

Abstract
A detachable sulfonate-silica hybrid strong cation-exchange monolith was synthesized in a fused silica capillary, and used for solid phase extraction with on-line pH gradient elution during capillary zone electrophoresis-tandem mass spectrometry (CZE-MS/MS) proteomic analysis. Tryptic digests were prepared in 50 mM formic acid and loaded onto the strong cation-exchange monolith. Fractions were eluted using a series of buffers with lower concentration but higher pH values than the 50 mM formic acid background electrolyte. This combination of elution and background electrolytes results in both sample stacking and formation of a dynamic pH junction, and allows use of relatively large elution buffer volumes while maintaining reasonable peak efficiency and resolution. A series of five pH bumps were applied to elute E. coli tryptic peptides from the monolith, followed by analysis using CZE coupled to an LTQ-Orbitrap Velos mass spectrometer; 799 protein groups and 3,381 peptides were identified from 50 ng of the digest in a 2.5 hour analysis, which approaches the identification rate for this organism that was obtained with an Orbitrap Fusion. We attribute the improved numbers of peptide and protein identifications to the efficient fractionation by the on-line pH gradient elution, which decreased the complexity of the sample in each elution step and improved the signal intensity of low abundance peptides. We also performed a comparative analysis using a nanoACQUITY UltraPerformance LCH system. Similar numbers of protein and peptide identifications were produced by the two methods. Protein identifications showed significant overlap between the two methods, whereas peptide identifications were complementary.
Introduction
Capillary zone electrophoresis-tandem mass spectrometry (CZE-MS/MS) has attracted interest for proteome analysis because it provides fast and high-resolution separations.1–5 However, CZE tends to produce relatively poor concentration detection limits and relatively short separation windows, which have discouraged routine its use for bottom-up proteomic analysis.
A number of sample preconcentration techniques have been coupled to CZE to improve concentration detection limits. Many techniques employ electrokinetic methods, which have been summarized in recent reviews.8–12 Busnel et al. coupled a porous sheathless interface with transient-isotachophoresis (tITP) for CZE-electrospray ionization mass spectrometry.13 Concentration detection limits in the subnanomolar range were produced by using solutions of 10% acetic acid as background electrolyte and ammonium acetate (pH 4 and various ionic strengths) as leading electrolyte. More importantly, the separation window of E. coli tryptic digest was increased to ~60 min and the peak capacity was improved to 327 by applying vacuum to the injection end of the capillary during the separation. No peptides were identified in that experiment.
Our lab recently reported the use of stacking conditions for the injection of large volumes of samples while retaining high peak capacities.14 Stacking was achieved by preparing the sample in a low ionic strength solution (0.04% formic acid containing 40% acetonitrile) and using a higher ionic strength separation buffer (1 M acetic acid). Over 10,000 peptides and 2,100 proteins were identified from a single-shot analysis of 400 ng of a HeLa tryptic digest in 110 min, which represents the state-of-the-art in bottom-up proteomics using CZE.
Chromatographic preconcentration, also known as micro solid-phase extraction (MSPE), is an alternative to electrokinetic sample preconcentration. In MSPE, analytes are adsorbed onto the solid-phase sorbents from a very large sample volume and eluted in a much smaller volume of solvent, leading to improved concentration detection limits. Sample volumes can be much larger than the capillary volume, providing dramatic levels of preconcentration. MSPE can be combined with multistep elution and electrophoretic preconcentration techniques to further increase the sensitivity and separation window of CZE. For example, Lee et al. reported a staggered multistep elution SPE system for analysis of the tryptic digest of a set of six standard proteins.15 Multistep elution SPE prior to CZE not only extends the separation capacity for analysis of peptide mixtures but also decreases the number of co-eluting peptide, producing an increased sequence coverage compared to single-step elution SPE-CZE/MS/MS. Wang et al. described a C8 SPE, multistep elution, tITP CZE-MS/MS procedure for the proteomic analysis of a Pyrococcus furiosus tryptic digest.16 The C8 SPE-tITP method identified more peptides with lower XCorr (<4) than with direct CZE analysis.
Chromatographic packing materials have been widely used as sorbents for the preparation of SPE capillary columns.17,18 However, most studies employ a frit to retain the packing material. Frit fabrication is time consuming, and the frit can cause bubble formation during CZE separation. In addition, the length of the packed bed usually is less than 0.5 cm due to the high backpressure of the packed column, which limits the loading capacity.19
Monolith-based SPE capillary columns are an attractive alternative to particle based devices.20,21 Monolithic SPE columns produce better permeability than pack columns, which allows use of long columns without significant backpressure. Monoliths can be covalently anchored to the wall, which eliminates frits.
Monoliths generally fall into three classes: organic polymer-based,22 silica-based, and organic-silica hybrid monoliths. Organic polymer-based monoliths have good pH stability, and their surface chemistry is easy to tune due to the wide range of monomers that are available. However, they tend to have low surface area, although nanoparticle templating can improve surface area. 23 In contrast, silica-based monoliths demonstrate high mechanical stability and larger surface area.24 Unfortunately, surface modification of silica-based monolith can be time-consuming. Organic-silica hybrid monoliths have attracted attention in recent few years. These hybrid devices combine the ease of preparation and pH stability of the organic polymer-based monolith with the mechanical stability of silica-based monoliths.25,26
We recently reported the preparation of a sulfonate-silica hybrid strong cation-exchange (SCX) monolith at the inlet of the separation capillary.27 We loaded a sample in an acidic buffer and eluted the sample with a basic buffer, while using an acidic separation buffer. This combination resulted in formation of a dynamic pH junction, which produced a dramatic improvement in peak profile and concentration detection limit. By loading 21 μL of a 1 × 10−7 M angiotensin II solution, an enrichment factor of 3,000 compared to standard electrokinetic injection was achieved while retaining efficient electrophoretic performance (N = 44,000 plates). The system was also applied to proteomic analysis. By loading 5.5 μL of a 10−3 mg/mL E. coli digest, 109 proteins and 271 peptides were identified in a single-shot analysis. This monolith was covalently bound to the capillary wall, which required the use of an uncoated separation capillary. This uncoated capillary produced significant electro-osmotic flow, speeding the separation, while reducing the separation window and reducing the number of peptide identifications.
In this work, a SCX SPE device was prepared in an uncoated capillary and coupled through a zero-dead volume connection to the coated separation capillary. We optimized the eluting conditions for on-line pH gradient elution and evaluated the performance of the SCX SPE CE-MS/MS platform for analysis of E. coli digests. We also compared the results with capillary LC-MS/MS. This report appears to be the first to employ gradient pH elution from a SPE with CZE-MS/MS for proteomic analysis and the first to use a detachable sample preconcentrator. Our results approach the state-of-the-art in peptide and protein identification rates from a prokaryote using CZE-MS/MS.
METHODS AND MATERIALS
Reagents and Chemicals
Bovine pancreas TPCK-treated trypsin, poly(ethylene glycol) (PEG, MW=10 000), urea, tetramethoxysilane (TMOS), vinyltrimethoxysilane (VTMS), ammonium bicarbonate (NH4HCO3), ammonium formate, ammonium acetate, dithiothreitol (DTT), iodoacetamide (IAA), 3-sulfopropyl methacrylate potassium salt, 2,2′-azobis(2-isobutyronitrile) (AIBN), and acetic acid (HAc) were purchased from Sigma-Aldrich (St. Louis, MO). Formic acid (FA) was purchased from Fisher Scientific (Pittsburgh, PA). Methanol was purchased from Honeywell Burdick & Jackson (Wicklow, Ireland). Uncoated and LPA-coated fused silica capillaries (50 μm i.d.×150 μm o.d.) were purchased from Polymicro Technologies (Phoenix, AZ). Water was deionized by a Nano Pure system from Thermo Scientific (Marietta, OH).
Preparation of the sulfonate-silica hybrid SCX monolithic column
Preparation of the sulfonate-silica hybrid SCX monolithic column employed a method similar to our original report.27 The fused silica capillary (50 μm i.d.×150 μm o.d.) was pretreated by washing with 0.1 M NaOH for 2 h, flushing with water until the outflow reached pH ~7.0, flushing with 0.1 M HCl for 24 h, and flushing again with water until the outflow reached pH ~7.0. The capillary was finally dried with a nitrogen stream for 24 h at room temperature. For the preparation of the sulfonate SCX hybrid monolithic column, a prehydrolyzed mixture was prepared by mixing and stirring acetic acid (10 mM, 5.0 mL), PEG (MW = 10,000, 540 mg), urea (450 mg), TMOS (1.8 mL), and VTMS (0.6 mL) for 1 h at 0 °C to form a homogeneous solution. Then, 37 mg of 3-sulfopropyl methacrylate potassium salt and 2 mg of AIBN were added into 0.5 mL of the hydrolyzed mixture with 10 min sonication. Next, the polymerization mixture was introduced into the pretreated capillary (50 μm i.d.×150 μm o.d., 15 cm long) using a syringe until the solution length reached 5 cm, as measured by eye. Both ends of the capillary were then sealed with pieces of rubber, and the capillary was incubated at 37 °C for 12 h for polycondensation and at 60 °C for 12 h for polymerization. The resulting sulfonate SCX hybrid column was then flushed with methanol and water to remove the porogen and other residual components. Finally, the sulfonate SCX hybrid monolithic column was cut to a length of 1~2 cm use in SCX SPE-CE-MS/MS analysis.
Preparation of E. coli sample
Solid lysogeny broth (LB) was used to make the agar plates for the E. coli culture. Solid LB was prepared by dissolving 3 g of NaCl, 3 g of tryptone, 1.5 g of yeast extract, and 6 g of agar in 300 mL of deionized water. Liquid LB medium (without agar) was also prepared for E. coli culture by mixing 10 g of NaCl, 10 g of tryptone, and 5 g of yeast extract in 1 L of deionized water. All media, plates, and other utensils and flasks were autoclaved before use. Frozen cultures of E. coli (Dh5-Alpha) were thawed and plated on the prepared agar plates. After incubation at 37 °C for 24 h, single colonies were isolated and grown in tubes with 4 mL of liquid LB medium and incubated in a shaker at 37 °C overnight. When the tubes’ contents turned opaque, the liquid medium was transferred into new flasks and shaken overnight at 37 °C. The liquid LB medium with E. coli was centrifuged, and the resulting E. coli pellets were washed with phosphate-buffered saline three times. Then, the E. coli pellets were suspended in 8 M urea and 100 mM Tris-HCl (pH 8.0) buffer supplemented with protease inhibitor and sonicated for 15 min on ice for cell lysis. The lysate was centrifuged at 18 000 g for 15 min, and the supernatant was collected. The protein concentration was measured by the BCA method. An aliquot of protein (900 μg) was precipitated by cold acetone overnight at −20 °C. After centrifugation, the pellet was washed again with cold acetone. The resulting protein pellet was dried at room temperature.
The dried E. coli proteins (120 μg) were dissolved in 100 μL of 100 mM NH4HCO3 (pH 8) with 8 M urea and denatured at 37 °C for 1 h. After addition of 10 μL of 1 M DTT, the mixture was incubated at 37 °C for 1 h to reduce disulfide bonds. Subsequently, 20 μL of 2 M IAA was added to the mixture, which was incubated in the dark at room temperature for 30 min. Then, 900 μL of 100 mM NH4HCO3 (pH 8) was added to reduce the concentration of urea below 1 M. Finally, the treated proteins were digested by incubation with trypsin at a trypsin/protein ratio of 1/25 (w/w) for 16 h at 37 °C. The digests were acidified with 10% TFA (v/v) to terminate the reaction. The tryptic digests were desalted with C18 SepPak columns (Waters, Milford, MA), followed by lyophilisation with a vacuum concentrator (Thermo Fisher Scientific, Marietta, OH). The dried protein digests were dissolved in 50 mM FA.
Elution buffers
Elution buffers with pH 3 and 4 were prepared from ammonium formate, and the pH was adjusted with formic acid. An elution buffer at pH 5 was prepared from ammonium acetate, and the pH was adjusted with acetic acid. Elution buffers with pH 7 and 8.2 were prepared from ammonium bicarbonate and the pH was adjusted with 10% ammonium hydroxide. The buffers were then diluted by water to the desired concentration.
Direct CZE-ESI-MS/MS analysis
The CZE system consists of two high-voltage power supplies (Spellman CZE 1000R) and an electrokinetically pumped electrospray interface.3,28 The interface was used to couple the CZE separation capillary to a LTQ-Orbitrap Velos mass spectrometer (Thermo Fisher Scientific). The electrospray emitter was made from borosilicate glass capillary (1.0 mm o.d. × 0.75 mm i.d., 10 cm long) pulled with a Sutter instrument P-1000 flaming/brown micropipette puller; the size of the emitter opening was 5–10 μm. The electrospray sheath flow buffer was 10% (v/v) methanol with 0.1%FA. A commercial LPA coated capillary (50 μm i.d.×150 μm o.d., 60 cm long) was used for the CZE separation. The separation buffer was 50 mM FA in water. A 20 nL injection volume was loaded by pressure. 19.5 kV was applied at the injection end of the capillary for separation and 1.5 kV on the sheath flow reservoir for electrospray. Voltage programming was controlled by LabView software.
The effect of the dead volume introduced by the connection
The SCX SPE monolithic column was connected to a LPA coated capillary by a home-made zero-volume connecter. This connecter used a single piece of fused silica sleeve tubing (Polymicro) with 180-μm i.d., which was held in place by a PEEK union (Upchurch).
To study the effect of the connector on the separation performance, we compared the CZE separation performance of a LPA coated capillary (50 μm i.d.×150 μm o.d., 60 cm long) before and after connection to an 8 cm length of uncoated fused capillary (50 μm i.d.×150 μm o.d.) at the inlet. The experiment was performed using an LTQ XL mass spectrometer and with 0.1 mg/mL BSA tryptic digests dissolved in 50 mM ammonium bicarbonate (pH 8.2) as a standard sample.
SCX SPE-CZE-ESI-MS/MS analysis with on-line pH gradient elution
The schematic diagram of the SCX SPE-CE-ESI-MS/MS system is shown in Figure 1. The sulfonate SCX hybrid monolith was connected to a LPA-coated capillary by a single piece of tubing sleeve with 180 μm i.d. and a PEEK union. The SCX column was first conditioned by 50 mM FA for 10 min. After the sample was loaded onto the SCX SPE column by pressure, the system was flushed with 50 mM FA to completely remove the unretained components. The on-line pH gradient elution was performed by sequentially injecting the elution buffers with different pH values by the pressure; each elution step is called a pH bump. After each pH bump, the background electrolyte was changed back to 50 mM FA and normal CE-ESI-MS/MS analysis was carried out under the same condition as direct CE-ESI-MS/MS.
Figure 1.
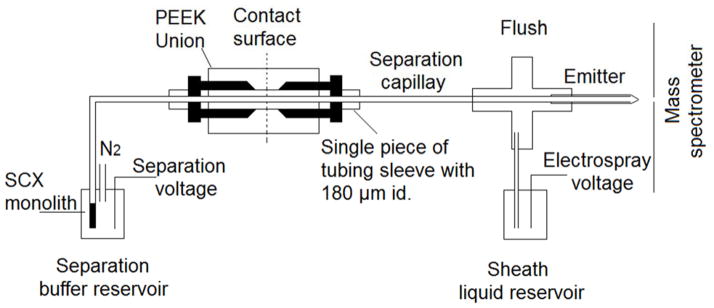
Schematic diagram of the SCX-SPE CZE-ESI-MS/MS system. Provisions are made to use N2 gas pressure to pump analyte and reagents through the monolith during its synthesis. The separation is driven by the difference between the separation voltage and the spray voltage. The sheath liquid is electrokinetically driven by the electrospray voltage.
UPLC-ESI-MS/MS analysis
A nanoACQUITY UltraPerformance LCH (UPLCH) system (Waters, Milford, MA, USA) with a UPLC BEH 130 C18 column (Waters, 100 μm × 100 mm, 1.7 μm) was coupled to an LTQ Orbitrap Velos mass spectrometer (Thermo Fisher Scientific) for peptide separation and identification. Buffer A (0.1%FA in water) and buffer B (0.1% FA in ACN) were used as mobile phases for gradient separation. Peptides were automatically loaded onto a commercial C18 reversed phase column (Waters, 100 μm ×100 mm, 1.7 μm particle, BEH130C18, column temperature 40°C) and flushed with 2% buffer B for 10 min at a flow rate of 1 μL/min, then followed by gradient: 10–11 min, 2–8% B; 11–71 min, 8–30% B; 71–72 min,30–80% B; 72–77 min, 80% B; 77–78 min, 80–2% B; 78–90 min, 2% B. The eluted peptides from the C18 column were pumped through a capillary tip for electrospray with the electrospray voltage of 1.5 kV, and the MS parameters were the same as that used for CE-ESI-MS/MS analysis.
Mass Spectrometer Operating Parameters
For experiments using an LTQ-XL mass spectrometer (Thermo Fisher Scientific), data dependent acquisition was applied. Full MS scans were acquired over the 380–1800 m/z range. The 10 most intense peaks were selected for fragmentation in the ion trap with normalized collision energy of 35%, activation q = 0.25, activation time of 30 ms, and one microscan. Peaks selected for fragmentation more than once within 45 s were excluded from selection for 60 s.
For experiments using an LTQ-Orbitrap Velos mass spectrometer (Thermo Fisher Scientific), Collision-Induced Dissociation (CID) mode was applied. The electrospray voltage was 1.5 kV, and the ion transfer tube temperature was held at 280 °C. The S-Lens RF level was 50.00. The mass spectrometer was programmed in data-dependent mode. Full MS scans were acquired in the Orbitrap mass analyzer over the m/z 380–1800 range with resolution of 30,000 (m/z 400) and the number of microscans set to 1. The target value was 1.00E+06, and maximum injection time was 500 ms. For MS/MS scans, the 20 most intense peaks with charge state ≥ 2 were sequentially isolated and further fragmented in the CID cell following one full MS scan. The normalized collision energy was 35%.
Database Searching
Database searching of the raw files was performed in Proteome Discoverer 1.4 with MASCOT 2.2.4. For standard proteins from bovine, ipi.bovin.v3.68.fasta database was used. For the E. coli cell lysate, a NCBI database of E. coli DH1 (4,160 sequences) was used. Database searching for the reversed database was also performed to evaluate the false discovery rate. The database searching parameters included full tryptic digestion and allowed up to two missed cleavages, precursor mass tolerance 10 ppm, and fragment mass tolerance 0.5 Da. Carbamidomethylation (C) was set as fixed modifications. Oxidation (M) and deamidated (NQ) were set as variable modifications. On the peptide level, peptide confidence value as high was used to filter the peptide identification, and the corresponding false discovery rate on peptide level was less than 1%. On the protein level, protein grouping was enabled.
RESULTS AND DISCUSSION
The effect of the dead volume introduced by the connection
We employed a simple zero-dead-volume connector to couple the monolith, which was prepared in an uncoated capillary, to a coated separation capillary, which provides long separation windows. To evaluate any degradation in separation performance due to the connector, we compared the separation of a BSA tryptic digest on a 60-cm long LPA coated capillary with and without coupling to an 8-cm segment of uncoated capillary. Electropherograms are shown in S-Figure 1 in Supporting information. A set of a dozen selected ion electropherograms were generated from both electropherograms, and a nonlinear least squares algorithm was used to fit a Gaussian function to the peaks. The migration times were highly correlated (r = 0.9997); the addition of the union added an average of 1.75 minutes to the migration time, due to the additional capillary length provided by the uncoated segment. Peak heights were highly correlated (r = 0.80). Median peak efficiencies were also very similar (120,000 without the union, 140,000 with the union). The increase in plate count that accompanied the use of the union is likely due to the increased capillary volume, which reduced the effect of injection volume on the separation efficiency.29
Protein coverage was essentially the same for the two systems (62% coverage for the coupled capillaries vs. 59% for the LPA capillary alone), as was the number of peptide identifications (51 peptide IDs for the coupled capillaries vs. 53 for the LPA capillary alone). These results demonstrate that the union preserves the separation performance of the LPA coated capillary.
On-line pH gradient elution analysis
Before each experiment, a 50 ng aliquot of an E. coli digest was loaded onto the SCX SPE column. The column was then flushed with 50 mM FA, followed by pH bumps using buffers at pH 3, 4, 5, 7 and 8.2. After each injection of the elution buffer, the separation electrolyte was changed back to 50 mM FA for CZE-ESI-MS/MS analysis.
We investigated a number of elution conditions. We first employed 90 nL aliquots of 10 mM elution buffers. After elution using five pH bumps, 528 protein groups and 1,567 peptides were identified. However, these experiments revealed that a large amount of sample was retained on the SCX SPE column. Therefore, we continued eluting using ammonium bicarbonate (pH 8.2) with concentrations of 40, 80, 120, 150, and 200 mM. Ultimately, 752 protein groups and 3,323 peptides were identified. However, only 2 peptides were identified in the 90 nL of 10 mM ammonium formate (pH 3) step. In contrast, many protein groups and peptides were identified in the 40 mM or higher concentration of ammonium bicarbonate (pH 8.2) elution steps.
To further investigate the eluting performance, the base peaks of seven randomly selected peptides were extracted from the electropherograms, S-Figure 2 in the Supporting information. Very low signal-to-noise ratios were observed for most of the peptides, which presumably were not eluted from the SCX SPE column by 90 nL of 10 mM elution buffer. In this case, the peptides were detected over many elution steps due to inefficient fractionation, and the overlaps between the adjacent gradient elution steps were very large, as shown in Supporting Figures 5(A).
To increase the eluting power, the eluting volume was increased to 150 nL. As shown in S-Figures 5(B) in the Supporting Information, the overlaps of peptide identifications between successive eluting steps decreased significantly when the eluting volume was increased to 150 nL. After five steps of pH bumps elution, 634 protein groups and 2,342 peptides were identified. We continued eluting using ammonium bicarbonate (pH 8.2) with concentrations of 40, 80, 120, 150, and 200 mM. Finally, 767 protein groups and 3,176 peptides were identified. Obviously, many more protein groups and peptides were identified at the first five pH gradient elution steps compared to the result obtained with 90 nL elution buffer. However, only 15 additional protein groups were identified in total and the eluted sample was greatly diluted by the large volume of elution buffer, which overwhelmed the effect of the pH junction.
We next increased the elution buffer concentration to 30 mM while using 90 nL of elution buffer in each elution step, S-Figure 6. After elution using five pH bumps, 799 protein groups and 3,381 peptides were identified in a 2.5 hour analysis. We performed additional elution using ammonium bicarbonate buffer (pH 8.2) with concentrations of 40, 80, 120, 150, and 200 mM, 877 protein groups and 3,949 peptides were identified in total. These ammonium bicarbonate elution steps remove residual peptides from the SCX monolith, minimizing carryover before reuse of the system.
Obviously, most of the protein groups (>91%) and peptides (>85%) could be identified during the pH bumps elution steps when the elution buffer concentration was increased to 30 mM. The number of identified protein groups approaches the results we reported using single shot CZE coupled with an Orbitrap Fusion mass spectrometer (956 protein groups and 4741 peptides),14 but our current results were obtained with a much less expensive Orbitrap Velos mass spectrometer. In addition, the results obtained in this paper were obtained from ~50 ng of protein digest, compared with ~250 ng used in reference 14. Further improvement of the identification performance of the SCX SPE-CZE-MS/MS platform is expected with an improved mass spectrometer.
The base peaks of seven randomly selected peptides were extracted from the electropherograms in different eluting steps using the 30 mM, 90 nL elution conditions, S-Figure 3. Most peptides were completely eluted from the SCX SPE column in a single elution step under these conditions, and overlaps between the adjacent eluting steps were very small, S-Figure 5(C). In this case, the complexity of the sample was greatly decreased in each pH gradient elution step. The detection of the low abundance peptides, which is usually difficult in the unfractionated digests due to the suppression caused by high abundance peptides, was greatly improved compared to single-shot analysis of an unfractionated sample.
Fewer identifications were made with a higher concentration elution buffer (50 mM); only 649 protein groups and 2,507 peptides were identified during the five pH bumps, S-Figures 5(D). Additional elution steps using ammonium bicarbonate (pH 8.2) at concentrations of 80, 120, 150, and 200 mM, resulted in the identification of 693 protein groups and 2,695 peptides in total. While the overlaps in identifications between adjacent steps were also very small, which suggested the efficient fractionation, we found that the peak intensity of the peptides were usually lower than with 30 mM elution buffer, especially for with low pH elution buffers, S-Figure 4. The decreased intensity may be due to the lack of sample stacking when a 50 mM elution buffer was used, which resulted in peak broadening. Therefore, five pH bumps with 30 mM elution buffers were used in the subsequent experiments.
Loading ability and reproducibility
Different amounts of E. coli digest in the range of 10 ng to 50 ng were loaded onto the SCX SPE column and analyzed by on-line pH gradient elution. As shown in Figure 2, the number of identified protein groups increased from 131 to 799 proteins and the number of identified peptides increased from 297 to 3,381 with increasing loading amounts.
Figure 2.

Analysis of the E. coli proteome digest using on-line pH gradient elution from a SCX SPE-CZE system. Experimental conditions: 50 μm i.d.×150 μm o.d.×1.5 cm sulfonate SCX SPE column; 50 μm i.d.×150 μm o.d.×60 cm long LPA coated capillary as separation capillary; 50 ng E. coli digests loading amount; 90 nL of 30 mM elution buffers at each elution step; 50 mM FA separation buffer; 19.5 kV separation voltage; and 1.5 kV spray voltage.
The run-to-run reproducibility of the proposed method was evaluated by duplicated analysis of 50 ng of E. coli digest with five pH bumps. 924 protein groups and 4,231 peptides were identified by duplicated runs. Among them, 514 protein groups and 1,361 peptides were identified by both runs. The overlaps of identified peptides and protein groups are shown in S-Figure 7 in the Supporting information.
Comparison with UPLC-MS/MS
We also analyzed the same E. coli digest using UPLC-MS/MS. The E. coli digests were loaded onto a reverse phase C18 analytical column followed by a 90 min gradient LC–MS/MS analysis, S-Figure 8 in the Supporting Information. The overlaps of peptides and protein groups identified by UPLC and SCX SPE-CE methods are shown in Figure 3. 957 protein groups and 4,995 unique peptides were identified with 50 ng E. coli digests by UPLC-MS/MS. 71% of the protein groups and 58% of the peptides identified in the UPLC runs were also identified by the SCX SPE-CE system. However, 14% of the protein groups and 14% of peptides that were identified by the SCX SPE-CE system were not identified by UPLC-MS/MS.
Figure 3.
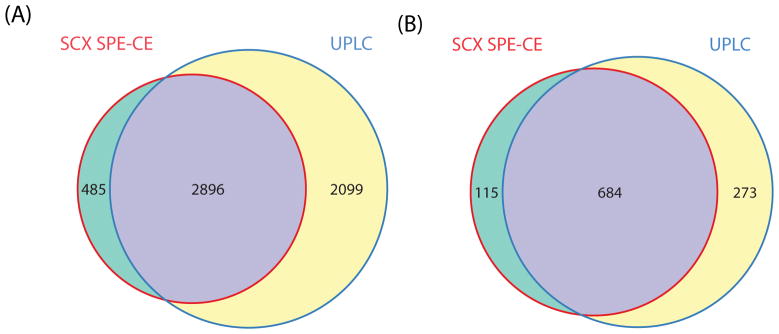
Peptides (A) and protein group (B) overlaps between the SCX SPE-CE and UPLC analysis of E. coli tryptic digests.
The pI distribution of the identified peptides from both methods was also investigated and the results are shown in Figure 4. In general, UPLC identified more acidic peptides. We assume that the most acidic peptides were poorly retained on the SCX resin during the initial loading step, and that the use of a lower pH loading buffer will improve their retention. In contrast, CZE generated slightly more identifications of basic peptides, which were very well behaved on the SCX monolith.
Figure 4.

pI distributions of the identified peptides by SCX SPE-CE and UPLC analysis of the trypic digest of the E. coli proteome.
Supplementary Material
Acknowledgments
We thank Dr. William Boggess and Dr. Matthew Champion in the Notre Dame Mass Spectrometry and Proteomics Facility for their help with this project. This work was funded by the National Institutes of Health (Grant R01GM096767).
Footnotes
Notes
The authors declare no competing financial interest.
References
- 1.Li Y, Champion MM, Sun L, Champion PAD, Wojcik R, Dovichi NJ. Anal Chem. 2011;84:1617–1622. doi: 10.1021/ac202899p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhao S, Zhong X, Tie C, Chen DDY. Proteomics. 2012;12:2991–3012. doi: 10.1002/pmic.201200221. [DOI] [PubMed] [Google Scholar]
- 3.Sun L, Zhu G, Zhao Y, Yan X, Mou S, Dovichi NJ. Angew Chem Int Ed Engl. 2013;52:13661–13664. doi: 10.1002/anie.201308139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhu G, Sun L, Yan X, Dovichi NJ. Anal Chem. 2013;85:2569–2573. doi: 10.1021/ac303750g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Moini M, Martinez B. Rapid Commun Mass Spectrom. 2014;28:305–310. doi: 10.1002/rcm.6786. [DOI] [PubMed] [Google Scholar]
- 6.Zhu G, Sun L, Yan X, Dovichi NJ. Anal Chem. 2014;86:6331–6336. doi: 10.1021/ac5004486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Saavedra L, Barbas C. J Biochem Biophys Methods. 2007;70:289–297. doi: 10.1016/j.jbbm.2006.11.005. [DOI] [PubMed] [Google Scholar]
- 8.Wu X. TrAC Trend Anal Chem. 2003;22:48–58. [Google Scholar]
- 9.Simpson SL, Jr, Quirino JP, Terabe S. J Chromatogr A. 2008;1184:504–541. doi: 10.1016/j.chroma.2007.11.001. [DOI] [PubMed] [Google Scholar]
- 10.Malá Z, Šlampová A, Gebauer P, Boček P. Electrophoresis. 2009;30:215–229. doi: 10.1002/elps.200800433. [DOI] [PubMed] [Google Scholar]
- 11.Breadmore MC, Thabano JRE, Dawod M, Kazarian AA, Quirino JP, Guijt RM. Electrophoresis. 2009;30:230–248. doi: 10.1002/elps.200800435. [DOI] [PubMed] [Google Scholar]
- 12.Kitagawa F, Otsuka K. J Chromatogr A. 2014;1335:43–60. doi: 10.1016/j.chroma.2013.10.066. [DOI] [PubMed] [Google Scholar]
- 13.Busnel JM, Schoenmaker B, Ramautar R, Carrasco-Pancorbo A, Ratnayake C, Feitelson JS, Chapman JD, Deelder AM, Mayboroda OA. Anal Chem. 2010;82:9476–9483. doi: 10.1021/ac102159d. [DOI] [PubMed] [Google Scholar]
- 14.Sun L, Hebert AS, Yan X, Zhao Y, Westphall MS, Rush MJP, Zhu G, Champion MM, Coon JJ, Dovichi NJ. Angew Chem Int Ed Engl. 2014;53:13931–13933. doi: 10.1002/anie.201409075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee WH, Wang C, Her GR. Rapid Commun Mass Spectrom. 2011;25:2124–2130. doi: 10.1002/rcm.5091. [DOI] [PubMed] [Google Scholar]
- 16.Wang Y, Fonslow BR, Wong CCL, Nakorchevsky A, Yates JR. Anal Chem. 2012;84:8505–8513. doi: 10.1021/ac301091m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Figeys D, Ducret A, Yates JR, Aebersold R. Nat Biotech. 1996;14:1579–1583. doi: 10.1038/nbt1196-1579. [DOI] [PubMed] [Google Scholar]
- 18.Figeys D, Ducret A, Aebersold R. J Chromatogr A. 1997;763:295–306. doi: 10.1016/s0021-9673(96)00847-3. [DOI] [PubMed] [Google Scholar]
- 19.Benavente F, Vescina MC, Hernández E, Sanz-Nebot V, Barbosa J, Guzman NA. J Chromatogr A. 2007;1140:205–212. doi: 10.1016/j.chroma.2006.11.092. [DOI] [PubMed] [Google Scholar]
- 20.Nema T, Chan ECY, Ho PC. J Pharm Biomed Anal. 2014;87:130–141. doi: 10.1016/j.jpba.2013.05.036. [DOI] [PubMed] [Google Scholar]
- 21.Huang X, Yuan D. CRC Crit Rev Anal Chem. 2012;42:38–49. [Google Scholar]
- 22.Lv Y, Alejandro FM, Fréchet JMJ, Svec F. J Chromatogr A. 2012;1261:121–128. doi: 10.1016/j.chroma.2012.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Thabano JRE, Breadmore MC, Hutchinson JP, Johns C, Haddad PR. J Chromatogr A. 2009;1216:4933–4940. doi: 10.1016/j.chroma.2009.04.012. [DOI] [PubMed] [Google Scholar]
- 24.Horie K, Kamakura T, Ikegami T, Wakabayashi M, Kato T, Tanaka N, Ishihama Y. Anal Chem. 2014 doi: 10.1021/ac4038625. [DOI] [PubMed] [Google Scholar]
- 25.Wu M, Wu R, Zhang Z, Zou H. Electrophoresis. 2011;32:105–115. doi: 10.1002/elps.201000349. [DOI] [PubMed] [Google Scholar]
- 26.Zhang Z, Wang F, Xu B, Qin H, Ye M, Zou H. J Chromatogr A. 2012;1256:136–143. doi: 10.1016/j.chroma.2012.07.071. [DOI] [PubMed] [Google Scholar]
- 27.Zhang Z, Sun L, Zhu G, Yan X, Dovichi NJ. Talanta. 2015;138:117–122. doi: 10.1016/j.talanta.2015.01.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wojcik R, Dada OO, Sadilek M, Dovichi NJ. Rapid Commun Mass Spectrom. 2010;24:2554–2560. doi: 10.1002/rcm.4672. [DOI] [PubMed] [Google Scholar]
- 29.Cheng YF, Wu SL, Chen DY, Dovichi NJ. Anal Chem. 1990;62:496–503. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.