

Abstract
The development path for antibody drug conjugates (ADCs) is more complex and challenging than for unmodified antibodies. While many of the preclinical considerations for both unmodified and antibody drug conjugates are shared, special considerations must be taken into account when developing an ADC. Unlike unmodified antibodies, an ADC must preferentially bind to tumor cells, internalize, and traffic to the appropriate intracellular compartment to release the payload. Parameters that can impact the pharmacological properties of this class of therapeutics include the selection of the payload, the type of linker, and the methodology for payload drug conjugation. Despite a plethora of in vitro assays and in vivo models to screen and evaluate ADCs, the challenge remains to develop improved preclinical tools that will be more predictive of clinical outcome. This review will focus on preclinical considerations for clinically validated small molecule ADCs. In addition, the lessons learned from Mylotarg®, the first in class FDA-approved ADC, are highlighted.
KEY WORDS: antibody drug conjugate, preclinical, tumor
INTRODUCTION
Antibody-drug conjugates (ADCs) consist of three basic components: the monoclonal antibody, the cytotoxic drug, from herein referred to as the payload, and the linker, which couples the payload to the antibody. The fundamental principle behind antibody drug conjugates is predicated on exploiting the exquisite specificity of monoclonal antibodies along with the cell-killing properties of cytotoxic drugs, with the ultimate goal of improving the therapeutic window for patients.
The first ADC was described over 50 years ago (1). Despite setbacks during the evolution of this class of therapeutics, the field has experienced a renaissance, as marked by the recent US Food and Drug Administration (FDA) approvals of Kadcyla® and Adcetris®. Moreover, about 30 new antibody drug conjugates are currently undergoing clinical evaluation (2). Equally important, while ADCs have historically been developed as agents for the treatment of cancer, the potential to employ this class of therapeutics to treat disease indications outside of oncology is also gaining momentum (3,4).
It is important to note that the development path for ADCs is more complex and challenging than for unmodified antibodies. Thus, while many of the preclinical considerations for both unmodified and antibody drug conjugates mirror one another, special considerations must be taken into account when developing an ADC. Unlike unmodified antibodies, an ADC must preferentially bind to tumor cells, internalize, and traffic to the appropriate intracellular compartment to release the payload (Fig. 1). Parameters that can impact the pharmacological properties of this class of therapeutics include the selection of the payload, the type of linker, and the methodology for payload drug conjugation (Fig. 1) (5).
Fig. 1.
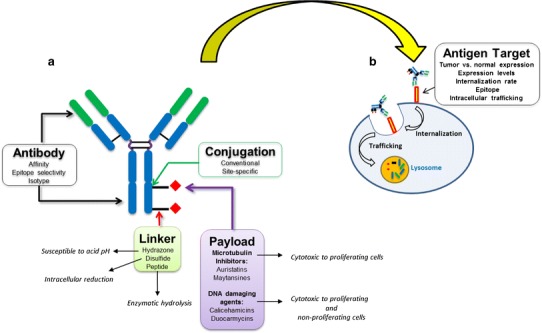
Schematic illustration of the parameters that impact the pharmacological properties of ADCs and considerations for antigen target selection. a The properties of the antibody, type of linker, payload class, and payload drug conjugation methodology are critical preclinical considerations. b Selection and characterization of the antigen target, confirmation of internalization, and intracellular trafficking to the lysosomal compartment are key factors
In spite of the available arsenal of in vitro assays and in vivo models, the challenge remains to develop improved preclinical tools that will be more predictive of clinical outcome. While the recent clinical successes of Adcetris® and Kadcyla® cannot be understated, we have yet to fully understand how these ADCs elicit their pharmacological effects in patients. The quest to develop more effective and less toxic ADCs continues. Suffice it to say, improved and more predictive preclinical studies, combined with clinical studies of next generation ADCs will no doubt augment our understanding and ability to develop agents with improved pharmacological properties, reduced toxicity, and enhanced efficacy, ultimately leading to more durable clinical responses in patients.
This review will address critical preclinical parameters to consider when developing an ADC. Furthermore, while different classes of payloads have been conjugated to an antibody, including protein toxins, radioisotopes, and small molecules, this review will exclusively focus on preclinical considerations for clinically validated small-molecule ADCs.
CONSIDERATIONS FOR TARGET SELECTION
The selection of the antigen target is a critical parameter for development of an ADC with an optimal safety and efficacy profile. The prototypical antigen target should exhibit a high level of tumor-specific or disease-specific expression and minimal to absent expression in normal tissues. In the context of cancer therapy, tractable antigen targets can be expressed on the tumor cell surface, tumor stem cells, in the tumor neovasculature or in the tumor stroma (6).
The level of antigen expression is a key parameter as it will determine how much of the ADC will bind to the target tissue and internalize. Thus, in the case of a putative antigen target that is tumor-specific, if the expression levels are low, limited binding and inefficient internalization of the ADC are likely, thereby restricting effective delivery of the cytotoxic payload and reducing the therapeutic window (7). By the same token, an antigen target that exhibits a high degree of expression on tumor cells should promote efficient binding and delivery of the cytotoxic payload. Importantly, there is also a strong correlation between elevated antigen target expression levels and clinical outcome. As a case in point, the FDA approval of Kadcyla® for Her2-positive breast cancer was based on data from the phase III EMILIA trial; data from this trial suggested that patients with increased breast tumor expression levels of Her2 exhibited improved progression-free survival and overall survival. More specifically, progression-free survival was 10.6 months for patients receiving therapy with tumors expressing higher Her2 levels versus 8.2 months for lower Her2 expression levels (8). Similarly, patients who received therapy harboring tumors with above median levels of Her2 had a median overall survival of 34.1 versus 26.5 months for patients with lower Her2 levels (8). These findings also have broad implications in formulating patient stratification strategies for a given ADC antigen target. Identification of an antigen-positive population is paramount to ensuring that the appropriate patient population receives treatment and will likely respond to therapy.
One of the key considerations for target selection is to also establish the type of normal tissue that express the antigen, the cell-cycle status of antigen-expressing cells in normal tissue, and whether there is a significant differential in expression in tumor (or disease) versus normal tissues (6). When profiling an antigen target, determining whether the antigen is expressed in vital organs or reproductive tissue is an important factor; reproductive tissues may be expendable, while vital organs are not. Suffice it to say, antigen expression in normal tissues may still be acceptable if expression in vital organs is minimal or non-existent. Once again, the FDA approval of Kadcyla® for Her2-positive breast cancer underscores this point; while Her2/neu is frequently amplified or overexpressed in a subset of human breast and ovarian cancers, it is also expressed in the heart, skin, breast, and on epithelial cells of the respiratory, gastrointestinal, urinary, and reproductive tract (9,10).
Another potential exception may include normal target tissues that are able to regenerate. For example, Rituximab, while not an ADC, is analogous as it is a depleting antibody. Rituximab targets the CD20 antigen that is expressed on B cells. Importantly, while Rituximab is employed to treat B cell non-Hodgkin lymphoma, this antibody depletes both malignant and normal B cells. Despite the ablation of normal B cells in this patient population, depletion of normal B cells is not a major safety issue (9,11).
An additional prototypical example of an antigen amenable to targeting with an ADC is CD30. CD30, a member of the TNF family, is upregulated on Reed-Sternberg cells of Hodgkin lymphoma and on systemic anaplastic large-cell lymphoma cells (ALCL) (12). In normal tissue, CD30 expression is restricted to activated T and B cells—as aforementioned, cell types that exhibit regenerative capacity (13,14). Adcetris® (brentuximab vedotin) is an ADC that targets the CD30 antigen and gained FDA approval in 2011 for the treatment of relapsed or refractory systemic ALCL and relapsed or refractory Hodgkin lymphoma.
As stated above, another consideration is the proliferative capacity of the cells expressing the antigen. Cells that are cycling are more sensitive to a class of payload known as tubulin inhibitors; (15,16) normal cells with an elevated proliferative capacity will therefore be more susceptible to this class of payload, leading to increased toxicity.
Lastly, it is worth noting that many classes of targets have been profiled for ADC-targeting. As an example, single and multiple transmembrane domain proteins, as well as glycosylphosphatidylinositol (GPI)-anchored proteins can mediate ADC internalization and anti-tumor activity in preclinical in vivo models (6,17).
Validation of Target Expression
Special consideration should be taken when validating putative antigens for targeting with an ADC. Identification of such antigens typically involves profiling messenger RNA (mRNA), protein, and assessing antibody binding for differences in expression across normal and tumor cell lines and tissue (18,19). Surveying appropriate tissues for validation of target expression by immunohistochemistry (IHC) with an antibody or panel of antibodies is the most commonly employed methodology toward establishing disease linkage and confirming that the antigen target is not abundantly expressed in normal tissues.
Despite the fact that IHC is the most widely accepted tool to assess antigen expression, this methodology does possess limitations. Firstly, IHC is a semi-quantitative measure of antigen expression levels. Equally important, IHC may not predict antibody uptake under physiological conditions—either in preclinical in vivo models or in the clinical setting (7). An alternate approach that may complement IHC data is to employ biodistribution or imaging studies. Such studies are typically performed using tumor-bearing mice and accumulation is evaluated via whole body imaging or measuring radioactivity in plasma and tissues; the ADC can be conjugated to a fluorescent tag or radioactively labeled.
In the context of preclinical animal models, a biodistribution study can provide direct confirmation that the ADC binds the target. Most importantly, biodistribution and imaging studies can enable one to assess antigen expression levels in the whole body or animal, evaluate antibody access, and confirm internalization—parameters that cannot be evaluated by IHC. Ultimately, one can establish whether the ADC is selectively binding the antigen and determine the amount of drug that has internalized and accumulated in the tumor relative to other tissue—measurements that directly impact the therapeutic window of an ADC.
INTERNALIZATION
Prior to selection of a drug payload and linker, confirmation of antibody-mediated internalization and trafficking to the appropriate intracellular compartment, namely the lysosome, are critical to elicit efficient cytotoxicity of antigen-expressing cells (Fig. 1). The antigen target should be rapidly internalized and ideally, exhibit efficient recycling on the cell surface to promote accumulation of the ADC into the cell, thereby eliciting robust cytotoxicity (7,20,21). It is also worth noting that the efficiency of internalization may vary between an unmodified mAb and an ADC (22). Experimental studies suggest that in some cases, both an ADC and an unconjugated mAb internalize at the same rate, and in other cases, the ADC may internalize with more rapid kinetics (23,24). Thus, in the context of a drug discovery program, profiling the internalization rate of both the unmodified mAb and subsequent confirmation with the ADC is paramount.
A number of parameters have been postulated to influence the rate of internalization—these include epitope selectivity, antibody affinity, and intracellular trafficking of the ADC (Fig. 1) (9,25–27).
As a case in point, Owen and colleagues generated a novel anti-Her2 mAb that maps to an epitope distinct from that of trastuzumab (Herceptin), the unmodified mAb which comprises Kadcyla®. These two mAbs, despite selectively binding to Her2, exhibit dissimilar cellular trafficking patterns and differential lysosomal accumulation (28).
With respect to antibody affinity, Rudnick and colleagues evaluated the impact of antibody binding affinity on solid tumor targeting and penetration with affinity variants that recognize the same epitope within Her2 (29). In brief, biodistribution studies profiling mAbs with moderate affinity exhibited the highest levels of tumor accumulation at 24 and 120 h after intravenous injection, whereas high-affinity mAbs exhibited the lowest levels of tumor accumulation. In addition, high-affinity antibodies exhibited greater internalization and degradation, thereby limiting their penetration into tumors. In contrast, lower-affinity antibodies penetrated tumors more efficiently, when rates of antibody-antigen dissociation were higher than those of antigen internalization (29). Thus, the learnings from the above studies should be weighed when embarking on an ADC discovery program.
PAYLOADS
As alluded to above, one major clinically validated payload class includes agents that inhibit tubulin polymerization. These agents comprise the auristatin and maytansine derivatives and exhibit preferential cytotoxicity to rapidly dividing cells (Fig. 1) (15,16,30). Auristatins are synthetic compounds while maytansinoids are derived from a natural product.
A number of maytansinoid ADCs have been characterized with demonstrated preclinical activity and more recently, clinical activity, targeting several different cell surface tumor antigens including CD19, PSMA, CD33, CD138, and CD56 (31–35). Importantly, Kadcyla® (ado-trastuzumab emtansine) carries an anti-mitotic maytansine derivative while Adcetris® (brentuximab vedotin) is conjugated to an auristatin derivative (36,37).
In addition, a second class of clinically validated small molecule payloads includes the calicheamicin and duocarmycin analogs (Fig. 1). These agents bind to the minor groove of DNA and are extremely cytotoxic, with picomolar potency across human tumor cell lines (38–40). Unlike tubulin inhibitors, DNA damaging payloads impact both proliferating and non-proliferating cells. This class of payload may be ideally suited for de-bulking solid tumors where a substantial proportion of the tumor mass consists of non-dividing cells. Moreover, this class of payload may be particularly effective in ablating tumor-initiating cells within a tumor mass; tumor-initiating cells have been postulated to be insensitive to standard chemotherapy at least partially due to their limited proliferative capacity (41,42).
It is worth highlighting that studies of a variety of human solid tumors suggest that approximately 10% to 20% of malignant cells within a tumor are actively dividing—a relatively small proportion of the total tumor cell population (43). Moreover, the average tumor volume doubling time of many human solid tumors in patients is on the order of months or years (44). In sharp contrast, the volume doubling time of human tumor xenografts in preclinical mouse models is on the order of days (45). In light of these observations, it is tempting to speculate that preclinical in vivo models may over-predict the efficacy of tubulin inhibitors due to the high proliferation rates of tumor xenografts. Suffice it to say, calicheamicin is an attractive payload for the treatment of many human solid tumors due to their slow doubling time.
Current ADCs employing calicheamicin are directed against liquid and solid tumor antigens including CD22 and 5T4 (46,47). Clinical demonstration of this payload is the antibody drug conjugate gemtuzumab ozogamicin (Mylotarg®; Pfizer), a humanized anti-CD33 IgG4 conjugated to calicheamicin (39,40,48). Mylotarg® was approved in 2000 by the FDA for the treatment of acute myeloid leukemia but was withdrawn in 2010 due to limited clinical activity and safety concerns owing to the rate of fatal toxicity observed in patients. Nonetheless, at least one additional targeted agent is under evaluation in the clinic that currently employs a calicheamicin payload (2,49).
LINKERS
Linkers are an important component which can impact the clinical efficacy and safety profiles of an ADC. Linkers should be stable in the circulation as pre-mature release of the payload can elicit toxicity; by the same token, the linker must deliver and release the payload into the cytosol of the target cell following internalization and trafficking into the requisite subcellular compartment (50).
The three general types of linkers employed for conjugating small molecule payloads to antibodies can be classified by their mode of cleavage (Fig. 1). The hydrazone linkers are susceptible to acid pH and thus release the payload under acidic conditions within the lysosomes of target cells (50,51). The disulfide linkers, which have been more widely used, undergo intracellular reduction. Their mode of action is based on the observation that the intracellular concentration of thiols, such as glutathione and cysteine, is much higher as compared to plasma (50,52). Thus, this class of linker is stable at physiological pH and releases the payload in the more reducing environment of the cytosol following internalization. The peptide linkers on the other hand, are hydrolyzed by lysosomal proteases (50–54).
Early antibody drug conjugates employed acid hydrazone linkers; however, the major pitfall of this class of linkers is their proclivity to undergo spontaneous cleavage (5). More recent linkers employ disulfide and peptidic moieties, as described above, which exhibit improved stability in circulation (5,55). The key strength of peptide-based linkers is that their hydrolysis is enzymatic; enzymes can be selected for preferential expression within tumor cells, thereby minimizing the likelihood of spontaneous drug release outside the cells and into the general circulation (52). Hence, because peptide linkers exhibit improved serum stability, they are also associated with improved anti-tumor activity (38,56). Furthermore, it is worth noting that conjugates coupled via a reducible disulfide bond linker demonstrate “bystander cytotoxicity” (38,57). With this class of disulfide-linked conjugates, the cytotoxic drug undergoes disulfide reduction followed by methylation (15). This modification of the drug, when released, is able to diffuse out of the cell and kill neighboring cells. Such a mechanism may prove particularly effective in eradicating tumor cells that do not express the antigen target within a tumor mass (58,59). However, by the same token, this class of linkers can contribute to the overall systemic toxicity of the ADC.
CONJUGATION METHODOLOGIES
Both cysteines and lysines are the two most common amino acids employed for conventionally conjugating the linker payload to the antibody. By way of example, lysine residues are the attachment point for Kadcyla® that is conjugated to a maytansine derivative (DM1) (60). On the other hand, cysteine residues are the attachment point for Adcetris® that is coupled to an auristatin derivative (MMAE) (60).
One important caveat with conventional conjugation approaches via cysteine and lysine residues is product heterogeneity. The conjugation yields a heterogeneous mixture of ADC species with a variable distribution in the number of payloads per antibody or drug loading. Product heterogeneity can potentially impact drug efficacy, especially if a significant proportion of the material is insufficiently conjugated. In other words, unconjugated or partially conjugated drug may compete for target binding of the ADC (with the appropriate antibody to drug stoichiometry), ultimately restricting efficient drug delivery to target cells. Importantly, studies also reveal that increased drug loading of an antibody results in reduced efficacy (61). Data demonstrate that antibody drug conjugates with high drug to antibody ratios are cleared more rapidly from the circulation, compromising efficacy and tolerability (61).
In order to circumvent this potential liability, IgGs have been recently engineered to contain predetermined sites for drug conjugation (i.e., site-specific conjugation) to yield uniform and more homogenous drug conjugates with defined stoichiometries. For example, McDonagh and colleagues describe the substitution of cysteine residues within the constant domains (that form the interchain disulfide bonds) with serine (62). A similar approach involves the incorporation of cysteines at defined sites available for drug conjugation; these mAbs, referred to as THIOMABs, contain two free cysteines in the antibody constant region (63). A recently published alternate strategy employs an enzymatic method for site-specific conjugation using bacterial transglutaminase. Strop and colleagues employed this approach to conjugate diverse payloads at multiple positions within an anti-EGFR antibody, and evaluated how the site of attachment impacts ADC stability, toxicity, and efficacy (64). In brief, they report that the conjugation site has a major impact on ADC stability and pharmacokinetics in a species-dependent manner (64). The authors conclude that this conjugation methodology can be employed to produce homogenous ADCs and also enables one to examine the role of position, linker, and payload to optimize the therapeutic index of an ADC candidate.
In sum, these novel approaches to better control drug loading are anticipated to generate highly cytotoxic drugs with increased tolerability, efficacy, and hopefully, more durable clinical responses.
ISOTYPE CONSIDERATIONS
The IgG1 isotype can potentially elicit antibody effector function, namely antibody-dependent cellular cytotoxicity (ADCC) and complement-dependent cytotoxicity (CDC). Effector function can prove advantageous as it provides an additional mode of action to ablate tumor cells and moreover, elicit a secondary immune response (65). Importantly, Kadcyla® has been demonstrated to elicit ADCC activity in vitro and in preclinical tumor models (66,67). An important caveat with an ADC that mediates effector function is the potential to elicit toxicity in normal tissues; it is worth mentioning that while an ADC conjugated to a tubulin inhibitor (e.g., auristatin or maytansine derivative) is likely to kill rapidly dividing cells, effector function may elicit cell death in quiescent tissue. In settings where effector function is undesirable, IgG2 or IgG4 may be preferred as these isotypes lack Fc-mediated effector activity.
OVERCOMING DRUG RESISTANCE
One significant limitation of Mylotarg® was that patients developed resistance to the drug when their tumors overexpressed P-glycoprotein (68,69). In fact, the majority of payloads employed in ADCs (70–72) are substrates for the P-glycoprotein transporter. When an antibody drug conjugate is internalized into the cell, the conjugate is processed and hydrophobic cytotoxic metabolites are generated; these hydrophobic metabolites can be substrates for P-glycoprotein and can therefore be susceptible to P-glycoprotein-mediated resistance. A new approach to circumvent this resistance has been reported through modifications in the linker conjugating the antibody to the payload (72) Kovtun and colleagues describe a maleimidyl-based hydrophilic linker coupled to antibody maytansinoid conjugates that appears to be a poor substrate for P-glycoprotein, thereby enabling the cytotoxic drug to remain inside the cell. Importantly, these conjugates are reported to bypass P-glycoprotein-mediated resistance in vitro and in vivo, with an improved therapeutic index (72). While the potential utility of this improved linker design is compelling, it remains to be seen whether such modifications will result in improved clinical responses in cancer patients.
BARRIERS TO EFFECTIVE TUMOR PENETRATION
Tumor and antigen accessibility is a critical factor and often a major hurdle for effective antibody drug delivery. Because of limited antibody tumor penetration, and hence reduced drug delivery, the requirement for highly potent payloads becomes paramount. It has been reported that only 0.001% to 0.01% of an injected unmodified antibody, and by analogy, an ADC, localizes to tumors in humans (73). This limitation is postulated to be a consequence of the architecture and physiology of solid tumors. The limited uptake of antibodies by tumors is the result of slow diffusion rates and the long distances required for diffusional transport in poorly vascularized tumors. This restricted uptake is further compromised by the fact that tumors often lack functional lymphatics (73–77), which can lead to increased levels of interstitial fluid pressure (IFP) (78,79). An increase in IFP is likely to reduce convection and thereby inhibit the uptake of antibodies (74,80). Small tumors (or micrometastases) on the other hand, tend to exhibit a more uniform circulatory system and lower IFP—as a result, ADC delivery should be more efficient under these conditions. In addition, many of the limitations in diffusion as described above can be circumvented by targeting liquid tumors.
Because solid tumors often exhibit a poorly organized vasculature, blood flow is sluggish and unstable. Therefore, delivery of oxygen and nutrients to cells that are not in close proximity to functional blood vessels is limited, resulting in hypoxia and accumulation of lactic and carbonic acid, which lower the pH in the tumor microenvironment (81,82). These oxygen- and nutrient-deprived regions are often necrotic and localized to the central regions of the solid tumor. As a consequence of the limited and abnormal blood flow rates in necrotic regions, efficient antibody drug delivery is severely restricted.
Improving tumor penetration and accumulation by using antibody fragments or protein scaffolds is a promising new avenue that is under investigation. As a case in point, preclinical studies with anti-CD30 diabodies conjugated to auristatin demonstrated efficacy in preclinical tumor models (83). However, whether such approaches yield improved tumor penetration and hence enhanced clinical responses in patients remains to be seen.
IN VIVO MODELS
Murine xenograft tumor models have proven instrumental in the selection and development of existing anti-neoplastic agents, including ADCs, for the treatment of human malignancies (84). Equally important, xenograft models have been shown to be clinically relevant as there is a correlation between activity in some animal models and Phase II clinical trials (85). As such, these models remain the model of choice for evaluation of novel therapeutic agents by the National Cancer Institute (86), academic institutions, and both the pharmaceutical and biotechnology industry.
In vivo models provide valuable information not only about efficacy, but also serve as an informative tool to predict safety and therapeutic index. However, one major caveat is that tumor xenograft models often do not fully recapitulate all stages of cancer progression (87–89). As in the case for any preclinical drug discovery program, the route of tumor implantation as well as selection of the tumor cell line(s) are both important parameters. As described earlier, human tumor xenografts adapted to grow in animals often exhibit a higher proliferative capacity as compared to the original patient tumor (88,89). The vascularity of the transplanted tumor may differ, with transplanted tumors typically exhibiting improved blood supply and reduced necrosis (89). While subcutaneous implantation is the most common route of tumor cell delivery in preclinical models, orthotopic implantation of tumor cell lines into their natural anatomic location may be more predictive and thus more clinically relevant. Orthotopic implantation allows human tumors to mimic clinical-like tumor growth and metastasis, in contrast to subcutaneous implantation, which is not a common site for human tumors (87,89). In addition, patient-derived xenograft (PDX) models, which employ primary tumor cells directly obtained from patients, as opposed to cell lines, are histologically intact, maintain tissue architecture, and preserve the original genetic lesions of the disease (90). While not broadly implemented, orthotopic implantation of patient-derived tumor fragments may ultimately prove more predictive of patient responses during the course of therapy with an ADC.
Lastly, an important consideration in the in vivo evaluation of an ADC is species cross-reactivity. In instances where the antibody component of an ADC exhibits poor or no cross-reactivity to the murine ortholog of the antigen target, a transgenic knock-in mouse may be necessary under certain settings. These animals are genetically engineered to express the human ortholog and enable one to better predict the impact of the antibody on tumor–host interactions (91). Equally important, the transgenic knock-in mouse enables one to circumvent the requirement for developing surrogate antibodies. These genetically engineered animals may be particularly informative if the ADC is directed against an antigen expressed in the host vasculature or stroma. Moreover, knock-in mice can also provide an early read on potential toxicities.
By the same token, if the antigen target is exclusively expressed on human tumor cells, a knock-in mouse model may not be required. In this setting, the ADC is expected to target the implanted human tumor that is expressing the antigen target.
SAFETY CONSIDERATIONS
Selection of the appropriate species for preclinical safety assessment must also be weighed. In similar fashion to an unmodified antibody program, the species should express the ortholog and the ADC should bind with similar affinity to the human target. ADC binding in human and animal species (e.g., rodent and non-human primate) should be evaluated by immunohistochemistry across a broad panel of tissues. Data to support species relevance should include a cross-species comparison of antigen tissue distribution, sequence similarity, epitope binding, and cytotoxicity assays with the ADC (92).
It is also important to highlight that unlike unmodified antibodies, ADCs can elicit on-target and off-target toxicities. On-target toxicity is the result of the ADC binding to and eliciting cytotoxicity of antigen-expressing normal cells and tissue. Off-target toxicities are mediated by the payload alone. These toxicities are elicited via pre-mature release of the payload in circulation, or alternatively, through endopinocytosis or FcR-mediated internalization by normal cells (6). Ultimately, the relative contribution of each of the aforementioned toxicities will determine the therapeutic index of an ADC candidate.
LESSONS LEARNED FROM MYLOTARG®
In 2000, Gemtuzumab ozogamicin, also referred to by the trade name Mylotarg® (Pfizer), was the first ADC approved by the US Food and Drug Administration (FDA) based on encouraging phase II data. More specifically, this agent was granted accelerated approval for patients with acute myeloid leukemia (AML) aged 60 years and older who were not eligible for cytotoxic chemotherapy. Mylotarg® targets CD33, which has been reported to be expressed on myeloid blasts of approximately 90% of AML patients (93). Because CD33 expression is largely restricted to immature cells of the myelomonocytic lineage and a high percentage of AML blasts, it is an antigen that is amenable to targeting with an ADC.
As previously described, the humanized anti-CD33 antibody is conjugated to calicheamicin. The antibody backbone is of the IgG4 isotype—this isotype was selected because it exhibits the longest circulating half-life of all isotypes and does not elicit antibody effector function. Importantly, the naked antibody is not cytotoxic (94). Mylotarg® also contains an acid labile hydrazone linker.
To support marketing approval, the FDA mandated completion of clinical studies in relapsed AML patients as well as randomized trials comparing Mylotarg® in combination with conventional chemotherapy versus conventional chemotherapy alone (95). Mylotarg® was voluntarily withdrawn from the market by Pfizer in 2010 after the post-approval trial that was devised to provide confirmatory evidence of clinical activity did not meet its endpoints. The S0106 trial conducted by the Southwest Oncology Group (SWOG) demonstrated that incorporating Mylotarg® to standard chemotherapy in previously untreated AML patients ages 18–60 years yielded no survival benefit and elicited significant toxicity.
What factors contributed to the early success and subsequent failure of Mylotarg® in clinical trials? What are the lessons learned from these trials?
As described earlier, one mechanism that is postulated to have contributed to the limited clinical efficacy of Mylotarg® is the elevated activity of P-glycoprotein in leukemic blasts. Interestingly, Walter and colleagues reported that patients responding to Mylotarg® exhibited significantly lower P-glycoprotein activity, namely reduced drug efflux, as compared to patients who did not respond (70). Thus, P-glycoprotein activity remained significantly associated with patient outcome. Recent preclinical data also corroborate these findings as Mylotarg® exhibited reduced cytotoxicity on cell lines that expressed P-glycoprotein in vitro; moreover, cells that were persistently exposed to low-dose Mylotarg® acquired resistance and expressed P-glycoprotein (96).
It is also hypothesized that the acid labile hydrazone linker may have been a contributing factor and hence a liability given its reported short half-life in plasma (97). In the context of Mylotarg®, a short half-life is likely to exacerbate toxicity in healthy tissue due to premature release of the active calicheamicin payload as well as impact potency by compromising efficient internalization of calicheamicin into leukemic blasts (98).
An added complication that is likely to have compromised efficacy and contributed to toxicity is the fact that Mylotarg® is a heterogeneous mixture of 50% conjugated antibody, with 0 to 8 calicheamicin moieties per IgG molecule with an average of 2 or 3, and 50% unconjugated material (95).
As highlighted above, significant toxicity was a key factor in the voluntary withdrawal of Mylotarg®. One of the safety concerns noted has centered on the reported symptoms of hepatotoxicity following administration with Mylotarg®. As a case in point, sinusoidal obstructive syndrome (SOS) or hepatic veno-occlusive disease (VOD), has been described and is a significant toxicity induced by Mylotarg® (99). In addition, a significant proportion of patients were reported to experience Grade 3 or 4 hyperbilirubinemia and Grade 3 or 4 alanine transaminase (ALT) level abnormalities (100). Deaths were also reported and associated with liver failure following treatment with Mylotarg®.
The exact cause of the liver-associated findings remains to be determined. However, it has been hypothesized that the toxicities associated with Mylotarg® treatment may relate to metabolism of unconjugated calicheamicin, leading to tissue damage, infiltration of leukemic blasts into the liver or non-specific uptake of the antibody drug conjugate by Kupffer cells (99–102). Interestingly, one recent report demonstrated that Kupffer cells and hepatocytes in the liver from a healthy individual highly express CD33, suggesting that the liver toxicities in AML patients following treatment with Mylotarg® may be directly related to antigen target expression in this tissue (102).
Despite the above factors, the most compelling data to support the utility of Mylotarg® is for the treatment of acute promyelocytic leukemia (APL), a subtype of AML; responses to Mylotarg® in this patient population have been promising. It is hypothesized that this clinical activity is due to the high and homogenous expression of CD33 on APL blasts and very low expression levels of P-glycoprotein (103,104).
In addition, subsequent to the withdrawal of Mylotarg®, emerging evidence indicates that other subsets of AML patients may benefit from the addition of this agent to conventional chemotherapy. Certain AML subtypes, namely patients with favorable risk cytogenetics, appear to benefit from Mylotarg®, while patients with adverse cytogenetics are highly unlikely to benefit (105,106).
The above learnings have contributed to our understanding and ability to develop ADCs with improved pharmacological properties, reduced toxicity, and enhanced efficacy. It is also worth noting that the lessons learned from Mylotarg® have been incorporated into the development and design of next-generation ADCs. The experience with Mylotarg® has fostered improvements in linker, payload, and conjugation technologies, as previously described in this review.
An additional lesson in the case of Mylotarg® is the importance of devising a robust patient stratification strategy. Moving forward, given that AML is a heterogeneous disease, identification of patients with well-defined molecular signatures will be critical to ensure this agent provides clinical benefit. Further trials to evaluate the optimal patient population(s) are warranted.
FUTURE DIRECTIONS
The recent FDA approvals of Adcetris® and Kadcyla® signify a new paradigm in cancer therapy. Despite these recent successes, the quest to develop improved linkers and payloads for ADCs will no doubt continue. Advances in linker and conjugation technologies should also yield ADCs with improved properties, ultimately yielding enhanced efficacy and safety profiles. ADCs that also employ effector function enhancement technologies hold tremendous promise in eliciting robust secondary immune responses in cancer patients and may ultimately elicit more durable remissions in the clinic. Efforts will also undoubtedly focus on identifying and targeting novel antigens, with the potential to treat not only malignancies but disease indications outside of oncology.
In the context of oncology, evaluation of combination therapies involving ADCs and other anti-neoplastic agents will continue to be explored and investigated. Combination therapies with recently FDA approved antibody checkpoint inhibitors (e.g., Yervoy®, Keytruda®) may hold tremendous promise in eliciting not only cellular cytotoxicity but also the ability to simultaneously elicit robust anti-tumor immune responses.
ADCs also hold tremendous potential to treat diseases outside of oncology. For example, this class of agents can be exploited to treat autoimmune diseases by targeting specific cell types to selectively suppress immune responses. Validation of this approach is evidenced by emerging clinical trial data with Ontak® (denileukin diftitox), a fusion protein combining IL-2 and diphtheria toxin. While this drug conjugate was originally approved by the FDA in 1999 for the treatment of cutaneous T cell lymphoma (CTCL), Ontak® has been evaluated in a variety of non-malignant disorders to ablate activated T lymphocytes with encouraging results (107,108). With the dramatic progress in linker, payload, and conjugation technologies since the original development of Ontak®, the potential remains to further improve the therapeutic window of ADCs for non-malignant disorders.
Moving forward, as ADCs become more broadly employed as therapeutic agents, the learnings from drug discovery efforts will enable the scientific community at large to further refine preclinical models. Moreover, as this field continues to mature, preclinical considerations will continue to be shaped and revised, with the expectation that more predictive tools will emerge and ultimately improve the likelihood of success in the clinic.
References
- 1.Decarvalho S, Rand HJ, Lewis A. Coupling of cyclic chemotherapeutic compounds to immune gamma-globulins. Nature. 1964;202:255–8. doi: 10.1038/202255a0. [DOI] [PubMed] [Google Scholar]
- 2.Mullard A. Maturing antibody-drug conjugate pipeline hits 30. Nat Rev Drug Discov. 2013;12(5):329–32. doi: 10.1038/nrd4009. [DOI] [PubMed] [Google Scholar]
- 3.Zheng B, Fuji RN, Elkins K, et al. In vivo effects of targeting CD79b with antibodies and antibody-drug conjugates. Mol Cancer Ther. 2009;8(10):2937–46. doi: 10.1158/1535-7163.MCT-09-0369. [DOI] [PubMed] [Google Scholar]
- 4.Graversen JH, Svendsen P, Dagnaes-Hansen F, et al. Targeting the hemoglobin scavenger receptor CD163 in macrophages highly increases the anti-inflammatory potency of dexamethasone. Mol Ther. 2012;20(8):1550–8. doi: 10.1038/mt.2012.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gerber HP, Senter PD, Grewal IS. Antibody drug-conjugates targeting the tumor vasculature: current and future developments. MAbs. 2009;1(3):247–53. doi: 10.4161/mabs.1.3.8515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sassoon I, Blanc V. Antibody-drug conjugate (ADC) clinical pipeline: a review. Methods Mol Biol. 2013;1045:1–27. doi: 10.1007/978-1-62703-541-5_1. [DOI] [PubMed] [Google Scholar]
- 7.Bander NH. Antibody-drug conjugate target selection: critical factors. Methods Mol Biol. 2013;1045:29–40. doi: 10.1007/978-1-62703-541-5_2. [DOI] [PubMed] [Google Scholar]
- 8.Baselga J, Verma S, Ro J, et al. Relationship between tumor biomarkers and efficacy in EMILIA, a phase III study of trastuzumab emtansine (T-DM1) in HER2-positive metastatic breast cancer. Cancer Res. 2013;73(8):LB–63. doi: 10.1158/1078-0432.CCR-15-2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Perez HL, Cardarelli PM, Deshpande S, et al. Antibody-drug conjugates: current status and future directions. Drug Discov Today. 2014;19(7):869–81. doi: 10.1016/j.drudis.2013.11.004. [DOI] [PubMed] [Google Scholar]
- 10.Press MF, et al. Expression of the HER-2/neu proto-oncogene in normal human adult and fetal tissues. Oncogene. 1990;5:953–62. [PubMed] [Google Scholar]
- 11.Solal-Celligny P. Safety of rituximab maintenance therapy in follicular lymphomas. Leuk Res. 2006;30(Suppl 1):S16–21. doi: 10.1016/s0145-2126(06)80004-4. [DOI] [PubMed] [Google Scholar]
- 12.Wahl AF, Klussman K, Thompson JD, et al. The anti-CD30 monoclonal antibody SGN-30 promotes growth arrest and DNA fragmentation in vitro and affects antitumor activity in models of Hodgkin’s disease. Cancer Res. 2002;62:3736–42. [PubMed] [Google Scholar]
- 13.Stein H, Foss HD, Durkop H, et al. CD30(+) anaplastic large cell lymphoma: a review of its histopathologic, genetic, and clinical features. Blood. 2000;96:3681–95. [PubMed] [Google Scholar]
- 14.Falini B, Pileri S, Pizzolo G, et al. CD30 (Ki-1) molecule: a new cytokine receptor of the tumor necrosis factor receptor superfamily as a tool for diagnosis and immunotherapy. Blood. 1995;85:1–14. [PubMed] [Google Scholar]
- 15.Chari RVJ. Targeted cancer therapy: conferring specificity to cytotoxic drugs. Acc Chem Res. 2008;41:98–107. doi: 10.1021/ar700108g. [DOI] [PubMed] [Google Scholar]
- 16.Doronina SO, Toki BE, Torgov MY, et al. Development of potent monoclonal antibody auristatin conjugates for cancer therapy. Nat Biotechnol. 2003;21(7):778–84. doi: 10.1038/nbt832. [DOI] [PubMed] [Google Scholar]
- 17.Teicher BA. Antibody-drug conjugate targets. Curr Cancer Drug Targets. 2009;9(8):982–1004. doi: 10.2174/156800909790192365. [DOI] [PubMed] [Google Scholar]
- 18.Carter P, Smith L, Ryan M. Identification and validation of cell surface antigens for antibody targeting in oncology. Endocrinol Relat Cancer. 2004;11(4):659–87. doi: 10.1677/erc.1.00766. [DOI] [PubMed] [Google Scholar]
- 19.Firer MA. Antibody-drug conjugates in cancer therapy—filling in the potholes that lie ahead. OA Cancer. 2013;1(1):8. [Google Scholar]
- 20.Alley SC, Zhang X, Okeley NM, et al. The pharmacologic basis for antibody-auristatin conjugate activity. J Pharmacol Exp Ther. 2009;330(3):932–8. doi: 10.1124/jpet.109.155549. [DOI] [PubMed] [Google Scholar]
- 21.Thurber GM, Schmidt MM, Wittrup KD, et al. Antibody tumor penetration: transport opposed by systemic and antigen-mediated clearance. Adv Drug Deliv Rev. 2008;60:1421–34. doi: 10.1016/j.addr.2008.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sievers EL, Senter PD. Antibody-drug conjugates in cancer therapy. Annu Rev Med. 2013;64:15–29. doi: 10.1146/annurev-med-050311-201823. [DOI] [PubMed] [Google Scholar]
- 23.Law CL, Cerveny CG, Gordon KA, et al. Efficient elimination of B-lineage lymphomas by anti-CD20-auristatin conjugates. Clin Cancer Res. 2004;10:7842–51. doi: 10.1158/1078-0432.CCR-04-1028. [DOI] [PubMed] [Google Scholar]
- 24.Smith LM, Nesterova A, Alley SC, Torgov MY, Carter PJ. Potent cytotoxicity of an auristatin-containing antibody-drug conjugate targeting melanoma cells expressing melanotransferrin/p97. Mol Cancer Ther. 2006;5:1474–82. doi: 10.1158/1535-7163.MCT-06-0026. [DOI] [PubMed] [Google Scholar]
- 25.Yoshikawa M, Mukai Y, Okada Y, et al. Robo4 is an effective tumor endothelial marker for antibody-drug conjugates based on the rapid isolation of the anti-Robo4 cell-internalizing antibody. Blood. 2013;121:2804–13. doi: 10.1182/blood-2012-12-468363. [DOI] [PubMed] [Google Scholar]
- 26.Ackerman ME, Pawlowski D, Wittrup KD, et al. Effect of antigen turnover rate and expression level on antibody penetration into tumor spheroids. Mol Cancer Ther. 2008;7(7):2233–40. doi: 10.1158/1535-7163.MCT-08-0067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Carter PJ. Potent antibody therapeutics by design. Nat Rev Immunol. 2006;6:343–57. doi: 10.1038/nri1837. [DOI] [PubMed] [Google Scholar]
- 28.Owen SC, Patel N, Logie J, et al. Targeting HER2+ breast cancer cells: lysosomal accumulation of anti-HER2 antibodies is influenced by antibody binding site and conjugation to polymeric nanoparticles. J Control Release. 2013;172(2):395–404. doi: 10.1016/j.jconrel.2013.07.011. [DOI] [PubMed] [Google Scholar]
- 29.Rudnick SI, Lou J, Shaller CC, et al. Influence of affinity and antigen internalization on the uptake and penetration of anti-HER2 antibodies in solid tumors. Cancer Res. 2011;71(6):2250–9. doi: 10.1158/0008-5472.CAN-10-2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Flygare JA, Pillow TH, Aristoff P. Antibody-drug conjugates for the treatment of cancer. Chem Biol Des. 2013;81:113–21. doi: 10.1111/cbdd.12085. [DOI] [PubMed] [Google Scholar]
- 31.Henry MD, Wen S, Silva MD, et al. A prostate-specific membrane antigen-targeted monoclonal antibody-chemotherapeutic conjugate designed for the treatment of prostate cancer. Cancer Res. 2004;64(21):7995–8001. doi: 10.1158/0008-5472.CAN-04-1722. [DOI] [PubMed] [Google Scholar]
- 32.Legrand O. An open label dose escalation study of AVE9633 administered as a single agent by intravenous (IV) infusion weekly for 2 weeks in 4-week cycle to patients with relapsed or refractory CD33-positive acute myeloid leukemia (AML) Blood. 2007;110:1850. [Google Scholar]
- 33.Polson AG, Calemine-Fenaux J, Chan P, et al. Antibody-drug conjugates for the treatment of non-Hodgkin’s lymphoma: target and linker-drug selection. Cancer Res. 2009;69(6):2358–64. doi: 10.1158/0008-5472.CAN-08-2250. [DOI] [PubMed] [Google Scholar]
- 34.Tassone P, Goldmacher VS, Neri P, et al. Cytotoxic activity of the maytansinoid immunoconjugate B-B4-DM1 against CD138+ multiple myeloma cells. Blood. 2004;104(12):3688–96. doi: 10.1182/blood-2004-03-0963. [DOI] [PubMed] [Google Scholar]
- 35.Tassone P, Gozzini A, Goldmacher VS, et al. In vitro and in vivo activity of the maytansinoid immunoconjugate huN901-N2’-deacetyl-N2’-(3-mercapto-1-oxopropyl)-maytansine against CD56+ multiple myeloma cells. Cancer Res. 2004;64(13):4629–36. doi: 10.1158/0008-5472.CAN-04-0142. [DOI] [PubMed] [Google Scholar]
- 36.Lewis Phillips GD, Li G, Dugger DL, et al. Targeting HER2-positive breast cancer with trastuzumab-DM1, an antibody-cytotoxic drug conjugate. Cancer Res. 2008;68(22):9280–90. doi: 10.1158/0008-5472.CAN-08-1776. [DOI] [PubMed] [Google Scholar]
- 37.Katz J, Janik JE, Younes A. Brentuximab vedotin (SGN-35) Clin Cancer Res. 2011;17:6428–36. doi: 10.1158/1078-0432.CCR-11-0488. [DOI] [PubMed] [Google Scholar]
- 38.Kovtun YV, Goldmacher VS. Cell killing by antibody-drug conjugates. Cancer Lett. 2007;255(2):232–40. doi: 10.1016/j.canlet.2007.04.010. [DOI] [PubMed] [Google Scholar]
- 39.Hamann PR, Hinman LM, Hollander I, et al. Gemtuzumab ozogamicin, a potent and selective anti-CD33 antibody-calicheamicin conjugate for treatment of acute myeloid leukemia. Bioconjug Chem. 2002;13(1):47–58. doi: 10.1021/bc010021y. [DOI] [PubMed] [Google Scholar]
- 40.Hamann PR, Hinman LM, Beyer CF, et al. An anti-CD33 antibody-calicheamicin conjugate for treatment of acute myeloid leukemia. Choice of linker. Bioconjug Chem. 2002;13(1):40–6. doi: 10.1021/bc0100206. [DOI] [PubMed] [Google Scholar]
- 41.Zhou BB, Zhang H, Damelin M, Geles KG, Grindley JC, Dirks PB. Tumour-initiating cells: challenges and opportunities for anticancer drug discovery. Nat Rev Drug Discov. 2009;8:806–23. doi: 10.1038/nrd2137. [DOI] [PubMed] [Google Scholar]
- 42.Gottesman MM. Mechanisms of cancer drug resistance. Annu Rev Med. 2002;53:615–27. doi: 10.1146/annurev.med.53.082901.103929. [DOI] [PubMed] [Google Scholar]
- 43.Frankfurt OS, Greco WR, Slocum HK, et al. Proliferative characteristics of primary and metastatic human solid tumors by DNA flow cytometry. Cytometry. 1984;5(6):629–35. doi: 10.1002/cyto.990050612. [DOI] [PubMed] [Google Scholar]
- 44.Friberg S, Mattson SJ. On the growth rates of human malignant tumors: implications for medical decision making. Surg Oncol. 1997;65(4):284–97. doi: 10.1002/(sici)1096-9098(199708)65:4<284::aid-jso11>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 45.Hlatky L, Olesiak M, Hahnfeldt P. Measurement of potential doubling time for human tumor xenografts using the cytokinesis-block method. Cancer Res. 1996;56:1660–3. [PubMed] [Google Scholar]
- 46.DiJoseph JF, Armellino DC, Boghaert ER, et al. Antibody-targeted chemotherapy with CMC-544: a CD22-targeted immunoconjugate of calicheamicin for the treatment of B-lymphoid malignancies. Blood. 2004;103(5):1807–14. doi: 10.1182/blood-2003-07-2466. [DOI] [PubMed] [Google Scholar]
- 47.Boghaert ER, Sridharan L, Khandke KM, et al. The oncofetal protein, 5T4, is a suitable target for antibody-guided anti-cancer chemotherapy with calicheamicin. Int J Oncol. 2008;32(1):221–34. doi: 10.3892/ijo.32.1.221. [DOI] [PubMed] [Google Scholar]
- 48.Bross PF, Beitz J, Chen G, et al. Approval summary: gemtuzumab ozogamicin in relapsed acute myeloid leukemia. Clin Cancer Res. 2001;7(6):1490–6. [PubMed] [Google Scholar]
- 49.Kantarjian H, Thomas D, Jorgensen J, et al. Inotuzumab ozogamicin, an anti-CD22-calecheamicin conjugate, for refractory and relapsed acute lymphocytic leukaemia: a phase 2 study. Lancet Oncol. 2012;13(4):403–11. doi: 10.1016/S1470-2045(11)70386-2. [DOI] [PubMed] [Google Scholar]
- 50.Senter PD. Potent antibody drug conjugates for cancer therapy. Curr Opin Chem Biol. 2009;13(3):235–44. doi: 10.1016/j.cbpa.2009.03.023. [DOI] [PubMed] [Google Scholar]
- 51.Kaneko T, Willner D, Monkovic I, et al. New hydrazone derivatives of adriamycin and their immunoconjugates—a correlation between acid stability and cytotoxicity. Bioconjug Chem. 1991;2(3):133–41. doi: 10.1021/bc00009a001. [DOI] [PubMed] [Google Scholar]
- 52.Ducry L, Stump B. Antibody-drug conjugates: linking cytotoxic payloads to monoclonal antibodies. Bioconjug Chem. 2010;21(1):5–13. doi: 10.1021/bc9002019. [DOI] [PubMed] [Google Scholar]
- 53.Toki BE, Cerveny CG, Wahl AF, Senter PD. Protease-mediated fragmentation of p-amidobenzyl ethers: a new strategy for the activation of anticancer prodrugs. J Org Chem. 2002;67(6):1866–72. doi: 10.1021/jo016187+. [DOI] [PubMed] [Google Scholar]
- 54.Dubowchik GM, Radia S, Mastalerz H, et al. Doxorubicin immunoconjugates containing bivalent, lysosomally-cleavable dipeptide linkages. Bioorg Med Chem Lett. 2002;12(11):1529–32. doi: 10.1016/s0960-894x(02)00194-4. [DOI] [PubMed] [Google Scholar]
- 55.Polakis P. Arming antibodies for cancer therapy. Curr Opin Pharmacol. 2005;5(4):382–7. doi: 10.1016/j.coph.2005.04.008. [DOI] [PubMed] [Google Scholar]
- 56.Wu AM, Senter PD. Arming antibodies: prospects and challenges for immunoconjugates. Nat Biotechnol. 2005;23(9):1137–46. doi: 10.1038/nbt1141. [DOI] [PubMed] [Google Scholar]
- 57.KovtunYV, Audette CA, Ye Y, et al. Antibody-drug conjugates designed to eradicate tumors with homogeneous and heterogeneous expression of the target antigen. Cancer Res. 66(6):3214–21. [DOI] [PubMed]
- 58.Erickson HK, Widdison WC, Mayo MF, et al. Tumor delivery and in vivo processing of disulfide-linked and thioether-linked antibody-maytansinoid conjugates. Bioconjug Chem. 2010;21:84–92. doi: 10.1021/bc900315y. [DOI] [PubMed] [Google Scholar]
- 59.Okeley NM, Miyamaoto JB, Zhang X, et al. Intracellular activation of SGN-35, a potent anti-CD30 antibody-drug conjugate. Clin Cancer Res. 2010;16:888–97. doi: 10.1158/1078-0432.CCR-09-2069. [DOI] [PubMed] [Google Scholar]
- 60.Flygare JA, Pillow TH, Aristoff P. Antibody-drug conjugates for the treatment of cancer. Chem Biol Drug Des. 2013;81:113–21. doi: 10.1111/cbdd.12085. [DOI] [PubMed] [Google Scholar]
- 61.Sanderson RJ, Hering MA, James SF, et al. In vivo drug-linker stability of an anti-CD30 dipeptide-linked auristatin immunoconjugate. Clin Cancer Res. 2005;11:843–52. [PubMed] [Google Scholar]
- 62.McDonagh CF, Turcott E, Westendorf L, et al. Engineered antibody-drug conjugates with defined sites and stoichiometries of drug attachment. Protein Eng Des Sel. 2006;19(7):299–307. doi: 10.1093/protein/gzl013. [DOI] [PubMed] [Google Scholar]
- 63.Hamblett KJ, Senter PD, Chace DF, et al. Effects of drug loading on the antitumor activity of a monoclonal antibody drug conjugate. Clin Cancer Res. 2004;10(20):7063–70. doi: 10.1158/1078-0432.CCR-04-0789. [DOI] [PubMed] [Google Scholar]
- 64.Strop P, Liu SH, Dorywalska M, et al. Location matters: site of conjugation modulates stability and pharmacokinetics of antibody drug conjugates. Chem Biol. 2013;20(2):161–7. doi: 10.1016/j.chembiol.2013.01.010. [DOI] [PubMed] [Google Scholar]
- 65.Adams GP, Weiner LM. Monoclonal antibody therapy of cancer. Nat Biotechnol. 2005;23:1147–57. doi: 10.1038/nbt1137. [DOI] [PubMed] [Google Scholar]
- 66.Lewis GD, Figari I, Fendly B, et al. Differential responses of human tumor cell lines to anti-p185HER2 monoclonal antibodies. Cancer Immunol Immunother. 1993;37(4):255–63. doi: 10.1007/BF01518520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Junttila TT, Li G, Parsons K, et al. Trastuzumab-DM1 (T-DM1) retains all the mechanisms of action of trastuzumab and efficiently inhibits growth of lapatinib insensitive breast cancer. Breast Cancer Res Treat. 2011;128(2):347–56. doi: 10.1007/s10549-010-1090-x. [DOI] [PubMed] [Google Scholar]
- 68.Linenberger ML, Hong T, Flowers D, et al. Multidrug-resistance phenotype and clinical responses to gemtuzumab ozogamicin. Blood. 2001;98(4):988–94. doi: 10.1182/blood.v98.4.988. [DOI] [PubMed] [Google Scholar]
- 69.Linenberger ML. CD33-directed therapy with gemtuzumab ozogamicin in acute myeloid leukemia: progress in understanding cytotoxicity and potential mechanisms of drug resistance. Leukemia. 2005;19(2):176–82. doi: 10.1038/sj.leu.2403598. [DOI] [PubMed] [Google Scholar]
- 70.Walter RB, Gooley TA, van der Velden VH, et al. CD33 expression and P-glycoprotein-mediated drug efflux inversely correlate and predict clinical outcome in patients with acute myeloid leukemia treated with gemtuzumab ozogamicin monotherapy. Blood. 2007;109(10):4168–70. doi: 10.1182/blood-2006-09-047399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tang R, Cohen S, Perrot JY, et al. P-gp activity is a critical resistance factor against AVE9633 and DM4 cytotoxicity in leukaemia cell lines, but not a major mechanism of chemoresistance in cells from acute myeloid leukaemia patients. BMC Cancer. 2009;9:199. doi: 10.1186/1471-2407-9-199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kovtun YV, Audette CA, Mayo MF, et al. Antibody-maytansinoid conjugates designed to bypass multidrug resistance. Cancer Res. 2010;70(6):2528–37. doi: 10.1158/0008-5472.CAN-09-3546. [DOI] [PubMed] [Google Scholar]
- 73.Espenetos AA, Snook D, Durbin H, et al. Limitations of radiolabeled monoclonal antibodies for localization of human neoplasms. Cancer Res. 1986;46(6):3183–91. [PubMed] [Google Scholar]
- 74.Jain RK. Barriers to drug delivery in solid tumors. Sci Am. 1994;271(1):58–65. doi: 10.1038/scientificamerican0794-58. [DOI] [PubMed] [Google Scholar]
- 75.The JRK, Eugene M. Landis Award Lecture 1996. Delivery of molecular and cellular medicine to solid tumors. Microcirculation. 1997;4(1):1–23. doi: 10.3109/10739689709148314. [DOI] [PubMed] [Google Scholar]
- 76.Jain RK, Munn LL, Fukumura D. Dissecting tumour pathophysiology using intravital microscopy. Nat Rev Cancer. 2002;2(4):266–76. doi: 10.1038/nrc778. [DOI] [PubMed] [Google Scholar]
- 77.Leu AJ, Berk DA, Lymboussaki A, Alitalo K, Jain RK. Absence of functional lymphatics within a murine sarcoma: a molecular and functional evaluation. Cancer Res. 2000;60(16):4324–7. [PubMed] [Google Scholar]
- 78.Heldin CH, Rubin K, Pietras K, Ostman A. High interstitial fluid pressure—an obstacle in cancer therapy. Nat Rev Cancer. 2004;4(10):806–13. doi: 10.1038/nrc1456. [DOI] [PubMed] [Google Scholar]
- 79.Milosevic MF, Fyles AW, Wong R, Pintilie M, Kavanagh MC, Levin W, et al. Interstitial fluid pressure in cervical carcinoma: within tumor heterogeneity, and relation to oxygen tension. Cancer. 1998;82(12):2418–26. doi: 10.1002/(sici)1097-0142(19980615)82:12<2418::aid-cncr16>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 80.Jain RK. Transport of molecules in the tumor interstitium: a review. Cancer Res. 1987;47(12):3039–51. [PubMed] [Google Scholar]
- 81.Gatenby RA, Gillies RJ. Why do cancers have high aerobic glycolysis? Nat Rev Cancer. 2004;4(11):891–9. doi: 10.1038/nrc1478. [DOI] [PubMed] [Google Scholar]
- 82.Tannock IF, Rotin D. Acid pH in tumors and its potential for therapeutic exploitation. Cancer Res. 1989;49(16):4373–84. [PubMed] [Google Scholar]
- 83.Kim KM, McDonagh CF, Westendorf L, et al. Anti-CD30 diabody—drug conjugates with potent antitumor activity. Mol Cancer Ther. 2008;7:2486–97. doi: 10.1158/1535-7163.MCT-08-0388. [DOI] [PubMed] [Google Scholar]
- 84.Newell DR. Flasks, fibres and flanks—pre-clinical tumour models for predicting clinical antitumor activity. Br J Cancer. 2001;84(10):1289–90. doi: 10.1054/bjoc.2001.1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Johnson JI, Decker S, Zaharevitz D, et al. Relationships between drug activity in NCI preclinical in vitro and in vivo models and early clinical trials. Br J Cancer. 2001;84(10):1424–31. doi: 10.1054/bjoc.2001.1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Plowman J, Dykes DJ, Hollingshead M, et al. Anticancer drug development guide. Cancer Drug Discov Dev. 1997:101-125.
- 87.Hoffman RM. Orthotopic metastatic mouse models for anticancer drug discovery and evaluation: a bridge to the clinic. Investig New Drugs. 1999;17:343–59. doi: 10.1023/a:1006326203858. [DOI] [PubMed] [Google Scholar]
- 88.Steel GG, et al. The response to chemotherapy of a variety of human tumour xenografts. Br J Cancer. 1983;47:1–13. doi: 10.1038/bjc.1983.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hood JD, Cheresh DA. Building a better trap. Proc Natl Acad Sci U S A. 2003;100:8624–5. doi: 10.1073/pnas.1633646100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Fichtner I, Rolff J, Soong R, et al. Establishment of patient-derived non-small cell lung cancer xenografts as models for the identification of predictive biomarkers. Clin Cancer Res. 2008;14:6456–68. doi: 10.1158/1078-0432.CCR-08-0138. [DOI] [PubMed] [Google Scholar]
- 91.Lute KD, May KF, Jr, Lu P, et al. Human CTLA4 knock-in mice unravel the quantitative link between tumor immunity and autoimmunity induced by anti-CTLA-4 antibodies. Blood. 2005;106:3127–33. doi: 10.1182/blood-2005-06-2298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Tabrizi MA, Bornstein GG, Klakamp SL, et al. Translational strategies for development of monoclonal antibodies from discovery to the clinic. Drug Discov Today. 2009;14(5–6):298–305. doi: 10.1016/j.drudis.2008.12.008. [DOI] [PubMed] [Google Scholar]
- 93.Griffin JD, Linch D, Sabbath K, et al. A monoclonal antibody reactive with normal and leukemic human myeloid progenitor cells. Leuk Res. 1984;8:521–34. doi: 10.1016/0145-2126(84)90001-8. [DOI] [PubMed] [Google Scholar]
- 94.Hamann PR, Hinman LM, Hollander I, et al. Gemtuzumab ozogamicin, a potent and selective anti-CD33 antibody-calicheamicin conjugate for treatment of acute myeloid leukemia. Bioconjug Chem. 2002;13:47–58. doi: 10.1021/bc010021y. [DOI] [PubMed] [Google Scholar]
- 95.Bross PF, Beitz J, Chen G, et al. Approval summary: gemtuzumab ozogamicin in relapsed acute myeloid leukemia. Clin Cancer Res. 2001;7:1490–6. [PubMed] [Google Scholar]
- 96.Matusmoto T, Jimi S, Hara S, et al. Importance of inducible multidrug resistance1 expression in HL-60 cells resistant to gemtuzumab ozogamicin. Leuk Lymphoma. 2012;53:1399–405. doi: 10.3109/10428194.2012.656102. [DOI] [PubMed] [Google Scholar]
- 97.van Der Velden VH, te Marvelde JG, Hoogeveen PG, et al. Targeting of the CD33-calicheamicin immunoconjugate Mylotarg (CMA-676) in acute myeloid leukemia: in vivo and in vitro saturation and internalization by leukemic and normal myeloid cells. Blood. 2001;97:3197–204. doi: 10.1182/blood.v97.10.3197. [DOI] [PubMed] [Google Scholar]
- 98.Polakis P. Arming antibodies for cancer therapy. Curr Opin Pharmacol. 2005;5:382–7. doi: 10.1016/j.coph.2005.04.008. [DOI] [PubMed] [Google Scholar]
- 99.McKoy JM, Angelotta C, Bennett CL, et al. Gemtuzumab ozogamicin-associated sinusoidal obstructive syndrome (SOS): an overview from the research on adverse drug events and reports (RADAR) project. Leuk Res. 2007;31:599–604. doi: 10.1016/j.leukres.2006.07.005. [DOI] [PubMed] [Google Scholar]
- 100.Larson RA, Sievers EL, Stadtmaeur EA, et al. Final report of the efficacy and safety of gemtuzumab ozogamicin (Mylotarg) in patients with CD33-positive acute myeloid leukemia in first recurrence. Cancer. 2005;104:1442–52. doi: 10.1002/cncr.21326. [DOI] [PubMed] [Google Scholar]
- 101.Maniecki MB, Hasle H, Fris-Hansen L, et al. Impaired CD163-mediated hemoglobin-scavenging and severe toxic symptoms in patients treated with gemtuzumab ozogamicin. Blood. 2008;112:1510–4. doi: 10.1182/blood-2007-09-114165. [DOI] [PubMed] [Google Scholar]
- 102.Maniecki MB, Hasle H, Bendix K, et al. Is hepatotoxicity in patients treated with gentuzumab ozogamicin due to specific targeting of hepatocytes? Leuk Res. 2011;35:e84–6. doi: 10.1016/j.leukres.2011.01.025. [DOI] [PubMed] [Google Scholar]
- 103.Lo Coco F, Ammatuna E, Noguera N. Treatment of acute promyelocytic leukemia with gemtuzumab ozogamicin. Clin Adv Hematol Oncol. 2006;4:57–62. [PubMed] [Google Scholar]
- 104.Candoni A, Damiani D, Michelutti A, et al. Clinical characteristics, prognostic factors and multidrug resistance related protein expression in 36 adult patients with acute promyelocytic leukemia. Eur J Haematol. 2003;71:1–8. doi: 10.1034/j.1600-0609.2003.00084.x. [DOI] [PubMed] [Google Scholar]
- 105.Burnett AK, Hills RK, Milligan D, et al. Identification of patients with acute myeloblastic leukemia who benefit from the addition of gemtuzumab ozogamicin: results of the MRC AML15 trial. J Clin Oncol. 2011;29:369–77. doi: 10.1200/JCO.2010.31.4310. [DOI] [PubMed] [Google Scholar]
- 106.Prebet T, Etienne A, Devillier R, et al. Improved outcome of patients with low- and intermediate-risk cytogenetics acute myeloid leukemia (AML) in first relapse with gemtuzumab and cytarabine versus cytarabrine: results of a retrospective comparative study. Cancer. 2011;117:974–81. doi: 10.1002/cncr.25554. [DOI] [PubMed] [Google Scholar]
- 107.Manoukian G, Hagemeister F. Denileukin diftitox: a novel immunotoxin. Expert Opin Biol Ther. 2009;9:1445–51. doi: 10.1517/14712590903348135. [DOI] [PubMed] [Google Scholar]
- 108.Martin A, Gutierrez E, Muglia J, et al. A multicenter dose-escalation trial with denileukin diftitox (ONTAK, DAB389IL-2) in patients with severe psoriasis. J Am Acad Dermatol. 2001;45:871–8. doi: 10.1067/mjd.2001.117852. [DOI] [PubMed] [Google Scholar]