

Abstract
A liquid chromatography-high resolution mass spectrometry (LC-HRMS) method was developed using three peptide drugs: salmon calcitonin, bivalirudin, and exenatide as model systems to assess the suitability of this approach for monitoring peptide drug product quality. Calcitonin and its related impurities displayed linear responses over the range from 0.1 to 10 μM (R2 values for calcitonin salmon, Glu14-calcitonin, and acetyl-calcitonin were 0.995, 0.996, and 0.993, respectively). Intra-assay precision in terms of relative standard deviation (%RSD) was less than 10% at all tested concentrations. The accuracy of the method was greater than 85% as measured by spiking 0.1, 0.3, and 1% of Glu14-calcitonin and acetyl-calcitonin into a stock calcitonin solution. Limits of detection for calcitonin, Glu14-calcitonin, and acetyl-calcitonin were 0.02, 0.03, and 0.04 μM, respectively, indicating that an impurity present at less than 0.1% (0.1 μM) of the drug product API concentration (107 μM) could be detected. Method validation studies analyzing bivalirudin and exenatide drug products exhibited similar results to calcitonin salmon in regard to high selectivity, sensitivity, precision, and linearity. Added benefits of using LC-HRMS-based methods are the ability to also determine amino acid composition, confirm peptide sequence, and quantify impurities, even when they are co-eluting, within a single experiment. LC-HRMS represents a promising approach for the quality control of peptides including the measurement of any peptide-related impurities. While the development work performed here is focus on peptide drug products, the principles could be adapted to peptide drug substance.
KEY WORDS: High resolution mass spectrometry, impurity profiling, LC-HRMS, peptide drug
INTRODUCTION
The number of peptide-related Abbreviated New Drug Applications (ANDAs) is dramatically increasing at the Office of Generic Drugs at the US Food and Drug Administration (FDA) (1,2). Peptides tend to have additional process impurities (e.g., amino acid (AA) deletion or insertion), and they are susceptible to a variety of degradation pathways (e.g., oxidation and/or deamidation; (3,4)), making these molecules more complex in terms of chemistry, manufacturing, and control than most small molecules. Many of the quality control methods (HPLC-UV) submitted for peptide drugs do not appear to be adequate for distinguishing and quantifying these process impurities or degradation products. A recent method validation study performed in our laboratory identified the presence of both an AA deletion and an AA insertion in a synthetic peptide drug product that could not be resolved using the HPLC-UV methods an applicant submitted. These data support the fact that HPLC-UV methods may be insufficient for ensuring the quality of peptide drug products.
The regulatory review of generic peptide products faces significant challenges not only due to the complexity of the drug substances but also because of a lack of general guidelines for establishing method validation and performing comparative analytical analysis for peptide drugs. Unlike small molecule drugs, therapeutic peptides are chains of amino acids and their impurities may contain partially identical sequences. For example, the solid-phase peptide synthesis method may result in the formation of numerous impurities due to incomplete coupling, truncations, and side reactions. Moreover, the structure can be further complicated with multiple degradation pathways (e.g., oxidation, deamidation, and/or truncation), possible secondary structure formation, and aggregation. All of these peptide-related impurities may significantly affect the quality, safety, and efficacy of peptide drugs. The current International Conference on Harmonisation (ICH) guidelines regarding the testing and quality of pharmaceutical products do not include peptides, and established method validation guidances may not be directly applicable to peptides due to the lack of suitable internal standards and/or reference materials. Generic drug applications of peptides primarily rely on Reference Listed Drugs (RLDs) for comparative testing and quality control assessments.
The suitability of advanced analytical techniques such as liquid chromatography-high resolution mass spectrometry (LC-HRMS) was explored to address this regulatory knowledge gap. LC-HRMS has been strongly favored by bioanalysis groups (5–9) due to its high selectivity and sensitivity. These properties are inherently advantageous when assessing larger molecules such as peptides and proteins (8–10). LC-HRMS methodologies are capable of detecting and identifying co-eluting impurities at low levels (1,11–15). Furthermore, the specificity of LC-HRMS technology typically allows peptides with changed sequences to be measured even if they are not separated chromatographically (4). Changes to the primary amino acid sequence due to degradation, truncation, insertion, substitution, or deletions typically result in detectable mass shifts. In this study, three different peptide drugs: calcitonin salmon (sCT), bivalirudin (BV), and exenatide (ET) were used as model systems to evaluate the suitability and validate the use of LC-HRMS for impurity detection and active pharmaceutical ingredient characterization.
Calcitonin salmon is used to treat metabolic bone diseases such as osteoporosis, Paget’s disease, and osteoarthritis (16). Two formulations, a nasal spray and an injectable, are available from both originator and follow-on manufacturers. All formulations of calcitonin salmon have the AA sequence of Cys-Ser-Asn-Leu-Ser-Thr-Cys-Val-Leu-Gly-Lys-Leu-Ser-Gln-Glu-Leu-His-Lys-Leu-Gln-Thr-Tyr-Pro-Arg-Thr-Asn-Thr-Gly-Ser-Gly-Thr-Pro-NH2. Known related peptides of the calcitonin salmon drug substance are listed in the USP monograph, including calcitonin salmon related compound A (RSA; N-acetyl-calcitonin salmon) and calcitonin salmon related compound B (RSB; calcitonin salmon-glycine) (17). While the acceptance criteria of the calcitonin salmon drug product assay for the nasal solution are between 80 and 115%, there is no specific requirement for impurities. For the calcitonin salmon drug substance, the assay acceptance criteria are between 90 and 105%, with individual impurities NMT 3.0% and total impurities NMT 5.0% of the total area of all (HPLC-UV) chromatographic peaks.
Bivalirudin (Angiomax or Angiox, manufactured by The Medicines Company) is a thrombin-specific anticoagulant (18), intended for administration by injection only. The sequence of bivalirudin is Phe-Pro-Arg-Pro-Gly-Gly-Gly-Gly-Asn-Gly-Asp-Phe-Glu-Glu-Ile-Pro-Glu-Glu-Tyr-Leu. Potential process impurities include the loss or addition of glycine between positions 4 and 9 (des-glycine (DG) or plus-glycine (PG)), deamidation of the asparagine at position 9 to form aspartic acid/iso-aspartic acid (beta-aspartic analog (BA)), and a truncated form, bivalirudin [12–20].
Exenatide (ET) is a regulator of glucose metabolism and insulin secretion, approved for the treatment of diabetes mellitus type 2 (19). It is a 39-amino acid synthetic peptide amide, with two different formulated products, Byetta and Bydureon, on the market. The amino acid sequence for exenatide is His-Gly-Glu-Gly-Thr-Phe-Thr-Ser-Asp-Leu-Ser-Lys-Gln-Met-Glu-Glu-Glu-Ala-Val-Arg-Leu-Phe-Ile-Glu-Trp-Leu-Lys-Asn-Gly-Gly-Pro-Ser-Ser-Gly-Ala-Pro-Pro-Pro-Ser-NH2. Potential process impurities include the addition or loss of glycine and double alanine.
As proof of concept, an LC-HRMS method was developed for calcitonin salmon, bivalirudin, exenatide, and their impurities to demonstrate the suitability of this approach for peptide drugs. Goals of this study included identification, characterization, separation, and quantification of the peptides and any peptide-related impurities present. The method was validated for selectivity, sensitivity, accuracy, precision, and linearity according to ICH Q2 analytical guidelines and serves as an analytical example for ensuring the quality of peptide drug products.
MATERIALS AND METHODS
Chemicals and Reagents
All drugs were purchased from a pharmacy or requested from the manufacturer unless otherwise noted. Calcitonin salmon and its related impurity standards were purchased from American Peptide Company, Inc (Sunnyvale, CA). The exenatide peptide standard was purchased from GenScript (Piscataway, NJ). All synthetic or reference standards were used as received after verifying the mass and sequence by using intact MS and tandem MS/MS. Chemical reagents including acetonitrile (ACN), methanol, water, acetone, formic acid, and dichloromethane (DCM) were purchased from Fisher (Fair Lawn, NJ, USA). All chemicals/solvents were of Optima LC-MS or ACS grade and used as received.
Methods
Preparation of Standards
Stock standard solutions of the peptides and their related impurities were prepared at 1 mg/mL in diluent (water:acetonitrile, 9:1, in a 100-mL Pyrex glass bottle). Stock solutions were aliquoted and stored at −80°C until use. Individual intermediate stock standards were prepared as needed. The highest working solution of peptide was prepared at 10 μM by mixing the individual intermediate stock standards with the appropriate amount of diluent. Working standards then were serial diluted to generate standards at 0.1, 0.25, 0.5, 1, 2.5, 5, and 10 μM. Stock and immediate solutions were prepared in 2 mL microcentrifuge tubes (low retention and siliconized). Samples for injection were in 2 mL HPLC glass vials.
Sample Preparation
Calcitonin salmon nasal solution samples (107 μM) were used as received. Bivalirudin for injection was prepared at concentration of 0.1 mg/mL (45.9 μM) in diluent. Exenatide solution was prepared at 2 mg/mL (478 μM) in diluent (20). Briefly, one vial of exenatide (2 mg dose) microspheres (about 45 mg) was dissolved in 0.3 mL of dichloromethane (DCM) in a 1.5-mL Eppendorf vial. Then, 0.7 mL of acetone was added to the mixture, and the vial was vortexed. After centrifugation at 11,752g for 5 min (Eppendorf centrifuge Model 5424), the supernatant was discarded. The residue was washed three times with 0.9 mL of mixed solvent (3:1 acetone:DCM) by repeating the above process. After 2 hours at room temperature, the residue was dissolved in diluent. The sample working solution (40 μM) was prepared by mixing 84 μL sample stock solution with 914 μL diluent.
Instrumentation
HPLC separation was achieved on an Agilent 1290 HPLC complex system consisting of a 1290 binary pump, thermostat, and autosampler (Agilent, Santa Clara, CA), coupled to an Agilent 6520B Q-TOF mass spectrometer which was used for HRMS analysis. Data were acquired with MassHunter Data Acquisition v B.04 and analyzed by MassHunter Qualitative Analysis v B.04 (Agilent, Santa Clara, CA) software.
LC-HRMS Analysis
HPLC separation was performed using a Waters ACQUITY UPLC BEH C18 column (100 mm × 2.1 mm, 130 Å, 1.7 μm). The HPLC mobile phase consisted of 0.1% formic acid in water (A) and 0.1% formic acid in acetonitrile (B). Gradient elution at a flow rate of 0.3 mL/min was performed with 5% B for 2.0 min, increased to 40% B over 37 min, then increased to 95% B at 40 min, and held for 2 min before decreasing to 5% B at 43 min. The column was re-equilibrated at 5% B for an additional 4 min for a total run time of 47 min. The column temperature was maintained at 30°C, and the autosampler was chilled at 4°C. A dual electrospray ionization source was operated in positive ion mode to acquire full scan mass spectra from 50 to 2000 m/z with a scan rate of 4.0 spectra/s. The source gas temperature was set at 350°C. Additional source parameters were set to Vcap at 4000 V, fragmentor at 210 V, skimmer1 at 80.0 V, and octopole RF peak at 750 V. The sample injection volume was 10 μL.
Data Analysis
Data analysis was performed using MassHunter Qualitative Analysis v B.04 (Agilent, Santa Clara, CA). A linear regression was used to generate calibration curve(s) from standards and calculate the concentrations of quality control and unknown samples. The fit equation of the standard curve was in the y = mx + b format, where y is the peak area of the analyte, x is the concentration of the analyte, m is the slope, and b is the intercept of the regression line. Peak areas obtained from extracted ion chromatograms (EICs) were used for linear regression. Peak areas obtained from total ion chromatograms (TICs) were used to investigate peptide drug impurities of calcitonin salmon and exenatide. Peak areas obtained from EIC were used to study peptide drug impurities of bivalirudin.
Method Validation
Linearity was established by generating a calibration curve with seven nonzero calibrators covering the expected concentration range. Each calibrator had five determinations. Each tested concentration in the linear range featured precision within ±15%. Limit of detection (LOD) was calculated using the following equation: LOD = 3.3 × SD of noise/slope. Limit of quantification (LOQ) was calculated using the equation following: LOQ = 10 × SD of noise/slope. The accuracy of the method for each peptide was determined by spiked recovery experiments at three different concentration spikes (low, middle, and high relative to the linear curve: 0.3, 0.9, 3.0 μM, respectively). Peak areas obtained from EIC were used for method validation.
Selection of Optimized HPLC Conditions
A LC-HRMS method can be optimized by modifying column temperature and gradient slope. LC-HRMS TIC analysis of exenatide at different column temperatures with gradient 5–40% B from 2 to 37 min was investigated. Column temperatures were tested at 35, 45, 50, 55, and 60°C. Increasing the column temperature improved both the peak shape and separation. A column temperature of 55°C was selected to improve column life without sacrificing resolution. For LC-HRMS analysis of exenatide, the slope of the elution gradient was varied as follows, while keeping the column temperature at 55°C: a, 5–40% B from 2 to 37 min; b, 10–40% B from 2 to 37 min; c, 5–30% B from 2 to 20 min, then 30–40% B from 20 to 40 min; d, 5–25% B from 2 to 15 min, then 25–40% B from 15 to 40 min. Gradient condition d was determined to yield the best peak shape and resolution.
Optimization of MS Parameters
MS parameters can be optimized for any specific peptide system. The higher molecular weight for exenatide compared to the other peptides studied necessitated a higher gas temperature. Nebulizer pressure and fragmentor voltage also were adjusted to achieve an appropriate response. The following settings were changed from the method previously used with bivalirudin and calcitonin: source parameters: gas temp 325 to 350°C, gas flow 12 to 13.0 L/min, nebulizer 20 to 40 psi; scan source parameters: Vcap 4000 V, fragmentor 175 to 210 V, skimmer1 65.0 to 80.0 V, octopole RF peak 750 V.
RESULTS
High Resolution Mass Spectra and Accurate Molecular Weight Determination
The LC-HRMS spectra of calcitonin salmon, bivalirudin, and their related impurities are shown in Fig. 1. The spectra on the left correspond to the +4 charge states of calcitonin salmon and its related impurities from the four synthetic standards: calcitonin salmon (sCT), N-acetyl-calcitonin (AC), Glu14-calcitonin (Glu14), and Glu20 -calcitonin (Glu20), of which the latter two species are isobaric. The experimental monoisotopic mass values were 3429.6841, 3471.6905, and 3430.6709 Da, which were manually calculated from multiply charged species in the mass spectra (Table I). Experimental values were consistent with theoretical values within 10 ppm error. The deamidated species (Glu14/20) and their corresponding +0.984 Da mass shifts relative to sCT are clearly distinguished by a positive shift in m/z from 858.4283 to 858.6750 for the +4 charge state (Fig. 1). The spectra on the right in Fig. 1. correspond to the +2 charge states of BV and its related impurities: BV, BA, DG, PG, and 12–20. The experimental monoisotopic mass values observed were 2178.9804, 2179.9666, 2121.9564, 2236.0018, and 1167.5282 Da, respectively, which were calculated from deconvolution of the mass spectra (Table I). Experimental values were consistent with theoretical values within 5 ppm error. The +0.984 Da mass shift associated with deamidation (BV to BA) can clearly be distinguished from the BV species at m/z (+2 charge) 1090.4975 to 1090.9906 (Fig. 1). The experimental monoisotopic mass for exenatide was 4184.0517 Da, which is consistent with the theoretical mass within 6 ppm error (Table I).
Fig. 1.
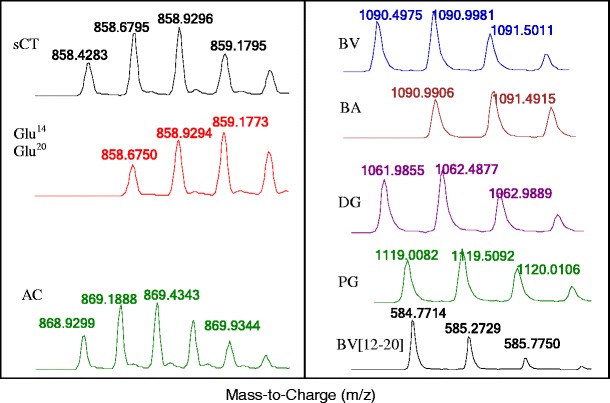
High resolution mass spectra of calcitonin salmon, bivalirudin, and their related impurities. Left: the +4 charge states of sCT and its related impurities are shown. Right: +2 charge states of BV and its related impurities are shown
Table I.
Monoisotopic Mass of Each Tested Peptide from LC-HRMS Experiment
| Compound name | Theoretical Monoisotopic Mass (Da) | Experimental Monoisotopic Mass (Da) | Δ Mass from theoretical (Da) | Mass error (ppm) |
|---|---|---|---|---|
| sCT | 3429.7133 | 3429.6841 | −0.0292 | −8.5 |
| Glu14/20 | 3430.6973 | 3430.6709 | −0.0264 | −7.7 |
| AC | 3471.7239 | 3471.6905 | −0.0334 | −9.6 |
| BV | 2178.9858 | 2178.9804 | −0.0054 | −2.5 |
| DG | 2121.9644 | 2121.9564 | −0.0079 | −3.8 |
| PG | 2236.0073 | 2236.0018 | −0.0054 | −2.5 |
| BV[12–20] | 1167.5336 | 1167.5282 | −0.0053 | −4.6 |
| BA | 2179.9698 | 2179.9666 | −0.0032 | −1.5 |
| ET | 4184.0273 | 4184.0517 | 0.0244 | 5.8 |
sCT calcitonin salmon, Glu 14/20 Glu14/20-calcitonin salmon, AC N-acetyl-calcitonin salmon, BV bivalirudin, DG des-glycine-bivalirudin, PG plus-glycine-bivalirudin, BV[12–20] 12–20 fragment of bivalirudin, BA beta-aspartic acid-bivalirudin, ET exenatide
Peptides Sequence Confirmation with LC-HRMS/MS
LC-HRMS/MS was used to confirm the sequences of calcitonin salmon and its related impurity standards (Fig. 2). Figure 2 shows the MS/MS fragmentation maps for each of the calcitonin compounds, with locations with observed fragmentation labeled in red and retained disulfide bonds shown in black. Assignment of MS/MS fragments was assisted by the use of ProSight PTM 2.0 (Northwestern University, Evanston, IL) and confirmed manually. MS/MS analysis in combination with accurate molecular weight determinations allows for both the AA composition and sequence to be confirmed. If any sequence issues such as a missing sulfide bond, deamination, or change in site of modification were to occur, this approach would be able to readily identify them.
Fig. 2.

Amino acid sequence confirmation for calcitonin salmon and its related impurities by LC-MS/MS. Black lines: intact disulfide bond. Red lines: MS/MS fragments generated by fragmentation of the +4 charge states. Accurate molecular weight determination in combination with MS/MS helps to determine AA composition and sequence confirmation which can help to identify any variation from the predicted peptide sequence
Separation of Peptide Impurities by Both LC and HRMS
The combination of LC chromatographic separation and selective HRMS detection was able to achieve baseline resolution of bivalirudin and its related impurities (Fig. 3). The top panel in Fig. 3 shows the total ion chromatogram (TIC) for bivalirudin and its related impurities. While BV, DG, and PG co-eluted, baseline separation from the BV[12–20] fragment and BA was observed in TIC. The bottom five chromatograms in Fig. 3 correspond to the LC-HRMS EICs of the +2 charge states of BV[12–20], DG, BV, BA, and PG, which were observed at m/z 584.7741, 1061.9895, 1090.5002, 1090.9922, and 119.0109, respetively. LC-HRMS EIC was able to identify and distinguish all bivalirudin and related peptides, including DG and PG which were observed to chromatographically co-elute with BV in LC only.
Fig. 3.
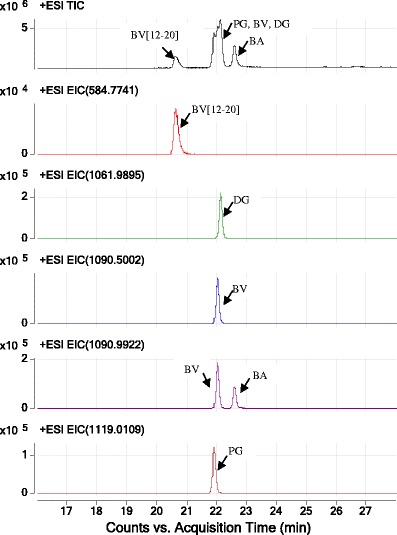
LC-HRMS TIC and EIC chromatograms of mixture of spiked synthetic bivalirudin and its related impurities reference standards. From top to bottom: TIC of BV and its related impurities, EIC of BV[12–20] (m/z at 584.7741), EIC of DG (m/z at 1061.9895), EIC of BV (m/z at 1090.5002), EIC of BA (m/z at 1090.9922), and EIC of PG (m/z at 1119.0109)
LC-HRMS Method Validation
Linearity
Peak areas obtained from extracted ion chromatograms (EICs) were used for quantitation of the peptide drugs and their related impurities. Two EIC mass extraction windows were used (20 and 100 ppm) in order to assess the impact of this parameter on the analysis.
Calcitonin Salmon and Its Related Peptides
The m/z value around which the mass extraction window was set for each calcitonin salmon related compound was determined based on selectivity and where the highest response was obtained, which were as follows: sCT at m/z 858.8681, Glu14 at m/z 859.1135, and AC at m/z 869.3684. Over the concentration range from 0.1 to 10 μM, calcitonin salmon and its related impurities (Glu14 and AC) featured linear responses using either mass extraction window (Table II). Sensitivity of each compound with a 100-ppm mass extraction window was several times higher (S2/S1 range from 2.6 to 2.79) than at the mass extraction window of 20 ppm. Because the wider mass extraction window did not compromise selectivity, precision, and accuracy, an EIC at a 100 ppm mass extraction window was used for subsequent sample analysis. Other peptides may require a narrower mass extraction window(s) to increase selectivity and specificity which may result in less sensitivity.
Table II.
Linearity (R 2) and Sensitivity (Linear Curve Slope) of Calcitonin Salmon, Bivalirudin, Exenatide, and Their Related Impurities
| Compounds | MEW 20 ppm | MEW 100 ppm | Sensitivity ratio | ||
|---|---|---|---|---|---|
| Slope (S1 × 106) | R 2 | Slope (S2 × 106) | R 2 | S2/S1 | |
| sCT | 1.905 | 0.995 | 5.320 | 0.996 | 2.79 |
| Glu14 | 1.649 | 0.996 | 4.284 | 0.997 | 2.60 |
| AC | 2.116 | 0.993 | 5.572 | 0.994 | 2.63 |
| BV | 1.234 | 1.000 | 3.107 | 0.999 | 2.52 |
| DG | 1.962 | 1.000 | 8.268 | 0.999 | 4.21 |
| PG | 1.032 | 0.999 | 4.868 | 0.999 | 4.72 |
| BV[12–20] | 8.424 | 0.999 | 2.586 | 0.999 | 3.07 |
| BA | 1.073 | 0.995 | 2.708 | 0.994 | 2.52 |
| ET | 1.234 | 0.999 | 3.116 | 0.999 | 2.53 |
MEW mass extraction window, sCT calcitonin salmon, Glu 14/20 Glu14/20-calcitonin salmon, AC N-acetyl-calcitonin salmon, BV bivalirudin, DG des-glycine-bivalirudin, PG plus-glycine-bivalirudin, BV[12–20] 12–20 fragment of bivalirudin, BA beta-aspartic acid-bivalirudin, ET exenatide
Bivalirudin and Its Related Peptides
The extracted ion m/z for each bivalirudin related compound was determined based on selectivity and highest response: BV at m/z 1090.5002, DG at m/z 1061.9895, PG at m/z 119.0109, BV[12–20] at m/z 584.7741, and BA at m/z 1090.9922. In the concentration range from 0.1 to 10 μM, bivalirudin and its related impurities (DG, PG, 12–20, and BA) featured a linear response using either mass extraction window (Table II). Sensitivity of each compound with a 100-ppm mass extraction window was several times higher (S2/S1 range from 2.5 to 4.7) than at the mass extraction window of 20 ppm.
Exenatide and Its Related Peptides
The extraction ion m/z at 1047.5150 was chosen for exenatide based on selectivity and highest response observed. Over the concentration range from 0.25 to 10 μM, exenatide featured a linear response using either mass extraction window (Table II). Sensitivity with a 100-ppm mass extraction window was 2.5 times higher (S2/S1 = 2.53) than at the mass extraction window of 20 ppm.
Precision
Table III summarizes the precision (relative standard deviation (%RSD)) results from quantitation of bivalirudin and its related impurities. All test concentrations featured %RSD values of less than 10% using data from either the 20 or 100 ppm mass extraction window. Similar results were obtained with the other two peptide drugs and their related impurities.
Table III.
Precision (%RSD) of Bivalirudin and Its Related Impurities
| Concentration (μM) | MEW 20 ppm | MEW 100 ppm | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| BV | DG | PG | 12–20 | BA | BV | DG | PG | 12–20 | BA | |
| 0.1 | 3.49 | 3.27 | 1.55 | 2.89 | 4.51 | 4.57 | 1.21 | 1.23 | 2.72 | 5.1 |
| 0.25 | 2.41 | 1.09 | 1.74 | 0.73 | 1.58 | 1.21 | 1.2 | 2.52 | 0.76 | 1.42 |
| 0.5 | 0.88 | 0.27 | 0.42 | 1.26 | 2.1 | 1.09 | 0.88 | 1.39 | 0.51 | 1.9 |
| 1 | 0.86 | 1.6 | 0.98 | 0.52 | 1.26 | 0.99 | 0.95 | 0.96 | 0.88 | 0.99 |
| 2.5 | 0.75 | 0.84 | 0.72 | 0.74 | 0.78 | 0.67 | 0.28 | 0.32 | 1.42 | 2.01 |
| 5 | 0.65 | 0.52 | 0.41 | 0.68 | 0.55 | 0.52 | 0.59 | 0.94 | 0.94 | 1.6 |
| 10 | 0.40 | 0.24 | 0.21 | 0.45 | 0.86 | 0.51 | 0.56 | 0.52 | 0.40 | 0.66 |
MEW mass extraction window, BV bivalirudin, DG des-glycine-bivalirudin, PG plus-glycine-bivalirudin, BA beta-aspartic acid-bivalirudin
Accuracy
Accuracy of related impurity measurements was determined based on spiked recovery using three different concentration spikes within the experimental linear range (Table IV). Related impurities of calcitonin salmon and bivalirudin were spiked into their respective API solutions at 0.1, 0.3, and 1% of the API concentration, which corresponded to spikes of 0.3 μM (low), 0.9 μM (middle), and 3.0 μM (high end of range). Spiked recoveries of sCT related impurities Glu14 and AC ranged between 85 and 114% at all spiked concentrations. Spiked recoveries of bivalirudin related impurities DG, PG, 12–20, and BA ranged from 96 to 110% at all spiked concentrations.
Table IV.
Accuracy (Spike Recovery, %) and Sensitivity (LOD, LOQ) of the Related Impurities of Calcitonin Salmon and Bivalirudin
| Compounds | Spike recovery (%) | Sensitivity (μM) | |||
|---|---|---|---|---|---|
| 0.3 μM | 0.9 μM | 3.0 μM | LOD | LOQ | |
| Glu14 | 85.1 | 88.6 | 97.9 | 0.03 | 0.08 |
| AC | 113.9 | 95.1 | 100.5 | 0.04 | 0.11 |
| DG | 102.4 | 98.7 | 100.1 | 0.01 | 0.04 |
| PG | 103.4 | 98.2 | 100.1 | 0.01 | 0.04 |
| BV[12–20] | 96.1 | 103.2 | 99.8 | 0.01 | 0.03 |
| BA | 110.3 | 98.2 | 105 | 0.04 | 0.11 |
LOD limit of detection, LOQ limit of quantification, Glu 14/20 Glu14/20-calcitonin salmon, AC N-acetyl-calcitonin salmon, DG des-glycine-bivalirudin, PG plus-glycine-bivalirudin, BV[12–20] 12–20 fragment of bivalirudin, BA beta-aspartic acid-bivalirudin
Limit of Detection and Limit of Quantification
The limits of detection and quantitation for the related impurities of calcitonin salmon and bivalirudin are summarized in Table IV. The LOD for Glu14-calcitonin and acetyl-calcitonin were 0.03 and 0.04 μM, equivalent to 0.03 or 0.04% of label claim, respectively. The LOQ for Glu14-calcitonin and acetyl-calcitonin were 0.08 and 0.11 μM, corresponding to 0.07 and 0.1% of label claim, respectively. The LOD for bivalirudin related impurities DG, PG, 12–20, and BA were 0.01, 0.01, 0.01, and 0.04 μM, respectively. The LOQ for bivalirudin related impurities DG, PG, 12–20, and BA were 0.04, 0.04, 0.03, and 0.11 μM, respectively. The LOD and LOQ values obtained indicate that impurities present at less than 0.1% of the peptide drug API concentration can be detected by LC-HRMS.
Peptide Drug Product Impurities Determination
Calcitonin Salmon Drug Products
A typical LC-HRMS TIC chromatogram of the calcitonin salmon nasal solution formulation is shown in Fig. 4. A magnified chromatogram from 22 to 36 min inset in Fig. 4 highlights the related impurities observed in the nasal solution product of calcitonin salmon. The material also was analyzed by using both this LC-HRMS method as well as the USP calcitonin salmon drug substance HPLC-UV method, which is used for both assay and impurity testing. The impurities detected using either method are summarized in Table V. The amount of total related impurities for the calcitonin salmon nasal solution using the LC-HRMS method was determined to be 2.64%. While the known related impurities were identified and quantitated, ten additional related impurities were also detected with LC-HRMS. When using the USP HPLC-UV method, the total related impurity of the calcitonin salmon nasal was determined to be 1.97%. In addition to the known related impurities that were identified and quantitated, four more related impurities were also found with LC-UV. More related impurities were detected in the calcitonin salmon nasal solution when using the LC-HRMS method than by using the USP HPLC-UV method. The amount of impurities detected for this drug product using either method was below the limit prescribed for the drug substance (total NMT 5%).
Fig. 4.
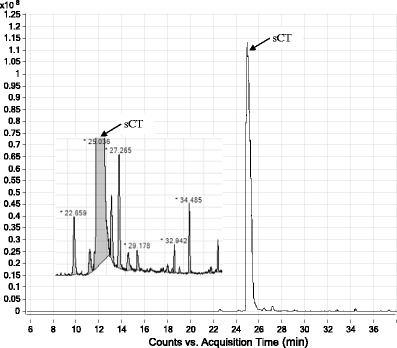
A LC-HRMS TIC chromatogram of peptide drug calcitonin salmon nasal solution. Inset: magnification from ~22 to 36 min highlights the impurities present in the calcitonin salmon product. Main peak (sCT) corresponds to the API. About ten related peptide impurities were observed and could be quantified
Table V.
Impurity Results (% Area) of LC-HRMS vs. USP sCT HPLC of the US Marketplace Calcitonin Salmon
| Methods | Peaks | RRT | % Area |
|---|---|---|---|
| LC-HRMS | Unknown1 | 0.91 | 0.39 |
| Unknown2 | 0.97 | 0.18 | |
| sCT | 1 | 97.36 | |
| Glu | 1.06 | 0.41 | |
| AC | 1.09 | 0.73 | |
| Unknown3 | 1.13 | 0.15 | |
| Unknown4 | 1.17 | 0.12 | |
| Unknown5 | 1.32 | 0.04 | |
| Unknown6 | 1.38 | 0.11 | |
| Unknown7 | 1.45 | 0.02 | |
| Unknown8 | 1.46 | 0.27 | |
| Unknown9 | 1.48 | 0.05 | |
| Unknown10 | 1.5 | 0.15 | |
| Total impurity | 2.64 | ||
| USP HPLC | Unknown1 | 0.4 | 0.14 |
| Unknown2 | 0.51 | 0.21 | |
| Unknown3 | 0.52 | 0.26 | |
| sCT | 1 | 98.03 | |
| Unknown4 | 1.06 | 0.28 | |
| Glu | 1.1 | 0.26 | |
| AC | 1.18 | 0.82 | |
| Total impurity | 1.97 |
RRT relative retention time, LC-HRMS liquid chromatography-high resolution mass spectrometry, USP United States Pharmacopeia, HPLC high-performance liquid chromatography, sCT calcitonin salmon, Glu glutamate, AC N-acetyl-calcitonin salmon,
Bivalirudin and Exenatide Drug Products
The impurity results by LC-HRMS of bivalirudin and exenatide products are summarized in Table VI. The total related impurities present in the bivalirudin for injection product was 1.5%. While the known impurities were identified and quantitated, three additional unknown related impurities were also detected. The total related impurity detected for the exenatide extended release microsphere product was 9.2%. While not shown here, the quantity of impurities detected in the exenatide extended release microsphere product is likely associated with the extraction protocol used because of the drug product formulation. The greater quantity of impurities in microsphere product is likely an artifact associated with degradation during the extraction protocol used to isolate the peptides from the drug product formulation.
Table VI.
Impurity Results (% Area) of LC-HRMS of the US Marketplace Bivalirudin and Exenatide
| Drugs | Peaks | RRT | % Area |
|---|---|---|---|
| Bivalirudin | Unknown1 | 20.3 | 0.2 |
| BV[12–20] | 20.64 | 0.1 | |
| PG | 21.85 | 0.7 | |
| BV | 21.92 | 98.5 | |
| DG | 22.1 | 0.2 | |
| Unknown2 | 22.4 | 0.1 | |
| BA | 22.47 | 0.1 | |
| Unknown3 | 23.4 | 0.1 | |
| Total impurity | 1.5 | ||
| Exenatide | Exenatide | 25.328 | 90.8 |
| Unknown1 | 26.794 | 3.8 | |
| Unknown2 | 27.854 | 5.4 | |
| Total impurity | 9.2 |
RRT relative retention time, BV[12–20] 12–20 fragment of bivalirudin, PG plus-glycine-bivalirudin, BV bivalirudin, DG des-glycine-bivalirudin, BA beta-aspartic acid-bivalirudin
DISCUSSION
TIC or EIC
TIC vs. EIC
Both LC-HRMS TICs and EICs can be used to calculate the percentage of the drug substance and potential impurities, provided that the API and its related impurities can be baseline separated chromatographically. For example, using TIC area normalization, the impurity content in exenatide was ~9.2%, while the EIC area normalization method determined an impurity content of ~8.8%. TIC and EIC calculations were consistent with each other within 1%. The choice to quantitate using either TIC and EIC should be made case by case in quality control environment with subsequent method validation studies.
If the API and its related impurities cannot be resolved with baseline separation via LC, as was observed in the case of bivalirudin and its related impurities, the EIC method should be used to circumvent the issue of co-eluting peptides.
EIC Mass Extraction Window
Different EIC mass extraction windows can be used to quantitate the drug product and any impurities present. Two EIC mass extraction windows were examined: 20 and 100 ppm. Over the concentration range from 0.1 to 10 μM, calcitonin salmon, bivalirudin, exenatide, and their related impurities had linear response with both mass extraction windows (Table II). Sensitivity with a 100-ppm mass extraction window was 2.5–4.7 (S2/S1) times higher than that with a 20-ppm mass extraction window. The mass extraction window can be optimized to any specific peptide with consideration to sensitivity, linearity, precision, accuracy, and robustness.
LC-HRMS over LC-UV
While some of the bivalirudin and its related impurity species could be baseline resolved using LC chromatographic separation alone, the use of a combined LC-HRMS EIC allowed for co-eluting peaks also to be detected selectively so that accurate quantitation could be performed on all peptide-related species detected. More peptide-related impurities were detected in the calcitonin salmon nasal solution than by using the USP HPLC-UV-based method. LC-HRMS has several inherent advantages over traditional HPLC-UV approaches, including the ability to provide both qualitative and quantitative data within a single experiment (6,9). Specific to peptide drugs, LC-HRMS allows for peptides to be easily detected with high sensitivity and over a wide linear dynamic range even in the absence of a UV chromophore. The information-rich HRMS data that can be used for accurate molecular weight determinations can be used in combination with online MS/MS to determine AA composition and confirm the AA sequence and also can be used to characterize any additional impurities that were detected.
CONCLUSION
A LC-HRMS method has been developed for the qualitative and quantitative identification and characterization of peptide drugs and related impurities with high selectivity, sensitivity, precision, and good linearity. Over the concentration range from 0.1 to 10 μM, calcitonin salmon and its related impurities featured a linear response (R2 value for calcitonin, Glu14-calcitonin, and acetyl-calcitonin were 0.995, 0.996, and 0.993, respectively). Intra-assay precision was acceptable with %RSD less than 10% for all standard concentrations used to construct the linear curve. Spiked recoveries of Glu14-calcitonin and acetyl-calcitonin ranged from 85 to 100% when spiked at 0.1, 0.3, and 1% of the concentration of the calcitonin drug product. Limits of detection for calcitonin, Glu14-calcitonin, and acetyl-calcitonin were 0.02, 0.03, and 0.04 μM, respectively, indicating that an impurity present at less than 0.1% of the API concentration could be detected. Validation studies of the method for analysis of bivalirudin and exenatide exhibited results consistent with those observed in the study of calcitonin salmon in regard to high selectivity, sensitivity, precision, and linearity of the methods.
LC-HRMS methods can be used to obtain quantitative results, with high selectivity, sensitivity, precision, and good linearity, as well as provide qualitative information, including characterization of peptide structure and sequence, within a single experiment, as demonstrated for the analysis of calcitonin salmon, bivalirudin, and exenatide. Furthermore, the LC-HRMS method was able to detect more impurities than those detected by using the USP HPLC-UV monograph method impurity test 1 for calcitonin salmon drug substance. LC-HRMS represents a promising approach that can be used as part of the analytical framework used to ensure proper quality control of peptide drug products.
Acknowledgments
Internal funding for this work was provided by the CDER Critical Path Program and the Office of Generic Drugs (MTB).
Disclaimer
The findings and conclusions of this article have not been formally disseminated by the Food and Drug Administration and should not be construed to represent any agency determination or policy.
Abbreviations
- AA
Amino acid
- AC
N-acetyl-calcitonin salmon
- API
Active pharmaceutical ingredient
- BA
Beta-aspartic acid-bivalirudin
- BV
Bivalirudin
- BV[12–20]
12–20 fragment of bivalirudin
- sCT
Calcitonin salmon
- DCM
Dichloromethane
- DG
Des-glycine-bivalirudin
- EIC
Extracted ion chromatogram
- ET
Exenatide
- Glu14
Glu14-calcitonin salmon
- Glu20
Glu20-calcitonin salmon
- LC-MS
Liquid chromatography mass spectrometry
- LC-HRMS
Liquid chromatography-high resolution mass spectrometry
- LOD
Limit of detection
- LOQ
Limit of quantification
- MEW
Mass extraction window
- MS/MS
Tandem mass spectrometry
- NDA/ANDA
New Drug Application/Abbreviated New Drug Application
- NMT
Not more than
- PG
Plus-glycine-bivalirudin
- SPPS
Solid-phase peptide synthesis
- TIC
Total ion chromatogram
References
- 1.Ermerand J, Vogel M. Applications of hyphenated LC-MS techniques in pharmaceutical analysis. Biomed Chromatogr. 2000;14:373–383. doi: 10.1002/1099-0801(200010)14:6<373::AID-BMC29>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 2.Kasparand AA, Reichert JM. Future directions for peptide therapeutics development. Drug Discov Today. 2013;18:807–817. doi: 10.1016/j.drudis.2013.05.011. [DOI] [PubMed] [Google Scholar]
- 3.Dorpe VS, Verbeken M, Wynendaele E, De Spiegeleer B. Purity profiling of peptide drugs. J Bioanal Biomed. 2011;15:1–15.
- 4.Gucinskiand AC, Boyne MT., 2nd Identification of site-specific heterogeneity in peptide drugs using intact mass spectrometry with electron transfer dissociation. Rapid Commun Mass Spectrom: RCMS. 2014;28:1757–1763. doi: 10.1002/rcm.6957. [DOI] [PubMed] [Google Scholar]
- 5.Ramanathan R, Jemal M, Ramagiri S, Xia YQ, Humpreys WG, Olah T, et al. It is time for a paradigm shift in drug discovery bioanalysis: from SRM to HRMS. J Mass Spectrom: JMS. 2011;46:595–601. doi: 10.1002/jms.1921. [DOI] [PubMed] [Google Scholar]
- 6.Ramanathan R, Korfmacher W. The emergence of high-resolution MS as the premier analytical tool in the pharmaceutical bioanalysis arena. Bioanalysis. 2012;4:467–469. doi: 10.4155/bio.12.16. [DOI] [PubMed] [Google Scholar]
- 7.Morin LP, Mess JN, Garofolo F. Large-molecule quantification: sensitivity and selectivity head-to-head comparison of triple quadrupole with Q-TOF. Bioanalysis. 2013;5:1181–1193. doi: 10.4155/bio.13.87. [DOI] [PubMed] [Google Scholar]
- 8.Wei H, Tymiak AA, Chen G. High-resolution MS for structural characterization of protein therapeutics: advances and future directions. Bioanalysis. 2013;5:1299–1313. doi: 10.4155/bio.13.80. [DOI] [PubMed] [Google Scholar]
- 9.Fung EN, Jemal M, Aubry AF. High-resolution MS in regulated bioanalysis: where are we now and where do we go from here? Bioanalysis. 2013;5:1277–1284. doi: 10.4155/bio.13.81. [DOI] [PubMed] [Google Scholar]
- 10.Dillenand L, Cuyckens F. High-resolution MS: first choice for peptide quantification? Bioanalysis. 2013;5:1145–1148. doi: 10.4155/bio.13.90. [DOI] [PubMed] [Google Scholar]
- 11.Ermerand J, Kibat P-G. A quality concept for impurities during drug development—use of the hyphenated LC–MS technique. Pharm Sci Technol Today. 1998;1:76–82. doi: 10.1016/S1461-5347(98)00025-X. [DOI] [Google Scholar]
- 12.Ermer J. The use of hyphenated LC–MS technique for characterisation of impurity profiles during drug development. J Pharm Biomed Anal. 1998;18:707–714. doi: 10.1016/S0731-7085(98)00267-2. [DOI] [PubMed] [Google Scholar]
- 13.Chopra S, Pendela M, Hoogmartens J, Van Schepdael A, Adams E. Impurity profiling of capreomycin using dual liquid chromatography coupled to mass spectrometry. Talanta. 2012;100:113–122. doi: 10.1016/j.talanta.2012.07.090. [DOI] [PubMed] [Google Scholar]
- 14.Chopra S, Van Schepdael A, Hoogmartens J, Adams E. Characterization of impurities in tylosin using dual liquid chromatography combined with ion trap mass spectrometry. Talanta. 2013;106:29–38. doi: 10.1016/j.talanta.2012.12.008. [DOI] [PubMed] [Google Scholar]
- 15.Gucinskiand AC, Boyne MT., 2nd Evaluation of intact mass spectrometry for the quantitative analysis of protein therapeutics. Anal Chem. 2012;84:8045–8051. doi: 10.1021/ac301949j. [DOI] [PubMed] [Google Scholar]
- 16.Chesnut CH, 3rd, Azria M, Silverman S, Engelhardt M, Olson M, Mindeholm L. Salmon calcitonin: a review of current and future therapeutic indications. Osteoporos Int. 2008;19:479–491. doi: 10.1007/s00198-007-0490-1. [DOI] [PubMed] [Google Scholar]
- 17.USP 36-NF 31 (United States Pharmacopeia Convention). Calcitonin salmon, calcitonin salmon injection, and calcitonin nasal solution. USP;2013. p. 2736-2741.
- 18.Reedand MD, Bell D. Clinical pharmacology of bivalirudin. Pharmacotherapy. 2002;22:105S–111S. doi: 10.1592/phco.22.10.105S.33616. [DOI] [PubMed] [Google Scholar]
- 19.Cvetkovicand RS, Plosker GL. Exenatide: a review of its use in patients with type 2 diabetes mellitus (as an adjunct to metformin and/or a sulfonylurea) Drugs. 2007;67:935–954. doi: 10.2165/00003495-200767060-00008. [DOI] [PubMed] [Google Scholar]
- 20.Liu B, Dong Q, Shi L, Wang M, Li C, Wu Y, et al. Development and validation of a reverse-phase high performance liquid chromatography method for determination of exenatide in poly(lactic-co-glycolic acid) microspheres. Chem Res Chin Univ. 2010;26:33–37. [Google Scholar]