

Abstract
International regulatory agencies have developed recommendations and guidances for bioequivalence approaches of orally inhaled drug products (OIDPs) for local action. The objective of this article is to discuss the similarities and differences among these approaches used by international regulatory authorities when applications of generic and/or subsequent entry locally acting OIDPs are evaluated. We focused on four jurisdictions that currently have published related guidances for generic and/or subsequent entry OIDPs. They are Therapeutic Goods Administration (TGA) in Australia, Health Canada (HC) in Canada, European Medicines Association (EMA) of European Union (EU), and the Food and Drug Administration (FDA) in the United States of America (USA). The comparisons of these bioequivalence (BE) recommendations are based on selection of reference products, formulation and inhaler device comparisons, and in vitro tests and in vivo studies, including pharmacokinetic (PK), pharmacodynamics (PD), and clinical studies. For the in vivo studies, the study design, choices of dose, subject inclusion/ exclusion criteria, study period, study endpoint, and equivalence criteria are elaborated in details. The bioequivalence on multiple-strength products and waiver options are also discussed.
Electronic supplementary material
The online version of this article (doi:10.1208/s12248-015-9733-9) contains supplementary material, which is available to authorized users.
KEY WORDS: bioequivalence, exhaled nitric oxide, generic, guidelines, orally inhaled drugs
INTRODUCTION
In the United States of America (USA), bioequivalence (BE) is defined as the absence of a significant difference in the rate and extent to which the active ingredient or active moiety in a pharmaceutical equivalent becomes available at the site of drug action when administered at the same molar dose under similar conditions in an appropriately designed study (1). BE is established in order to infer therapeutic equivalence (TE) between the generic drug product and corresponding reference drug product in the abbreviated new drug applications (ANDAs). It also plays an important role in supporting formulation modifications during the new drug product development phase, as well as for post-approval changes in drug applications.
At present, the use of pharmacokinetic studies to determine BE for systemically acting drug products is common across regions (2). These drugs reach their sites of action through systemic circulation. However, such approach may not always be considered sufficient to establish BE of locally acting drugs since their intended actions and deliveries are at the local sites and consequently do not rely on the systemic circulation. Therefore, alternative approaches are considered for BE of locally acting drugs.
Orally inhaled drug products (OIDPs) are a class of products that includes dry powder inhaler (DPI), metered-dose inhaler (MDI), and nebulization products. Unlike the traditional dosage forms, most inhalation products are designed as locally acting in the lungs, and their drug delivery does not solely or necessarily directly rely on the systemic circulation. It is also challenging to find a clinically relevant biomarker that is sensitive in detecting potential differences in local drug delivery. Furthermore, the majority of inhalation products are complex dosage forms that integrate formulation and device adding complexity in their BE establishment.
Establishing BE standards for locally acting OIDPs has been a long-lasting challenge for regulatory agencies around the world. Currently, regulatory guidelines on BE standards are available only in limited international jurisdictions. The recommended BE standards of these international jurisdictions have some variations. This paper summarizes the similarities and differences on the BE guidelines for orally inhaled drugs from four organizations, Therapeutic Goods Administration (TGA) in Australia, Health Canada (HC) in Canada, European Medicines Association (EMA) of European Union (EU), and the Food and Drug Administration (FDA) in the USA, with the focus on the OIDPs intended for local sites for action. The majority of these OIDPs are indicated for treatment of asthma and chronic obstructive pulmonary disease (COPD). The inhaled antibiotics and other drugs intended to have systemic exposure (e. g., inhaled insulin) are not within the scope of discussion.
METHODS
The objective of this review is to compare the regulatory approaches for establishing BE of generic and/or subsequent entry OIDPs to their corresponding reference drugs across various international jurisdictions and organizations. The jurisdictions and organizations discussed in this paper are those that currently have published related BE guidances on OIDPs. Table I lists the international jurisdictions discussed in this review, along with their corresponding regulatory agencies. The title of the related guidances published and the scopes covered by these guidelines are also included.
Table I.
International Regulatory Authorities and Respective Bioequivalence Guidelines on Inhalation Drug Products
| International regulatory authority | Agency | BE guidelines referenced | Scope | Date posted/effective |
|---|---|---|---|---|
| Australia | Therapeutic Goods Administration (TGA) | Guidance 19: Inhalation and nasal medicines/19.2 Generic MDI (13) | Support applications to register new inhalation and nasal medicine; applications to register generic inhalation and nasal medicines; requests to vary registered inhalation and nasal medicines Include medicines for treating asthma; chronic obstructive pulmonary disease (COPD), other conditions of the lungs |
August 9, 2013 |
| Also refer to EMA guidance on bioequivalence guideline for abridged orally inhalation drugs | See EMA guidance | See EU | ||
| Canada | Health Canada (HC) | Release of the Draft Guidance Document Data Requirements for Safety and Effectiveness of Subsequent Market Entry Inhaled Corticosteroid Products for Use in the Treatment of Asthma for Industry (2) | Support Abbreviated New Drug Submissions (ANDS); Supplemental New Drug Submissions (SNDS) Include medicines for: corticosteroid for asthma treatment (not COPD) |
September 19, 2011 |
| Guidance to Establish Equivalence or Relative Potency of Safety and Efficacy of a Second Entry Short-Acting Beta2-Agonist Metered-Dose Inhaler (MDI) (7) | Short-acting beta2-agonist metered-dose inhaler only | February 1, 1999 | ||
| Guidance For Industry Pharmaceutical Quality of Inhalation and Nasal Products (section Regional information) (8) | Addresses quality aspects of new marketing authorization applications (including for generic products) and does not outline expected quality aspects related to changes in existing inhalation and nasal products Includes products for administration of the drug substance to the lungs, such as pressurized MDI, DPI, products for nebulization, and non-pressurized MDIs, as well as pressurized metered-dose nasal sprays, nasal powders, and nasal liquids |
October 1, 2006 | ||
| European Union (EU) | European Medicines Agency (EMA) | Doc. Ref. CPMP/EWP/4151/00 Rev. 1—Guideline on The Requirements for Clinical Documentation for Orally Inhaled Products (OIP) Including the Requirements for Demonstration of Therapeutic Equivalence Between Two Inhaled Products for Use in the Treatment of Asthma And Chronic Obstructive Pulmonary Disease (COPD) in Adults and for Use in the Treatment of Asthma in Children and Adolescents (4) | Recommendations for demonstration of therapeutic equivalence between two inhaled products, in the context of abridged applications or variations/extensions to a marketing authorization, used in the management and treatment of adult patients with asthma and/or COPD and children and adolescents with asthma Include dosage forms: pressurized MDI; breath-operated MDI; pressurized MDI with spacers or holding chambers; non-pressurized MDI; solution and suspensions for nebulization; DPIs |
January 22, 2009 |
| United States of America (USA) | Food and Drug Administration (FDA) | Draft Bioequivalence Recommendations for Specific Product: Fluticasone Propionate/Salmeterol Xinafoate Dry Powder Inhaler (FP-SX DPI) (14) | For FP-SX DPI | September 2013 |
| Draft Bioequivalence Recommendations for Specific Product: Nebulized Budesonide Inhalation Suspension (15) | For nebulized budesonide inhalation suspension | September 2012 | ||
| Draft Bioequivalence Recommendations for Specific Product: Albuterol Sulfate Metered-Dose Inhaler (16) | For albuterol sulfate MDI | June 2013 |
BE bioequivalence, MDI metered-dose inhaler, EMA European Medicines Association
We compared the general BE approaches from different regulatory authorities and delineated the details of their similarities and differences categorized as follows:
General BE approaches from the international jurisdictions
Considerations on the selection of the reference products
Formulation and device considerations related to bioequivalence
In vitro BE studies
- In vivo BE studies:
- Pharmacokinetic (PK) studies
- Pharmacodynamic (PD) studies
- Clinical studies
Multiple-strength products and waiver option
Sources of information were the BE guidelines published by the aforementioned regulatory agencies. Notices should be made that Australia references the guidelines published by the EMA on certain recommended tests, meanwhile enforcing its own considerations on other aspects such as the PD and clinical studies.
RESULTS
General Regulatory Approaches to Establish BE on Orally Inhaled Drug Products
Recent published draft/finalized guidances on BE recommendations for OIDPs include the guidelines from Australia, Canada, EMA, and the USA. EMA guidelines have been widely adopted in European countries and referenced by some other regions such as Australia.
It should be noted that there is a difference in BE definition between the USA and that of European jurisdiction. The term BE in the USA, as defined in the introduction section, emphasizes equivalence of drug available at its site of action. Under such definition, PD and clinical studies which are usually direct indicatives of drug at its action sites, as well as PK study which is a reflective of drug availability at its action site, are all included under the US BE context. US FDA uses BE as the term for both systemically and locally acting drugs. In EMA definition, the term bioequivalence emphasizes primarily on the systemic exposure, i.e., PK study (3). For locally acting orally inhaled drug products, where PD, PK, and/or in vitro options are possible assessments, EMA guideline uses a term therapeutic equivalent (TE) (4). With this clarification, the authors simply adopt the corresponding terms from different jurisdictions in the subsequent text.
Although the ultimate goal appears to be the same, i.e., to establish appropriate equivalence approaches for OIDPs, the routes adopted by these jurisdictions to reach this goal are distinctly different. The recommended approaches for generic and/or subsequent entry orally inhaled drug products by these regulatory authorities are outlined below.
EMA recommends a step-wise approach to establish TE between the test and reference products (5): step 1—in vitro equivalence test; step 2—comparison of lung deposition and systemic exposure; and step 3—PD and clinical studies to demonstrate the local bioequivalence. The demonstration of bioequivalence at step 1 or step 2 precludes the need for further bioequivalence studies. The diagram of the step-wise BE approach recommended in EMA guidance is demonstrated in Fig. 1.
Fig. 1.
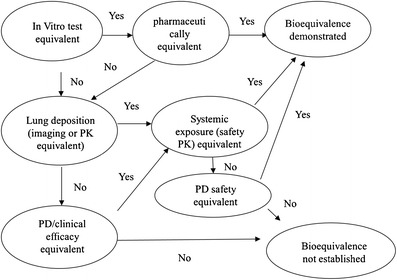
Diagram illustrating EMA’s step-wise approach to establish TE for orally inhaled drug products (adopted from (17))
Differing from the TE approach taken by EMA, the US FDA’s BE recommendations for locally acting OIDPs are based on the aggregated weight-of-evidence (6). This approach utilizes appropriate in vitro studies, pharmacokinetic (or pharmacodynamic) studies, and pharmacodynamic (or clinical efficacy) studies to demonstrate equivalence in local delivery. Formulation and device similarities are also taken into account in ensuring equivalence as well as the patient compliance in terms of exchangeability. To be concluded equivalent to a reference listed drug (RLD) product, the generic OIDPs were recommended to meet all these criteria (Fig. 2). US FDA has published several BE recommendations for certain specific inhalation drug products, including nebulized budesonide inhalation suspension, albuterol MDI, and fluticasone propionate (FP)-salmeterol xinafoate (SX) DPI products.
Fig. 2.
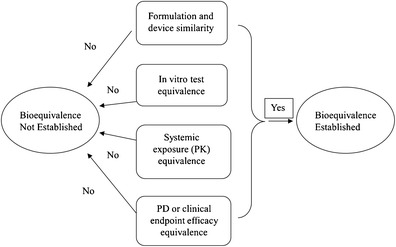
Diagram illustrating US FDA’s approach to establish BE for generic orally inhaled drug products: weight-of-evidence approach
Although not explicitly illustrated in its guidelines, Health Canada adopts an approach, similar to US FDA’s, to establish equivalence for inhaled corticosteroids and bronchodilators (7, 8, 9). In this pathway, aggregated evidences, including comparison of in vitro testing, clinical efficacy results, and systemic exposure of PK profiles, are to demonstrate equivalence.
Australia advocates the clinical efficacy studies primarily for test MDI products containing drugs such as beta-2 agonists, anticholinergics, non-corticosteroid prophylactics, and corticosteroids. Additional considerations, such as product performance tests (i.e., comparable aerodynamic characteristics of the product) as well as formulation and device factors (i.e., excipients, valve and metering system), are also recommended, provided that the clinical efficacy equivalence is established.
Selection of the Reference Products
Three jurisdictions, Health Canada, Australia TGA, and US FDA, recommend the reference drug be selected from the marketed innovator products of its own respective countries (Table II). US FDA has the publication “Approved Drug Products with Therapeutic Equivalence Evaluations” (10) (Orange Book) where the applicant can reference for a specific RLD for its submission (10).
Table II.
Selection of Reference Listed Products for Orally Inhaled Drug Products
| Jurisdiction or organization | Reference drug product selection for orally inhaled drug product |
|---|---|
| Therapeutic Goods Administration | Preferable to select an Australian marketed innovator medicine with which to compare the generic medicine (13) |
| Heath Canada | Canadian reference product |
| European Medicines Agency | Authorized innovator product if this product is still available |
| The choice of reference product should be justified | |
| Food and Drug Administration | USA marketed reference product |
EMA recommends the authorized innovator product as reference product if this product is available. Reference sourced any European Union member state or Norway, Iceland, and Lichtenstein is justifiable.
Formulation and Device Recommendations Related to Bioequivalence
Most orally inhaled drug products are combination products with the co-development of formulation and device. In formulation, there are many factors that potentially influence the aerodynamic performance of the drugs. The polymorphism, crystal habit of drug substances, drug-to-carrier ratio (DPI), types of propellant, surfactants (MDI), and physiochemical properties including viscosity, buffer capacity, drug particle size distribution of the drug and the carrier, etc. likely influence the regional deposition of the drug in the lungs and further the clinical efficacy.
US FDA, EMA, and HC each has its respective recommendations with regard to drug substance, formulation, device, and physicochemical properties of the proposed OIDPs. The Australia agency adopts the EMA bioequivalence guideline for abridged oral inhalation drugs with respect to pharmaceutical quality criteria. The similarities and differences of the related properties between the aforementioned jurisdictions are summarized in Table III.
Table III.
Comparison of the Recommendations for Drug Substance, Formulation, and Device of Orally Inhaled Product Equivalence
| Active pharmaceutical ingredients (API) | Similarities: contain the same active substance Differences: EMA, TGA: • Same form of active substance (i.e., same salt, ester, hydrate or solvate, etc.) • For the active substance in the solid state (powder, suspension): any differences in crystalline structure and/or polymorphic form should not influence the dissolution characteristics, the performance of the product, or the aerosol particle behavior FDA—nebulized budesonide inhalation suspension • Sameness of polymorphic form of the drug substance • Sameness of shape (crystalline habit) of the drug substance FDA—albuterol sulfate MDI and FP-SX DPI: not mentioned |
| Inactive ingredients | HC: qualitatively (Q1) the same and quantitatively (Q2) essentially the same (within ±10% difference) |
| EMA, TGA: | |
| • For nebulization solution with the same qualitative and quantitative composition as the reference products, the clinical study may be waived | |
| • Any qualitative and/or quantitative differences in excipients should not influence the performance of the product, aerosol particle behavior and/or be likely to affect the inhalation behavior of the patient | |
| • Any qualitative and/or quantitative differences in excipients should not change the safety profile of the product | |
| • In case of new propellants and excipients, more studies are recommended: clinical efficacy, safety profile, toxicology, local tolerability | |
| FDA: | |
| • Albuterol MDI and nebulized budesonide inhalation suspension: Q1 the same /Q2 essentially the same (within ±5% difference from RLD) | |
| • FP-SX DPI: If Q2 different from RLD, the sponsor should justify and provide pharmaceutical development data, involving in vitro testing of multiple drug-to-excipient ratios that encompass combinations below and above the ratios used in the T and R products | |
| Device | HC: recommends qualitative and quantitative analysis of |
| • The physical attributes (e.g., dimensions, materials used) | |
| • Operating characteristics of the delivery devices - functionality of the system | |
| EMA, TGA: | |
| • Similar inhaled volume through the device (within ±15% difference) | |
| • Similar handling of the inhalation devices for the test and the reference products | |
| • Similar resistance to airflow (within ±15% difference) | |
| FDA: | |
| A sponsor is encouraged to submit a working model and engineering drawings of the product to the Office of Generic Drugs (OGD) prior to ANDA submission | |
| • For FP-SX DPI, the device of the test product should have the following characteristics: passive, pre-metered multi-dose format, with 60 doses, external operating procedures consisting of 4 steps as per RLD labeling, similar size and shape to the RLD product device, comparable device resistance to the RLD product and with dose counter. | |
| • For albuterol MDI, the device should be similar in shape and size to the RLD product device. The test product should have a dose counter if the RLD product has one | |
| Physicochemical properties of drug product | HC: Physicochemical properties are recommended |
| • For aqueous product: description, osmolality (or osmolarity), surface tension, viscosity, pH, buffering capacity and specific gravity | |
| • MDI: surface tension, viscosity, specific gravity, vapor pressure, freezing point, and refractive index | |
| • DPI: particle size distribution of the carrier (if present), bulk and tapped density, particle morphology (shape, texture, and surface properties), melting point, electrostatic charge, porosity, specific surface area, hygroscopicity, and moisture content | |
| Acceptance criteria: they are should be essentially the same, within ±10% difference | |
| Other jurisdictions do not have these criteria |
EMA European Medicines Association, TGA Therapeutic Goods Administration, FDA Food and Drug Administration, DPI dry powder inhaler, MDI metered-dose inhaler, RLD reference listed drug
For drug substance, all four jurisdictions indicate that the same drug substance should be present in the test drug product. However, the scope of “sameness” is seemingly defined slightly differently with respect to the physical forms of drug substance. Canada’s guideline does not specify the recommendations for drug substances in terms of physical forms and polymorphism. EMA and Australia indicates that the same form of active substances, i.e., same salt, ester, hydrate or solvate, etc., should be present in the test products. Meanwhile, EMA and Australia accept different crystalline and/or polymorphic form provided that any difference in this does not influence the dissolution and performance of the product. In the USA, guidances on albuterol sulfate MDI and FP-SX DPI do not specify the drug substance physical forms. For the US nebulized budesonide inhalation suspension product, it is recommended for an in vitro bioequivalence approach that the same polymorphic form and crystal habit of the drug substance are present in the generic drug formulation.
These jurisdictions introduce different guidelines on the inactive ingredients in the formulation. Health Canada and the USA recommend the inactive ingredients be qualitatively (Q1) and quantitatively (Q2) the same as the reference products in general, but these two agencies have different definition in terms of Q2 sameness. Health Canada considers a difference within ±10% from the reference product as Q2 the same, whereas the USA uses “±5%” as its boundary. Specified in the BE recommendation for FP-SX DPI, the USA would consider accepting a quantitatively (Q2) different (from RLD) generic product, given that the sponsors are able to provide acceptable pharmaceutical development data, as well as bioequivalence evidence. Australia and EMA have rules for Q1 and Q2 sameness only in case of pure in vitro approvals. They also allow Q1 and/or Q2 difference in excipients concurrently enforcing that such difference should not influence the performance of the drug product, aerosol particle behavior, and/or likely the inhalation behavior of the patients. Additionally, change in the safety profile of the product should not be introduced by this difference. In case of new propellants and excipients, EMA recommends the applicants conduct the clinical efficacy and safety studies in addition to toxicological and preclinical program and mandates the applicants assess the local tolerability and investigate the evidence of increased bronchial irritability or paradoxical bronchospasm. EMA also stresses the necessity of assessing the effect of the new propellant or excipient on mucociliary clearance.
Health Canada emphasizes the physiochemical property comparisons between the test and reference products. The physiochemical properties of the nebulization aqueous products, MDI, and DPI products include but are not limited to osmolality, surface tension, viscosity, particle size distribution of the carrier, etc. Health Canada sets acceptance criteria of ±10% for these physiochemical property comparisons. These highlights of physicochemical property comparisons are not specified in other jurisdictions in the published guidances.
Regarding the inhalation devices for the device and formulation integrated products, international jurisdictions have different regulatory details. Health Canada recommends the applicants submit the results of a qualitative and quantitative analysis of the physical attributes (such as the dimension and material used in the device) and operating characteristics of the test and reference product devices. However, there are no specific acceptance criteria imposed for such device comparison. EMA recommends comparison of device-related parameters such as the inhaled volume through the device (to enable a sufficient drug substance into the lung) and device resistance, meanwhile setup acceptance criteria be within ±15% difference for such comparisons. In addition, EMA stresses that the handling of the device to release the required amount of drug substance be similar.
US FDA recommends the generic product device possess the same mechanism of function, similar operating and handling procedures as the RLD device. The USA encourages the applicants to submit working models and engineering drawings of the generic product to the agency prior to the official submission. In BE recommendations for FP-SX DPI and albuterol MDI, FDA indicates that in order to ensure the interchangeability of the generic and reference products in patients’ hands, the test and reference product devices are expected to be similar in size and shape. With regard to the dose counter, the guidance recommends that the test product have a dose counter if the RLD has one.
In Vitro Studies
In vitro performance is one of the important approaches for bioequivalence assessment of test inhalation drug products. Although in vitro results are not always well correlated with in vivo performance, it is generally accepted that the in vitro methods are usually less variable and more sensitive in detecting differences in product performance. All the aforementioned jurisdictions recommend in vitro tests for test inhalation product bioequivalence, but the respective recommendations are somewhat different in terms of the types of in vitro tests requested, statistical approaches employed, and equivalence criteria applied. The similarities and differences of these in vitro tests are summarized in Table IV.
Table IV.
Comparison of the Regulatory Recommendations for In Vitro Tests of Inhalation Drug Product Equivalence
| Delivered dose | Similarities: delivered dose uniformity are recommended Difference: HC: a statistical comparison is encouraged EMA: the targeted delivered dose should be similar (within ±15% difference) FDA: • Nebulized budesonide inhalation suspension: Test: mean delivered dose Equivalence criteria: population bioequivalence (PBE) • FP-SX DPI and albuterol MDI: Test: single actuation content at beginning (B), middle (M), and end (E) life stages using 3 flow rates Equivalence criteria: PBE |
| Particle/droplet size distribution profiles | Similarities: • Particle size distribution profile should be provided • Cascade impactor/impinger is recommended HC: a statistical comparison is encouraged EMA: • Data should be obtained with a range of clinically relevant flow rates • Fine particle mass and mass of upper stage are recommended • Comparison can be performed per impactor stage or justified group of stages • Equivalence criteria: the maximum allowable in vitro difference should be indicated and justified, e.g., ± 15% may be justifiable FDA: • Nebulized budesonide inhalation suspension: Tests: 1. Comparative drug particle and agglomerate particle size distribution in nebulized aerosol at the specified flow rate per RLD labeling. 2. Aqueous droplet size distribution by a laser diffraction method Equivalence criteria: PBE • FP-SX DPI and albuterol MDI: Test: APSD at the B and E life stages at three flow rates for DPI and one flow rate for MDI |
| Others | FDA: Nebulized budesonide inhalation suspension: • Comparative mean nebulization time • Comparative drug particle and agglomerate particle size distribution in the suspension • Comparative unit dose content of drug in the ampules Albuterol MDI: • Spray pattern at B life stage at two distances • Plume geometry at B life stage • Priming and repriming HC: content uniformity/ uniformity of dosage units is recommended for nebulization inhalation product |
HC Health Canada, MDI metered-dose inhaler, DPI dry powder inhaler, APSD aerodynamic particle size distribution
Generally speaking, two in vitro tests, delivered dose and particle size distribution upon aerosolization, are commonly recommended by all the aforementioned jurisdictions.
Delivered Dose
Delivered dose refers to the quantity of drug substance that is available to the user, ex-device, on a per dose basis. Health Canada recommends the applicants justify the difference in the delivered dose and the potential impact on the safety and efficacy of the drug product. Potential underdose and overdose of the test product should be discussed. The testing data are encouraged to be analyzed in a statistical approach, although no specific statistical method is referenced in its guidance. EMA recommends the targeted delivered dose between the test and reference product be similar, specifically within ±15% difference. US FDA accepts the single actuation content (SAC) test at beginning (B), middle (M), and end (E) life stages for albuterol MDI and FP-SX DPI. Each SAC determination is based on one actuation of the product. The SAC is recommended to be tested at one flow rate as per the RLD labeling for MDI and three flow rates for DPI. In DPI, the labeled flow rates of the DPI device are to be selected as one of the flow rates and the other two equal to ±50% of that labeled flow rate. Population bioequivalence (PBE) statistical approach is recommended as a tool for data analysis to determine equivalence. The detailed procedures for PBE analysis are published in FDA’s drug-specific bioequivalence recommendation for nebulized budesonide inhalation suspension.
Particle Size Distribution Upon Aerosolization
Characterizing the in vitro particle size distribution of inhalation products using cascade impaction has become a standard approach to describe the aerodynamic particle size distribution (APSD) performance of these products. This technique relies on separating the emitted aerosol into a series of size ranges based on their aerodynamic diameter. The relevant size range for lung deposition is typically expected to be in the range of 1−5 μm, which is frequently described as the fine particle mass (FPM). Scientists at Pharmaceutical Quality Research Institute (PQRI) workgroup indicated that the addition of the parameter of impactor stage mass (ISM), defined as a sum of the drug mass on all stages of the cascade impactor (CI) plus the terminal filter, but excluding the top CI stage, as part of the in vitro profile, enhances the discrimination of the test and reference inhalation products in some statistical tests (11). Mass median aerodynamic diameter (MMAD) and geometric standard deviation (GSD) are important parameters in determination of the aerodynamic behavior of the aerosols from the inhalation products.
All jurisdictions recommend that a cascade impactor be used for this test and the particle size distribution profile of each individual stage be submitted.
Health Canada does not provide any details for this test procedure but encourages a statistical comparison for this test. EMA poses more specific recommendations for this test and analysis procedure. It recommends this test be conducted at a certain range of flow rates related to the intended patient population, and the flow rate dependence be compared. The sponsors are encouraged to investigate the minimum (e.g., 10th percentile), median, and maximum (e.g., 90th percentile) achievable flow rate in the patient population(s) and compare for the stages that represent the fine particle mass as well as the upper stages of the impactor which are relevant to the efficacy and safety of the medicinal product in vivo. EMA accepts the particle size distribution data per impactor stage or justified group of stages. If grouped stage data are submitted, at least four groups of stages are expected, which should be justified based on the expected deposition sites in the lungs. Though no fixed acceptance criteria for APSD test were set, it is mentioned that the maximum allowable in vitro difference should be indicated and justified, e.g., ±15% may be justifiable. A decision regarding equivalence is made based on the preestablished protocol and maximum allowable differences.
US FDA has different recommendations for nebulized products, MDIs, and DPIs. For nebulized products, the APSD tests are to be conducted per the RLD labeling and using the same nebulizer(s) as specified in the RLD label. For MDIs, the APSD tests are tested at one flow rate. For DPI, three flow rates are recommended. In terms of life stages, doses at the beginning and end for MDI and DPI are to be tested. For nebulized budesonide suspension, additionally, the aqueous droplet size distribution by laser diffraction method is also recommended. FDA employs a statistical method, PBE, for equivalence analysis on majority of in vitro BE tests. The sponsors are encouraged to submit data on individual stages, as well as key parameters describing particle sizing such as ISM, MMAD, GSD, and FPM, for the agency’s evaluation.
Content Uniformity/Uniformity of Dosage Units
The current recommendations for content uniformity test from different jurisdictions are only intended for nebulization inhalation products with unit or ampule. Health Canada recommends this test only for inhalation single dose nebulization product. US FDA recommends this in vitro test for nebulized budesonide inhalation suspension.
Other In Vitro Testing
For nebulized budesonide inhalation suspension product, US FDA recommends comparative mean nebulization time test and drug particle size distribution in the ampoule. For albuterol MDI, FDA recommends spray pattern, plume geometry, and priming and repriming tests. These tests are not recommended by other jurisdictions.
Human In Vivo Bioequivalence Studies
The criteria for human in vivo BE studies differ across jurisdictions. EMA guideline indicates that if the product does not satisfy all of the pharmaceutical criteria for equivalence, in vivo studies should be performed to substantiate equivalence. Australia emphasizes in vivo clinical studies. US FDA recommends in vivo studies in addition to in vitro BE studies for albuterol MDI and FP-SX DPI. Health Canada mentions that since in vivo and in vitro correlations have not been established, reliance on only in vitro data is not enough for MDI test products. Thus, the in vivo studies are recommended to demonstrate the product equivalence.
There are three types of in vivo BE studies applicable for BE determination of OIDPs: PK, PD, and clinical studies. For locally acting OIDPs, the primary purposes of human PK studies are to examine the systemic exposure of the inhaled active substances or, alternatively, to monitor the systemic safety of the test inhalation drugs compared with the reference drugs. If the blood concentrations of the active substances are too low to be reliably quantified in a PK study, a human PD study can also be conducted for this purpose. In addition, PK studies are also believed to provide indirect evidence in supporting local delivery to the appropriate site of action. PD and clinical studies are often used as approaches to assess equivalence in clinical effect of the locally acting OIDPs. Details of these in vivo studies are illustrated below.
Human PK Studies Evaluating Systemic Exposure
All aforementioned jurisdictions recommend measurement of the systemic exposure of the active substance in the biological fluid, primarily in blood. Health Canada requests that a PK study be carried out to examine the safety of a subsequent market entry inhaled corticosteriods (ICS) product in comparison with a reference product. Similarly, US FDA recommends a PK study to compare the systemic exposure of the test and reference products. In EMA guidance, PK studies for inhalation drugs are intended to serve two purposes: (1) to assess pulmonary deposition, in which the absorption of the active moiety from the gastrointestinal (GI) tract should be excluded (for example by using charcoal blockage) and (2) to investigate the systemic safety, where the total systemic exposure including via the lungs and the GI tract should be investigated.
Not much detailed information is available on study design of the PK BE studies from these jurisdictions, which leaves much latitude in study design to the pharmaceutical industry. Health Canada simply recommend a single dose study design; US FDA’s-specific BE recommendations for albuterol MDI and FP-SX DPI recommend a single dose, crossover PK study under the fasting condition.
Recommendations related to the choice of dose for the PK studies vary among the different jurisdictions. Health Canada prefers a single, maximum labeled adult dose, while US FDA recommends the minimum number of inhalations of the test and reference products, which is sufficient to characterize a PK profile by using a sensitive analytical method. EMA indicates a single dose in this regard.
With regard to study subjects, all aforementioned jurisdictions recommend recruiting adult healthy subjects only for systemic exposure PK BE study except EMA. EMA points out adults of the intended patient population should be included for PK studies. US FDA specifies that a population of healthy male and non-pregnant healthy female subjects is preferred.
As for the PK equivalence acceptance criteria, all jurisdictions accept that 90% confidence interval (CI) of test (T) to reference (R) ratio of the geometric means of area under the curve (AUC) should be, in general, within the range of 80.00–125.00%. For maximum concentration (Cmax) parameter, Health Canada recommends the mean of Cmax ratio (T/R) be within 80.0–125.0% for orally inhaled corticosteroid products (12). Health Canada does not put these criteria for other types of OIDPs (7). EMA considers tighter, yet not specified, limits of 90% CIs for drugs with narrow therapeutic window and widened (i.e., 75–133%) 90% CIs for Cmax ratio for highly variable drugs (3). The similarity and differences in PK study recommendation are listed in Table V.
Table V.
Comparison of Pharmacokinetic Bioequivalence Study Recommendations of International Regulatory Authorities for Locally Acting Orally Inhaled Drug Products
| Study design | HC: single dose study EMA: • For purpose of pulmonary deposition, the PK study has to be able to exclude absorption of the active moiety from the GI tract • For safety purpose, the PK study should include the measurement of that amount via the lung and GI tract FDA: • The PK study is recommended in the choice of combination of in vivo and in vitro BE studies, but there is no details on study design. • FP-SX DPI and albuterol MDI: fasting, single dose, two-way crossover |
| Dose | HC: maximum labeled adult dose EMA: single dose FDA: FP-SX DPI: minimum number of inhalations that is sufficient to characterize a PK profile |
| Subjects | Similarities: for systemic exposure purpose only, adult healthy subjects are recommended Differences: HC: no additional recommendations EMA: for safety purpose, adult intended patient population FDA: normal healthy males and non-pregnant females, general population |
| Equivalence acceptance criteria | Similarities: 90% CIs of geometric mean T/R ratio of AUC generally should be within 80.0–125.0% Differences: HC: relative mean of C max (T/R) is within 80.0–12.5.0% for inhaled corticosteroids (12) EMA: T max are also compared; 90% CI for C max ratio (T/R) is within 80.00–125.00%; 90% CIs may recommend tighter limits for narrow therapeutic window; for highly variable drugs, 90% CIs for C max ratio may be widened to 75–133% FDA: 90% CI for C max ratio is within 80.00–125.00%; reference scaled approach can be considered for highly variable drugs |
HC Health Canada, EMA European Medicines Association, FDA Food and Drug Administration, PK pharmacokinetic, GI gastrointestinal tract, DPI dry powder inhaler, MDI metered-dose inhaler, CI confidence interval, AUC area under the curve
It has been widely accepted that systemic PK study provides safety information for OIDPs in terms of systemic exposure. Currently, PK study alone to establish BE of OIDPs is not universally accepted by regulatory jurisdictions. Joint efforts from US FDA, industry, and academic are being made in investigating using PK study in conjunction of in vitro tests as means to establish bioequivalence of OIDPs.
Human PD Studies Evaluating Systemic Safety
Pharmacodynamic studies for safety purpose are only specified in EMA and Health Canada guidelines. FDA considers PD studies as a means of demonstrating equivalent safety only when the measurement of PK study is infeasible, i.e., drug level in blood is too low to be characterized in PK study. The comparison of the PD study for safety purpose, in the context below, is only limited to EMA and Health Canada.
Recommendations are also available from Health Canada and EMA to accommodate situations when blood or plasma levels are too low to be quantified reliably in a routine PK BE study, a circumstance not often seen nowadays with the advancement of analytical technology. Under such occasions, both Health Canada and EMA offer that the systemic exposure be determined using a PD study by assessment of the effect on the hypothalamic–pituitary–adrenal axis (HPA). Furthermore, EMA provides different recommendations for different age groups. Per EMA guideline, assessment of the effect on HPA axis is recommended only for adults. In children, safety data cannot be extrapolated from data generated in adults with asthma or from a surrogate adult population. Instead, systemic safety in children should be demonstrated through pharmacodynamic equivalence using two different but relevant tests: an assessment of the systemic effects of ICS on the HPA axis and an assessment of lower leg bone growth rate as a surrogate marker for growth. As for the acceptance criteria of HPA axis PD test, Health Canada recommends 90% CIs of T/R ratio of serum cortisol area under the 24-h curve (SCO-24 AUC) be within 80–125%. EMA does not specify acceptance criteria. Other details of PD studies for safety purpose of these two jurisdictions are outlined in Table VI.
Table VI.
Comparison of Bioequivalence Recommendations on Pharmacodynamics Studies for Systemic Safety of European Medicines Agency and Health Canada for Locally Acting Orally Inhaled Drug Products
| Similarities PD marker | To assess the effect on the hypothalamic–pituitary adrenocortical (HPA) axis in adults |
| Dose | HC: single- or multiple-dose study to compare T and R EMA: no information provided |
| Study details | Similarities: serum cortisol area under the 24-h curve Differences: HC: • Serum cortisol is measured every two hours for 24 h. EMA: • Adults: C max during 24 h measurement. • Steady state has to be reached. • There are special considerations for children |
| Acceptance criteria | HC: log or non-log data; 90% CIs of T/R SCO-24 AUC should be within 80–125% EMA: no information provided |
HC Health Canada, EMA European Medicines Association
PD and Clinical Studies Demonstrating Bioequivalence in Efficacy
For locally acting drugs, PD and clinical studies offer direct evidence to demonstrate their equivalence in efficacy. The following section discusses the regulatory approaches for BE efficacy studies on two main classes of locally acting inhaled medications for asthma and COPD treatment: bronchodilators and corticosteroids.
PD and Clinical Bioequivalence Studies on Bronchodilators
The bronchodilators include long-acting beta-2 agonist bronchodilators (LABA), short-acting beta-2 agonist bronchodilators (SABA), and anticholinergics. Australia and EMA’s guidances cover LABA, SABA, and anticholinergics. Health Canada provides guidance on SABA MDI only. US FDA’s drug-specific BE recommendation uses albuterol MDI as an example for this category. It is found that for bronchodilators, two studies—bronchodilation and bronchoprotection studies—are commonly recommended by the aforementioned international jurisdictions.
Australia advocates the bronchodilator effect comparison study for establishing bioequivalence of test beta-2 agonists and anticholinergic MDIs. A randomized, double-blind, crossover study in stabilized mild to moderate asthmatic patient is recommended. Two parameters, forced expiratory volume in 1 s (FEV1) and/or the area under the curve for FEV1 measured for a sufficient time to fully define each response curve, are considered as primary endpoints. Two products are deemed clinically equivalent if their effect does not differ by more than 20%; in other words, the chosen confidence interval of the effect ratio of test to the reference product falls within the range of 80–120%. It is noted that Australia considers challenge tests alone, such as histamine provocation or exercise, insufficient to confirm equivalence of beta-2 agonist performance; however, these can serve as supporting studies.
For locally acting short-acting beta-agonist MDI products, Health Canada recommends two PD measurements, bronchodilation and bronchoprotection bioequivalence studies. For both studies, in order to ensure the sensitivity of the study, more than one dose, i.e., a minimum of four treatments—one and two puffs of both the reference and test products, is recommended. Thus, the study data are collected according to a four-sequence, four-period, and four-treatment crossover design. For the bronchodilation study, equivalence is established based on criteria that the 90% confidence intervals for relative potency for maximum FEV1 and area under the FEV1 curve (AUFC) be contained within 80–125%; for the bronchoprotection study, the 90% confidence interval for the relative potency for test to reference PC20, which is the concentration of provocative agent that produces a 20% decrease in FEV1, should be contained within 80–125%.
Similar to other agencies, EMA favors bronchodilation and bronchoprotection studies as a means for assessing TE of SABAs, LABAs, and anticholinergics. For both studies, double-blind, double-dummy, and two-dose level study designs are adopted. The duration of the study and the choice of primary and secondary endpoints are dependent on the therapeutic class of the test product. In general, an airway function indicator, FEV1, and concentration/dose of the provocation agent (PC20 or PD20) are expected to be measured.
A method using either bronchoprovocation or bronchodilation study is also recommended by FDA for the in vivo efficacy equivalence of albuterol MDI. Single-dose, double-blind, double-dummy, randomized, crossover study designs are adopted for both types of studies. The FDA suggests a placebo dose, two reference doses, and one test dose as the study dose design, in order to demonstrate dose–response and study sensitivity. PD endpoints are post-dose PC20 or PD20 for a bronchoprovocation study and areas under the effect curve (AUEC0–4 h and AUEC0–6 h) and maximum FEV1 (FEV1max) for bronchodilation study. Dose-scale analysis, which is not emphasized in any other jurisdictions, of the PD data on the endpoints is adopted by FDA for such a study. The 90% CI for the relative bioavailability (F) falling within 67.00–150.00% concludes equivalence of the PD study of such design.
PD and Clinical Bioequivalence Studies on Corticosteroids
Unlike bronchodilators, demonstrating BE of locally acting corticosteroids based on clinical studies, appears rather challenging due to the shallower dose–response and longer time to effect. Different PD or clinical studies with various endpoints are recommended by international jurisdictions, some are seemingly at the investigational stage at present, thus needing to be further explored.
For BE of inhaled corticosteroid product intended for asthma treatment, an airway inflammation biomarker, quantitative sputum eosinophil counts, is the preferred biomarker for Health Canada. An alternative biomarker—exhaled nitric oxide (eNO)—is also mentioned as an alternative. A double-blind, randomized, three-arm (Canadian reference product, test product, and placebo) study in steroid-naïve with stable mild asthmatic patients is encouraged. While a parallel design is preferred, a crossover design could be considered provided the washout period is long enough to eliminate carryover effect. Two co-primary endpoints, an inflammatory marker (e.g., sputum eosinophil counts or eNO) as well as change in pre-bronchodilator FEV1, are recommended, and both need to meet clinical efficacy criteria as well as clinical equivalence criteria. Clinical efficacy is defined as a reduction of at least a 50% in eosinophils compared with placebo. Meanwhile, mean FEV1 of test and reference products should show an improvement of at least 10% compared with placebo. To meet the therapeutic equivalence criteria, the 90% confidence interval of the relative mean FEV1 and eosinophil count of the test to the reference product should be within 80–125%, based on log transformed data. There is no emphasis on dose–response in the clinical equivalency study. The lowest single dose of Canadian reference product ICS is to be used for a therapeutic equivalence trial.
The approaches for demonstrating equivalent therapeutic efficacy of ICS proposed by EMA are the two models bronchodilation and bronchoprotection studies, which are the same as that for BE of bronchodilators. Specifically, a double-blind, randomized, parallel study is recommended, and a crossover study can be used as an alternative. A significant dose–response relationship with at least two doses of the test product compared with two doses of the reference product is recommended, and the doses are to be chosen at the steep portion of the dose–response curve. Recognizing the challenges in demonstrating equivalent efficacy of inhaled corticosteroids, EMA guidance also mentions, in brief, that other efficacy variables including eNO and sputum eosinophils validated quality of life (QoL) questionnaires and validated patient-reported outcome measures (PROMs) could also be investigated.
Generally, reproducible, clinically relevant and sensitive PD biomarkers are prerequisite for the PD bioequivalence study approach. In US FDA, the current draft BE guidance for albuterol sulfate MDI recommends to include zero dose together with two other different dose levels in the bronchodilatation PD study. If a dose–response relationship is evident (i.e., the high dose shows superior response to the low dose and the low dose–response is in the steep part of the dose–response curve), then this method is expected to be sensitive. A dose–response relationship of a biomarker is recommended for PD studies. When the dose–response relationship is not evident, a clinical endpoint BE study can be as an alternative approach, for example, the clinical endpoint study in US FDA-specific BE guidance for fluticasone propionate and salmeterol xinafoate DPI product. In EU, a sensitive PD endpoint is one where nonzero dose levels show a dose–response, and sensitivity is not shown with placebo (4).
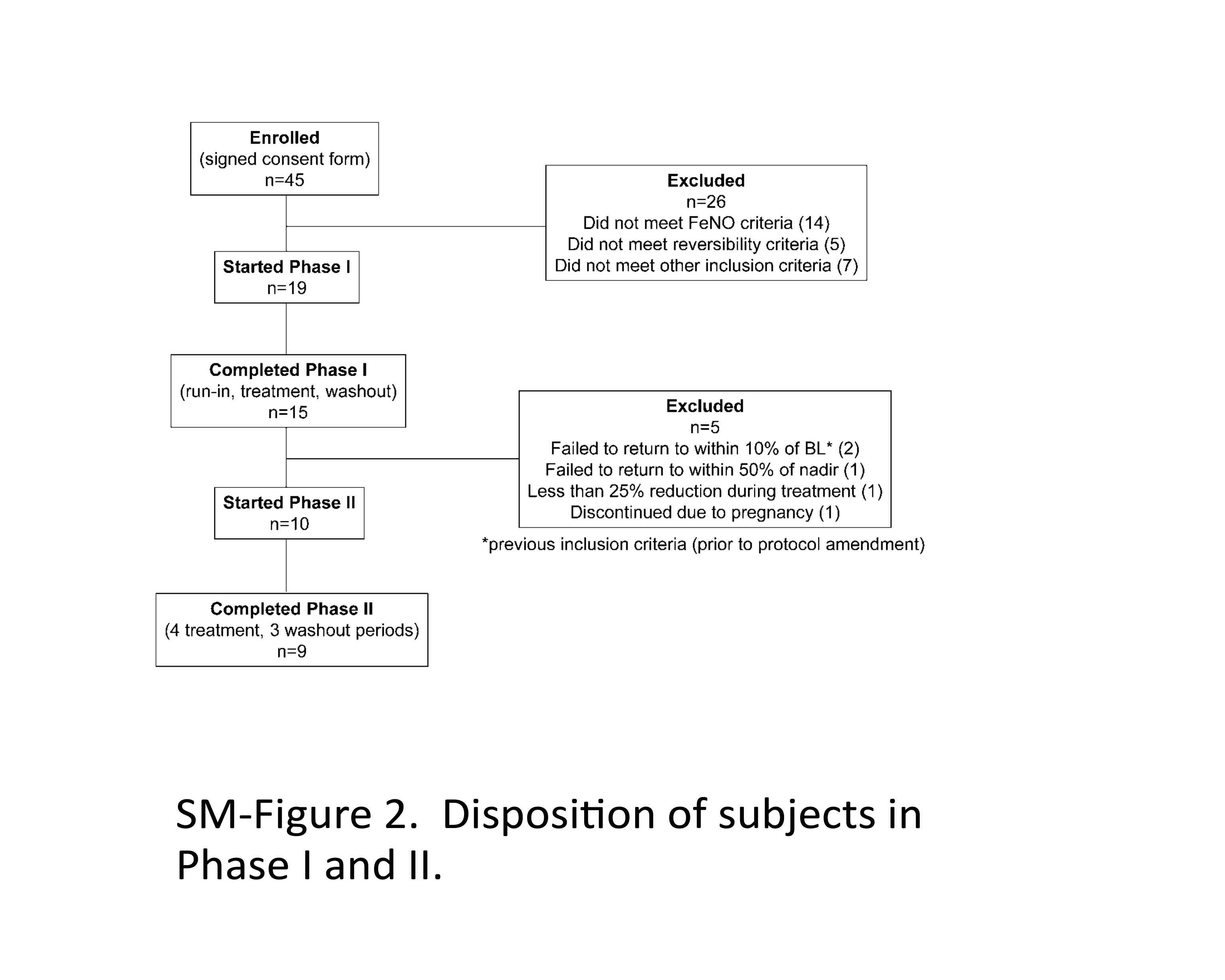
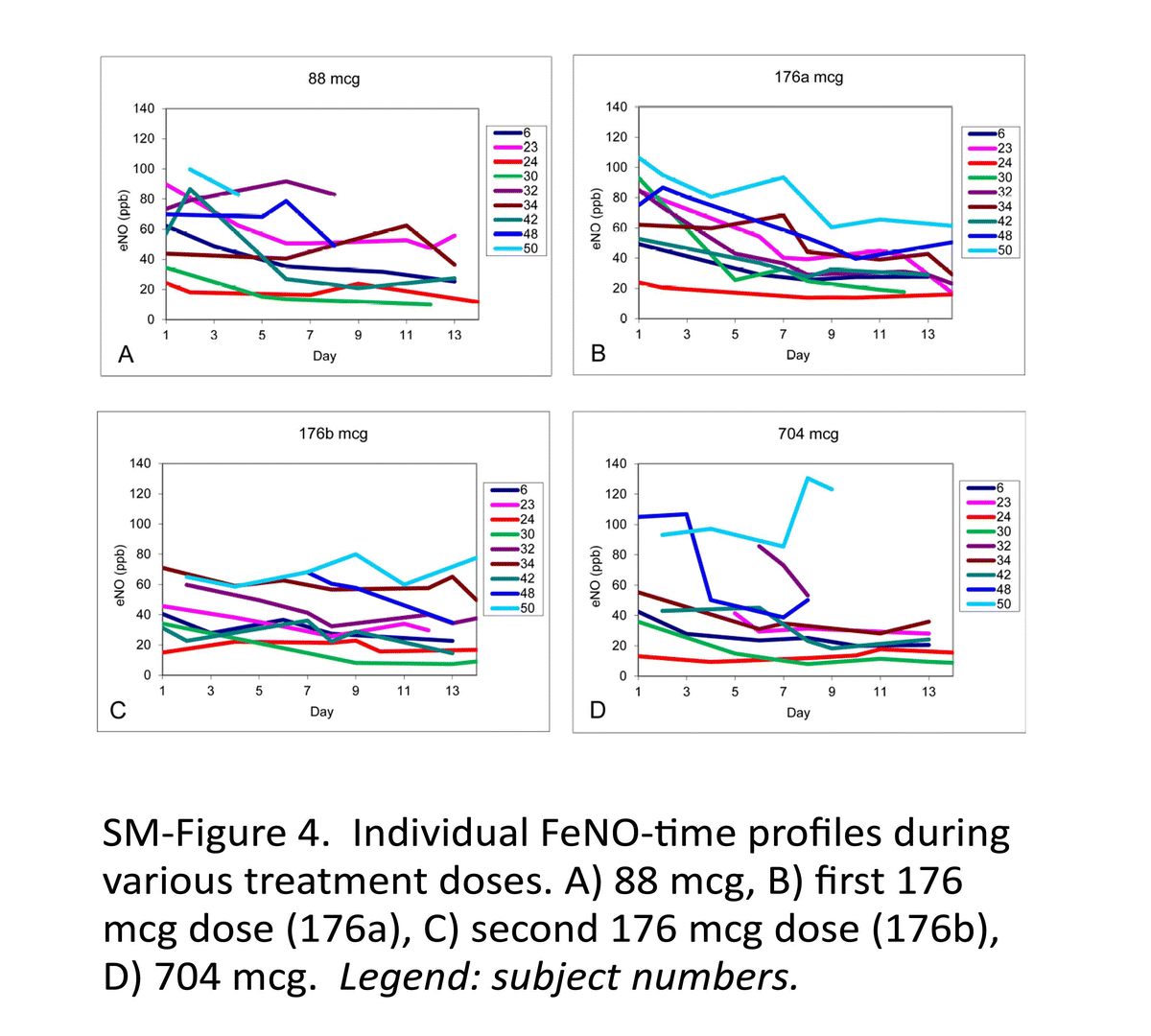
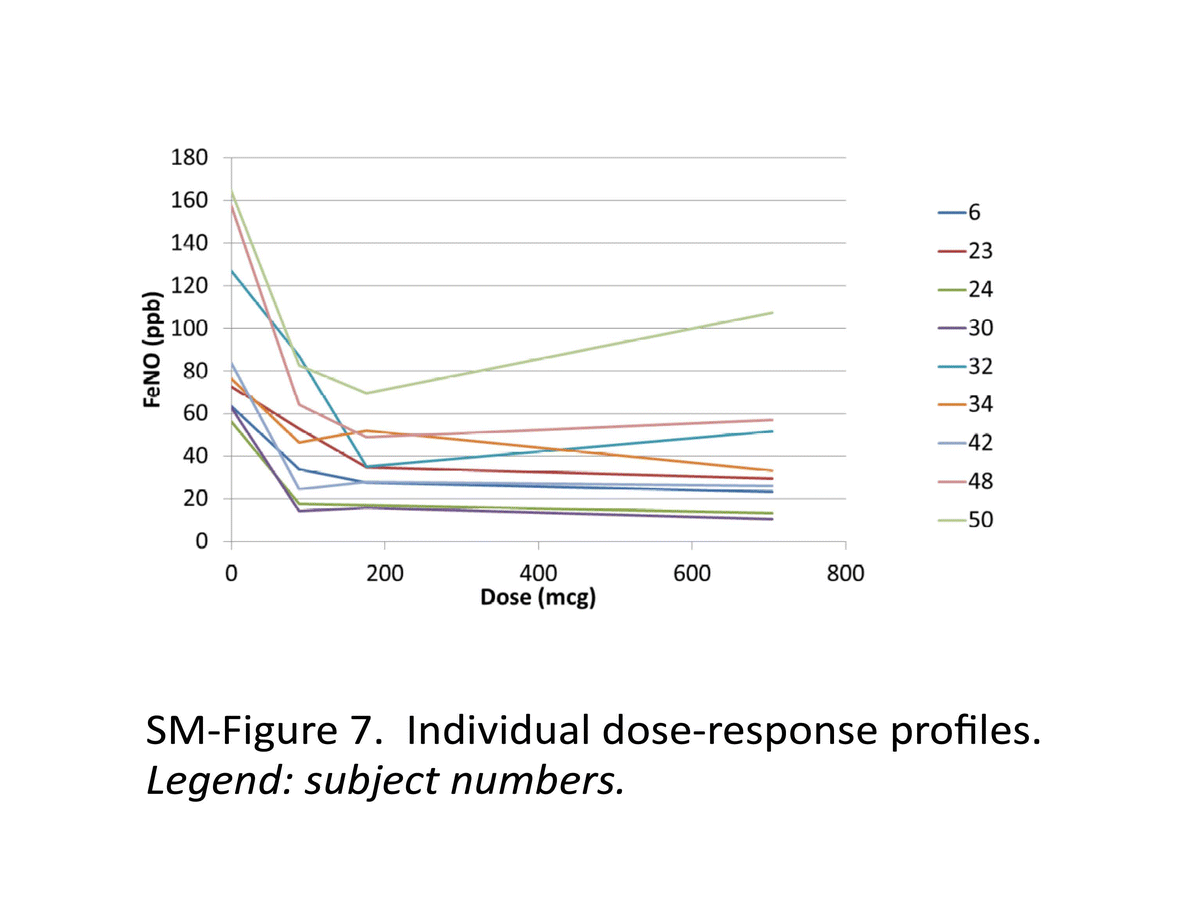
Currently, the explored PD biomarker for ICS, such as eNO, has its own limitations. US FDA has conducted investigations on bioequivalence study of corticosteroid using fractional eNO (FeNO) as the biomarker. This study was to explore the dose–response of FeNO after inhalation of fluticasone propionate (Flovent HFA). The details of the study are included in the Supplementary Materials of this paper. The PD study results indicated that a dose–response relationship for FeNO was not evident for different doses of fluticasone propionate HFA administered twice daily, suggesting that these dose levels are near or on the plateau region of the dose–response curve. Therefore, the study designs based on FeNO as a biomarker to demonstrate a dose–response relationship for ICS such as fluticasone need further evaluation.
Recently, FDA has recommended a clinical endpoint study to confirm the clinical efficacy equivalence of FP-SX DPI. In this confirmatory study, a randomized, multiple-dose, placebo-controlled, parallel group design, consisting of a 2-week run-in period and a 4-week double-blind treatment period on the lowest strength of the test and reference products, is recommended. For this particular combination drug product, the BE study endpoints depend on (i) baseline-adjusted area under the serial FEV1–time curve calculated from time zero to 12 h (AUC0–12 h) on the first day of the treatment and (ii) baseline-adjusted FEV1 measured in the morning prior to the dosing of inhaled medications on the last day of a 4-week treatment. Equivalence criteria are set as “the 90% CIs for the T/R ratios for the primary endpoints should fall within the limits of 80.00–125.00%.” It should be noted that this particular study design is not intended to demonstrate dose–response; rather, it serves as confirmatory evidence to support the equivalence in efficacy of the generic to the reference product, at a given dose. The outcome of this study, once meeting the equivalence criteria, adds weight from the clinical perspective, supporting the equivalence of a generic product to the reference listed product.
The Australian guidance does not provide specific clinical recommendations for corticosteroid MDI. The guidance merely recommends a comparative efficacy study that incorporates appropriate monitoring of adverse effects. Table VII summarizes the PD and clinical studies recommended by international jurisdictions for ICS and bronchodilators.
Table VII.
Comparisons of the Bioequivalence Recommendation for Clinical Efficacy Studies for Inhaled Corticosteroids and Bronchodilators from International Jurisdictions
| International jurisdictions | Clinical studies for inhaled ICS | Clinical efficacy studies for bronchodilators |
|---|---|---|
| Therapeutic Goods Administration | No detail provided | Bronchodilation study is preferred Bronchoprotection study alone is not sufficient but could serve as supporting study |
| Health Canada | Quantitative sputum eosinophil counts Exhaled nitric oxide (eNO) is also suggested |
Bronchodilation and bronchoprotection studies |
| European Medicines Agency | Bronchodilation and bronchoprotection studies EMA guidance also mentioned in brief that other efficacy variables including expired nitric oxide (eNO) and sputum eosinophils, validated quality of life (QoL) questionnaires, and validated patient-reported outcome measures (PROMs) could also be considered |
Bronchodilation and bronchoprotection studies |
| Food and Drug Administration | A confirmatory clinical study measuring lung function, no dose–response recommendation | Bronchodilation and bronchoprotection studies |
ICS inhaled corticosteriods, EMA European Medicines Association
Multiple Strengths and Waiver Options for Orally Inhaled Drug Products
For multiple-strength inhalation products, EMA recommends that dose linearity be investigated in vitro for both test and reference products across all proposed strengths. If there is dose linearity in vitro, then it may be sufficient to establish bioequivalence clinically with only one strength of the active substance. It is usually appropriate to study the lowest strength, at more than one dose level, to enhance the sensitivity of the study. If the linearity relationship does not exist for either of the test or reference product, bioequivalence of the test product to the reference product will have to be established with more than one product strength and possibly with all product strengths.
According to the drug-specific BE recommendations for DPI FP-SX by US FDA, in vitro BE studies and PK BE studies are to be conducted on all strengths of test products with no waiver granted on any strengths. The rational for such a recommendation was based on the current evidence that dose proportionality of PK response is not clearly established. In addition, the in vitro and in vivo correlation is currently not well understood. The PD or clinical BE study is to be conducted with the lowest strength of product, the strength that is most sensitive to the PD study, and waived for higher strengths.
Others
There are some other considerations, such as sample retention for in vitro and in vivo BE studies (US FDA—Budesonide Inhalation Suspension BE guidance) and special considerations for studies in children and adolescent subjects (EMA), which are uniquely mentioned by one jurisdiction but not others. Since these recommendations are only described by one jurisdiction, therefore there is no basis for comparison. The readers are referred to the respective guidances for further information.
CONCLUSIONS
Currently, only a limited number of jurisdictions from worldwide regulatory agencies have published draft or finalized guidance to establish bioequivalence of the generic and/or subsequent entry OIDPs. These guidelines possess commonalities and differences. From a detailed comparison of these guidances, it can be concluded that the key difference among these agencies is the general pathways taken to arrive at bioequivalence: EMA advocates a stepwise approach, US FDA and Health Canada recommend an aggregated weight of evidence approach, and Australia primarily focuses on in vivo clinical efficacy approach. The itemized comparison reveals that there are many similarities in regulatory recommendations for formulation, in vitro testing, in vivo PK studies, and in vivo PD and clinical studies on bronchodilators. More differences are observed in the area of in vivo PD and clinical studies with corticosteroids. Identifying the commonalities and differences of bioequivalence recommendations of locally acting inhalation drug products provides insights to understanding the approval standards of different agencies around the world and furthermore helps to set the scope for future global harmonization of bioequivalence recommendations in this important drug category.
Electronic supplementary material
Below is the link to the electronic supplementary material.
{kind=link}
(GIF 44 kb)
{kind=link}
{kind=link}
(GIF 50 kb)
{kind=link}
{kind=link}
(GIF 78 kb)
{kind=link}
{kind=link}
(GIF 152 kb)
{kind=link}
{kind=link}
(GIF 115 kb)
{kind=link}
{kind=link}
(GIF 81 kb)
{kind=link}
{kind=link}
(GIF 66 kb)
{kind=link}
(DOC 38 kb)
(DOC 33 kb)
(DOC 33 kb)
(DOC 35 kb)
(DOC 71.5 kb)
Acknowledgments
Disclaimer
This article reflects the views of the authors and should not be construed to represent FDA’s views or policies.
Footnotes
Wallace Adam has been retired from the agency Office of Research and Standards, Office of Generic Drugs, Center for Drug Evaluation and Research, Food and Drug Administration, Silver Spring, Maryland, USA.
REFERENCES
- 1.Code of Federal Regulations Title 21, 320.1.
- 2.Davit B, Braddy AC, Conner DP, Yu LX. International guidelines for bioequivalence of systemically available orally administered generic drug products: a survey of similarities and differences. AAPS J. 2013;15(4):974–90. doi: 10.1208/s12248-013-9499-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.EMA. Guideline on the investigation of bioequivalence. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2010/01/WC500070039.pdf. 2010.
- 4.EMA. Guideline on the requirements for clinical documentation for orally inhaled products (OIP) including the requirements for demonstration of therapeutic equivalence between two inhaled products for use in the treatment of asthma and chronic obstructive pulmonary disease (COPD) in adults and for use in the treatment of asthma in children and adolescents. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003504.pdf. 2009.
- 5.Fuglsang A. The US, and EU regulatory landscapes for locally acting generic/hybrid inhalation products intended for treatment of asthma and COPD. J Aerosol Med Pulm Drug Deliv. 2012;25(4):243–7. doi: 10.1089/jamp.2012.0970. [DOI] [PubMed] [Google Scholar]
- 6.Lee SL, Adams WP, Li BV, Conner DP, Chowdhury BA, Yu LX. In vitro considerations to support bioequivalence of locally acting drugs in dry powder inhalers for lung diseases. AAPS J. 2009;11(3):414–23. doi: 10.1208/s12248-009-9121-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Health-Canada. Guidance to establish equivalence or relative potency of safety and efficacy of a second entry short-acting beta2-agonist metered dose inhaler. http://www.hc-sc.gc.ca/dhp-mps/alt_formats/hpfb-dgpsa/pdf/prodpharma/mdi_bad-eng.pdf. 1999.
- 8.Health-Canada. Guidance for industry: pharmaceutical quality of inhalation and nasal products. http://www.hc-sc.gc.ca/dhp-mps/alt_formats/hpfb-dgpsa/pdf/prodpharma/inhalationnas-eng.pdf. 2006.
- 9.Mayers I. Introduction to the Canadian scientific advisory committee on respiratory and allergy therapies: in vivo evaluation for clinical testing in COPD and asthma therapy using generics. J Aerosol Med Pulm Drug Deliv. 2012;25:1–5. doi: 10.1089/jamp.2011.0913. [DOI] [PubMed] [Google Scholar]
- 10.US-FDA. Orange Book. http://www.accessdata.fda.gov/scripts/cder/ob/default.cfm.
- 11.Christopher D, Adams WP, Lee DS, Morgan B, Pan Z, Singh GJ, et al. Product quality research institute evaluation of cascade impactor profiles of pharmaceutical aerosols: part 2—evaluation of a method for determining equivalence. AAPS PharmSciTech. 2007;8(1):E39–48. doi: 10.1208/pt0801005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Health-Canada. Draft guidance document data requirements for safety and effectiveness of subsequent market entry inhaled corticosteroid products for use in the treatment of asthma. http://www.hc-sc.gc.ca/dhp-mps/alt_formats/pdf/consultation/drug-medic/draft_inhal_ebauche_corticost-eng.pdf. 2011.
- 13.Australia. Australian guidance 19: inhalation and nasal medicines. Section 19.2.1: selecting a reference medicine. http://www.tga.gov.au/industry/pm-argpm-guidance-19-02.htm. 2013.
- 14.US-FDA. Draft guidance on fluticasone propionate; salmeterol xinafoate. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM367643.pdf. 2013.
- 15.US-FDA. Draft guidance on budesonide. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM319977.pdf. 2012.
- 16.US-FDA. Draft guidance on albuterol sulfate. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM346985.pdf. 2013.
- 17.Health-Canada. Guidance document: comparative bioavailability standards: formulations used for systemic effects. www.hc-sc.gc.ca/dhp-mps/prodpharma/applic-demande/guide-ld/bio/gd_standards_ld_normes-eng.php. 2012.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(GIF 44 kb)
(GIF 50 kb)
(GIF 78 kb)
(GIF 152 kb)
(GIF 115 kb)
(GIF 81 kb)
(GIF 66 kb)
(DOC 38 kb)
(DOC 33 kb)
(DOC 33 kb)
(DOC 35 kb)
(DOC 71.5 kb)