

Abstract
CONSPECTUS
In recent years, carbon–hydrogen bond functionalization has evolved from an organometallic curiosity to mainstream applications in the synthesis of complex natural products and drugs. The use of C–H bonds as a transformable functional group is advantageous because these bonds are the most abundant functionality in organic molecules. One-step conversion of these bonds to the desired functionality shortens synthetic pathways, saving reagents, solvents, and labor. Less chemical waste is generated as well, showing that this chemistry is environmentally beneficial. This Account describes the development and use of bidentate, monoanionic auxiliaries for transition-metal-catalyzed C–H bond functionalization reactions. The chemistry was initially developed to overcome the limitations with palladium-catalyzed C–H bond functionalization assisted by monodentate directing groups. By the use of electron-rich bidentate directing groups, functionalization of unactivated sp3 C–H bonds under palladium catalysis has been developed. Furthermore, a number of abundant base-metal complexes catalyze functionalization of sp2 C–H bonds. At this point, aminoquinoline, picolinic acid, and related compounds are among the most used and versatile directing moieties in C–H bond functionalization chemistry. These groups facilitate catalytic functionalization of sp2 and sp3 C–H bonds by iron, cobalt, nickel, copper, ruthenium, rhodium, and palladium complexes. Exceptionally general reactivity is observed, enabling, among other transformations, direct arylation, alkylation, fluorination, sulfenylation, amination, etherification, carbonylation, and alkenylation of carbon–hydrogen bonds. The versatility of these auxilaries can be attributed to the following factors. First, they are capable of stabilizing high oxidation states of transition metals, thereby facilitating the C–H bond functionalization step. Second, the directing groups can be removed, enabling their use in synthesis and functionalization of natural products and medicinally relevant substances. While the development of these directing groups presents a significant advance, several limitations of this methodology are apparent. The use of expensive second-row transition metal catalysts is still required for efficient sp3 C–H bond functionalization. Furthermore, a disadvantage is the need to install and subsequently remove the relatively expensive directing group.
1. INTRODUCTION
Carbon–hydrogen bond functionalization methodology has undergone explosive growth in recent years, evolving from an organometallic curiosity to applications in the synthesis of complex natural products.1 The use of C–H bonds as a transformable functional group is advantageous because these bonds are typically the most abundant functionality in organic molecules. Direct conversion of these bonds to the desired functionality shortens synthetic pathways, saving reagents, solvents, and labor. Less chemical waste is generated as well. These advantages were realized some time ago.2 However, obvious difficulties have prevented the widespread use of C–H bond functionalization methodology. First, most organic molecules contain many different C–H bonds. Achieving selective reactivity of the desired bond is often challenging. Second, alkane C–H bonds are unreactive because of their high bond energy and low acidity. Consequently, relatively few methods for regioselective functionalization of unactivated (not benzylic or α to heteroatom) C–H bonds have been developed. Third, the most common catalysts for C–H bond functionalization reactions are scarce second- and third-row transition metals. It is advantageous to replace them with abundant first-row transition metal catalysts. Fourth, reported methods for C–H bond functionalization often have limited substrate scope and require extensive optimization of the reaction conditions for every substrate class. Consequently, chemists applying these methodologies to the synthesis of natural products or medicinally relevant structures might choose to employ classical cross-coupling methodologies, which, while lengthening the synthetic schemes, follow predictable reactivity patterns and afford high yields. This Account summarizes our efforts to address these problems and develop user-friendly, general auxiliaries for C–H bond functionalization that could be widely used for synthetic purposes.
2. BRIEF HISTORICAL BACKGROUND
Scientists need to acknowledge and critically analyze the work of early investigators to avoid duplications and to ensure that new methodology is improved relative to already published chemistry. Solving previously solved problems and duplication or minor modifications of earlier work should be avoided if we wish to develop useful synthetic methods. Presumably, the first reactions that can be deemed “C–H activation” by transition metals were published in 1858 by Vogel Jr.3a and Quet.3b These papers report the formation of metal acetylides in the reaction of silver and copper salts with acetylene (lighting gas). Copper-promoted dimerization of alkynes, reported by Glaser, is likely the first example of C–H activation followed by C–H functionalization.3c These examples involve reactivity of comparatively acidic alkyne C–H bonds. In 1892, Volhard showed that thiophene reacts with HgCl2 to afford 2-thienylmercuric chloride.4 The arylmercury complex reacts with acid chlorides to produce 2-acetyl- and benzoylthiophenes in excellent yields. Thus, the C–H bond functionalization field is over a century old. Kharasch showed in 1931 that arenes are aurated by gold(III) chloride, demonstrating that transition-metal complexes can participate in C–H activation chemistry.5 Kharasch also reported that arene chlorination is catalyzed by AuCl3, which presumably is the first example of transition-metal catalysis in C–H bond functionalization. In these early reports, the regioselectivity of C–H activation/functionalization was determined by the acidity of the C–H bond, the electrophilic reactivity of the arene, or factors that influence the concerted metalation–deprotonation (CMD) pathway.6 Currently, the most widely used methods in C–H functionalization rely on directed C–H activation. A functional group coordinates to the transition metal and directs C–H activation to the ortho position of the substrate, resulting in regioselective C–H bond functionalization.7,8 This concept originates in cyclometalation reactions discovered by Cope and Kleiman and cobalt-catalyzed imine carbonylation reported by Murahashi in 1955.9 Transition-metal-catalyzed alkane functionalization in which both C–H bond cleavage and functionalization occur at the metal center was disclosed in 1969 by Shilov.10 He showed that the reaction of alkanes with platinum salts affords a mixture of alcohols and alkyl halides. Intermolecular examples of C–H bond activation by well-defined late-transition-metal complexes were disclosed in the 1980s when Bergman11 and Graham12 reported the oxidative addition of unactivated alkane C–H bonds to Cp*(PMe3)Ir fragments to afford stable alkyl hydride products. These discoveries formed the basis for the achievements in C–H bond functionalization that have occurred in the last 15 years. The ultimate test of chemical methodology is its successful application in the synthesis of complex molecules. Examples of total syntheses employing C–H bond functionalization show that the methodology is maturing and may become complementary to traditional cross-coupling procedures.1c
3. DESIGN OF BIDENTATE, MONOANIONIC AUXILIARIES AND PALLADIUM CATALYSIS
3.1. Early Work
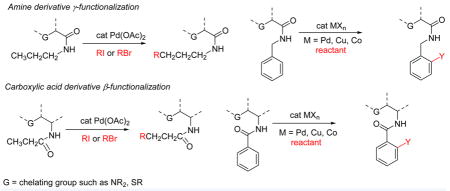
Starting in 2005, we developed a number of palladium-catalyzed arylations of directing-group-containing arenes.13 This chemistry is based on early observations of Tremont and Liebeskind describing palladium-promoted ortho alkylation of imine sp2 C–H bonds.14 The methodology is remarkably general, and under nearly identical conditions anilides, benzylamines, benzoic acid amides, benzoic acids, and 2-substituted pyridines can be ortho-arylated under palladium catalysis. However, attempts to develop directed sp3 C–H bond functionalization were not successful, and at best low conversions were observed, showing the need for new, more efficient directing groups. The following issues were taken into auxiliary design considerations. First, cyclometalations are more facile if a stronger directing group is used or bidentate coordination of the metal is possible.15 Second, a removable directing group is required in order to increase the synthetic applicability of C–H bond funtionalization reactions. Third, since the arylations described above likely proceed through high-valent palladium complexes, an anionic auxiliary would help in stabilizing high-energy palladium(III) or -(IV) species.16 Consequently, bidentate coordination in which one ligand is pyridine may facilitate the C–H activation reaction, and an electron-rich anionic auxiliary would facilitate the C–H bond functionalization step proceeding via a high-valent metal intermediate. These considerations led to development of aminoquinoline and picolinic acid auxiliaries for the arylation of carboxylic acid amine and derivatives (Scheme 1).17 The arylation regiochemistry is dictated by the formation of a five-membered palladated chelate (1-2 or 1-5).
Scheme 1.

Auxiliary Design
Our initial work showed that the picolinic acid auxiliary is useful for amine γ-arylation (1-10 to 1-11), and the aminoquinoline auxiliary effects carboxylic acid β-arylation (1-7 to 1-8). The structurally similar 2-aminomethylpyridine derivative 1-9 was degraded under the reaction conditions. These arylations were among the first examples of palladium-catalyzed transformations of unactivated sp3 C–H bonds into C–C bonds with no adjacent tertiary centers. The next steps were to explore the generality of the approach, to expand the synthetic scope of C–H bond functionalization, and to investigate the organometallic chemistry of the bidentate auxiliaries. A bidentate, monoanionic auxiliary was used in copper-promoted sp2 C–H bond functionalization in 1990.18a
3.2. New Auxiliaries and Palladium Catalysis
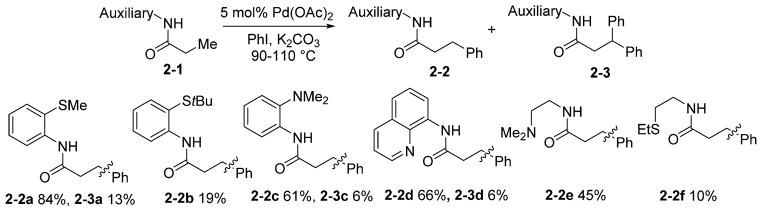
Further experiments were directed at understanding the structural requirements of the bidentate auxiliaries and finding bases other than AgOAc to promote arylation.19 After a short optimization, it was possible to replace AgOAc with simple inorganic salts such as K2CO3 or CsOAc. However, in several cases AgOAc is superior, and some optimization of the reaction conditions is required for every substrate class to obtain high yields of arylated products. The successful auxiliary requires a coordinating group and an acidic NH group that can coordinate to palladium in a bidentate fashion to form a five-membered-ring chelate (Scheme 2). 2-Thiomethylaniline, 2-dimethylami-noaniline, and aminoquinoline auxiliaries were efficient and afforded products 2-2 in high yields with acceptable selectivities for monoarylation. Thioethers more hindered than 2-thiomethylaniline gave low arylation yields (2-2b). Aliphatic auxiliaries were less efficient, and low conversions were obtained (2-2e and 2-2f).
Scheme 2.
Auxiliaries for sp3 C–H Bond Arylation
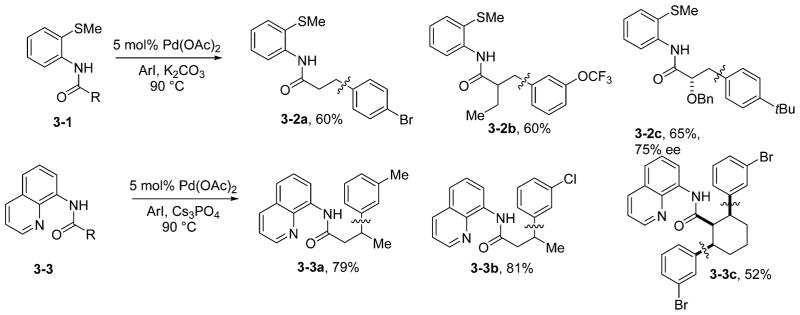
Monoarylation of methyl groups is best accomplished with amides containing the 2-thiomethylaniline auxiliary. If mono-arylation of methylene or diarylation of methyl groups is required, the aminoquinoline auxiliary can be used (Scheme 3). The reactions are highly regioselective, with one regioisomer being obtained in nearly all cases. For cyclic compounds such as 3-3c, high diastereoselectivity for the formation of the all-cis isomer is observed. Electron-rich (3-2c and 3-3a) and electron-poor (3-2a, 3-2b, 3-3b, and 3-3c) aryl iodides can be used. In most instances, ortho substitution on the aryl iodide is not tolerated. Arylation of sp2 C–H bonds in aminoquinoline benzamides was not pursued further since unprotected benzoic acids can be reacted with both aryl iodides and chlorides under palladium catalysis.13d Direct arylation of benzoic acids is preferable to functionalization of aminoquinoline benzamides since the two additional steps to introduce and remove the directing group are not needed. Nickel- and iron-catalyzed arylation of sp3 bonds in aminoquinoline amides has been reported.18b,c While these reactions employ abundant first-row transition metal catalysts, they appear to be limited to substrates possessing quaternary centers adjacent to a carbonyl group, and arylation of most secondary C–H bonds is inefficient.
Scheme 3.
Arylation of sp3 C–H Bonds in Carboxamides
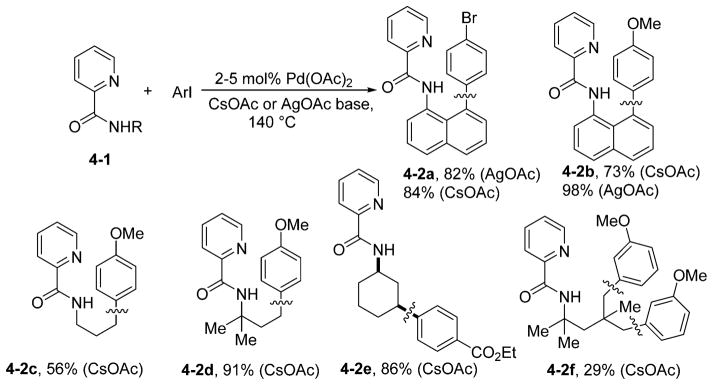
Arylation of C–H bonds in picolinamides can be accomplished under conditions that do not require AgOAc as an additive (Scheme 4).20 Since free benzylamines can be arylated by ArI under palladium catalysis,13c the use of the picolinic acid directing group in these substrates was not extensively investigated. However, Ag(I) oxidizes 1-naphthylamine. Therefore, the arylation of 1-naphthylamine picolinamide, which is stable in the presence of Ag(I), was investigated. Both AgOAc and CsOAc can be used as the base (4-2a and 4-2b). However, higher yields are obtained with AgOAc, and lower catalyst loadings can be used. Imines derived from 8-aryl-1-naphthylamines are useful as ligands for late-transition-metal-catalyzed olefin polymerization.22 Aliphatic C–H bonds can be arylated using CsOAc as the base (4-2c to 4-2f). Even methylene groups are reactive (4-2e), and δ-arylation is possible if the γ-positions are unavailable, although the yield is lower (4-2f).
Scheme 4.
Picolinamide-Directed Arylation of C–H bonds
Our early reaction conditions that employed AgOAc as the base were not successful for alkylation of C–H bonds because of the background reaction between alkyl iodides and Ag(I). The new conditions using simple alkali-metal bases allowed for the alkylation of both sp2 and sp3 C–H bonds since the competitive destruction of alkyl iodides is slow (Scheme 5).19,20 We determined that the optimal auxiliary for C–H bond alkylation of carboxylic acid derivatives is 8-aminoquinoline and that for alkylation of amine derivatives the picolinamide auxiliary can be employed. Benzamide sp2 C–H bonds can be alkylated by simple (5-2a) and functionalized (5-2b) alkyl iodides as well as benzyl bromides (5-2c). Primary sp3 C–H bonds can be allylated (5-2d), benzylated, and alkylated (5-2f and 5-2g). Secondary sp3 C–H bonds are reactive, but the alkylations proceed in low yield (5-2h). Benzylamine picolinamides are alkylated (5-4a) and benzylated (5-4b) in excellent yields. Alkylation of 1-naphthylamine picolinamide affords the product in 49% yield (5-4c). Alkylation by employing a secondary alkyl iodide gives the product in modest yield (5-4d). Alkylation of sp3 C–H bonds in picolinamides occurs in low yield (5-4e). Iron- and nickel-catalyzed alkylation of sp2 C–H bonds in aminoquinoline and other bidentate benzamides has been reported, showing that abundant first-row transition metals participate in sp2 C–H bond functionalization.18d,e,24c Chen has shown that amino-quinoline amide methylene C–H bonds can be alkylated by activated electrophiles.23c
Scheme 5.

Alkylation of C–H Bonds
3.3. Amino Acid Functionalization
Bidentate-auxiliary-directed C–H functionalization is applicable to the synthesis of complex structures. Thus, amino acid derivative functionalization is feasible (Scheme 6).21 Alanine derivative 6-1 possessing a 2-thiomethylaniline directing group can be selectively monoarylated with a number of different aryl iodides, affording phenylalanine derivatives 6-2. Simple aryl groups such as 2-naphthyl can be introduced in high yield (6-2a). Even azido functionality is tolerated (6-2d). Heterocyclic iodides give arylated products in good yields (6-2b and 6-2c). The directing group can be cleaved to afford benzthiophenylalanine derivative 6-3. Arylation of 6-1 is perhaps the most convergent method for the synthesis of modified phenylalanines, as nearly any modification can be achieved using a single starting material.
Scheme 6.

Amino Acid Functionalization
Arylation of methylene groups requires the presence of an aminoquinoline directing group. Thus, phenylalanine derivative 6-4 can be arylated with 2-iodothiophene, affording the product 6-5a as a single diastereomer. Diastereoselective arylation was achieved with lysine and leucine derivatives (6-5b and 6-5c). Removal of the directing group with BF3·Et2O in MeOH affords protected amino acid 6-6. Alkylation of 6-7 gives lipidic amino acid derivative 6-8 in modest yield.
Several groups have reported the synthesis of modified amino acids by directed arylation or alkylation.23 Corey disclosed the arylation and acetoxylation of amino acid aminoquinoline amides in 2006.23a Others have developed methods for aminoquinoline-directed alkylation and arylation of amino acid derivatives.23b,c,e Other directing moieties have also been used for functionalization of amino acid derivatives, including an interesting 2-pyridylsulfonyl group.23d,24a,b,d
3.4. Mechanistic Considerations
Several potential reaction intermediates and related organometallic complexes were studied (Scheme 7).19 The reaction of 8-aminoquinoline pivalamide with palladium acetate in acetonitrile affords the cyclometalated complex 7-2. The cyclometalated complex is formed under catalytic arylation conditions. Upon dissolution of 7-2 in CD3CO2D at room temperature, complete H/D exchange of the aliphatic hydrogens was observed within minutes. The loss of alkyl hydrogen resonances in 7-2 as a function of time was measured by NMR spectroscopy at −35 °C in CD2Cl2. A first-order rate constant (k) value of 2.0 × 10−4 s−1 was obtained, corresponding to ΔG‡= 18 kcal/mol. Consequently, the C–H activation step in aminoquinoline amides is very efficient even for sp3 C–H bonds. Complex 7-2 was reacted with p-tolyl iodide in acetone at room temperature. Following treatment with HI, a mixture of mono- and disubstitution products was obtained. This result shows that monomeric palladated complexes are capable of reacting with aryl iodides under mild conditions and are competent intermediates in the catalytic cycle. The reaction of related 7-5 with bromine at −78 °C in CH2Cl2 afforded green palladium(IV) dibromide 7-6 in good yield. Complex 7-6 is a rare example of a relatively stable palladium(IV) alkyl halide. The dianionic, tridentate NNC ligand stabilizes the Pd(IV) oxidation state, allowing efficient catalytic functionalization of C–H bonds and isolation of typically unstable high-valent Pd complexes.16
Scheme 7.

Mechanistic Considerations
A possible reaction mechanism is presented in Scheme 8. The formation of palladium amide 8-2 is followed by the C–H activation to afford 8-3. Substrates that do not have the N–H group are inactive, showing the importance of the anionic amide ligand. A κ2N,N′ palladium acetate–benzylpicolinamide complex was isolated in a related system.17 The diastereoselectivity in the amino acid arylation is set during the C–H activation step (8-3 to 8-3), as evidenced by H/D exchange experiments.21 Complex 8-3 then undergoes reaction with the aryl iodide to afford Pd(IV) species 8-4. While Pd(III) intermediacy cannot be excluded, the mild reaction conditions for the reaction of 7-2 with ArI and successful arylations in coordinating solvents such as CH3CN that could break up Pd(III) dimers argue for a Pd(IV) intermediate. Oxidative addition is followed by reductive elimination from 8-4 that proceeds with retention of configuration. Ligand exchange affords 8-6 and regenerates 8-2.
Scheme 8.

Possible Reaction Mechanism
4. COPPER CATALYSIS
4.1. General Considerations
We hypothesized that bidentate auxiliaries would promote copper-catalyzed functionalization of sp2 C–H bonds on the basis of the following considerations: (1) copper-promoted C–H activation has been reported in a macrocyclic polyamine system,25 (2) conversion of Cu(III)–C bonds to various functionalities has been demonstrated in the same system,26a–c and (3) both macrocyclic amine and bidentate auxiliaries stabilize highvalent copper intermediates. Furthermore, an early report by Reinaud showed that copper(II) mediates aromatic hydroxylation by trimethylamine N-oxide.18a Additionally, in an early work, Yu showed that 2-phenylpyridine can be functionalized under copper catalysis.27 Presumably, these reactions would be viable for substrates possessing removable directing groups.
4.2. Sulfenylation of sp2 C–H Bonds
Since disulfides can oxidize Cu(I) to Cu(III),28 we decided to investigate directed sulfenylation of sp2 C–H bonds.29 At the time of our report, no examples of transition-metal-catalyzed C–H bond trifluoromethylsulfenylation had been reported. The reasoning proved fruitful, as bis(trifluoromethyl) disulfide was shown to be an effective reagent in aminoquinoline-directed sp2 C–H bond sulfenylation. Amides containing electron-donating and -withdrawing groups on the benzamide ring are suitable substrates, providing the products in good yields (Scheme 9). Moreover, the reaction shows excellent functional group tolerance that is characteristic for most copper-catalyzed C–H bond functionalizations. Heterocyclic substrates are also reactive. 3-Thiophenecarboxylic acid amino-quinoline amide was converted to disubstituted product 9-2a in synthetically useful yield. Other disulfides, such as di-n-butyl disulfide, di-tert-butyl disulfide, and bis(p-nitrophenyl)disulfide are also active sulfenylating reagents. Benzylamine derivatives can also be sulfenylated by employing Cu(OAc)2, disulfide reagents, and a picolinic acid directing group. However, these substances are less reactive than aminoquinoline amides, requiring an excess of copper(II) acetate and higher reaction temperatures. Compounds lacking substitution at the α-position of the benzylamine afforded the products in low yields.
Scheme 9.

Sulfenylation of sp2 C–H Bonds
4.3. Amination and Etherification of sp2 C–H Bonds
Well-defined Cu(III) complexes react with N-nucleophiles to form C–N bonds.26a According to the considerations described earlier, directed amination of sp2 C–H bonds should be feasible. Gratifyingly, aminoquinoline-directed, copper-catalyzed sp2 C–H bond amination is possible (Scheme 10).30 The amination is successful for both electron-rich and electron-poor amides, and the reaction tolerates most common substituents. In contrast to copper-promoted sulfenylation, where clean monofunctionalization was impossible, amination selectively delivers monofunctionalization products at the less sterically demanding position. Secondary amines possessing many functionalities, such as ester, ether, cyano, and NHBoc, afford coupling products in excellent yields. Interestingly, selective coupling with amine NH in the presence of an amide NH was observed (10-2g). Reactions with primary amines are low-yielding (10-2h). Direct coupling of a C–H bond with an N–H bond to afford C–N functionality is perhaps the most direct method for amine synthesis. Iron-catalyzed, aminoquinoline-directed amination of sp2 C–H bonds by N-chloroamines was later disclosed by Nakamura.26d Furthermore, Yu has reported copper-promoted, bidentate-auxiliary directed amination of arene C–H bonds.26e
Scheme 10.
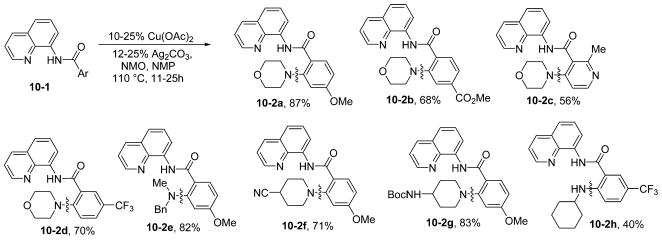
Amination of sp2 C–H Bonds
Stahl has reported methoxylation of aminoquinoline benzamide by employing methanol solvent and excess Cu(OAc)2.26f We have shown that auxiliary-assisted alkoxylation and phenoxylation of β-sp2 C–H bonds of benzoic acid derivatives and γ-sp2 C–H bonds of amine derivatives are possible (Scheme 11).31 The reaction employs (CuOH)2CO3 as the catalyst; air as the oxidant; phenol or alcohol as the coupling partner; DMF, pyridine, or DMPU as the solvent; and K2CO3, tetramethylguanidine, or K3PO4 as the base at 90–130 °C. The reactions are operationally simple, and in most cases they are run simply in open vessels under air. The method is advantageous compared with other sp2 C–H bond etherification procedures because it utilizes an inexpensive malachite catalyst, air as the oxidant, and a removable directing group. Both aminoquinoline and picolinamide directing groups can be employed, but aminoquinoline-directed reactions are more efficient. The method shows very high generality and functional group tolerance, with ester, amine, nitro, nitrile, and halogen (including iodide) functionalities being compatible with the reaction conditions.
Scheme 11.
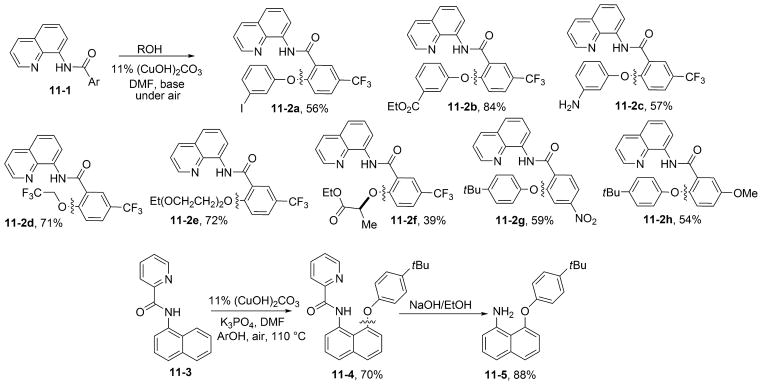
Etherification of sp2 C–H Bonds
4.4. Fluorination of sp2 C–H Bonds
Ribas has reported copper-catalyzed nucleophilic aryl fluorination and aryl halide exchange in a highly geometrically constrained system that stabilizes Cu(III) intermediates.26c Consequently, C–F reductive elimination from Cu(III) occurs under mild conditions. Since aminoquinoline amides stabilize high oxidation states in transition metals, we speculated that copper-catalyzed, aminoquinoline-directed sp2 C–H bond fluorination should be possible. On the basis of these considerations, we developed a method for direct, copper-catalyzed, auxiliary-assisted fluorination of β-sp2 C–H bonds of aminoquinoline benzamides and γ-sp2 C–H bonds of benzylamine picolinamides (Scheme 12).32 The reaction employs CuI as the catalyst and AgF as the nucleophilic fluoride source. The method allows for selective mono- or difluorination of benzamide substrates depending on the reaction temperature and the amounts of CuI catalyst, oxidant, and AgF. As shown before, picolinamides show decreased reactivity compared with aminoquinoline derivatives. The reaction shows excellent functional group tolerance and provides a straightforward way for the preparation of ortho-fluorinated benzoic acids via C–H functionalization under copper catalysis.
Scheme 12.

Fluorination of C–H Bonds
5. COBALT CATALYSIS
Coupling of sp2 C–H bonds with carbon–carbon multiple bonds under second-row transition metal catalysis is well-known.1h,24h Replacement of expensive second-row metal catalysts with readily available cobalt complexes would be beneficial. Catalysis by low-valent cobalt has been demonstrated by Yoshikai.1f However, the use of terminal alkynes or simple alkenes that can be isomerized by metal hydride species is rare. Furthermore, the reductive conditions of low-valent metal catalysis limit the functional group tolerance. High-valent cobalt-catalyzed coupling of alkynes with directing-group-containing indoles is known.33 We speculated that 8-amino-quinoline and picolinic acid auxiliaries would promote cobalt-catalyzed coupling of sp2 C–H bonds with alkynes since Co(III) activates sp2 C–H bonds and carbon–carbon multiple bonds insert into Co(III)–C bonds.34 The coupling of alkynes with aminoquinoline benzamides proceeds in trifluoroethanol solvent and requires simple Co(II) acetate as the catalyst, Mn(OAc)2 as a cocatalyst, and oxygen from air as a terminal oxidant (Scheme 13).35 The reaction is successful for both internal (13-2a to 13-2e, 13-2g, and 13-2h) and terminal (13- 2f) alkynes. Many functionalities on alkynes are compatible with the reaction, including free hydroxyl groups (13-2e). The reaction also has excellent functional group tolerance with respect to the benzamide coupling component. Picolinamides are also reactive (13-4), and in the case of 1-naphthylamine picolinamide, an acyclic product was isolated.
Scheme 13.

Coupling with Alkynes
It is likely that alkenes would also participate in similar coupling reactions because of their ability to insert into Co(III)–C bonds.34 On the basis of previous results, we decided to use a readily available cobalt(II) catalyst in trifluoroethanol solvent with sodium pivalate as the base, manganese acetate as a cooxidant, and oxygen from air as a terminal oxidant.36 The reaction scope is presented in Scheme 14. Ethylene (14-2e), mono- (14-2a to 14-2d, 14-2f, and 14-2g), and disubstituted (14-2h) alkenes are reactive. The reaction possesses excellent funtional group tolerance on both the alkene and amide coupling partners. Vinylamides are also reactive (14-4). Isomerizable alkenes such as 1-hexene give products in high yields (14-2f). The reactions proceed at room temperature.
Scheme 14.

Coupling with Alkenes
Success with cobalt-catalyzed alkyne and alkene couplings led us to investigate directed carbonylation of aminoquinoline benzamides (Scheme 15).37 Carbonylation of aminoquinoline benzamide C–H bonds proceeded at room temperature in trifluoroethanol solvent using oxygen from air as an oxidant and required Mn(OAc)3 as an additive. High functional group tolerance is observed. Bromo (15-2b), cyano (15-2e), and ester (15-2f) substituents are tolerated. Scale-up of the reaction to at least 5 mmol is feasible. The directing group can be easily removed by treatment with ammonia, affording a high yield of 4-methylphthalimide (15-6). The reaction mechanism likely includes aminoquinoline-directed C–H activation by cobalt-(III) species followed by migratory insertion of alkyne, alkene, or CO. Reductive elimination and reoxidation of Co(I) to Co(III) releases the product and regenerates the active catalyst.
Scheme 15.
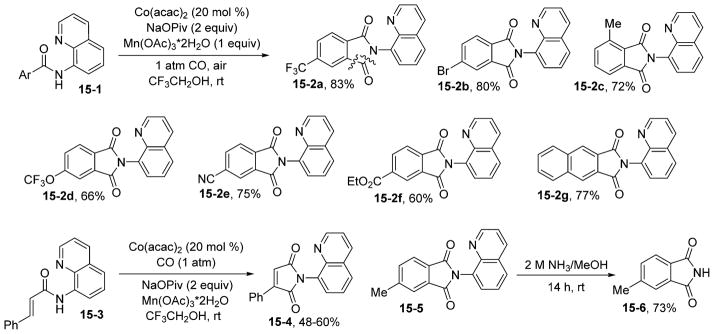
Carbonylation of sp2 C–H Bonds
6. SUMMARY AND OUTLOOK
This Account has described the development and use of bidentate, monoanionic auxiliaries for transition-metal-catalyzed C–H bond functionalization reactions. The chemistry was developed to overcome the limitations with palladium-catalyzed C–H bond functionalization assisted by relatively weak monodentate directing groups, namely, their inability to promote reactivity of unactivated sp3 C–H bonds.13 By the use of stronger, electron-rich bidentate directing groups, a general method for palladium-catalyzed arylation and alkylation of sp3 C–H bonds was developed. This chemistry has proven to be remarkably general and has been extensively used in the total synthesis of complex natural products.23b,24e–g On the basis of mechanistic considerations of palladium-catalyzed reactions, C–H functionalization directed by the bidentate coordinating groups has been extended to first-row transition metals. At this point, aminoquinoline, picolinic acid, and their derivatives are among the most used and versatile directing groups in C–H bond functionalization chemistry, allowing the use of sp2 and sp3 C–H bonds and catalysis by iron, cobalt, nickel, copper, ruthenium, rhodium, and palladium. The general limitations and potential improvements of this chemistry are as follows. First, the use of expensive second-row transition metal catalysts is still required for sp3 C–H bond functionalization. First-row transition metals are mostly employed in sp2 C–H bond functionalization. Their use in conversions of sp3 C–H bonds has severe limitations, such as the inability to functionalize secondary C–H bonds, the requirement of quaternary centers adjacent to reactive positions, and the need to use over-stoichiometric amounts of metal promoter and second-row transition metal (silver) additives. New auxiliaries or new reaction concepts are required for the efficient use of first-row-metal catalysis in transformations of unactivated sp3 C–H bonds. Second, a serious synthetic issue is the need to install and then remove the relatively expensive aminoquinoline (or other) auxiliary. It would be far more convenient if carboxylic acid or amine C–H bonds could be functionalized directly. While palladium-catalyzed functionalization of sp2 C–H bonds in benzoic acids and benzylamines is known,13c,d thus negating the need to develop aminoquinoline- and picolinamide-directed functionalization of aromatic C–H bonds by second-row transition metals, the corresponding reactions of aliphatic compounds are rare.38 Thus, the use of simpler directing groups under second- or first-row transition metal catalysis would be highly beneficial.
Acknowledgments
We thank the Welch Foundation (E-1571, E-0044) and NIGMS (R01GM077635) for supporting this research.
Biographies
Olafs Daugulis received an undergraduate degree from Riga Technical University in Latvia. After earning a Ph.D. from the University of Wisconsin under the guidance of Prof. Edwin Vedejs in 1999, he worked as a postdoctoral associate with Prof. Maurice Brookhart at the University of North Carolina at Chapel Hill. He joined the chemistry faculty at the University of Houston in 2003. He was promoted to Full Professor and awarded the Robert A. Welch Chair in Chemistry in 2014. He is interested in developing methods for carbon–hydrogen bond functionalization and the application of organometallic chemistry to organic chemistry problems.
James Roane earned a B.S. degree in Chemistry at the University of New Hampshire in 2011. He is currently a fourth-year Ph.D. candidate in Prof. Daugulis’ lab. He contributed to the development of the sp2 C–H amination methodology and discovered the method for copper-catalyzed C–H bond etherification.
Ly Dieu Tran graduated from the University of Technology in Ho Chi Minh City, Vietnam, with a B.S. degree in Chemical Engineering. She then moved to Texas and earned her Ph.D. in Organic Chemistry at the University of Houston, focusing on C–H bond functionalization using transition metal catalysis. She is currently a postdoctoral researcher in Prof. Adam Matzger’s group at the University of Michigan.
Footnotes
The authors declare no competing financial interest.
References
- 1.(a) Engle KM, Mei TS, Wang X, Yu JQ. Bystanding F+ Oxidants Enable Selective Reductive Elimination from High-Valent Metal Centers in Catalysis. Angew Chem, Int Ed. 2011;50:1478–1491. doi: 10.1002/anie.201005142. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Lyons TW, Sanford MS. Palladium-Catalyzed Ligand-Directed C–H Functionalization Reactions. Chem Rev. 2010;110:1147–1169. doi: 10.1021/cr900184e. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Gutekunst WR, Baran PS. C–H Functionalization Logic in Total Synthesis. Chem Soc Rev. 2011;40:1976–1991. doi: 10.1039/c0cs00182a. [DOI] [PubMed] [Google Scholar]; (d) Ackermann L. Carboxylate-Assisted Transition-Metal-Catalyzed C–H Bond Functionalizations: Mechanism and Scope. Chem Rev. 2011;111:1315–1345. doi: 10.1021/cr100412j. [DOI] [PubMed] [Google Scholar]; (e) Baudoin O. Transition Metal-Catalyzed Arylation of Unactivated C(sp3)–H Bonds. Chem Soc Rev. 2011;40:4902–4911. doi: 10.1039/c1cs15058h. [DOI] [PubMed] [Google Scholar]; (f) Ge K, Yoshikai N. Low-Valent Cobalt Catalysis: New Opportunities for C–H Functionalization. Acc Chem Res. 2014;47:1208–1219. doi: 10.1021/ar400270x. [DOI] [PubMed] [Google Scholar]; (g) Rouquet G, Chatani N. Catalytic Functionalization of C(sp2)–H and C(sp3)–H Bonds by Using Bidentate Directing Groups. Angew Chem, Int Ed. 2013;52:11726–11743. doi: 10.1002/anie.201301451. [DOI] [PubMed] [Google Scholar]; (h) Colby DA, Bergman RG, Ellman JA. Rhodium-Catalyzed C–C Bond Formation via Heteroatom-Directed C–H Bond Activation. Chem Rev. 2010;110:624–655. doi: 10.1021/cr900005n. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Zhang F, Spring DR. Arene C–H Functionalisation Using a Removable or Traceless Directing Group Strategy. Chem Soc Rev. 2014;43:6906–6919. doi: 10.1039/c4cs00137k. [DOI] [PubMed] [Google Scholar]
- 2.Arndtsen BA, Bergman RG, Mobley TA, Peterson TH. Selective Intermolecular Carbon–Hydrogen Bond Activation by Synthetic Metal Complexes in Homogeneous Solution. Acc Chem Res. 1995;28:154–162. [Google Scholar]
- 3.(a) Vogel A., Jr . Kunst-und Gewerbeblatt des Polytechnischen Vereins für das Konigreich Bayern. 1858. Chemisch-technische Mittheilungen; pp. 23–33. [Google Scholar]; (b) Quet M. Note sur un phénomène de polarité dans la décomposition des gaz par l’étincelle électrique. C R Hebd Seances Acad Sci. 1858;46:903–905. [Google Scholar]; (c) Glaser C. Beiträge zur Kenntniss des Acetenylbenzolsenylbenzols. Ber Dtsch Chem Ges. 1869;2:422–424. [Google Scholar]
- 4.Volhard J. Ueber Verbindungen des Thiophens, seiner Homologen und einiger Ketone mit Quecksilberchlorid. Liebigs Ann Chem. 1892;267:172–185. [Google Scholar]
- 5.Kharasch MS, Isbell HS. The Chemistry of Organic Gold Compounds. III. Direct Introduction of Gold into the Aromatic Nucleus. J Am Chem Soc. 1931;53:3053–3059. [Google Scholar]
- 6.Gorelsky SI, Lapointe D, Fagnou K. Analysis of the Palladium-Catalyzed (Aromatic)C–H Bond Metalation–Deprotonation Mechanism Spanning the Entire Spectrum of Arenes. J Org Chem. 2011;77:658–668. doi: 10.1021/jo202342q. [DOI] [PubMed] [Google Scholar]
- 7.Ryabov AD. Mechanisms of Intramolecular Activation of Carbon–Hydrogen Bonds in Transition-Metal Complexes. Chem Rev. 1990;90:403–424. [Google Scholar]
- 8.For another mechanistic possibility, see: Zhang X, Kanzelberger M, Emge TJ, Goldman AS. Selective Addition to Iridium of Aryl C–H Bonds Ortho to Coordinating Groups. Not Chelation-Assisted. J Am Chem Soc. 2004;126:13192–13193. doi: 10.1021/ja046476q.
- 9.(a) Kleiman JP, Dubeck M. The Preparation of Cyclo-pentadienyl [o-(Phenylazo)phenyl]nickel. J Am Chem Soc. 1963;85:1544–1545. [Google Scholar]; (b) Cope AC, Siekman RW. Formation of Covalent Bonds from Platinum or Palladium to Carbon by Direct Substitution. J Am Chem Soc. 1965;87:3272–3273. [Google Scholar]; (c) Murahashi S. Synthesis of Phthalimidines from Schiff Bases and Carbon Monoxide. J Am Chem Soc. 1955;77:6403–6404. [Google Scholar]
- 10.Gol’dshleger NF, Tyabin MB, Shilov AE, Shteinman AA. Activation of Saturated Hydrocarbons. Deuterium–Hydrogen Exchange in Solutions of Transition Metal Complexes. Russ J Phys Chem. 1969;43:1222–1223. [Google Scholar]
- 11.Janowicz AH, Bergman RG. Carbon–Hydrogen Activation in Completely Saturated Hydrocarbons: Direct Observation of M + R–H → M(R)(H) J Am Chem Soc. 1982;104:352–354. [Google Scholar]
- 12.Hoyano JK, Graham WAG. Oxidative Addition of the Carbon–Hydrogen Bonds of Neopentane and Cyclohexane to a Photochemically Generated Iridium(I) Complex. J Am Chem Soc. 1982;104:3723–3725. [Google Scholar]
- 13.(a) Daugulis O, Zaitsev VG. Anilide ortho-Arylation Using C–H Activation Methodology. Angew Chem, Int Ed. 2005;44:4046–4048. doi: 10.1002/anie.200500589. [DOI] [PubMed] [Google Scholar]; (b) Shabashov D, Daugulis O. Catalytic Coupling of C–H and C–I Bonds Using Pyridine as a Directing Group. Org Lett. 2005;7:3657–3659. doi: 10.1021/ol051255q. [DOI] [PubMed] [Google Scholar]; (c) Lazareva A, Daugulis O. Direct Palladium-Catalyzed ortho-Arylation of Benzylamines. Org Lett. 2006;8:5211–5213. doi: 10.1021/ol061919b. [DOI] [PubMed] [Google Scholar]; (d) Chiong HA, Pham QN, Daugulis O. Two Methods for Direct ortho-Arylation of Benzoic Acids. J Am Chem Soc. 2007;129:9879–9884. doi: 10.1021/ja071845e. [DOI] [PubMed] [Google Scholar]
- 14.McCallum JS, Gasdaska JR, Liebeskind LS, Tremont SJ. Palladium-Mediated 2,6-Dialkylation of N-Benzylidene Imines: Preparation of 2,6-Dialkylbenzaldehydes. Tetrahedron Lett. 1989;30:4085–4088. [Google Scholar]
- 15.Rimml H, Venanzi LM. The Facile Cyclometallation Reaction of 1,3-Bis[(diphenylphosphino)methyl]benzene. J Organomet Chem. 1983;259:C6–C7. [Google Scholar]
- 16.Canty AJ. Organopalladium and Platinum Chemistry in Oxidizing Milieu as Models for Organic Synthesis Involving the Higher Oxidation States of Palladium. Dalton Trans. 2009:10409–10417. doi: 10.1039/b914080h. [DOI] [PubMed] [Google Scholar]
- 17.Zaitsev VG, Shabashov D, Daugulis O. Highly Regioselective Arylation of sp3 C–H Bonds Catalyzed by Palladium Acetate. J Am Chem Soc. 2005;127:13154–13155. doi: 10.1021/ja054549f. [DOI] [PubMed] [Google Scholar]
- 18.(a) Reinaud O, Capdevielle P, Maumy M. Copper(II) Mediated Aromatic Hydroxylation by Trimethylamine N-Oxide. J Chem Soc, Chem Commun. 1990:566–568. [Google Scholar]; (b) Aihara Y, Chatani N. Nickel-Catalyzed Direct Arylation of C(sp3)–H Bonds in Aliphatic Amides via Bidentate-Chelation Assistance. J Am Chem Soc. 2014;136:898–901. doi: 10.1021/ja411715v. [DOI] [PubMed] [Google Scholar]; (c) Shang R, Ilies L, Matsumoto A, Nakamura E. β-Arylation of Carboxamides via Iron-Catalyzed C(sp3)–H Bond Activation. J Am Chem Soc. 2013;135:6030–6032. doi: 10.1021/ja402806f. [DOI] [PubMed] [Google Scholar]; (d) Song W, Lackner S, Ackermann L. Nickel-Catalyzed C–H Alkylations: Direct Secondary Alkylations and Trifluoroethylations of Arenes. Angew Chem, Int Ed. 2014;53:2477–2480. doi: 10.1002/anie.201309584. [DOI] [PubMed] [Google Scholar]; (e) Fruchey ER, Monks BM, Cook SP. A Unified Strategy for Iron-Catalyzed ortho-Alkylation of Carboxamides. J Am Chem Soc. 2014;136:13130–13133. doi: 10.1021/ja506823u. [DOI] [PubMed] [Google Scholar]
- 19.Shabashov D, Daugulis O. Auxiliary-Assisted Palladium-Catalyzed Arylation and Alkylation of sp2 and sp3 Carbon–Hydrogen Bonds. J Am Chem Soc. 2010;132:3965–3972. doi: 10.1021/ja910900p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nadres ET, Santos GIF, Shabashov D, Daugulis O. Scope and Limitations of Auxiliary-Assisted, Palladium-Catalyzed Arylation and Alkylation of sp2 and sp3 C–H Bonds. J Org Chem. 2013;78:9689–9714. doi: 10.1021/jo4013628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tran LD, Daugulis O. Nonnatural Amino Acid Synthesis by Carbon–Hydrogen Bond Functionalization Methodology. Angew Chem, Int Ed. 2012;51:5188–5191. doi: 10.1002/anie.201200731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang D, Nadres ET, Brookhart M, Daugulis O. Synthesis of Highly Branched Polyethylene Using “Sandwich” (8-p-Tolyl naphthyl α-diimine)nickel(II) Catalysts. Organometallics. 2013;32:5136–5143. [Google Scholar]
- 23.(a) Reddy SBV, Reddy LR, Corey EJ. Novel Acetoxylation and C–C Coupling Reactions at Unactivated Positions in α-Amino Acid Derivatives. Org Lett. 2006;8:3391–3394. doi: 10.1021/ol061389j. [DOI] [PubMed] [Google Scholar]; (b) Feng Y, Chen G. Total Synthesis of Celogentin C via Stereoselective C–H Activation. Angew Chem, Int Ed. 2010;49:958–961. doi: 10.1002/anie.200905134. [DOI] [PubMed] [Google Scholar]; (c) Zhang SY, Li Q, He G, Nack WA, Chen G. Stereoselective Synthesis of β-Alkylated α-Amino Acids via Palladium-Catalyzed Alkylation of Unactivated Methylene C(sp3)–H Bonds with Primary Alkyl Halides. J Am Chem Soc. 2013;135:12135–12141. doi: 10.1021/ja406484v. [DOI] [PubMed] [Google Scholar]; (d) Poveda A, Alonso I, Fernández-Ibáñez MA. Experimental and Computational Studies on the Mechanism of the Pd-Catalyzed C(sp3)–H γ-Arylation of Amino Acid Derivatives Assisted by the 2-Pyridylsulfonyl Group. Chem Sci. 2014;5:3873–3882. [Google Scholar]; (e) Chen K, Hu F, Zhang SQ, Shi BF. Pd(II)-Catalyzed Alkylation of Unactivated C(sp3)–H Bonds: Efficient Synthesis of Optically Active Unnatural α-Amino Acids. Chem Sci. 2013;4:3906–3911. [Google Scholar]
- 24.(a) Gong W, Zhang G, Liu T, Giri R, Yu JQ. Site-Selective C(sp3)–H Functionalization of Di-, Tri-, and Tetrapeptides at the N-Terminus. J Am Chem Soc. 2014;136:16940–16946. doi: 10.1021/ja510233h. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ye X, He Z, Ahmed T, Weise K, Akhmedov NG, Petersen JL, Shi X. 1,2,3-Triazoles as Versatile Directing Group for Selective sp2 and sp3 C–H Activation: Cyclization vs Substitution. Chem Sci. 2013;4:3712–3716. [Google Scholar]; (c) Gu Q, Al Mamari HH, Graczyk K, Diers E, Ackermann L. Iron-Catalyzed C(sp2)–H and C(sp3)–H Arylation by Triazole Assistance. Angew Chem, Int Ed. 2014;53:3868–3871. doi: 10.1002/anie.201311024. [DOI] [PubMed] [Google Scholar]; (d) Fan M, Ma D. Palladium-Catalyzed Direct Functionalization of 2-Aminobutanoic Acid Derivatives: Application of a Convenient and Versatile Auxiliary. Angew Chem, Int Ed. 2013;52:12152–12155. doi: 10.1002/anie.201306583. [DOI] [PubMed] [Google Scholar]; (e) Gutekunst WR, Baran PS. Total Synthesis and Structural Revision of the Piperarborenines via Sequential Cyclobutane C–H Arylation. J Am Chem Soc. 2011;133:19076–19079. doi: 10.1021/ja209205x. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Gutekunst WR, Gianatassio R, Baran PS. Sequential Csp3–H Arylation and Olefination: Total Synthesis of the Proposed Structure of Pipercyclobutanamide. Angew Chem, Int Ed. 2012;51:7507–7510. doi: 10.1002/anie.201203897. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Gutekunst WR, Baran PS. Applications of C–H Functionalization Logic to Cyclobutane Synthesis. J Org Chem. 2014;79:2430–2452. doi: 10.1021/jo4027148. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Roquet G, Chatani N. Ruthenium-catalyzed ortho-C–H bond alkylation of aromatic amides with α,β-unsaturated ketones via bidentate-chelation assistance. Chem Sci. 2013;4:2201–2208. [Google Scholar]
- 25.Ribas X, Jackson DA, Donnadieu B, Mahía J, Parella T, Xifra R, Hedman B, Hodgson KO, Llobet A, Stack TDP. Aryl C–H Activation by CuII To Form an Organometallic Aryl–CuIII Species: A Novel Twist on Copper Disproportionation. Angew Chem, Int Ed. 2002;41:2991–2994. doi: 10.1002/1521-3773(20020816)41:16<2991::AID-ANIE2991>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 26.(a) Huffman LM, Stahl SS. Carbon–Nitrogen Bond Formation Involving Well-Defined Aryl-Copper(III) Complexes. J Am Chem Soc. 2008;130:9196–9197. doi: 10.1021/ja802123p. [DOI] [PubMed] [Google Scholar]; (b) King AE, Huffman LM, Casitas A, Costas M, Ribas X, Stahl SS. Copper-Catalyzed Aerobic Oxidative Functionalization of an Arene C–H Bond: Evidence for an Aryl-Copper(III) Intermediate. J Am Chem Soc. 2010;132:12068–12073. doi: 10.1021/ja1045378. [DOI] [PubMed] [Google Scholar]; (c) Casitas A, Canta M, Solà M, Costas M, Ribas X. Nucleophilic Aryl Fluorination and Aryl Halide Exchange Mediated by a CuI/CuIII Catalytic Cycle. J Am Chem Soc. 2011;133:19386–19392. doi: 10.1021/ja2058567. [DOI] [PubMed] [Google Scholar]; (d) Matsubara T, Asako S, Ilies L, Nakamura E. Synthesis of Anthranilic Acid Derivatives through Iron-Catalyzed Ortho Amination of Aromatic Carboxamides with N-Chloroamines. J Am Chem Soc. 2014;136:646–649. doi: 10.1021/ja412521k. [DOI] [PubMed] [Google Scholar]; (e) Shang M, Sun SZ, Dai HX, Yu JQ. Cu(II)-Mediated C–H Amidation and Amination of Arenes: Exceptional Compatibility with Heterocycles. J Am Chem Soc. 2014;136:3354–3357. doi: 10.1021/ja412880r. [DOI] [PubMed] [Google Scholar]; (f) Suess AM, Ertem MZ, Cramer CJ, Stahl SS. Divergence between Organometallic and Single-Electron-Transfer Mechanisms in Copper(II)-Mediated Aerobic C–H Oxidation. J Am Chem Soc. 2013;135:9797–9804. doi: 10.1021/ja4026424. [DOI] [PubMed] [Google Scholar]
- 27.Chen X, Hao XS, Goodhue CE, Yu JQ. Cu(II)-Catalyzed Functionalizations of Aryl C–H Bonds Using O2 as an Oxidant. J Am Chem Soc. 2006;128:6790–6791. doi: 10.1021/ja061715q. [DOI] [PubMed] [Google Scholar]
- 28.Willert-Porada MA, Burton DJ, Baenziger NC. Synthesis and X- ray Structure of Bis (tri fluoromethyl) (N, N-diethyldithiocarbamato)copper, a Remarkably Stable Perfluoroalkylcopper(III) Complex. J Chem Soc, Chem Commun. 1989:1633–1634. [Google Scholar]
- 29.Tran LD, Popov I, Daugulis O. Copper-Promoted Sulfenylation of sp2 C–H Bonds. J Am Chem Soc. 2012;134:18237–18240. doi: 10.1021/ja3092278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tran LD, Roane J, Daugulis O. Directed Amination of Non-Acidic Arene C–H Bonds by a Copper–Silver Catalytic System. Angew Chem, Int Ed. 2013;52:6043–6046. doi: 10.1002/anie.201300135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Roane J, Daugulis O. Copper-Catalyzed Etherification of Arene C–H Bonds. Org Lett. 2013;15:5842–5845. doi: 10.1021/ol402904d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Truong T, Klimovica K, Daugulis O. Copper-Catalyzed, Directing Group-Assisted Fluorination of Arene and Heteroarene C–H Bonds. J Am Chem Soc. 2013;135:9342–9345. doi: 10.1021/ja4047125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ikemoto H, Yoshino T, Sakata K, Matsunaga S, Kanai M. Pyrroloindolone Synthesis via a Cp*CoIII-Catalyzed Redox-Neutral Directed C–H Alkenylation/Annulation Sequence. J Am Chem Soc. 2014;136:5424–5431. doi: 10.1021/ja5008432. [DOI] [PubMed] [Google Scholar]
- 34.Schmidt GF, Brookhart M. Implications of Three-Center, Two-Electron M–H–C Bonding for Related Alkyl Migration Reactions: Design and Study of an Ethylene Polymerization Catalyst. J Am Chem Soc. 1985;107:1443–1444. [Google Scholar]
- 35.Grigorjeva L, Daugulis O. Cobalt-Catalyzed, Aminoquinoline-Directed sp2 C–H Bond Alkenylation by Alkynes. Angew Chem, Int Ed. 2014;53:10209–10212. doi: 10.1002/anie.201404579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Grigorjeva L, Daugulis O. Cobalt-Catalyzed, Aminoquinoline-Directed Coupling of sp2 C–H Bonds with Alkenes. Org Lett. 2014;16:4684–4687. doi: 10.1021/ol502005g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Grigorjeva L, Daugulis O. Cobalt-Catalyzed Direct Carbonylation of Aminoquinoline Benzamides. Org Lett. 2014;16:4688–4690. doi: 10.1021/ol502007t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.(a) Giri R, Maugel N, Li JJ, Wang DH, Breazzano SP, Saunders LB, Yu JQ. Palladium-Catalyzed Methylation and Arylation of sp2 and sp3 C–H Bonds in Simple Carboxylic Acids. J Am Chem Soc. 2007;129:3510–3511. doi: 10.1021/ja0701614. [DOI] [PubMed] [Google Scholar]; (b) Dangel BD, Johnson JA, Sames D. Selective Functionalization of Amino Acids in Water: A Synthetic Method via Catalytic C–H Bond Activation. J Am Chem Soc. 2001;123:8149–8150. doi: 10.1021/ja016280f. [DOI] [PubMed] [Google Scholar]; (c) McNally A, Haffemayer B, Collins BSL, Gaunt MJ. Palladium-catalysed C–H activation of aliphatic amines to give strained nitrogen heterocycles. Nature. 2014;510:129–133. doi: 10.1038/nature13389. [DOI] [PubMed] [Google Scholar]