

Abstract
Rationale
Matrix metalloproteinases (MMPs) regulate remodeling of the left ventricle (LV) post-myocardial infarction (MI). MMP-12 has potent macrophage-dependent remodeling properties in the atherosclerotic plaque; however, post-MI roles have not been examined.
Objective
The goal was to determine MMP-12 post-MI mechanisms.
Methods and Results
Male C57BL/6J mice (3–6 months old) were subjected to left coronary artery ligation. Saline or the RXP 470.1 MMP-12 inhibitor (MMP-12i; 0.5 mg/kg/day) were delivered by osmotic mini-pump beginning 3h post-MI, and mice were sacrificed at days (d)1, 3, 5 or 7 post-MI and compared to d0 controls (mice without MI; n=6–12/group/time). MMP-12 expression increased early post-MI, and contrary to expected, neutrophils were a surprising early cellular source for MMP-12. MMP-12i reduced MMP-12 activity 33±1% at d1 post-MI. Despite similar infarct areas and survival rates, MMP-12i led to greater LV dilation and worsened LV function. At d7 post-MI, MMP-12i prolonged pro-inflammatory cytokine upregulation (IL1r1, IL6ra, IL11, and Cxcr5) and decreased CD44 (both gene and protein levels). Hyaluronan (HA), a CD44 ligand, was elevated at d1 and d7 post-MI with MMP12i, as a result of decreased fragmentation. Because CD44-HA regulates neutrophil removal, apoptosis markers were evaluated. Caspase 3 increased, while cleaved caspase 3 levels decreased in MMP-12i group at d7 post-MI, indicating reduced neutrophil apoptosis. In isolated neutrophils, active MMP-12 directly stimulated CD44, caspase 3, and caspase 8 expression.
Conclusion
Our results reveal a novel protective mechanism for MMP-12 in neutrophil biology. Post-MI, MMP-12i impaired CD44-HA interactions to suppress neutrophil apoptosis and prolong inflammation, which worsened LV function.
Keywords: MMP-12, proteomics, neutrophil, apoptosis, LV remodeling, CD44
1. Introduction
Myocardial Infarction (MI) occurs as a result of prolonged coronary artery occlusion that blocks the blood supply to the downstream myocardium in the left ventricle (LV).[1] A prolonged reduction in LV blood flow leads to myocyte necrosis, which impairs the ability of the heart to contract.[2] Myocyte necrosis is followed by release of inflammatory mediators and infiltration of leukocytes.[2] This inflammatory phase clears dead myocytes and matrix debris, which is crucial for cardiac wound healing and infarct scar formation.[3] For infarct healing to progress, the inflammatory phase needs to be resolved in a timely manner that includes removal of apoptotic polymorphonuclear leukocytes (neutrophils). This occurs during the proliferative phase characterized by robust extracellular matrix (ECM) synthesis.[3] Coordination and regulation of the cellular events during LV remodeling is required for appropriate wound healing and scar formation.[2–4]
Matrix metalloproteinases (MMPs) are zinc dependent enzymes that cleave both ECM and non-ECM substrates and play key roles in LV remodeling.[5] Multiple MMPs and their inhibitors (the tissue inhibitor of metalloproteinases; TIMPs) are altered post-MI.[6, 7] MMP-12 (macrophage metalloelastase; 55 kDa pro-enzyme) is activated to form a 22 kDa mature form.[8] MMP-12 plays a critical role in acute and chronic inflammatory diseases, including chronic obstructive pulmonary disease and atherosclerosis, but MMP-12 has not been evaluated in the post-MI LV.[9] Macrophages are major MMP-12 producers, and macrophages are key cells in the post-MI wound healing response.[10] MMP-12 expression by other cell types, including neutrophils, and MMP-12 secretion profiles and functions post-MI have not yet been studied.
First generation MMP inhibitors lacked specificity and selectivity, making their in vivo use problematic.[11, 12] The Dive lab has developed a potent and highly selective MMP-12 inhibitor, RXP 470.1, which successfully reduced atherosclerotic progression and beneficially altered plaque phenotype in apolipoprotein E (apoE) null mice.[13] This inhibitor has a Ki of 0.2 nmol/L against human MMP-12 and a Ki of 4 nmol/L against mouse MMP-12.[13] We used RXP 470.1 to elucidate the roles of MMP-12 in LV remodeling post-MI. Our study revealed an important regulatory link between MMP-12 and LV remodeling.
2. Methods
2.1 Mice
All animal procedures were performed based on the Guide for the Care and Use of Laboratory Animals (Eighth edition, 2011) and were approved by the Institutional Animal Care and Use Committees at the University of Texas Health Science Center at San Antonio and the University of Mississippi Medical Center. The C57BL/6J WT male mice used were 3–6 months old, an age that is homogeneous in physiological maturation.[14] All mice were kept in the same room in a light-controlled environment with a 12:12 hour light-dark cycle and with free access to standard mouse chow and water. All of the MI mice were subjected to permanent occlusion of the left coronary artery as previously described [15]. At 3 h post-MI, infarction was confirmed by echocardiography, and osmotic pumps with saline or MMP-12 inhibitor (RXP 470.1; 0.5mg/kg/day; Alzet 1007D) were inserted subcutaneously. The MMP-12 inhibitor group (MMP-12i) was given the RXP470.1 compound. The dose given was selected based on calculation of the RXP 470.1 dose needed to reach a plasma concentration of 100 nmol/L [13]. Mice were sacrificed at days 1, 3, 5 or 7 post-MI (n=6–12 surviving mice/time point/group). Day 0 mice (n=6–12 for each assay) served as d0 controls.
2.2 Echocardiography
Transthoracic echocardiography was performed using a Vevo 2100™ system (VisualSonics, Toronto, Ontario, Canada) with a 30 MHz image transducer. Mice were anesthetized with 0.5–2% isoflurane in an oxygen mix. Electrocardiogram, body temperature, and heart rate were monitored throughout the imaging procedure. Measurements were taken from the LV parasternal long axis (B-mode) and short axis (M-mode) views. For each variable, three images from consecutive cardiac cycles were measured and averaged. All images were acquired at heart rates >400 bpm to achieve physiologically relevant measurements.
2.3 Survival analysis and autopsy
The mice were checked daily for survival, and all mice that were found dead were analyzed by autopsy. At autopsy, cardiac rupture was confirmed if LV rupture was seen and a blood clot was observed in the thoracic cavity.
2.4 Tissue harvest and infarct area evaluation
At the sacrifice time point, mice were anesthetized with 0.5–2% isoflurane in an oxygen mix. Heparin was administered (i.p., 4U/g body weight) and 5 min later, the blood was collected from the common carotid artery. The blood was centrifuged for plasma collection, mixed with proteinase inhibitor, and snap frozen at −80°C. The heart was collected as described previously.[7] The heart was flushed with cardioplegic solution (NaCl, 69 mM; NaHCO3, 12 mM; glucose, 11 mM; 2,3-butanedione monoxime, 30 mM; EGTA, 10 mM; Nifedipine, 0.001 mM; KCl, 50 mM) to arrest hearts at diastole. The hearts were excised and the LV and right ventricle (RV) were separated and weighed individually.
The LV was sliced into apex, middle, and base sections and stained with 1% 2, 3, 5-triphenyltetrazolium chloride (TTC, Sigma) for evaluation of infarct area. The LV infarct region (LVI) was separated from non-infarcted remote region (LVC), and lung weights and tibia lengths were collected. The LVI was calculated using Photoshop (Adobe) and is presented as percentage of infarct area to total LV area. [16] The LVI and LVC were individually snap frozen and stored at −80°C for real time RT2-PCR analysis (n=6/ group) or immunoblotting analysis (n=6/ group). The LV middle section was fixed in 10% zinc formalin (Fisher Scientific), paraffin-embedded, and sectioned for histological examination (n=12/ group).
2.5 MMP-12 activity assay
A standard colorimetric kit (Enzo life sciences, Framingdale, NY) was used per manufacturer instructions to measure MMP-12 activity in d1 plasma samples from saline or MMP-12i treated mice. As a secondary method to confirm MMP-12i presence in the plasma, a standard fluorogenic substrate was used to measure MMP-12 activity in d7 plasma samples from saline or MMP-12i treated mice. The assays were performed in triplicate.
2.6 Protein extraction and immunoblotting
The LV was homogenized in 1x phosphate-buffered saline (PBS) with 1x proteinase inhibitors cocktail (PI, Roche; 16 μL for every mg of wet LV mass) using the Power Gen 1000 Homogenizer (Fisher Scientific). The homogenate was centrifuged at 4700 rpm. The supernatant (soluble protein fraction) was extracted and stored at −80°C. The remaining pellet (insoluble protein fraction) was homogenized in Protein Extraction Reagent 4 (Sigma) with 1x protease inhibitor. The insoluble protein fraction was stored at −80°C. Protein levels were quantified by Bradford assay (BioRad, Hercules, CA).
Total protein (10 μg) from each fraction (soluble and insoluble) were run on 4–12% criterion Bis–Tris gels (Bio-Rad, Hercules, CA), transferred onto nitrocellulose membranes (Bio-Rad) and stained with MemCode™ Reversible Protein Stain Kit (Thermo Scientific, Waltham, MA) to verify loading accuracy. Membranes were blocked with 5% non-fat milk (Bio-Rad) and incubated overnight at 4°C with primary antibodies against MMP-12 (Millipore AB2964, 1:1000), TGF-β (R&D systems MAB1835, 1:1000), CD44 (Abcam ab119863, 1:1000), Hyaluronic Acid (Abcam ab53842, 1:1000), CD18 (Abcam ab157146, 1:1000), Caspase 3 (Cell signaling 9662, 1:1000), Galectin-3 (R&D systems VPG802; 1:1000) and neutrophils (anti-neutrophil mouse monoclonal, Cedarlane CL8993AP; 1:1000) followed by incubation at room temperature with secondary antibodies. Signal detection was done using Amersham ECL Substrate (GE Healthcare, Waukesha, WI). Protein levels were quantified by densitometry using the IQ-TL image analysis software (GE Healthcare, Waukesha, WI). Densitometric units were normalized to the densitometry of the total protein stain for the entire lane. For the time course analysis, n=6 samples per group for each time point were pooled and used as a representative time course.
2.7 Real Time RT2-PCR
RNA extraction was performed using TRIzol® Reagent (Invitrogen Life Technologies, Grand Island, NY, USA) according to manufacturer instructions. RNA levels were quantified using the NanoDrop ND-1000 Spectrophotometer (Thermo Scientific,Waltham, MA, USA). Reverse transcription of RNA (0.4 μg) was performed using the RT2 First Strand Kit (Qiagen, Valencia, CA, USA). Real Time RT2-PCR gene array for Inflammatory Cytokines and Receptors (Qiagen, Valencia, CA, USA; PAMM-011A) and for Extracellular Matrix and Adhesion Molecules (Qiagen, Valencia, CA, USA; PAMM-013A) were performed to quantify gene expression levels.[7] The gene levels were normalized to the reference gene hypoxanthine guanine phosphoribosyl transferase 1 (Hprt1), and the data reported as 2-ΔCt values × 100 for extracellular matrix genes and 2-ΔCt values × 1000 for inflammatory genes. The experiments were performed according to the MIQE guidelines with one exception. Hprt1, was the only reference gene showing no change in expression (GusB, Hsp90ab1, Actb and Gapdh were all significantly changed post-MI).
2.6 Blood neutrophil isolation and stimulation
Neutrophils were isolated from the blood as previously described [17]. Briefly, blood was collected in heparinized tubes from the common carotid arteries of 5 mice and pooled, and neutrophils were separated by density gradient centrifugation at 250 × g for 35 min using lympholyte poly cell separating media (Cedarlane, CL5070) followed by hypotonic lysis of erythrocytes using red cell lysis buffer (Miltenyi biotec). A single cell suspension was achieved with pre-separation filters (Miltenyi Biotec 130-041-407, 30 μm) and the cellular pellet resuspended in PEB buffer (PBS containing 0.5% BSA and 2 mM EDTA).
The cell suspension was serially incubated with anti-Ly-6G MicroBead Kit mouse (10 min, Miltenyi Biotec, 130-092-332) and CD11b MicroBeads mouse/human (Miltenyi Biotec, 130-049-601) and isolated using a MiniMACS magnetic separator column. The effluent containing Ly-6G+ neutrophils was resuspended in 1 mL of RPMI 1640 media and plated in a 6-well plate (1.5×105 cells/ well). The neutrophils were treated with active MMP-12 (500 U, Enzo Life Sciences, BML-SE138) and incubated at 37°C for 3 h. Cells incubated with fresh media were used as the unstimulated negative controls. MMP-12i or RXP470.1 can only inhibit transition of pro to active MMP-12. Since we are using active MMP-12 to stimulate the neutrophils and RXP 470.1 does not inhibit already activated MMP-12, we did not include an active MMP-12 + RXP 470.1 group. The cells were harvested for RNA isolation to perform RT2-PCR. Mmp12, Cd44, Casp3, and Casp8 gene expression levels were assessed using specific primers (Mmp12, Mm00500554_m1; Cd44, Mm01277163_m1; Caspase 3, Mm01195085_m1, and Caspase 8, Mm00802247_m1). The gene levels were normalized to Hprt1 and expressed as 2-ΔCt values±SEM.
2.7 Immunohistochemistry
The middle section of the LV was paraffin-embedded and sectioned at 5 μm for immunohistochemical staining as described previously [7]. Heat mediated antigen retrieval (Target retrieval solution, Dako) was performed to expose antigen epitopes. Sections were blocked with rabbit blocking serum of Vectastain elite ABC Kit (Vector Laboratories, Marion, IA, USA). A primary antibody specific for macrophages (Mac-3, Cedarlane CL8943AP; 1:100), neutrophils (anti-neutrophil mouse monoclonal, Cedarlane CL8993AP; 1:100) or cleaved caspase 3 (Cell signaling 9661, 1:100) was used at 4°C overnight. Later, the sections were incubated with respective secondary antibodies. Positive staining was visualized by HistoMark Black (KPL 54-75-00) and eosin was used as a counterstain. Images were captured at 40× magnification with Image-Pro software (Media Cybernetics, Bethesda, MD, USA) and quantification was performed as the percentage of positively stained area to total area.
2.8 Statistical analyses
All analyses were performed by investigators blinded to treatment groups and time post-MI. Data are presented as mean±SEM. Survival rates were analyzed by Kaplan-Meier survival analysis and compared by the log-rank test. Rupture rates were analyzed by Fisher’s exact test. Two group comparisons were analyzed by Students t-test. Multiple group comparisons were analyzed using one-way ANOVA, followed by the Student Newman-Keuls when the Bartlett’s variation test was passed, or using the nonparametric Kruskal-Wallis test, followed by Dunn post-hoc test when the Bartlett’s variation test did not pass. Statistical significance was set at p<0.05.
3. Results
3.1 MMP-12 protein expression increased early post-MI
Figure 1A shows that following MI MMP-12 protein levels in the LV infarct (LVI) increased at d1 and remained elevated through d7 compared to d0 control (without MI) LV. Figure 1B shows that MMP-12 gene levels were elevated at d1 post-MI in saline treated mice (p<0.05) and returned towards baseline by d7 (p=0.23 for d7 saline vs d0), while gene levels were increased at both d1 and d7 time points in the MMP-12i (both p<0.05). Figure 1C demonstrates that MMP-12 protein levels were elevated at both times compared to d0 LV for both the saline and MMP-12i groups in the soluble fraction (all p<0.05). In Figure 1C, the d7 MMP-12i protein level was significantly increased compared to saline treated d7 post-MI LV (p<0.05). These results indicate that RXP 470.1 did not inhibit MMP-12 protein expression early post-MI but rather prolonged MMP-12 gene upregulation to increase MMP-12 protein levels at day 7 post-MI. The fact that MMP-12 gene remains upregulated at d7 post-MI suggested a compensatory response to reduced MMP-12 activity.
Figure 1. MMP-12 gene and protein expression increased early post-MI; neutrophils were a surprising early source; and RXP470.1 inhibited active MMP-12.

(A) MMP-12 protein expression increased in the LV infarct (LVI) region at d1 post-MI and remained upregulated until d7. n=6/group pooled for each time point. The gene and protein expression of MMP-12 was analyzed in the saline and MMP-12 inhibitor (MMP-12i) treated groups. Both (B) RT-PCR and (C) immunoblot analysis showed increased MMP-12 expression levels in saline and MMP-12i LVI compared to day 0. n=6/group. *p<0.05 vs. day 0, #p<0.05 vs. d7 saline. (D) Neutrophils isolated from LVI at d1 post-MI showed higher MMP-12 mRNA expression compared to control neutrophils isolated from blood of unoperated WT mice. n=3/group. *p<0.05 vs. control. (E) The MMP-12i, RXP 470.1 reduced MMP-12 activity post-MI. Plasma samples at d1 post-MI were analyzed for MMP-12 activity using a colorimetric kit. MMP-12 activity was reduced by 33±1% in the inhibitor treated group compared to the saline group. n=10/group, #p<0.05 vs. d1 saline
3.2 Neutrophils were a surprising early source of MMP-12 in the post-MI infarct
Previous studies have shown that macrophages are the main producers of MMP-12.[10] Because MMP-12 was 4-fold increased at d1 post-MI, we evaluated if neutrophils were also a source of MMP-12 in the post-MI LV. Figure 1D illustrates that neutrophils isolated from the d1 LV infarct showed high expression of MMP-12, which was absent in neutrophils isolated from control d0 blood.
3.3 MMP-12i reduced MMP-12 activity post-MI
Since MMP-12 expression increased very early, we inhibited MMP-12 at 3 h post-MI, a time that would be clinically relevant. As shown in Figure 1E at d1 post-MI, MMP-12i plasma showed 33±1% reduction in MMP-12 recruitable activity. A secondary enzyme inhibition assay showed that at d7 post-MI, MMP-12i recruitable activity in plasma remained 22.8±0.1% reduced, indicating prolonged inhibition. The average RXP 470.1 concentration in the plasma was 10.7±2.2 nM. These results demonstrate that RXP 470.1 inhibited MMP-12 activity but not gene or protein expression early post-MI.
3.4 MMP-12 inhibition did not affect survival rate, cardiac rupture rate, or infarct area
As shown in Figure 2A, 12 out of 36 saline treated mice survived 7 days after MI (33%), and 12 out of 24 MMP-12i treated mice survived 7 days after MI (50%; p=0.29). Cardiac rupture occurred between 3–7 days post-MI as shown in figure 2B, consistent with previous reports.[18, 19] Autopsy revealed that 80% of non-survivor saline treated mice died of cardiac rupture (19/24), while 100% of the non-survivor MMP-12i mice died of cardiac rupture (12/12; Figure 2B; p=0.15). Figure 2C shows that infarct areas were similar between saline (57±2% at d1, 57±2% at d7) and MMP-12i (57±2% at d1, 58±3% at d7) MI groups (ANOVA p=0.98). All mice, therefore, received similar ischemic injury, and MMP-12 inhibition had no effect on the initial ischemic response. This was expected since inhibition began at 3 h post-MI, a time when myocyte salvage would not be expected in a permanent occlusion model.
Figure 2. MMP-12 inhibition does not affect survival rate, cardiac rupture, or infarct area.

(A) Survival rate was similar between saline and MMP-12 inhibitor (MMP-12i) treated mice. (B) Cardiac rupture percentages were similar between saline and MMP-12i treated mice. In the saline group, 19 out of 24 non-surviving mice had ruptured hearts; whereas in the MMP-12i group, all 12 of the non-surviving mice had ruptured hearts. (C) Infarct areas were similar between saline and MMP-12i treated mice at d1 and d7 post-MI. n=12/group.
3.5 MMP-12 inhibition exacerbated LV dysfunction and wall thinning at d7 post-MI
MMP-12i increased infarct wall thinning compared to saline treatment (Table 1). The LV remodeling index (end diastolic volume (EDV)/LV mass),[16] increased from d0 to d7 post-MI in both saline and MMP-12i groups, and the MMP-12i mice displayed a 28% higher remodeling index than saline controls, indicating exacerbated remodeling. The LV hypertrophy index (end diastolic diameter/average systolic wall thickness) was 27% higher in the MMP-12i mice compared to saline (Table 1).
Table 1.
MMP-12 inhibition worsens LV geometry and function post-MI.
| Day 0 | Day 7 MI Saline | Day 7 MI MMP-12i | |
|---|---|---|---|
| Body Weight; g | 30±1 | 28±1 | 26±1* |
| LV Mass/ Body Weight; mg/g | 3.0±0.1 | 4.0±0.2* | 4.0±0.2* |
| RV Mass/Body Weight; mg/g | 0.60±0.05 | 1.00±0.07* | 1.00±0.07* |
| Wet Lung Weight/ Body Weight; mg/g | 5.0±0.5 | 10±1* | 9±1* |
| Infarct Wall Thickness (diastolic; mm) | 0.90±0.03 | 0.66±0.04* | 0.56±0.04*# |
| LV Remodeling Index | 0.84±0.08 | 1.50±0.15* | 1.91±0.15*# |
| LV Hypertrophy Index | 4.0±0.1 | 10.0±0.8* | 13±1*# |
| End Diastolic Volume; μl | 85±5 | 163±11* | 201±18*# |
| End Systolic Volume; μl | 16±3 | 144±11* | 186±17*# |
| Ejection Fraction; % | 81±3 | 13±1* | 8±1*# |
Values are presented as mean±SEM, n=12/group.
p<0.05 vs. day 0, and
p<0.05 vs. saline treated mice.
Remodeling Index = End diastolic volume/LV mass. Hypertrophy index = End diastolic diameter/Average Systolic Wall Thickness. LV - Left ventricle; MI - Myocardial infarction.
Despite similar infarct areas, MMP-12i induced more LV dilation, as end systolic and end diastolic volumes were significantly elevated compared to saline treated LV (both p<0.05, Table 1). The post-MI reduction in ejection fraction (EF) was 43% greater with MMP-12i (p<0.05). The increased volumes and decreased ejection fractions suggested a protective role for MMP-12 in cardiac remodeling.
3.6 MMP-12 inhibition increased ECM degradation at d7 post-MI
Since significant differences in LV geometry and function were observed, we evaluated LV scar quality and composition. Table 2 summarizes ECM gene expression changes between infarcted (saline or MMP-12i groups) and no MI (d0 controls) as well as between MMP-12 inhibitor treated and vehicle control groups (MMP-12i vs saline). The qualitative increase, decrease, or no change results shown summarize quantitative measurements that were statistically different (all p<0.05). Out of 84 ECM and adhesion molecules analyzed in LVI at days 0, 1, and 7 post-MI, 63 genes at d1 and 53 genes at d7 were statistically different in saline and MMP-12i group collectively compared to day 0.(Table 2; all p<0.05). At d7 post-MI, MMP-12i LVI showed increased ECM gene upregulation compared to d0 (38 genes for MMP-12i versus 25 genes for saline MI), indicating that during LV remodeling multiple ECM genes are MMP-12 dependent.
Table 2.
ECM and adhesion gene expression changes between infarcted (saline or MMP-12i groups) and no MI (day 0 controls) as well as between MMP-12 inhibitor treated and vehicle control groups (MMP-12i vs saline). The increase, decrease, and no change were based on quantitative measurements that were statistically different based on RT2-PCR analysis for n=6 samples/ group. Qualitative directions of change and not average Ct values for each group are shown here. Out of 84 genes measured, 63 genes at d1 and 53 genes at d7 shown below were all significantly different among groups (all p<0.05).
| Saline vs Day 0 | MMP-12i vs Day 0 | MMP-12i vs Saline | |
|---|---|---|---|
| Day 1 post-MI | |||
| Cdh1, Ctnna2, Hc, Itgae, Lama1, Thbs1 | ↑ | ||
| Adamts1 | ↑ | ||
| Adamts2, Cd44, Cntn1, Col2a1, Ctgf, Hapln1, Itga3, Itga5, Itgam, Itgav, Itgb1, Itgb3, Mmp1a, Mmp3, Mmp7, Mmp8, Mmp9, Mmp10, Mmp14, Ncam2, Sele, Sell, Selp, Spp1, Spock1, Syt1, Timp1, Tnc, Vcan | ↑ | ↑ | |
| Fbln1, Thbs3 | ↓ | ||
| Itga4, Lamb3 | ↓ | ||
| Adamts5, Ctnna1, Ctnnb1, Cdh2, Cdh3, Cdh4, Col3a1, Col4a3, Col6a1, Itgax, Lama2, Lama3, Lamb2, Lamc1, Mmp2, Mmp11, Mmp15, Sgce, Sparc, Thbs2, Timp2, Vcam1, Vtn | ↓ | ↓ | |
| Day 7 post-MI | |||
| Cdh3, Ctnna2, Hapln1, Hc, Lama1, Mmp10, Mmp14, Mmp1a, Mmp7, Ncam2, Syt1 | ↑ | ||
| Adamts2, Cdh1, Col1a1, Col2a1, Col3a1, Col5a1, Vcan, Ctgf, Emilin1, Fn1, Itgb2, Itgae, Itgam, Itgav, Ncam1, Postn1, Sparc, Spp1, Thbs1, Thbs2, Thbs3, Timp1, Timp2, Tnc | ↑ | ↑ | |
| Mmp8, Spock1 | ↑ | ↑ | |
| Cd44 | ↑ | ↑ | ↓ |
| Icam1 | ↓ | ||
| Lama2, Lamb2 | ↓ | ||
| Adamts1, Ctnna1, Ctnnb1, Cdh2, Cdh4, Col4a3, Itgax, Lamb3, Mmp15, Pecam1, Sele, Vtn | ↓ | ↓ | |
Arrows depict direction of change; n=6/group. MI - Myocardial Infarction. Genes discussed in manuscript are shown in bold.
Gene names in alphabetical order: Adamts - A disintegrin-like and metallopeptidase (reprolysin type) with thrombospondin type 1 motif, Cdh1 – Cadherin 1, Cntn1 – Contactin 1, Col2a1 – Collagen type II alpha I, Ctgf – Connective tissue growth factor, Ctnna1 - Catenin (cadherin associated protein) alpha 1, Ctnna2 - Catenin (cadherin associated protein) alpha 2, Ctnnb1 – Catenin (cadherin associated protein) beta 1, Emilin1 - Elastin microfibril interfacer 1, Fbln1 – Fibulin 1, Fn1 – Fibronectin 1, Hapln I – Hylauronan and proteoglycan link protein I, Hc - Hemolytic complement, Icam1 - Intercellular adhesion molecule 1, Itga – Integrin alpha, Itgae – Integrin alpha E, epithelial-associated, Itgae - Integrin alpha E, epithelial-associated, Itgam – Integrin alpha M, Itgav - Integrin alpha V, Itgax –Integrin alpha X, Itgb – Integrin beta, Lama1 – Laminin alpha 1, Lamb – Laminin beta, Lamc – Laminin gamma, Ncam – Neural cell adhesion molecule, Pecam1 - Platelet/endothelial cell adhesion molecule 1, Postn – Periostin, Sele – Selectin, endothelial cell, Sell – Selectin lympohocyte, Selp – Selectin platelet, Sgce – Sarcoglycan epsilon, Sparc - Secreted acidic cysteine rich glycoprotein, Spock1 - Sparc/osteonectin, cwcv and kazal-like domains proteoglycan 1, Spp1 – Osteopontin, Syt1 - Synaptotagmin I, Thbs - Thrombospondin, Timp - Tissue inhibitor of metalloproteinase, Tnc – Tenascin C, Vcam1 – Vascular cell adhesion molecule 1, Vcan – Versecan, Vtn - Vitronectin
Collagen I (Col 1a1), collagen III (Col 3a1), and fibronectin (Fn1) were not different between saline and MMP-12i LVI, indicating that these particular ECM genes were not MMP-12 regulated (Table 2). Mmp8, Mmp10, and Mmp14 were increased with MMP-12i, demonstrating increased degradative capacity (Table 2). This result provides a mechanism for the elevated LV wall thinning seen with MMP-12i. Of note, these 3 MMPs are secreted by neutrophils and share common substrates with MMP-12, and their enhanced expression indicates a compensatory mechanism operating with MMP-12 inhibition and indirectly provides secondary confirmation of MMP-12 inhibition. Previous studies have also shown that MMP-8 and MMP-14 contribute to the adverse LV remodeling post-MI, indicating that some of the MMP-12i effects seen were due to upregulation of these MMPs.[20–22]
3.7 MMP-12 inhibition did not affect leukocyte infiltration but increased inflammatory cytokine expression
The inflammatory response is a key factor dictating cardiac remodeling, and the inflammatory response directly regulates the ECM response. We therefore evaluated neutrophil and macrophage numbers at days 1 and 7 post-MI. There were no significant differences between the saline and MMP-12i groups in terms of neutrophil (Figure 3) and macrophage numbers (Figure 4). The fact that leukocyte infiltration values did not change but LV function was worse with MMP-12 inhibition suggested that leukocyte function may be different.
Figure 3. MMP-12 inhibition does not affect neutrophil infiltration at day 1 post-MI.

(A) Protein expression of neutrophil was not significantly different between the saline and MMP-12i group. (B) Neutrophil infiltration into infarct area at day 1 post-MI showed no difference between saline and MMP-12i mice. Arrows depict positive staining. n=6–12/group, Scale bar is 200 μm.
Figure 4. MMP-12 inhibition does not affect macrophage infiltration at day 7 post-MI.

(A) Galectin-3 expression was not significantly different between the saline and MMP-12i group. Peritoneal macrophages were used as positive control (labeled as C) (B) Macrophage infiltration into infarct area at day 7 post-MI showed no difference between saline and MMP-12i mice. Arrows depict positive staining. n=6–12/group, Scale bar is 200 μm.
To assess profile of cytokines expressed by leukocytes as a method of evaluating leukocyte function, we measured 84 inflammatory molecules in the LVI at days 1 and 7 post-MI and in the LV of d0 controls. Table 3 summarizes inflammatory gene expression changes between infarcted (saline or MMP-12i groups) and no MI (d0 controls) as well as between MMP-12 inhibitor treated and vehicle control groups (MMP-12i vs saline). The increase, decrease and no change were based on quantitative measurements that were statistically different (all p<0.05). Compared to saline treated LVI, 45 genes at d1 and 41 genes at d7 were statistically different, as listed in Table 3 (all p<0.05). Saline d1 post-MI LVI had 30 upregulated genes and 13 downregulated genes (all p<0.05). MMP-12 inhibition attenuated the inflammatory cytokine upregulation, leading to only 17 upregulated genes and 15 downregulated genes at d1 post-MI (all p<0.05), indicating a delayed inflammatory response early post-MI. At d7 post-MI, the saline LVI had 15 upregulated genes and 11 downregulated genes; whereas the MMP-12i LVI had 29 upregulated genes and 9 downregulated genes (all p<0.05), indicating delayed inflammation resolution with MMP-12 inhibition. Of the upregulated genes in the MMP-12i LVI, the pro-inflammatory interleukins (IL)-18, -1r1, and 6ra are secreted by neutrophils, which signified prolonged neutrophil presence.[23–25]
Table 3.
Inflammatory gene expression changes between infarcted (saline or MMP-12i groups) and no MI (day 0 controls) as well as between MMP-12 inhibitor treated and vehicle control groups (MMP-12i vs saline). The increase, decrease, and no change were based on quantitative measurements that were statistically different based on RT2-PCR analysis for n=6 samples/ group. Qualitative directions of change and not average Ct values for each group are shown here. Out of 84 genes measured, 45 genes at d1 and 41 genes at d7 were significantly different among groups (all p<0.05).
| Saline vs Day 0 | MMP-12i vs Day 0 | MMP-12i vs Saline | |
|---|---|---|---|
| Day 1 post-MI | |||
| (a): Il10, Il11, Tgfb1 (p): Ccl9, Ccl20, Ccr8, Cd40lg, Cxcl1, Cxcl10, Il3, Lta, Xcr1 (both): Il4 |
↑ | ||
| (a): Il1r2, (p): Ccl2, Ccl3, Ccl4, Ccl7, Ccr1, Cxcl5, Il1b, Il17b, Il1r1, Il18rb, Il20, Il1f6, Il1f8, Il2rg, Itgam, Spp1 |
↑ | ↑ | |
| (p): Ccl25 | ↓ | ||
| (p): Cxcl11, Il2rb | ↓ | ||
| (a): Abcf1, Bcl6, Il10rb, Tollip (p): C3, Ccl11, Ccr10, Cxcl9, Il15, Il6st, Mif, Scye1 |
↓ | ↓ | |
| Day 7 post-MI | |||
| (p): Cxcl10 | ↑ | ||
| (a) Il4, Tgfb1 (p) Casp1, Ccl3, Ccl4, Ccl9, Ccl17, Ccr2, Ccr3, Il18, Il1f6, Il1f8, Il20, Lta |
↑ | ||
| (a): Il1r2 (p) Ccl20, Ccr1, Cd40lg, Cx3cl1, Il2rg, Il3, Itgb2, Itgam, Spp1, Xcr1 |
↑ | ↑ | |
| (p): Il6ra | ↑ | ↑ | |
| (a): Il11 (p): Cxcr5, Il1r1 |
↑ | ↑ | ↑ |
| (p): Pf4, Tnf | ↓ | ||
| (a): Abcf1, Tollip (p): Ccl11, Cxcl9, Ccr10, Il15, Il6st, Mif, Scye1 |
↓ | ↓ | |
Arrows depict direction of change; n=6/group. (a)- anti-inflammatory; d-day; MI- Myocardial infarction; (p)-pro-inflammatory. The genes discussed in manuscript are shown in bold.
Gene description in alphabetical order: Abcf1 – ATP-binding cassette, sub-family F (GCN20) member 1, Bcl – B-cell leukemia/lymphoma 6, C3 – Complement component 3, Casp1 – Caspase 1, Ccl – Chemokine (C-C) ligand, Ccr – Chemokine (C-C) receptor, Cd40lg – Cd40 ligand, Cx3cl – Chemokine (C-X3-C motif) ligand 1, Cxcl – Chemokine (C-X-C) ligand, Cxcr – Chemokine (C-X-C motif) receptor 3, Il – Interleukin, Il10rb – Interleukin 10 receptor beta, Il1f - Interleukin 1 family, member, Il1r – Interleukin 1 receptor type I, Il1r2 – Interleukin 1 receptor type I, Il2rb – Interleukin 2 receptor beta chain, Il2rg – Interleukin 2 receptor gamma chain, Il6st – Interleukin 6 signal transducer, Itgam – Integrin alpha M, Itgb – Integrin beta 2, Lta – Lymphotoxin, Mif – Macrophage migration inhibitory factor, Pf4 – Platelet factor 4, Scye1 – Small inducible cytokine subfamily E member 1, Spp1 – Osteopontin, Tgfb – Transforming growth factor beta, Tnf – Tumor necrosis factor, Tollip – Toll interacting protein, Xcr1 – Chemokine (C motif) receptor 1
Transforming growth factor β (TGFβ) is a regulator of inflammation that is robustly expressed at d7 post-MI.[26] All TGFβ isoforms are upregulated under ischemic conditions, and direct administration of TGFβ attenuates pro-inflammatory cytokine levels.[26] Figure 5A illustrates that TGFβ levels were increased 120±25% (for TGFβ2) and 83±8% (for TGFβ3) in the saline group compared to d0 in the soluble fraction (both p<0.05). However, in the MMP-12i group this upregulation was inhibited, and TGFβ densitometries were lower compared to saline LVI (40±6% for TGFβ2 and 20±10% for TGFβ3; both p<0.05). TGFβ inhibition leads to exacerbated LV dilation, and increased cytokine levels [27]. Our data indicate that TGFβ may act as a downstream stimulator of resolution of inflammation, through direct regulation by MMP-12.
Figure 5. MMP-12 inhibition reduced transforming growth factor β (TGFβand CD44 levels at d7 post-MI.

(A) Protein expression of TGF-β isoforms increased in the saline group at d7 post-MI compared to day 0. This increase was attenuated in the MMP-12i group. Lung tissue lysate was used as a positive TGFβ control (labeled as C). n=6/group, *p<0.05 vs. day 0 and #p<0.05 vs. saline. (B) CD44 gene expression increased at d1 post-MI in both saline and MMP-12i group. CD44 gene expression increased at d7 post-MI in both groups but at reduced levels in the MMP-12i group. CD44 protein expression in saline and MMP-12i treated groups at (C) d1 increased in both groups and at (D) d7 increased in both groups but at reduced levels in the MMP-12i group. n=6/group or time point. *p<0.05 vs. day 0, #p<0.05 vs. respective saline.
3.8 MMP-12 inhibition decreased expression of CD44 at d7 post-MI
TGFβ has been shown to stimulate MMP-14 induced cleavage of CD44 in cancer cells.[28] Absence of CD44 enhances dilative LV remodeling post-MI, demonstrating a significant post-MI role for CD44 [3]. In the saline group, Cd44 expression was enhanced at both d1 and 7 post-MI (Figure 5B and Table 2, p<0.05 versus day 0). MMP-12 inhibition did not affect CD44 gene (Figure 5B) and protein levels (Figure 5C) at d1 post-MI, but at d7 post-MI, CD44 levels were 54±4% reduced at the gene level (Figure 5B) and 50±1% reduced at the protein level (Figure 5D) compared to saline LVI (soluble fraction).
3.9 MMP-12 inhibition disrupted the CD44 – hyaluronic acid axis post-MI
Hyaluronic acid or hyaluronate (HA) is a principal CD44 ligand.[29] HA is a ubiquitously distributed polysaccharide that organizes ECM and increases during inflammation and tissue remodeling.[29] Full-length HA undergoes fragmentation, and fragment clearance is a stimulus for inflammation resolution during wound healing.[3] Both HA fragmentation and clearance are modulated by CD44.[3] Figure 6A shows that at day 1 post-MI, full-length HA increased 278±92% in saline LVI compared to day 0 LV, and this increase had returned to control levels by d7 post-MI (soluble fraction). In contrast, MMP-12i resulted in an 118±42% increase of HA at d1 post-MI that was 200±62% higher at d7 post-MI compared to time matched saline groups (soluble fraction, Figure 6A), demonstrating amplified and extended HA accumulation due to increased synthesis or low fragmentation and clearance rates. These data are consistent with a previous report in keratinocytes showing that CD44 abrogation was accompanied by abnormal prolonged HA accumulation.[29] Our data reveals that MMP-12 inhibition negatively impacted the CD44-HA axis, leading to HA accumulation and impaired inflammation resolution.
Figure 6. MMP-12 inhibition resulted in higher hyaluronic acid (HA) levels at d1, prolonged accumulation at d7, and reduced apoptosis at d7 post-MI.

(A) The CD44 ligand, HA, was elevated at d1 post-MI in the MMP-12 inhibitor (MMP-12i) group compared to the saline group, and these levels were sustained through d7 post-MI. n=6/group. *p<0.05 vs. day 0, #p<0.05 vs. respective saline. (B) MMP-12i increased protein expression of caspase 3, an apoptosis marker. Peritoneal macrophages were used as a positive control (labeled as C). (C) Immunohistochemistry analysis showed that cleaved caspase 3 expression was reduced in the infarct area of MMP-12i group at d7 post-MI compared to the saline MI group. (D) CD18, an adhesion molecule that suppresses apoptosis, was upregulated in the MMP-12i group compared to saline at d7 post-MI. Peritoneal macrophages were used as a positive control (labeled as C). n=5–10/ group, *p<0.05 vs. day 0 and #p<0.05 vs. d7 saline. Scale bar is 200 μm.
3.10 MMP-12 inhibition reduced neutrophil apoptosis at d7 post-MI
CD44 regulates apoptosis by interacting with HA,[3] a critical mechanism in wound healing to clear inflammatory cells from injury sites.[30] Neutrophils isolated from rat blood showed that CD44-HA interaction triggers neutrophil apoptosis.[31] Because the CD44-HA interaction was impaired with MMP-12i, we investigated if neutrophil apoptosis was prevented. Figure 6B shows that protein levels of intact full-length caspase 3 were upregulated in the saline group compared to day 0 (soluble fraction). With MMP-12i, these levels further increased an additional 58% compared to saline, suggesting accumulation due to reduced caspase 3 cleavage. Indeed, cleaved caspase 3 was decreased 50% in MMP-12i LVI (Figure 6C). CD18 (β2 integrin; Itgb2) is a cell adhesion molecule that suppresses neutrophil apoptosis during endothelial transmigration,[32] and CD18 levels were elevated at d7 post-MI in the MMP-12i LVI as shown in figure 6D. While d7 saline LVI showed no significant change in CD18 levels compared to day 0, CD18 levels were 140% increased with MMP-12i in the soluble fraction.
3.11 MMP-12 directly regulated CD44 and MMP-12i stimulated pro-apoptotic gene expression in neutrophils in vitro
The above results indicated that a role of MMP-12 is to stimulate neutrophil apoptosis. To verify this, neutrophils were isolated from the blood of WT mice and stimulated with active MMP-12. Stimulation resulted in a 4.5-fold increase in CD44 gene levels compared to unstimulated cells (Figure 7A). Caspase 3 (Figure 7B) and caspase 8 (Figure 7C) were 2-fold increased. This revealed a direct role for MMP-12 in instigating neutrophil exit from sites of inflammation.
Figure 7. Active MMP-12 directly stimulated CD44, Caspase 3, and Caspase 8 gene expression in neutrophils.
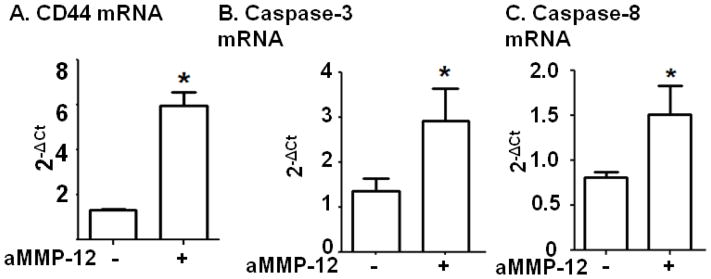
(A) Neutrophils isolated from mouse blood stimulated with active MMP-12 showed increased CD44 expression compared to unstimulated cells. n=3–5/group. (B) Stimulation of isolated blood neutrophils with active MMP-12 leads to significant increase in apoptotic gene expression (Caspase 3 and Caspase 8). n=3–5/group, *p<0.05 vs. unstimulated. aMMP-12 - active MMP-12.
4. Discussion
The goal of this study was to dissect MMP-12 roles in the post-MI setting. The major findings were: 1) MMP-12 expression increased early post-MI, with neutrophils being an early source of MMP-12, 2) MMP-12 inhibition worsened LV dysfunction, increased ECM degradation, and prolonged neutrophil presence, leading to sustained pro-inflammatory cytokine levels at d7 post-MI, 3) MMP-12 inhibition disrupted the CD44-HA interaction to reduce neutrophil apoptosis post-MI and impair inflammation resolution. Taken together, these findings reveal direct roles for MMP-12 in regulating the post-MI inflammatory response, such that MMP-12 inhibition resulted in exacerbated cardiac remodeling.
Consistent with previous reports, MMP-12 expression was very low in the day 0 LV, indicating MMP-12 is not a major player in cardiac homeostasis under normal conditions. Day 7 post-MI MMP-12 protein expression was robustly increased in MMP-12i group compared to saline treated mice. RXP 470.1 inhibited MMP-12 activity but not gene or protein expression early post-MI. This result was as expected since RXP 470.1 is a transition state analog, which binds to the transition state structure of MMP-12 when it converts from a pro to active form. MMP-12 has been extensively studied in chronic obstructive pulmonary disease patients and has been shown to be highly expressed in lung macrophages.[33] The known cellular sources of MMP-12 include macrophages, endothelial cells and vascular smooth muscle cells. [33–35] Interestingly, we found that the cell source for the early d1 post-MI MMP-12 expression was predominantly the neutrophil. This is the first report to show that neutrophils also express MMP-12. Expression in this new cell types raises the possibility that MMP-12 may play context dependent roles.
We observed worsened cardiac function post-MI with MMP-12 inhibition, revealing a protective role for MMP-12 in the post-MI setting. This is in contrast to an atherosclerotic model, where Johnson et al showed that MMP-12 inhibition, using the same RXP 470.1 used in this study, reduces atherosclerosis progression and beneficially alters plaque phenotype in apo-E null mice.[13] The differences observed between these two studies indicate MMP-12 has both detrimental and beneficial mechanisms that are dependent on injury stimuli and which MMP-12 substrates are present. In the atherosclerotic plaque study, RXP 470.1 inhibits MMP-12 activity in macrophages to yield a benefit, while it inhibited neutrophil functions to impair cardiac wound healing in our study. The possibility that MMP-12i also targets the macrophage pool at later stage post-MI cannot be excluded; although if this occurs, the net result at day 7 is detrimental remodeling. In our study, the MMP-12 inhibitor was infused at 3 hours after coronary artery ligation; whereas in the atherosclerotic study, the MMP-12 inhibitor was delivered to the mice 8 weeks later, after they developed stable plaques. This raises the fascinating possibility that MMP-12i in an acute inflammation model is harmful while MMP-12i in a chronic inflammation model is helpful. MMP-12 has in fact been characterized as an inflammatory mediator associated with both acute and chronic inflammatory diseases.[9]
ECM turnover regulates cardiac remodeling post-MI, and alterations that disturb the balance between synthesis and breakdown of ECM may worsen LV function. Excessive ECM degradation by MMPs leads to excessive wall thinning, which can result in rupture or LV dilation by disrupting cardiomyocyte alignment.[4, 36] MMP-12 inhibition, therefore, disturbed the ECM balance in favor of increased degradation to result in greater LV dilation. Additionally, we observed that MMP-12 inhibition did not delay neutrophil infiltration but rather delayed its removal. This was evidenced by increased presence of neutrophil secreted cytokines (IL)-18, -1r1, and 6ra in LVI at d7 post-MI. The fact that day 1 neutrophil numbers were not different indicates that the reduced inflammatory cytokine profile seen in day 1 LVI was not sufficient to yield an effect on leukocyte entry. A recent report by Marchant et al showed that inhibition of MMP-12 by RXP470.1 in wild type A/J mice elevated plasma IFN-γ levels.[37] This is consistent with our study where increased inflammation was observed with MMP-12 inhibition.
CD44 plays a critical role in several inflammatory diseases and is expressed on neutrophils, macrophages, and endothelial cells.[3, 38, 39] HA is a space filling ECM component responsible for structural maintenance of tissue and is a direct regulator of inflammation.[39] However, whether HA acts as a pro- or anti-inflammatory mediator is still contradictory, and the answer is likely both, as HA functions are mediated by HA fragment length, cell source, type of receptor binding it, type of tissue injury, and the presence of enzymes that cleave HA to particular fragments.[39] Using a mouse model of ischemia-reperfusion, Huebner et al showed critical functions for CD44 in wound healing and LV remodeling.[3] In a lung injury model, CD44 deficiency prolonged inflammatory gene expression and prevented low molecular weight HA clearance.[30] HA aggregation alters CD44 conformation, leading to less CD44-HA binding and less HA clearance.[40] Low molecular weight HA fragments can induce MMP-12 expression in an alveolar macrophage cell line, indicating a positive feedback loop.[41] We demonstrated in vitro that MMP-12 directly stimulated neutrophil CD44. Our results reveal that MMP-12 regulate HA clearance through CD44 dependent and independent mechanisms.
Neutrophils are the predominant leukocyte recruited during the first wave of inflammation.[42] Circulating neutrophils in the blood have a short life span and rapidly undergo apoptosis in the absence of an extravasation signal.[43] Neutrophil apoptosis in the post-MI LV is an important mechanism to turn off the inflammatory signal.[44, 45] MMP-12 activity has been associated with apoptosis in atherosclerosis, fibrosis, and cancer models.[13] MMP-12 inhibition with RXP 470.1 promotes macrophage apoptosis in atherosclerosis.[13] In our study, we found that MMP-12 inhibition with RXP 470.1 attenuated neutrophil apoptosis.
MMP-12 belongs to a family of metal endopeptidases, of which neutral endopeptidase (NEP) 24-11 is also included. NEP 24-11 is a plasma and endothelial membrane-bound zinc metalloprotease that cleaves bradykinin. [46] Treatment with bradykinin improved LV remodeling by reducing both the heart weight to body weight ratio and LV wall thickness in renovascular hypertensive rats. Since bradykinin is an NEP 24-11 substrate, it is possible that NEP24-11 may also play a role in post-MI remodeling.
In conclusion, MMP-12 inhibition exacerbated cardiac dysfunction by disrupting the CD44-HA axis to increase and prolong inflammation and reduce neutrophil apoptosis. Therefore, MMP-12 has a net protective role in the MI setting, and inhibiting MMP-12 early post-MI would not likely be a favorable approach to improve LV remodeling.
Highlights.
MMP-12 expression increased early post-MI, and the neutrophils are a previously unknown early source.
MMP-12 inhibition worsened LV dysfunction, increased ECM degradation, and increased pro-inflammatory cytokine levels at d7 post-MI
MMP-12 inhibition disrupted the CD44-HA interaction and reduced neutrophil apoptosis to impair resolution of inflammation at d7 post-MI.
Our results reveal a unique and protective role of matrix metalloproteinase-12 (MMP-12) in the early post-myocardial infarction (MI) left ventricle.
Acknowledgments
We acknowledge technical support from Dustin R. Bratton.
6. Source of Funding. We acknowledge funding support from the American Heart Association for 14POST18770012, 14SDG18860050, and 15SDG22930009. We acknowledge support from NIH HHSN 268201000036C (N01-HV-00244) for the San Antonio Cardiovascular Proteomics Center, HL075360, HL051971, and GM104357 and from the Biomedical Laboratory Research and Development Service of the Veterans Affairs Office of Research and Development Award 5I01BX000505.
Footnotes
7. Disclosures. None
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Jourdan-Lesaux C, Zhang J, Lindsey ML. Extracellular matrix roles during cardiac repair. Life sciences. 2010;87:391–400. doi: 10.1016/j.lfs.2010.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lambert JM, Lopez EF, Lindsey ML. Macrophage roles following myocardial infarction. International journal of cardiology. 2008;130:147–58. doi: 10.1016/j.ijcard.2008.04.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Huebener P, Abou-Khamis T, Zymek P, Bujak M, Ying X, Chatila K, et al. CD44 Is Critically Involved in Infarct Healing by Regulating the Inflammatory and Fibrotic Response. J Immunol. 2008;180:2625–33. doi: 10.4049/jimmunol.180.4.2625. [DOI] [PubMed] [Google Scholar]
- 4.Sutton MG, Sharpe N. Left ventricular remodeling after myocardial infarction: pathophysiology and therapy. Circulation. 2000;101:2981–8. doi: 10.1161/01.cir.101.25.2981. [DOI] [PubMed] [Google Scholar]
- 5.Lindsey ML, Zamilpa R. Temporal and spatial expression of matrix metalloproteinases and tissue inhibitors of metalloproteinases following myocardial infarction. Cardiovasc Ther. 2012;30:31–41. doi: 10.1111/j.1755-5922.2010.00207.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zamilpa R, Lindsey ML. Extracellular matrix turnover and signaling during cardiac remodeling following MI: causes and consequences. Journal of molecular and cellular cardiology. 2010;48:558–63. doi: 10.1016/j.yjmcc.2009.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ma Y, Chiao YA, Zhang J, Manicone AM, Jin YF, Lindsey ML. Matrix metalloproteinase-28 deletion amplifies inflammatory and extracellular matrix responses to cardiac aging. Microsc Microanal. 2012;18:81–90. doi: 10.1017/S1431927611012220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lagente V, Le Quement C, Boichot E. Macrophage metalloelastase (MMP-12) as a target for inflammatory respiratory diseases. Expert opinion on therapeutic targets. 2009;13:287–95. doi: 10.1517/14728220902751632. [DOI] [PubMed] [Google Scholar]
- 9.Nenan S, Boichot E, Lagente V, Bertrand CP. Macrophage elastase (MMP-12): a pro-inflammatory mediator? Memorias do Instituto Oswaldo Cruz. 2005;100 (Suppl 1):167–72. doi: 10.1590/s0074-02762005000900028. [DOI] [PubMed] [Google Scholar]
- 10.Xu J, Xu F, Barrett E. Metalloelastase in lungs and alveolar macrophages is modulated by extracellular substance P in mice. American journal of physiology Lung cellular and molecular physiology. 2008;295:L162–70. doi: 10.1152/ajplung.00282.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Le Quement C, Guenon I, Gillon JY, Valenca S, Cayron-Elizondo V, Lagente V, et al. The selective MMP-12 inhibitor, AS111793 reduces airway inflammation in mice exposed to cigarette smoke. British journal of pharmacology. 2008;154:1206–15. doi: 10.1038/bjp.2008.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Churg A, Wang R, Wang X, Onnervik PO, Thim K, Wright JL. Effect of an MMP-9/MMP-12 inhibitor on smoke-induced emphysema and airway remodelling in guinea pigs. Thorax. 2007;62:706–13. doi: 10.1136/thx.2006.068353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Johnson JL, Devel L, Czarny B, George SJ, Jackson CL, Rogakos V, et al. A selective matrix metalloproteinase-12 inhibitor retards atherosclerotic plaque development in apolipoprotein E-knockout mice. Arterioscler Thromb Vasc Biol. 2011;31:528–35. doi: 10.1161/ATVBAHA.110.219147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Flurkey KCJ, Harrison DE. The Mouse in Biomedical Research. 2. Burlington, MA: American College Laboratory Animal Medicine (Elsevier); 2007. [Google Scholar]
- 15.Salto-Tellez M, Yung Lim S, El-Oakley RM, Tang TP, ZAAL, Lim SK. Myocardial infarction in the C57BL/6J mouse: a quantifiable and highly reproducible experimental model. Cardiovascular pathology : the official journal of the Society for Cardiovascular Pathology. 2004;13:91–7. doi: 10.1016/S1054-8807(03)00129-7. [DOI] [PubMed] [Google Scholar]
- 16.Zamilpa RKR, Cigarroa J, IV, Dai Q, Escobar GP, Martinez H, Jimenez F, Ahuja SS, Lindsey ML. CC chemokine receptor 5 deletion impairs macrophage activation and induces adverse remodeling following myocardial infarction. American Journal of Physiology Heart and Circulatory Physiology. 2011;300:H1418–26. doi: 10.1152/ajpheart.01002.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Oh H, Siano B, Diamond S. Neutrophil isolation protocol. J Vis Exp. 2008 doi: 10.3791/745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gao X-M, Xu Q, Kiriazis H, Dart AM, Du X-J. Mouse model of post-infarct ventricular rupture: time course, strain- and gender-dependency, tensile strength, and histopathology. Cardiovasc Res. 2005;65:469–77. doi: 10.1016/j.cardiores.2004.10.014. [DOI] [PubMed] [Google Scholar]
- 19.Gao XM, Ming Z, Su Y, Fang L, Kiriazis H, Xu Q, et al. Infarct size and post-infarct inflammation determine the risk of cardiac rupture in mice. International journal of cardiology. 2010;143:20–8. doi: 10.1016/j.ijcard.2009.01.019. [DOI] [PubMed] [Google Scholar]
- 20.Spinale FG, Janicki JS, Zile MR. Membrane-associated matrix proteolysis and heart failure. Circulation research. 2013;112:195–208. doi: 10.1161/CIRCRESAHA.112.266882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yarbrough WM, Mukherjee R, Stroud RE, Rivers WT, Oelsen JM, Dixon JA, et al. Progressive induction of left ventricular pressure overload in a large animal model elicits myocardial remodeling and a unique matrix signature. The Journal of thoracic and cardiovascular surgery. 2012;143:215–23. doi: 10.1016/j.jtcvs.2011.09.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fertin M, Lemesle G, Turkieh A, Beseme O, Chwastyniak M, Amouyel P, et al. Serum MMP-8: A Novel Indicator of Left Ventricular Remodeling and Cardiac Outcome in Patients after Acute Myocardial Infarction. PloS one. 2013;8:e71280. doi: 10.1371/journal.pone.0071280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McColl SR, Paquin R, Menard C, Beaulieu AD. Human neutrophils produce high levels of the interleukin 1 receptor antagonist in response to granulocyte/macrophage colony-stimulating factor and tumor necrosis factor alpha. The Journal of experimental medicine. 1992;176:593–8. doi: 10.1084/jem.176.2.593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Paulsson JM, Moshfegh A, Dadfar E, Held C, Jacobson SH, Lundahl J. In-vivo extravasation induces the expression of interleukin 1 receptor type 1 in human neutrophils. Clinical and experimental immunology. 2012;168:105–12. doi: 10.1111/j.1365-2249.2011.04548.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Robertson SE, Young JD, Kitson S, Pitt A, Evans J, Roes J, et al. Expression and alternative processing of IL-18 in human neutrophils. European journal of immunology. 2006;36:722–31. doi: 10.1002/eji.200535402. [DOI] [PubMed] [Google Scholar]
- 26.Rainer PP, Hao S, Vanhoutte D, Lee DI, Koitabashi N, Molkentin JD, et al. Cardiomyocyte-Specific Transforming Growth Factor beta Suppression Blocks Neutrophil Infiltration, Augments Multiple Cytoprotective Cascades, and Reduces Early Mortality After Myocardial Infarction. Circulation research. 2014;114:1246–57. doi: 10.1161/CIRCRESAHA.114.302653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bujak M, Frangogiannis NG. The role of TGF-[beta] signaling in myocardial infarction and cardiac remodeling. Cardiovasc Res. 2007;74:184–95. doi: 10.1016/j.cardiores.2006.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kuo YC, Su CH, Liu CY, Chen TH, Chen CP, Wang HS. Transforming growth factor-beta induces CD44 cleavage that promotes migration of MDA-MB-435s cells through the up-regulation of membrane type 1-matrix metalloproteinase. International journal of cancer Journal international du cancer. 2009;124:2568–76. doi: 10.1002/ijc.24263. [DOI] [PubMed] [Google Scholar]
- 29.Kaya G, Rodriguez I, Jorcano JL, Vassalli P, Stamenkovic I. Selective suppression of CD44 in keratinocytes of mice bearing an antisense CD44 transgene driven by a tissue-specific promoter disrupts hyaluronate metabolism in the skin and impairs keratinocyte proliferation. Genes & development. 1997;11:996–1007. doi: 10.1101/gad.11.8.996. [DOI] [PubMed] [Google Scholar]
- 30.Teder P, Vandivier RW, Jiang D, Liang J, Cohn L, Pure E, et al. Resolution of lung inflammation by CD44. Science. 2002;296:155–8. doi: 10.1126/science.1069659. [DOI] [PubMed] [Google Scholar]
- 31.Takazoe K, Tesch GH, Hill PA, Hurst LA, Jun Z, Lan HY, et al. CD44-mediated neutrophil apoptosis in the rat. Kidney international. 2000;58:1920–30. doi: 10.1111/j.1523-1755.2000.00364.x. [DOI] [PubMed] [Google Scholar]
- 32.El Kebir D, Jozsef L, Pan W, Filep JG. Myeloperoxidase delays neutrophil apoptosis through CD11b/CD18 integrins and prolongs inflammation. Circulation research. 2008;103:352–9. doi: 10.1161/01.RES.0000326772.76822.7a. [DOI] [PubMed] [Google Scholar]
- 33.Molet S, Belleguic C, Lena H, Germain N, Bertrand CP, Shapiro SD, et al. Increase in macrophage elastase (MMP-12) in lungs from patients with chronic obstructive pulmonary disease. Inflammation research : official journal of the European Histamine Research Society [et al] 2005;54:31–6. doi: 10.1007/s00011-004-1319-4. [DOI] [PubMed] [Google Scholar]
- 34.Serrati S, Cinelli M, Margheri F, Guiducci S, Del Rosso A, Pucci M, et al. Systemic sclerosis fibroblasts inhibit in vitro angiogenesis by MMP-12-dependent cleavage of the endothelial cell urokinase receptor. The Journal of pathology. 2006;210:240–8. doi: 10.1002/path.2048. [DOI] [PubMed] [Google Scholar]
- 35.Wu L, Tanimoto A, Murata Y, Sasaguri T, Fan J, Sasaguri Y, et al. Matrix metalloproteinase-12 gene expression in human vascular smooth muscle cells. Genes to cells : devoted to molecular & cellular mechanisms. 2003;8:225–34. doi: 10.1046/j.1365-2443.2003.00628.x. [DOI] [PubMed] [Google Scholar]
- 36.Sam F, Sawyer DB, Chang L-F, Eberli FR, Ngoy S, Jain M, et al. Progressive left ventricular remodeling and apoptosis late after myocardial infarction in mouse heart. Am J Physiol Heart Circ Physiol. 2000;279:H422–H8. doi: 10.1152/ajpheart.2000.279.1.H422. [DOI] [PubMed] [Google Scholar]
- 37.Marchant DJ, Bellac CL, Moraes TJ, Wadsworth SJ, Dufour A, Butler GS, et al. A new transcriptional role for matrix metalloproteinase-12 in antiviral immunity. Nature medicine. 2014;20:493–502. doi: 10.1038/nm.3508. [DOI] [PubMed] [Google Scholar]
- 38.Kawana H, Karaki H, Higashi M, Miyazaki M, Hilberg F, Kitagawa M, et al. CD44 suppresses TLR-mediated inflammation. J Immunol. 2008;180:4235–45. doi: 10.4049/jimmunol.180.6.4235. [DOI] [PubMed] [Google Scholar]
- 39.Petrey AC, de la Motte CA. Hyaluronan, a Crucial Regulator of Inflammation. Frontiers in immunology. 2014;5:101. doi: 10.3389/fimmu.2014.00101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yu Q, Stamenkovic I. Localization of matrix metalloproteinase 9 to the cell surface provides a mechanism for CD44-mediated tumor invasion. Genes & development. 1999;13:35–48. doi: 10.1101/gad.13.1.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Horton MR, Shapiro S, Bao C, Lowenstein CJ, Noble PW. Induction and regulation of macrophage metalloelastase by hyaluronan fragments in mouse macrophages. J Immunol. 1999;162:4171–6. [PubMed] [Google Scholar]
- 42.Ma Y, Yabluchanskiy A, Lindsey ML. Neutrophil roles in left ventricular remodeling following myocardial infarction. Fibrogenesis & tissue repair. 2013;6:11. doi: 10.1186/1755-1536-6-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Maugeri N, Rovere-Querini P, Evangelista V, Godino C, Demetrio M, Baldini M, et al. An intense and short-lasting burst of neutrophil activation differentiates early acute myocardial infarction from systemic inflammatory syndromes. PloS one. 2012;7:e39484. doi: 10.1371/journal.pone.0039484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Krijnen PA, Nijmeijer R, Meijer CJ, Visser CA, Hack CE, Niessen HW. Apoptosis in myocardial ischaemia and infarction. Journal of clinical pathology. 2002;55:801–11. doi: 10.1136/jcp.55.11.801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Anversa P, Cheng W, Liu Y, Leri A, Redaelli G, Kajstura J. Apoptosis and myocardial infarction. Basic research in cardiology. 1998;93 (Suppl 3):8–12. doi: 10.1007/s003950050195. [DOI] [PubMed] [Google Scholar]
- 46.Tanaka Y, Nagai M, Date T, Okada T, Abe Y, Seki S, et al. Effects of bradykinin on cardiovascular remodeling in renovascular hypertensive rats. Hypertension research : official journal of the Japanese Society of Hypertension. 2004;27:865–75. doi: 10.1291/hypres.27.865. [DOI] [PubMed] [Google Scholar]