Cheema FH, Takayama H, Harris C, Reyes-Soffer G, Kn+¦ll R, Milting H, Naka Y, Mancini D, Schulze PC. Adipose Tissue Inflammation and Adiponectin Resistance in Patients With Advanced Heart Failure / Clinical Perspective. Circ Heart Fail. 2012;5:340–348. doi: 10.1161/CIRCHEARTFAILURE.111.964031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kadowaki T, Yamauchi T. Adiponectin and Adiponectin Receptors. Endocr Rev. 2005;26:439–451. doi: 10.1210/er.2005-0005. [DOI] [PubMed] [Google Scholar]
- 18.Tobin AB. G-protein-coupled receptor phosphorylation: where, when and by whom. Br J Pharmacol. 2008;153(Suppl 1):S167–S176. doi: 10.1038/sj.bjp.0707662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Raake PW, Vinge LE, Gao E, Boucher M, Rengo G, Chen X, DeGeorge BR, Jr, Matkovich S, Houser SR, Most P, Eckhart AD, Dorn GW, Koch WJ. G protein-coupled receptor kinase 2 ablation in cardiac myocytes before or after myocardial infarction prevents heart failure. Circ Res. 2008;103:413–422. doi: 10.1161/CIRCRESAHA.107.168336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yi W, Sun Y, Yuan Y, Lau WB, Zheng Q, Wang X, Wang Y, Shang X, Gao E, Koch WJ, Ma XL. C1q/Tumor necrosis factor-related protein-3, a newly identified adipokine, is a novel antiapoptotic, proangiogenic, and cardioprotective molecule in the ischemic mouse heart. Circulation. 2012;125:3159–3169. doi: 10.1161/CIRCULATIONAHA.112.099937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang Y, Wang XL, Zhao J, Wang YJ, Lau WB, Yuan YX, Gao EH, Koch WJ, Ma XL. Adiponectin inhibits oxidative/nitrative stress during myocardial ischemia and reperfusion via PKA signaling. Am J Physiol Endocrinol Metab. 2013;305:E1436–E1443. doi: 10.1152/ajpendo.00445.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang Y, Gao E, Tao L, Lau WB, Yuan Y, Goldstein BJ, Lopez BL, Christopher TA, Tian R, Koch W, Ma XL. AMP-activated protein kinase deficiency enhances myocardial ischemia/reperfusion injury but has minimal effect on the antioxidant/antinitrative protection of adiponectin. Circulation. 2009;119:835–844. doi: 10.1161/CIRCULATIONAHA.108.815043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang Y, Wang X, Lau WB, Yuan Y, Booth DM, Li J, Scalia R, Preston KJ, Gao E, Koch WJ, Ma XL. Adiponectin Inhibits TNF-alpha-Induced Vascular Inflammatory Response via Caveolin-Mediated Ceramidase Recruitment and Activation. Circ Res. 2014;114:792–805. doi: 10.1161/CIRCRESAHA.114.302439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ciccarelli M, Chuprun JK, Rengo G, Gao E, Wei Z, Peroutka RJ, Gold JI, Gumpert A, Chen M, Otis NJ, Dorn GW, II, Trimarco B, Iaccarino G, Koch WJ. G Protein-Coupled Receptor Kinase 2 Activity Impairs Cardiac Glucose Uptake and Promotes Insulin Resistance After Myocardial Ischemia. Circulation. 2011;123:1953–1962. doi: 10.1161/CIRCULATIONAHA.110.988642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang Y, Wang X, Jasmin JF, Lau WB, Li R, Yuan Y, Yi W, Chuprun K, Lisanti MP, Koch WJ, Gao E, Ma XL. Essential role of caveolin-3 in adiponectin signalsome formation and adiponectin cardioprotection. Arterioscler Thromb Vasc Biol. 2012;32:934–942. doi: 10.1161/ATVBAHA.111.242164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Goldstein BJ, Scalia R. Adipokines and vascular disease in diabetes. Curr Diab Rep. 2007;7:25–33. doi: 10.1007/s11892-007-0006-6. [DOI] [PubMed] [Google Scholar]
- 27.Zhu W, Cheng KK, Vanhoutte PM, Lam KS, Xu A. Vascular effects of adiponectin: molecular mechanisms and potential therapeutic intervention. Clin Sci (Lond) 2008;114:361–374. doi: 10.1042/CS20070347. [DOI] [PubMed] [Google Scholar]
- 28.Tao L, Gao E, Jiao X, Yuan Y, Li S, Christopher TA, Lopez BL, Koch W, Chan L, Goldstein BJ, Ma XL. Adiponectin cardioprotection after myocardial ischemia/reperfusion involves the reduction of oxidative/nitrative stress. Circulation. 2007;115:1408–1416. doi: 10.1161/CIRCULATIONAHA.106.666941. [DOI] [PubMed] [Google Scholar]
- 29.Mazumder PK, O’Neill BT, Roberts MW, Buchanan J, Yun UJ, Cooksey RC, Boudina S, Abel ED. Impaired cardiac efficiency and increased fatty acid oxidation in insulin-resistant ob/ob mouse hearts. Diabetes. 2004;53:2366–2374. doi: 10.2337/diabetes.53.9.2366. [DOI] [PubMed] [Google Scholar]
- 30.Doehner W, Rauchhaus M, Ponikowski P, Godsland IF, von HS, Okonko DO, Leyva F, Proudler AJ, Coats AJ, Anker SD. Impaired insulin sensitivity as an independent risk factor for mortality in patients with stable chronic heart failure. J Am Coll Cardiol. 2005;46:1019–1026. doi: 10.1016/j.jacc.2005.02.093. [DOI] [PubMed] [Google Scholar]
- 31.Semeniuk LM, Kryski AJ, Severson DL. Echocardiographic assessment of cardiac function in diabetic db/db and transgenic db/db-hGLUT4 mice. Am J Physiol Heart Circ Physiol. 2002;283:H976–H982. doi: 10.1152/ajpheart.00088.2002. [DOI] [PubMed] [Google Scholar]
- 32.Schram K, Sweeney G. Implications of myocardial matrix remodeling by adipokines in obesity-related heart failure. Trends Cardiovasc Med. 2008;18:199–205. doi: 10.1016/j.tcm.2008.10.001. [DOI] [PubMed] [Google Scholar]
- 33.Liu Q, Yuan B, Lo KA, Patterson HC, Sun Y, Lodish HF. Adiponectin regulates expression of hepatic genes critical for glucose and lipid metabolism. Proc Natl Acad Sci U S A. 2012;109:14568–14573. doi: 10.1073/pnas.1211611109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kadowaki T, Yamauchi T, Kubota N, Hara K, Ueki K, Tobe K. Adiponectin and adiponectin receptors in insulin resistance, diabetes, and the metabolic syndrome. J Clin Invest. 2006;116:1784–1792. doi: 10.1172/JCI29126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gable DR, Hurel SJ, Humphries SE. Adiponectin and its gene variants as risk factors for insulin resistance, the metabolic syndrome and cardiovascular disease. Atherosclerosis. 2006;188:231–244. doi: 10.1016/j.atherosclerosis.2006.02.010. [DOI] [PubMed] [Google Scholar]
- 36.Mullen KL, Pritchard J, Ritchie I, Snook LA, Chabowski A, Bonen A, Wright D, Dyck DJ. Adiponectin resistance precedes the accumulation of skeletal muscle lipids and insulin resistance in high-fat-fed rats. Am J Physiol Regul Integr Comp Physiol. 2009;296:R243–R251. doi: 10.1152/ajpregu.90774.2008. [DOI] [PubMed] [Google Scholar]
- 37.Gulli RA, Tishinsky JM, MacDonald T, Robinson LE, Wright DC, Dyck DJ. Exercise restores insulin, but not adiponectin, response in skeletal muscle of high-fat fed rodents. Am J Physiol Regul Integr Comp Physiol. 2012;303:R1062–R1070. doi: 10.1152/ajpregu.00176.2012. [DOI] [PubMed] [Google Scholar]
- 38.Ding Q, Wang Z, Chen Y. Endocytosis of adiponectin receptor 1 through a clathrin- and Rab5-dependent pathway. Cell Res. 2009;19:317–327. doi: 10.1038/cr.2008.299. [DOI] [PubMed] [Google Scholar]
- 39.Mao X, Kikani CK, Riojas RA, Langlais P, Wang L, Ramos FJ, Fang Q, Christ-Roberts CY, Hong JY, Kim RY, Liu F, Dong LQ. APPL1 binds to adiponectin receptors and mediates adiponectin signalling and function. Nat Cell Biol. 2006;8:518–523. doi: 10.1038/ncb1404. [DOI] [PubMed] [Google Scholar]
- 40.Wang C, Xin X, Xiang R, Ramos FJ, Liu M, Lee HJ, Chen H, Mao X, Kikani CK, Liu F, Dong LQ. Yin-Yang Regulation of Adiponectin Signaling by APPL Isoforms in Muscle Cells. J Biol Chem. 2009;284:31608–31615. doi: 10.1074/jbc.M109.010355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ouchi N, Kihara S, Arita Y, Okamoto Y, Maeda K, Kuriyama H, Hotta K, Nishida M, Takahashi M, Muraguchi M, Ohmoto Y, Nakamura T, Yamashita S, Funahashi T, Matsuzawa Y. Adiponectin, an adipocyte-derived plasma protein, inhibits endothelial NF-kappaB signaling through a cAMP-dependent pathway. Circulation. 2000;102:1296–1301. doi: 10.1161/01.cir.102.11.1296. [DOI] [PubMed] [Google Scholar]
- 42.Tang YT, Hu T, Arterburn M, Boyle B, Bright JM, Emtage PC, Funk WD. PAQR proteins: a novel membrane receptor family defined by an ancient 7-transmembrane pass motif. J Mol Evol. 2005;61:372–380. doi: 10.1007/s00239-004-0375-2. [DOI] [PubMed] [Google Scholar]
- 43.Thomas P, Pang Y. Membrane progesterone receptors: evidence for neuroprotective, neurosteroid signaling and neuroendocrine functions in neuronal cells. Neuroendocrinology. 2012;96:162–171. doi: 10.1159/000339822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gesty-Palmer D, Chen M, Reiter E, Ahn S, Nelson CD, Wang S, Eckhardt AE, Cowan CL, Spurney RF, Luttrell LM, Lefkowitz RJ. Distinct +¦-Arrestin- and G Protein-dependent Pathways for Parathyroid Hormone Receptor-stimulated ERK1/2 Activation. J Biol Chem. 2006;281:10856–10864. doi: 10.1074/jbc.M513380200. [DOI] [PubMed] [Google Scholar]
- 45.Iaccarino G, Barbato E, Cipolletta E, De AV, Margulies KB, Leosco D, Trimarco B, Koch WJ. Elevated myocardial and lymphocyte GRK2 expression and activity in human heart failure. Eur Heart J. 2005;26:1752–1758. doi: 10.1093/eurheartj/ehi429. [DOI] [PubMed] [Google Scholar]
- 46.Gurevich EV, Tesmer JJ, Mushegian A, Gurevich VV. G protein-coupled receptor kinases: more than just kinases and not only for GPCRs. Pharmacol Ther. 2012;133:40–69. doi: 10.1016/j.pharmthera.2011.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cipolletta E, Campanile A, Santulli G, Sanzari E, Leosco D, Campiglia P, Trimarco B, Iaccarino G. The G protein coupled receptor kinase 2 plays an essential role in beta-adrenergic receptor-induced insulin resistance. Cardiovasc Res. 2009;84:407–415. doi: 10.1093/cvr/cvp252. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.