

Abstract
Metabotropic glutamate receptors (mGluR) are mainly expressed in the central nervous system (CNS) and contain eight receptor subtypes, named mGluR1 to mGluR8. The crystal structures of mGluR1 and mGluR5 that are bound with the negative allosteric modulator (NAM) were reported recently. These structures provide a basic model for all class C of G-protein coupled receptors (GPCRs) and may aid in the design of new allosteric modulators for the treatment of CNS disorders. However, these structures are only combined with NAMs in the previous reports. The conformations that are bound with positive allosteric modulator (PAM) or agonist of mGluR1/5 remain unknown. Moreover, the structural information of the other six mGluRs and the comparisons of the mGluRs family have not been explored in terms of their binding pockets, the binding modes of different compounds, and important binding residues. With these crystal structures as the starting point, we built 3D structural models for six mGluRs by using homology modeling and molecular dynamics (MD) simulations. We systematically compared their allosteric binding sites/pockets, the important residues, and the selective residues by using a series of comparable dockings with both the NAM and the PAM. Our results show that several residues played important roles for the receptors’ selectivity. The observations of detailed interactions between compounds and their correspondent receptors are congruent with the specificity and potency of derivatives or compounds bioassayed in vitro. We then carried out 100 ns MD simulations of mGluR5 (residue 26-832, formed by Venus Flytrap domain, a so-called cysteine-rich domain, and 7 trans-membrane domains) bound with antagonist/NAM and with agonist/PAM. Our results show that both the NAM and the PAM seemed stable in class C GPCRs during the MD. However, the movements of “ionic lock,” of trans-membrane domains, and of some activation-related residues in 7 trans-membrane domains of mGluR5 were congruent with the findings in class A GPCRs. Finally, we selected nine representative bound structures to perform 30 ns MD simulations for validating the stabilities of interactions, respectively. All these bound structures kept stable during the MD simulations, indicating that the binding poses in this present work are reasonable. We provided new insight into better understanding of the structural and functional roles of the mGluRs family and facilitated the future structure-based design of novel ligands of mGluRs family with therapeutic potential.
Electronic supplementary material
The online version of this article (doi:10.1208/s12248-015-9742-8) contains supplementary material, which is available to authorized users.
KEY WORDS: allosteric, class C of G protein-coupled receptors, homology model, metabotropic glutamate receptor family, molecular dynamics simulation
INTRODUCTION
Glutamate is an important excitatory neurotransmitter in the central nervous system (CNS) of mammalian. It can trigger a fast excitatory response mediated through ligand-gated ion channels receptors (ionotropic receptors; 1). It can also trigger slower-acting and longer-lasting metabotropic glutamate receptors (mGluR; 2). These receptors are the potential targets for CNS disorders, such as Alzheimer’s diseases and epilepsy (3–6).
mGluRs belong to class C of G-protein coupled receptors (GPCRs; 5,7). The mGluRs play key roles in the central and peripheral nervous systems (3). They are involved in memory, learning, anxiety, and pain. The drugs for mGluRs can be used to treat CNS disorders and drug abuse (8). According to their sequence similarities, these eight subtypes are divided into 3 groups (I–III; 9): mGluR1 and mGluR5 belong to group I, mGluR2 and mGluR3 belong to group II, and mGluR4, mGluR6, mGluR7, and mGluR8 belong to group III. Group I mGluRs are mainly postsynaptic, signal through the Gq protein signaling pathway, and increase N-methyl-D-aspartate receptor (NMDA) receptor activity and risk of excitotoxicity (10,11). The group I mGluRs have been considered promising therapeutic targets to treat diseases including cancer, chronic pain, schizophrenia, Alzheimer’s disease, and anxiety (4,5). Group II and III mGluRs, on the other hand, are mainly presynaptic, signal through the Gi/Go protein signaling pathway, and decrease NMDA receptor activity and risk of excitotoxicity (3). All mGluRs form stable disulphide-linked dimers (3,12). Each monomer contains an extracellular Venus Flytrap domain (VFTD, orthosteric glutamate binding site; 13), 7 trans-membrane domains (7TMD, contains the allosteric binding site), and a so-called cysteine-rich domain (CRD, bridged the VFTD and 7TMD; 14). The 7TMD of mGluRs is analogous to the same domains in class A/B GPCRs (15).
Drug development that targets mGluRs first focuses on the orthosteric site (16). Orthosteric ligands have the advantages of better lipid solubility, less interactions with other brain proteins, and no metabolism by CYP450 (10,17). However, most of the orthosteric ligands lack sub-type selectivity. Christina et al. (3) recently published a review for mGluR5 about the orthosteric and allosteric ligand binding pockets for drug discovery. They reported that the residues within 5 Å of the orthosteric L-glutamate binding sites of the two group I mGluRs (mGluR1 and mGluR5) are 100% conserved, including Tyr64, Arg68, Trp100, Gly150, Ser151, Ser152, Ser173, Ala174, Thr175, Tyr223, Asp305, Gly306, Arg310, Lys328, Asp394, and Lys396. Thus, it seems impossible to develop orthosteric agonists selective for identical pockets of mGluR1/mGluR5. They also found that only one residue (Leu310/Gln310) was different for two group II mGluRs. Similarly, the subtype selectivity was very difficult to achieve. Only 12 of 16 residues were the same for group III mGluRs (3). This indicated that potential selectivity can be found in group III. Goudent et al. (18) recently reported an orthosteric bitopic mGluR4 agonist with more than 30-fold selectivity over other group III mGluRs.
However, discovering allosteric compounds exhibiting subtype selectivity is an attractive field of drug research. Allosteric binding sites contain better receptor subtype selectivity when compared to the orthosteric site (10,12,19,20). Moreover, allosteric ligands can reduce the risk of receptor oversensitization, as strict allosteric modulators (those with no intrinsic agonist activity) only modulated the natural response to the endogenous ligand instead of activating the receptor directly. By binding to the allosteric sites, these ligands can act as either negative allosteric modulators (NAMs) or positive allosteric modulators (PAMs) to inhibit or activate the receptor, respectively. A NAM is inactive in the absence of the orthosteric ligand. Substances that occupy the allosteric binding site and are functionally neutral are called silent allosteric modulators (SAMs).
Two allosteric modulators of GPCRs have appeared on the market, including Cincalet (a PAM of extracellular calcium-sensing receptor for hyperparathyroidism) and Maraviroc (a NAM of chemokine CC-motif receptor 5 for human immunodeficiency virus). Allosteric modulators of mGluRs for treating human CNS disorders are new approaches for the development of therapeutic targets. Lately, several allosteric modulators entered clinical development (8). mGluR5 NAMs have been demonstrated promising effects of in fragile X syndrome, Parkinson’s disease, anxiety, and affective disorder in the Phase II clinical trials (21,22). At the same time, a novel mGluR2 PAM also entered into the Phase II testing for anxious depressions and schizophrenia (23,24). Additional allosteric modulators are in late discovery or pre-clinical development, including mGluR5 PAMs for schizophrenia (25) and mGluR4 PAMs for Parkinson’s disease (26).
The 7TMDs of mGluR1 (27) and mGluR5 (28) in group I were reported in 2014. These structures provided detailed insight into the architecture of the trans-membrane domains of class C GPCRs, including the accurate location of the allosteric binding site within the trans-membrane domain and key micro-switched residues that regulate receptor signaling. So far, the structures only combined with NAMs. Many dynamics processes and underlying mechanism of mGluR1/5 remain unknown.
In the present work, we built 3D homology models of the other six mGluRs by using homology modeling and molecular dynamics (MD) simulations based on the X-ray crystal structure of mGluR5 (PDB entry: 4OO9; resolution, 2.6 Å) (28). We systematically reported the predicted allosteric binding sites/pockets and the important residues, including the residues specific for ligand selectivity and functionality. In our 100 ns, MD simulations of mGluR5 bound with antagonist/NAM or agonist/PAM, the findings of “ionic lock” and some activation-related residues in mGluR5 were congruent with the reports in class A of GPCRs. The representative bound-structures of each mGluR were also validated by MD simulations.
MATERIALS AND METHODS
Preparation of Crystal Structures of mGluR1 and mGluR5
For mGluRs family, the X-ray crystal structures of extracellular VFTD (orthosteric glutamate binding site) of four subtypes have been reported, including mGluR1 (PDB entry: 3KS9; resolution, 1.9 Å), mGluR3 (PDB entry: 3SM9; resolution, 2.26 Å), mGluR5 (PDB entry: 3LMK; resolution, 2.44 Å), and mGluR7 (PDB entry: 3MQ4; resolution, 2.8 Å). However, only mGluR1 (27) and mGluR5 (28) have the X-ray crystal structures of 7TMD contains the allosteric binding site, which are reported in 2014. The detailed comparisons of sequence identities for 7TMD were listed in Table S1, while the detailed sequence alignments of 7TMD were shown in Figure S1. Both the X-ray crystal structures of 7TMDs for mGluR1 (PDB entry: 4OR2; resolution, 2.8 Å; 27) and mGluR5 (PDB entry: 4OO9; resolution, 2.6 Å; 28) were considered as the templates for constructing the 7TMDs of other members. However, after comparing the sequence identities (Table S1), we chose mGluR5 as the template to construct the structures of other six mGluRs as the sequence identities of 7TMD among the mGluR family members were higher than 44%.
Structures of mGluR1 and mGluR5 were downloaded from the Protein Data Bank (http://www.pdb.org/pdb/). SYBYL-X 1.3 (29) was used to prepare their X-ray crystal structures, including residual repair and energy minimization.
In the present study, we mainly focused on the 7TMDs, so the sequences and structures were mainly related to the 7TMDs for the followings.
Homology Modeling of 7TMDs of Other Six Members and the Structure of mGluR5
The sequences of 7TMDs of mGluR2 (Q14416), mGluR3 (Q14832), mGluR4 (Q14833), mGluR6 (O15303), mGluR7 (Q14831), mGluR8 (O00222), and the whole sequence of mGluR5 (P41596) were downloaded from UniProtKB (http://www.uniprot.org/uniprot/). The X-ray crystal structure of 7TMD (PDB entry: 4OO9; resolution, 2.6 Å; 28) of mGluR5 was used as the template to construct the 3D structures of other six mGluRs, by using Modeller9.12 (30). Especially, each mGluR had one disulfide bond. We patched the disulfide bond between cysteine in extracellular loop (ECL) 2 and cysteine in trans-membrane domain 3 (TM3). The detailed information of disulfide bond of each mGluR can be found in Table S1.
Moreover, we also used Modeller9.12 to construct the entire structure of mGluR5 (residue 26-832), including the VFTD (PDB entry: 3LMK; resolution, 2.44 Å), a so-called CRD (bridged the VFTD and 7TMD), and 7TMD (PDB entry: 4OO9; resolution, 2.6 Å) (28). The protocols of building homology models can be found in our recent publications (17,31).
Energy Minimization and Structural Validation
After obtaining the 3D structures of 7TMDs, SYBYL-X 1.3 was used for the energy minimizations. The detailed parameters were described in our previous publications (17,31). ProSA-web Z-scores (32) and ProCheck Ramachandran plots (33,34) were used to analyze the structural stereo chemical and evaluation of all the models.
Conformational Sampling and Secondary Energy Minimization for mGluR Models
For selecting the most reasonable structures of mGluRs 7TMDs, a 10-ns MD simulation was conducted for each mGluR. Briefly, each mGluR was embedded into a water-lipid box. We then performed two-step minimizations. We first fixed the backbone-atoms of each mGluR to perform 5 ns MD simulations, and then performed another 5 ns simulations with the flexible protein. Water molecules and lipids were included without constraints. NAMD package (version 2.9b1) with CHARMM27 force field was used to carry out the MD simulations. Detailed protocols and parameters can be found in the following section—“Molecular Dynamics Simulation” (17,31). The conformation of each mGluR with the lowest energy was chosen as the optimal model, followed by the secondary energy minimization by SYBYL-X 1.3.
Molecular Docking for the Studies of Ligands / mGluR Interactions
A series of dockings were conducted for each mGluR model. The MOLCAD module in SYBYL-X 1.3 was used for predicting the potential binding pocket for each mGluR. Surflex-Dock GeomX, a docking program that implemented in SYBYL, was used to generate the detailed ligand-receptor interactions, in which the Total Score was expressed as −log10 (Kd; 35). The detailed parameters (36) of docking can be found in our recent publications (17).
Molecular Dynamics Simulation
After docking, we chose the whole structures of mGluR5 bound with antagonist-NAM and agonist-PAM for performing the MD simulations.
Special caution was applied to the protonation states of histidine residues due to its ionized state at pH 7.40. Two software programs, PROPKA 3.1 (37) and VEGA ZZ 2.4.0 (38) were used to calculate the pK values of protein. In mGluR5 model, all histidine were not protonated, due to the predicted pK values ranging from 0.79 to 6.50 (<7.40). Four residues were charged in the present work, including Asp−, Glu−, Lys+, and Arg+.
VMD (39) program was used to embed the protein-ligands complexes into a pre-equilibrated and periodic structure of 1-palmytoyl-2-oleoyl-sn-glycero-3-phosphatidylcholine. The lipid molecules within 3 Å of the mGluR5 were eliminated. Then we inserted them into a water-box (TIP3P (40) water model), and eliminated the waters molecules within 3 Å of the protein.
The systems (mGluR5-antagonist-NAM or mGluR5-agonist-PAM, respectively) included the whole structure of mGluR5 model with ligands, 85/85 lipid molecules, 15,667/15,667 water molecules, 0/0 sodium ions, and 4/4 chloride ions for a total of 71,162/71,195 atoms per periodic cell. The sizes of the water-lipid box were 60 × 70 × 160 Å3/60 × 70 × 160 Å3. Then two-step minimizations were conducted, in which each minimization was running 50,000 steps. The first one was running with the protein backbone fixed, while the second one was running with flexible protein with released constraints.
Treating the last frame of the second minimization as the starting point, 100 ns MD simulations were conducted by using the following parameters. NAMD package (41; version 2.9b1) with CHARMM27 (42,43) force field was used to carry out the MD simulations. Particle mesh Ewald (44) method were used to calculated the electrostatics, which was with a 12 Å non-bonded cutoff and a grid spacing of 1 Å per grid point in each dimension. The van der Waals energies were calculated by using two cutoff values. Switching radius was set to 10 Å. Cutoff radius was set to 12 Å. The temperature and pressure were maintained constant by a Langevin thermostat (310 K) and Langevin barostat (1 atm), respectively. The time step of MD simulations was set to 1 fs. The data was saved every 10 ps for analysis. VMD software was used to analyze the trajectory from MD simulation (45).
Additionally, we used the same protocols to perform 30 ns MD for the other nine representative complexes for validating the stabilities of binding poses, including mGluR1-YM-202074, mGluR1-YM-230888, mGluR2-analog of BINA, mGluR3-RO-4491533, mGluR4-VU0364770, mGluR4-ADX88178, mGluR5-CDPPB, mGluR7-AMN082, and mGluR8-AZ12216052 (pose 1). Only the 7TMDs of each mGluR were used to perform the MD. These systems included the corresponding structure of the mGluR model with ligands, 67 /67 /67 /70 /68 /68 /68 /71 /71 lipid molecules, 3131 /3131 /3148 /3129 /3109 /3109 /3160 /3134 /3136 water molecules, 0 /0 /0 /0 /0 /0 /0 /0 /0 sodium ions, and 15 /15 /13 /11 /5 /5 /14 /5 /5 chloride ions for a total of 22638 /22632 /22621 /22249 /22661 /22669 /22914 /23083 /23062 atoms per periodic cell. All these sizes of the water-lipid box were 66 × 66 × 70 Å3.
RESULTS
3D Structure of mGluRs Family
The comparisons of 3D structures between mGluR1 and mGluR5 were conducted as shown in Figure S2, and the only difference was the conformation of ECL2. We compared the sequences of the other six mGluRs with these two reported structures in Table S1. Our results show that mGluR5 has better sequence identities with the other six receptors and has a higher resolution than mGluR1. So, mGluR5 was chosen as the template to construct the other six subtypes.
After obtaining the initial 3D structural models of each mGluR, we carried out structural refinement by the conformational sampling using 10 ns MD simulation. We selected the conformation with the lowest energy as the optimal model for each mGluR and then performed the energy minimization. We compared the conformations between before-MD and after-MD for each mGluR, and found that there were few changes for both the backbone and side chains. The root mean square deviations (RMSD) of each mGluR after MD were 2.15, 2.23, 2.36, 2.44, 2.04, 2.56, 2.87, and 2.77 Å. These RMSDs were correlated well with the results from Doré et al. (28) Ramachandran plots of other six mGluRs were shown in Figure S3, including mGluR2, mGluR3, mGluR4, mGluR6, mGluR7, and mGluR8.
Based on the sequence identities, the conserved structural configuration of GPCRs, and the models of mGluRs, we illustrated a representative 3D model for the mGluR family, as shown in Figure S4. The main difference was the conformation of ECL2. Moreover, the length of intracellular loop 3 (loop between TM5 and TM6) was shorter than class A or B. Finally, the ECL3 of four group III mGluRs were longer, as well as the ECL2 in group III.
Residues Involved in the Allosteric Binding Site of Three Groups
We then compared the allosteric binding site/pocket among three groups of mGluRs, as shown in Fig. 1. The allosteric binding sites of mGluR1 and mGluR5 were defined based on the crystal structure bound with NAM. The allosteric binding sites in both mGluR1 and mGluR5 were formed by the residues from TM2, TM3, TM5, TM6, and TM7. Figure S5 shows the detailed information of two negative allosteric modulators in group I. 4-fluoro-N-(4-(6-(isopropylamino)pyrimidin-4-yl)thiazol-2-yl)-N-methylbenzamide and its analogs, NAM of mGluR1 (27), was found to be higher in the mGluR1 allosteric site, a position more analogous to that of ligands bound in the orthosteric site of the class A receptor. However, for mavoglurant (28), a NAM of mGluR5, was oriented with an approximate 30° tilt (relative to the axis of the trans-membrane bundle) in the allosteric site. Importantly, mavoglurant directly interacted with three residues in TM2 in the bottom part of the allosteric site, including Gly6242.45, Ile6252.46, and Gly6282.49, which the FITM did not interact. The mGluRs in group II and III shared the similar allosteric sites, as shown in Fig. 1c to h.
Fig. 1.
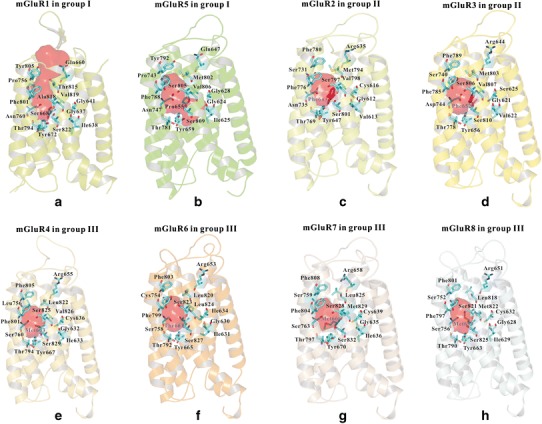
Binding pockets of mGluR family. The binding pocket of a mGluR1 and b mGluR5 in group I. The binding pocket of c mGluR2 and d mGluR3 in group II. The binding pocket of e mGluR4, f mGluR6, g mGluR7, and h mGluR8 in group III
For the residues involved in the allosteric binding sites, the crystal structures of mGluR1 and mGluR5 show more details than the other receptors in the mGluR family. Figure 1a, b also shows some important residues (for clarification) involved in the allosteric binding site of mGluR1 and mGluR5. These residues were congruent with the co-crystal structures of these two receptors.
Important residues involved in the mGluRs family are listed in Table I, based on our models. The twenty-two important residues involved in the present work may directly interact with the ligands. The residues listed in Table I were consistent with the results from Doré et al. (28) In Table I, seven out of twenty-two were the identical residues in the mGluRs family. The residues highlighted in bold were the unique residues. For example, Val6643.36, Ser6683.40, Thr8157.32, and Ala8187.35 were unique for mGluR1. Importantly, we found that Pro6553.40 in mGluR5 made the allosteric binding site different from the seven other mGluRs. Phe643/Phe6523.40, the same residue in two group II mGluRs (mGluR2 /mGluR3, in Table I), differed from the other mGluRs. We performed 30 ns MD simulations for both mGluR2 and mGluR3 for conformational sampling. Our results show that Phe643/Phe6523.40 was rigid during MD simulation.
Table I.
Sequence Alignments of the Allosteric Binding Site in the Eight mGluRs Divided by Groups
| Residues | I | II | III | ||||||
|---|---|---|---|---|---|---|---|---|---|
| mGluR1 | mGluR5 | mGluR2 | mGluR3 | mGluR4 | mGluR6 | mGluR7 | mGluR8 | ||
| 2.45 | G637 | G624 | G612 | G621 | G632 | G630 | G635 | G628 | |
| 2.46 | I638 | I625 | V613 | V622 | I633 | I631 | I636 | I629 | |
| 2.49 | G641 | G628 | C616 | S625 | C636 | I634 | C639 | C632 | |
| 2.56 | L648 | L635 | F623 | F632 | M643 | M641 | M646 | M639 | |
| 3.32 | Q660 | Q647 | R635 | R644 | R655 | R653 | R658 | R651 | |
| 3.36 | V664 | I651 | L639 | L648 | L659 | L657 | L662 | L655 | |
| 3.40 | S668 | P655 | F643 | F652 | M663 | T661 | M666 | M659 | |
| 3.44 | Y672 | Y659 | Y647 | Y656 | Y667 | Y665 | Y670 | Y663 | |
| 5.40 | V753 | V740 | M728 | M737 | L753 | L751 | I756 | L749 | |
| 5.43 | P756 | P743 | S731 | S740 | L756 | C754 | S759 | S752 | |
| 5.44 | L757 | L744 | L732 | L741 | L757 | L755 | L760 | L753 | |
| 5.47 | N760 | N747 | N735 | D744 | S760 | S758 | S763 | S756 | |
| 5.51 | I764 | I751 | I739 | V748 | M764 | M762 | M767 | M760 | |
| 6.46 | T794 | T781 | T769 | T778 | T794 | T792 | T797 | T790 | |
| 6.49 | I797 | I784 | I772 | I781 | V797 | I795 | V800 | I793 | |
| 6.50 | W798 | W785 | W773 | W782 | W798 | W796 | W801 | W794 | |
| 6.53 | F801 | F788 | F776 | F785 | F801 | F799 | F804 | F797 | |
| 6.57 | Y805 | Y792 | F780 | F789 | F805 | F803 | F808 | F801 | |
| 7.32 | T815 | M802 | M794 | M803 | L822 | L820 | L825 | L818 | |
| 7.35 | A818 | S805 | S797 | S806 | S825 | S823 | S828 | S821 | |
| 7.36 | V819 | V806 | V798 | V807 | V826 | L824 | M829 | M822 | |
| 7.39 | S822 | S809 | S801 | S810 | S829 | S827 | S832 | S825 | |
The allosteric binding site was defined according to the structural details from two crystal structures of mGluRs (5 Å within ligands), including mGluR1 (PDB entry, 4OR2; resolution, 2.8 Å) and mGluR5 (PDB entry, 4OO9; resolution, 2.6 Å). Seven residues that involved in the binding pocket were the identical residues in the mGluRs family, as shown in Table I. The residues highlighted in bold were the unique residues in the relative positions, and one residue-Pro6553.40 in mGluR5 distinguished the binding pocket from other mGluRs. Three residues in mGluR5 (Gly6242.45, Ile6252.46, and Gly6282.49) are available in its allosteric binding site due to the conformation of Pro6553.40, while residues in the same position in other mGluRs may not be available
In summary, based on the higher sequence identities among the mGluR family, the mGluRs shared similar 3D structures. The allosteric binding sites in group II and III shared large similarities while the allosteric binding sites in group I were very different. mGluR1/5 in group I had the X-ray crystal structures, so they provided much finer details about the allosteric sites. It is noted that mGluR1 and mGluR5 share 78.5% sequence identity. However, the sequence identities between group I and the other two groups (group II and III) are about 41–44% (see Table S1). Although it is reasonable to construct the druggable homolog models of the other six mGluRs by using the crystal structure of mGluR5, it is still possible that there are some inaccuracies during homology modeling. Moreover, the sequence identities of trans-membrane domain between group II and III are larger than 65%. More identical residues involved in the binding pocket of group II/III can be found in Table I. These may explain the reasons why allosteric binding sites in group II and III shared large similarities while the allosteric binding sites in group I were different.
DISCUSSION
Structural Differences Between mGluRs (Class C) and Other Three Classes of GPCRs (Class A, B, and F)
Since the first crystal structure of rhodopsin was reported in 2000 (46), more than 25 crystal structures of GPCRs have been reported. Each class of GPCRs has its own represented crystal structure (s). Comparisons with mGluR5 (class C) were carried out by using the dopamine 3 receptor (D3R, PDB entry: 3PBL; 47) from class A, the corticotropin-releasing factor receptor 1 (CRF1R, PDB entry: 4K5Y; 48) from class B, and the smoothened receptor (SMO, PDB entry: 4JKV; 49) from class F, with all structures in the inactive state. Here, we selected D3R as the template for class A, because similar alignments between CRF1R and D3R have been conducted by Hollenstein and co-workers (48). Moreover, we removed all the loops for clarification, including extracellular loops and intracellular loops.
Figure S6 shows the superposition and comparison of mGluR5 (class C) with the D3R (class A), CRF1R (class B), and SMO receptor (class F). Significantly, the extracellular side of TM5 of mGluR5 was shifted closer to the central axis of the receptor in comparison to other three classes of GPCRs (27,28). Furthermore, TM2 and TM7 also moved closer to the central axis of the receptor. Due to these movements, the top of the allosteric site formed by TM1, TM2, and TM7 was not available in mGluR5. The intracellular part of TM5 also shifted closer to the central axis of the receptor in comparison to D3R (class A) and CRF1R (class B). However, in comparison to SMO (class F), no significant movement of TM5 in mGluR5 was observed. Moreover, intracellular sides of other TMs of mGluR5 also showed similar movements. All these movements were congruent with the findings of mGluR1 and mGluR5 reported recently (27,28).
mGluR1 Bound with Selective Negative Allosteric Modulators
As far as we know, there have been some docking studies focused on NAMs for mGluR1, with a similar allosteric binding site as class A GPCR. Based on the recent co-crystal structure of mGluR1 with FITM (PDB entry: 4OR2; 27), we docked two highly selective NAMs (YM-202074 (50) and YM-230888 (51)) into mGluR1, which might be more accurate. Pharmacological properties of the two compounds can be found in Table II, while the structural information can be found in Figure S7.
Table II.
Pharmacological Properties of Important Drugs Used in the Present Work
| Compound | Potency (EC50/IC50 uM) | Selectivity | Ref. | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| mGluR1 | mGluR2 | mGluR3 | mGluR4 | mGluR5 | mGluR6 | mGluR7 | mGluR8 | |||
| YM-202074 | 0.0086 ± 0.0009 | >10 | >10 | >10 | 68% inhibition at 10 uM | >10 | >10 | N.R | mGluR1(NAM) | (50) |
| YM-230888 | 0.013 ± 0.0024 | >10 | >10 | >10 | >10 | >10 | >10 | N.R | mGluR1(NAM) | (51) |
| MTEP | >100 | >10 | >10 | >100 | 0.005 | N.R | >10 | N.R | mGluR5(NAM) | (52) |
| Mavoglurant | >10 | >10 | >10 | >10 | 0.03 | >10 | >10 | >10 | mGluR5(NAM) | (28) |
| Basimglurant | N.R | N.R | N.R | N.R | 0.039 | N.R | N.R | N.R | mGluR5(NAM) | - |
| VU0405386 | N.R | N.R | N.R | N.R | 0.0026 | N.R | N.R | N.R | mGluR5(PAM) | (53) |
| CDPPB | N.A | N.A | N.A | N.A | 0.027 | N.A | N.A | N.A | mGluR5(PAM) | (54) |
| VU0403602 | N.A | N.A | N.A | N.A | 0.007 | N.A | N.A | N.A | mGluR5(ago-PAM) | (55,56) |
| Analog of BINA | >10 | 0.05 | >10 | >10 | >10 | >10 | >10 | >10 | mGluR2(PAM) | (57) |
| RO-4491533 | N.A | 0.002 | 0.002 | N.A | N.A | N.A | N.A | N.A | mGluR2/3(NAM) | (58) |
| VU0155041 | N.R | N.R | N.R | 0.7 | N.R | N.R | >15 | >15 | mGluR4(PAM) | (10) |
| VU0364770 | N.R | N.R | N.R | 0.3 | N.R | 7 | >10 | >10 | mGluR4(PAM) | (10) |
| ADX88178 | N.R | N.R | N.R | 0.004 | N.R | >3 | >3 | >3 | mGluR4(PAM) | (10) |
| AMN082 | N.R | N.R | N.R | >10 | N.R | >10 | 0.09 | >10 | mGluR7(PAM) | (10) |
| AZ12216052 | N.R | N.R | N.R | >30 | N.R | N.R | N.R | 1 | mGluR8(PAM) | (10) |
N.R. not reported, N.A. not active
Figure 2a, b shows the detailed interactions between these two ligands and mGluR1. Many similar interactions could be found between these two compounds. Thr8157.32 (the amine group) was observed to form strong hydrogen bonds (<3.5 Å) with these two compounds. This important residue also formed a strong hydrogen bond with FITM in the crystal structure (PDB entry: 4OR2; 27). Moreover, Gln6603.32 formed a strong hydrogen bond both with YM-202074 (∼3.8 Å) and YM-230888 (∼3.0 Å). Several residues formed strong hydrophobic interactions with these two ligands, including Leu6482.56, Val6643.36, Pro7565.43, Trp7986.50, Phe8016.53, and Ala8187.35. Most of these residues were consistent with mutagenesis studies (3,9,27). However, some differences could also be found. Ser6683.40 and Tyr8056.57 (Fig. 2a) in mGluR1 also formed hydrogen bonds with YM-202074. Moreover, YM-202074 could extend to the bottom of the pocket and interacted tightly with Tyr6723.44, Thr7946.46, and Ser8227.39, while no similar interactions could be observed for YM-230888. We suggest that these differences may lead YM-202074 to seem more potent than YM-230888.
Fig. 2.
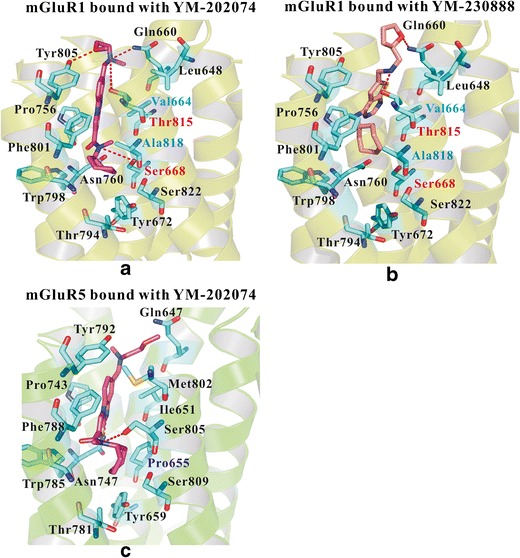
Detailed interactions between mGluR1 (group I) and its selective NAM. a The detailed interactions between mGluR1 and YM-202074 (K i = 4.8 ± 0.37 nM), b the detailed interactions between mGluR1 and YM-230888, and c the binding pose of YM-202074 in mGluR5, comparing the selectivity between mGluR1 and mGluR5. Gln6603.32 and Thr8157.32 played important roles for the bindings in mGluR1. Val6643.36, Ser6683.40, Thr8157.32, and Ala8187.35 in mGluR1 played key roles for its selectivity
We compared different residues between mGluR1 and mGluR5 to explore their selectivity. Figure 2a, c show very similar binding poses of YM-202074 in these two receptors. In mGluR1, two residues including Ser6683.40 and Thr8157.32 formed hydrogen bonds with YM-202074. However, in mGluR5, the corresponding residues were the hydrophobic residues—Pro6553.40 and Met8027.32, which lacked a strong hydrogen bond with this compound. Other different hydrophobic residues in mGluR1 may also contribute to the selective binding of YM-202074, including Val6643.36 and Ala8187.35. The residues in the same positions at mGluR5 were Ile6513.36 and Ser8057.35. Based on the detailed binding interactions, we suggest that two hydrophilic and two hydrophobic residues in mGluR1 contributed to its selectivity, including Val6643.36, Ser6683.40, Thr8157.32, and Ala8187.35. Moreover, the corresponding residues in mGluR5 may also contribute to selectivity of mGluR5, including Ile6513.36, Pro6553.40, Met8027.32, and Ser8057.35. Importantly, we suggest that YM-202074 was not active in mGluR5, because this compound did not bind to the “recognition area” of mGluR5 (formed by Gly6242.45, Ile6252.46, Gly6282.49, Pro6553.40, and Ser8097.39). More details can be found in the following section.
mGluR5 Bound with Both Negative and Positive Allosteric Modulators
mGluR5 is considered as potential target for anti-addictive and antidepressant effects in clinical application (3,9,21,22,28). With the help of allosteric modulators, selective antagonists have shown hepatoprotective and inflammatory effect. In contrast, mGluR5 agonists may be used as inotropic.
Compared with the structures of CRF1R (PDB entry: 4K5Y) (48) and rhodopsin (PDB entry: 1F88; 46), the position of TM6 in mGluR5 may mediate specific binding and function of the receptor due to the basic structure of class C GPCR. Trp7856.50 did not point toward the allosteric binding site, which allowed the binding of small molecules. We found that one part of the allosteric binding pocket of mGluR5 had a restrict entrance, which was different from the cavity in the crystal structure of mGluR1. Gly6242.45, Ile6252.46, Gly6282.49, Pro6553.40, Ser8057.35, Val8067.36, and Ser8097.39 were directly involved in the pocket. This allosteric site was important for the recognition of ligands of both the NAMs and PAMs.
We chose two NAMs (MTEP (52) and Basimglurant) and two PAMs (CDPPB (54) and VU0403602 (55,56)), and docked them into mGluR5, as shown in Fig. 3a–d. Among the four allosteric modulators, three shared chemical similarity (MTEP, Basimglurant, and VU0403602). Alkyne, the special part of the aromatic nucleus, linked with carbon-carbon triple bond consisted of a narrower shape to fit the allosteric binding site. The chemical structure of CDPPB did not contain alkyne. However, the amide group in CDPPB played a similar role as alkyne, as shown in Fig. 3c.
Fig. 3.
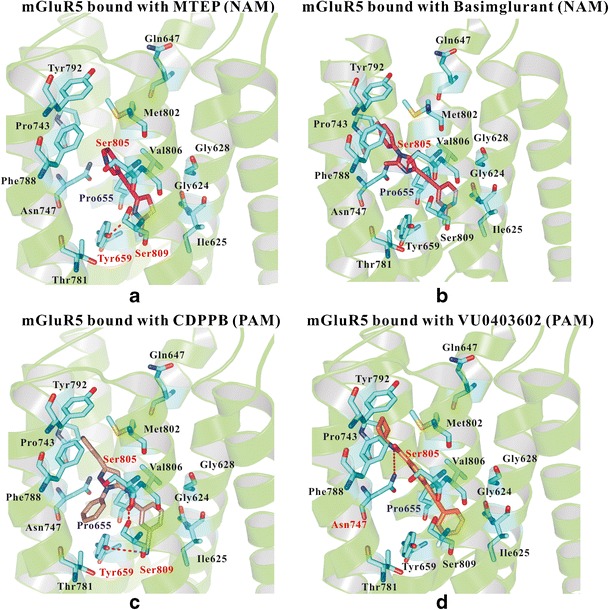
Detailed interactions between mGluR5 (group I) and its NAMs and PAMs. a The binding of MTEP (NAM) in mGluR5, b the binding of Basimglurant (NAM) in mGluR5, c the detailed interactions of CDPPB (PAM) in mGluR5, and d the detailed interactions of VU0403602 (PAM) in mGluR5. Several important residues may form hydrogen bonds with the ligands, including Tyr6593.44, Asn7475.47, Ser8057.35, and Ser8097.39. A region formed by Gly6242.45, Ile6252.46, Gly6282.49, Pro6553.40, and Ser8097.39 played important roles for recognizing the active compounds of mGluR5
The hydrogen bonds between allosteric modulators and several specific residues (Asn7475.47, Ser8057.35, or Ser8097.39) formed important connections between receptors and ligands. The co-crystal structure of mGluR5 indicated that the modulators link to mGluR5 by using at least one of the hydrogen bonds. The Ser8057.35 was involved in most of the cases. In the structures with PAMs, the small molecules also formed hydrogen bonds with Asn7475.47. Importantly, Pro6553.40, a unique residue in mGluR5, may play a key role in its selectivity.
3-((2-Methyl-4-thiazolyl) ethynyl) pyridine (MTEP) was designed as a selective NAM (Fig. 3a). The small molecule was considered as a lead compound for new drugs specifically effective on mGluR5. As a derivate of MPEP, MTEP showed similar neuroprotection effect in lower concentration. We compared the detailed interactions between MPEP and MTEP as shown in Figure S8 and found almost the same binding poses. However, a new hydrogen bond was found in the interaction between MTEP (Figure S8) and Ser8057.35 in mGluR5 due to the replacement of pyridine with benzene in MTEP, which contributed to the higher potency and more selectivity of this NAM. Previous studies presented homology models with predicted binding ligands such as MPEP. The models published by Mølck et al. (59), Gregory et al. (60), and Dalton et al. (12) were conducted on the template of class A GPCRs. The possibility of horizontal binding was mentioned in these previous studies. However, our co-crystal based model was better to explain the interaction of MTEP with Ser8057.35, Ser8097.39 and other related residues.
VU0403602, identified a potent and selective PAM, also displayed as an intrinsic partial agonist in vitro. It was classified as ago-PAM because of its ability to act as an enhancer for endogenous agonists and to increase agonist potency. The hydrogen bond (4.9 Å) with Ser8057.35 was defined in Fig. 3d. Interestingly, the binding of CDPPB and mGluR5 was not connected with the alkyne on the benzene ring. The Ser8057.35 formed a hydrogen bond to the acyl amino, the flexible structure linked to the aromatic ring. Another hydrogen bond of Ser8097.39 and the nitrogen of the cyan in meso-position were found in the binding receptor as well.
In summary, our results show that both NAMs and PAMs bound to the "recognition area" of mGluR5 (formed by Gly6242.45, Ile6252.46, Gly6282.49, Pro6553.40, and Ser8097.39). Ser8057.35 and Ser8097.39 played key roles in the binding of small molecules, and Pro6553.40 may be the key residue in its selectivity.
mGluR2 and mGluR3 in Group II Bound with Their Allosteric Modulators
Compared to group I (mGluR1 and mGluR5), group II (mGluR2 and mGluR3) has less sequence similarity and pharmacology mechanism. However, mGluR2 and mGluR3 share sequence homology, common pharmacology and negative coupling to cAMP. Group II mGluR agonists not only have robust activity in a range of animal models that predict anxiolytic and antipsychotic activities, but also have efficacy in patients suffering from schizophrenia (61). On the other hand, antagonists and NAMs of mGluR2/3 have potential antidepressant efficacy (62).
In order to explore the selectivity of mGluR2/3 and compare the different binding residues between NAM and PAM, we built the homology models of mGluR2/3 on the basis of the crystal structure of mGluR5 (Table S1), and performed the dockings with existing ligands (a highly selective mGluR2 PAM, an analog of BINA (57); a non-selective mGluR2/3 NAM, RO-4491533(58)). As shown in Fig. 4a to d, the potential allosteric binding pockets of mGluR2/3 were mainly formed by TM3, TM5, TM6, and TM7, and defined by the reported key residues (as shown in Table I). The allosteric binding pocket of the mGluR2 model was very similar to that of mGluR3, and both of them were verified by recent docking results and mutagenesis studies (63), such as Phe643/Phe6523.40, Tyr647/Tyr6563.44, Asn735/Asp7445.47, Thr769/Thr7786.46, Phe776/ Phe7856. 53, and Ser801/Ser8107.39.
Fig. 4.

Comparisons of the binding modes and the selective residues between mGluR2 and mGluR3 in group II. The binding poses of an analog of BINA in mGluR2/3 a and c and the binding modes of RO-4491533 in mGluR2/3 b and d. Hydrogen bond interactions between the ligands and residues were shown as a dashed red line
Figure 4a, b show that the NAM and PAM of mGluR2 shared overlapping binding sites (formed by Phe6433.40, Tyr6473.44, Ser7315.43, Leu7325.44, Ile7395.51, Thr7696.46, Phe7766.53, Ser7977.35, Val7987.36, and Ser8017.39). However, the positions and the key residues for the NAM and PAM in mGluR2 were very different. The PAM (an analog of BINA) can interact tightly with the upper part of the allosteric binding pocket. Its two benzenes on the top of the binding pocket formed hydrogen bonds with Arg6353.32 (∼3.2 Å) and Ser7315.43 (∼4.8 Å). It also formed a strong hydrophilic interaction with Ser7977.35 (∼3.7 Å). However, the NAM (RO-4491533) mainly formed hydrophobic interactions with mGluR2/3. Differently, it can extend to the bottom of the binding pocket, forming strong hydrophobic interactions with Leu6092.42, Val6132.46, Phe6433.40, and Ser8017.39. This difference might provide us with some information to understand the differences between the NAM and the PAM.
Our results show many similarities and differences by comparing the binding pocket of mGluR2 (Fig. 4a) and mGluR3 (Fig. 4c) for the analog of BINA (biphenyl-indanone A, which is a well-known PAM for mGluR2, with an EC50 of 50 nM). Two residues formed hydrogen bonds with this analog of BINA, including Arg635/Arg6443.32 and Ser797/Ser8067.35, as shown in Fig. 4a, c. The PAM shared very similar binding poses in mGluR2 and mGluR3. Two residues, including Asn735/Asp7445.47 and Ile739/Val7485.51, caused the main differences between models. The high selectivity of the PAM might be induced by Asn7355.47 and Ile7395.51 in mGluR2 (Asp7445.47 and Val7485.51 in mGluR3 may also contribute to the selectivity). Asn7355.47 in mGluR2 had an acid amide group, which could donate hydrogen to form a hydrogen bond with the nucleophiles’ group. Ile7395.51 in mGluR2 had a longer hydrocarbon chain, which might interact tightly with the five-membered ring in the analog of BINA. As shown in Fig. 4c, the five-membered ring turned over and could not form a strong π-π interaction with Tyr6563.44 in mGluR3. These may explain the fact that the PAM had nearly no binding affinity in mGluR3.
Moreover, as displayed in Fig. 4b, d, RO-4491533, a NAM with high binding affinity for both the mGluR2 and mGluR3, shared almost the same binding poses and interactions. RO-4491533 pointed deep into a narrow cavity between TM3 and TM7, constituting intensive hydrophobic interactions with several residues, including Phe643/ Phe6523.40, Tyr647/Tyr6563.44, and Ser801/Ser8107.39. Additionally, the strong π-π interaction with Phe643/Phe6523.40 may also contribute to the high binding affinity.
Based on the docking results, we suggest that Asn735/Asp7445.47 and Ile739/Val7485.51 play important roles for the selectivity for mGluR2/3. Different binding residues and binding sites may also play key roles for the recognitions of PAM and NAM.
mGluR4 Bound with Its Highly Selective PAMs
Herein, we chose to compare mGluR4 with mGluR6, and to compare mGluR7 with mGluR8 based on sequence identity. Sequence identity between mGluR4 and mGluR6 was more than 85 percent, while the sequence identity between mGluR4 and mGluR7/8 was less than 80 percent. We docked three PAMs (VU0155041 (10,64; Fig. 5a), VU0364770 (10; Fig. 5b), and ADX88178 (10; Fig. 5c)) into the same binding pocket of mGluR4, which had relatively higher selectivity on mGluR4 (Table II). As shown in Fig. 5a to c, the three PAMs are located closely to the hydroxyl in Ser7605.47 and Ser8257.35. So the hydrogen acceptor groups in the PAMs formed two hydrogen bonds with Ser7605.47 and Ser8257.35, respectively, indicating the importance of these two residues, which was consistent with former reports (18,26,64). Other residues also contributed to the binding and selectivity due to their hydrophobic nature, including Met6633.40, Leu7565.43 (selective residue), Phe8016.53, Phe8056.57, Leu8227.32, and Val8267.36 (selective residues). Interestingly, the binding pose of ADX88178 was slightly different from the others. Unlike other PAMs, it extended deeply into the bottom of the narrow pocket, and formed strong hydrophobic interactions with the residues around its pyrazole group (Tyr6673.44, Thr7946.46, and Ser8297.39), which might be responsible for its higher binding affinity comparing to the other two PAMs.
Fig. 5.

The detailed interactions of three highly selective PAMs: a VUP155041, b ADX88178, and c VU0364770 in the allosteric binding pocket of mGluR4, and d VU0364770 in mGluR6
As far as we know, there was no available highly selective PAM for mGluR6. We used VU0364770 (10), with 7 uM binding affinity in mGluR6, to explore the potential binding pose, and to explore the reason that VU3604770 had a better affinity with mGluR4. As shown in Fig. 5a, d, the pyridine group in VU0364770 cannot form the hydrogen bond with Ser8237.35 in mGluR6 as mGluR4 was, caused by different residues, including Thr6613.40 and Leu8247.36 in mGluR6 (the corresponding residues were Met6633.40 and Val8267.36 in mGluR4). In the homology model of mGluR4, with a longer chain of Met6633.40 in the lower position and a shorter chain of Val8267.36 near Ser8257.35, the pyridine of VU0364770 was more likely to stay near Ser8257.35, without falling down and inserting into the lower position of the binding pocket as in the homology model of mGluR6. Additionally, Leu7565.43 in mGluR4 (the corresponding residue was Cys7545.43 in mGluR6), may contribute to the selectivity of mGluR4.
Our results show that both Ser7605.47 and Ser8257.35 played key roles in mGluR4 for the binding of PAMs. Moreover, Thr6613.40 in mGluR6, Leu756/ Cys7545.43, Val826/Leu8247.36, may be involved in the selectivity of mGluR4/6.
mGluR7/8 Bound with Highly Selective PAMs
mGluR7 remained the most elusive of the eight known proteins. We built the models of mGluR7/8 and docked them with their PAMs. AMN082 (10) was discovered as the first mGluR7 selective positive allosteric molecule which had oral activity and the ability to cross the blood brain barrier. It also had potential therapeutic application on the CNS disorder by mimicking the effect of glutamate. According to our results, Ser7595.43 formed a strong hydrogen bond with the amidogen of AMN082, as shown in Fig. 6a. Additionally, the π-π interaction between benzenes in the Phe8046.53 and AMN082 may enhance their binding affinity. Importantly, Ile7565.40, a unique residue in mGluR7, formed strong hydrophobic interaction with AMN082, which may contribute to its selectivity.
Fig. 6.

The detailed interactions of highly selective PAM a AMN082 in mGluR7 and two potential binding modes (b and c) of selective PAM AZX88178 in mGluR8
Similarly, we also predicted possible binding poses and the interactions between mGluR8 and its PAM-AZ12216052 (10). One of the possible pose (Fig. 6c) was similar to mGluR7 (Fig. 6a). The shape of the pocket entrance was not exactly the same due to the different residues (Ile7565.40/Leu7495.40). Ile7565.40 in mGluR7 was unique, which may contribute to the selectivity of the receptor. The PAM bound to almost an identical site as in mGluR7, but a little bit deeper. Two residues including Ser7565.47 (∼3.9 Å) and Ser8217.35 (a weaker or no hydrogen bond, ∼5.3 Å) in the mGluR8 formed hydrogen bonds with AZ12216052. However, according to the docking score, it was highly possible that the PAM was able to display in another pocket, as shown in Fig. 6b. This pocket was narrower and formed by Leu6252.42, Met6593.40, Tyr6633.44, Leu6663.47, Thr7906.46, Met8227.36, Ser8297.43, and Leu8337.47. The small molecule fitted the pocket well by hydrogen bonds interacting, including Tyr6633.44 and Ser8297.43. We will further validate this in the following section by using MD simulations.
According to the docking results, we propose Ser759/Ser7525.43 and Ser763/Ser7565.47 may play key roles for the binding of ligands, and Ile7565.40 in mGluR7 may contribute to the selectivity of mGluR7. Moreover, two different binding poses of AZ12216052 could be observed in mGluR8, indicating that different compounds may prefer different parts of the allosteric binding site in mGluR8.
Comparisons of the VFTD and CRD in mGluR5 after MD
We carried out the 100 ns MD simulations for the mGluR5 (residue 26-832, formed by Venus Flytrap domain, a so-called cysteine-rich domain, and 7 trans-membrane domains) bound with (a) antagonist-LY-344545 (65) (an epimer of LY-341495, mGluR2/3 selective antagonist) and NAM-mavoglurant (28) and (b) agonist-glutamate and PAM-VU0405386 (53). The original binding pose of agonist-glutamate in mGluR5 was the same as it was in the crystal structures (mGluR5 complexes with glutamate-PDB entry: 3LMK), as shown in Fig. 7b. Moreover, the binding pose of antagonist-LY-344545 in mGluR5 differed from that of LY-341495 in the crystal structure of mGluR1 (mGluR1 complexes with LY341495-PDB entry: 3KS9), as shown in Fig. 7a, which was caused by the different conformations of VFTD between mGluR1 and mGluR5. However, the important interactions were very similar. Both the LY-344545 (in our mGluR5 model) and LY341495 (in mGluR1) formed strong interactions (or hydrophobic interaction) with the same residues, includingTyr74/Tyr64, Trp110/Trp100, Tyr236/Tyr223, and Lys409/Lys396 in mGluR1/mGluR5.
Fig. 7.

Conformational change of a antagonist-LY-344545 and b agonist-glutamate in the Venus Flytrap domain (VFTD) during 100 ns MD simulation. The structure and residues before MD are highlighted in green, the structure and residues in mGluR5 bound with antagonist and NAM are highlighted in blue, while the structure and residues in mGluR5 bound with agonist and PAM are highlighted in purple
Figure S9a shows the alignment of mGluR5 (residue 26-832) bound with antagonist-LY-344545 and NAM-mavoglurant between before MD (green) and after MD (blue). The root mean square deviation (RMSD) of Cα atom in this mGluR5 was 7.85 Å, where the RMSD is a measure of the average distance between the backbones of superimposed proteins. Given two sets of vectors V and W, the RMSD is defined as: . Figure S9b shows the alignment of mGluR5 (residue 26-832) bound with agonist-glutamate and PAM-VU0405386 between before MD (green) and after MD (purple). The RMSD of this mGluR5 was 10.04 Å. Our results show that both the VFTD and CRD deviated greatly. Moreover, the antagonist-LY-344545 in the orthosteric binding site of VFTD kept stable during the MD simulation, as shown in Fig. 7a (blue). However, large conformational changes of agonist-glutamate bound within the orthosteric binding site of VFTD, could be observed during our simulation, as shown in Fig. 7b. The glutamate moved about 4.5 Å during the MD simulation. We believed it was caused by the big orthosteric binding pocket of mGluR5 and flexible conformation of glutamate compared to the rigid structure of the antagonist LY344545. Meanwhile, the CRD in mGluR5 bound with antagonist-LY-344545 and NAM-mavoglurant kept stable, especially the three beta-sheets, as shown in Figure S9a. However, three beta-sheets of CRD in mGluR5 bound with agonist-glutamate and PAM-VU0405386 broke into the random coils, as shown in Figure S9b. These different changes caused by the VFTD and CRD may lead to the differences of 7TMD.
Comparisons of 7TMD after MD Simulation
he RMSD and root mean square fluctuation (RMSF) of Cα atom in 7TMD were shown in Fig. 8a, b. When a dynamical system fluctuates about some well-defined average position the RMSD from the average over time can be referred to as the RMSF, where the mean square fluctuation (MSF) is a measure of the deviation between the position of the particle i and some reference positions, (where T is the time that one wants to average, and is the reference position of the particle i). The main difference between RMSD and RMSF is that the average is taken over time with RMSF, giving a value for each particle i. Our results show both the RMSD of 7TMD in mGluR5 bound with antagonist-NAM (∼4.5 Å) and bound with agonist-PAM (∼5.5 Å) were equilibrated within 20 ns, as shown in Fig. 8a, so 100 ns was a reasonable time-scale. Figure 8b indicates qualitatively similar profiles of RMSF for the corresponding residues between these two simulations. Moreover, the residues (Met714-Val729) from ECL2 fluctuated greatly (according to RMSF), as shown in Fig. 8b.
Fig. 8.

Conformational changes of mGluR5 (formed by VFTD, CRD, and 7TMD). a The RMSD of 7TMD in mGluR5, b the RMSF of 7TMD in mGluR5, c conformation changes of mavoglurant (NAM), d conformation changes of VU0405386 (PAM), e “ionic lock” in mGluR5-mavoglurant (NAM), f “ionic lock” in mGluR5-VU0405386 (PAM), g conformation comparisons of 7TMD (viewed from intracellular side), and h conformational comparisons of activation-related residues, including Trp7856.50, Lys8217.51, and Tyr8237.53
Conformational Changes of Ligands
As we can see in Fig. 8c, d, the conformations of both mavoglurant (NAM, highlighted in orange) and VU0405386 (PAM, highlighted in orange) did not endure large changes, compared with their conformations before MD simulation (highlighted in white). Three residues in 7TMD of mGluR5 formed strong hydrogen bonds with the NAM-mavoglurant during our MD simulation, including Asn7475.47, Ser8057.35, and Ser8097.39. Ser8057.35 also formed a strong hydrogen bond with the PAM-VU0405386 during the simulation. Both Pro6553.40 and Tyr6593.44 formed strong hydrophobic interactions with NAM and PAM.
"Ionic lock" in Class C
Importantly, the "ionic lock" in 7TMD of mGluR5 differed greatly with the different ligands. The “ionic lock” motif in class C was formed by Lys6653.50 in TM3 and highly conserved residue Glu7706.35 in TM6. Figure 8e shows details of "ionic lock" in mGluR5 bound with mavoglurant (NAM). The distance between Lys6653.50 and Glu7706.35 after MD simulation remained within 3.0 Å. However, for mGluR5 bound with VU0405386 (PAM), the observed distance between Lys6653.50 and Glu7706.35 after MD simulation extended to 7.1 Å, as shown in Fig. 8f. These results of “ionic lock” in class C GPCR were congruent with the findings in class A GPCR (17,45,66,67). However, there were some similarities between these two types of simulations. Both the distances between Ser6132.34/Ser6142.35 and Lys6653.50/Glu7706.35 were not stable during the simulations. Moreover, our results show that Arg6683.53 endured large conformational changes.
Movements of TMs
Figure 8g and Figure S10 show the alignments of 7TMD for these two type of simulations. Viewed from the extracellular side, no changes were observed for the trans-membrane domains. However, our results show the intracellular side of TM5 moved against the allosteric binding site (moved outward) when mGluR5 bound with PAM. This result was also congruent with the finding of class A GPCRs (17,45,66,67). We suggest that the outward-movement of TM5 facilitated the coupling of G-protein. The similar conformational changes and movements of TM5 could be observed in the crystal structure of beta-2 active structure (PDB entry: 3SN6), coupling with G-protein (68).
Conformational Changes of Activation-Related Residues
Figure 8h shows the comparisons of conformational changes of important residues. Several residues (highlighted in purple) in mGluR5 bound with PAM (also with agonist) and endured large conformational changes, including Trp7856.50, Lys8217.51, and Tyr8237.53. These residues were reported to play key roles for the activation and their results were also congruent with the conclusions in class A GPCRs (17,45,66,67).
Most findings of mGluR5 in class C remained congruent with previous studies of class A GPCRs (17,45,66,67).
Validations of the Bound-Structures by Using MD Simulations
Additionally, we selected other nine representative structures to perform 30 ns MD simulations for validating the stabilities of binding poses, respectively, including mGluR1-YM-202074, mGluR1-YM-230888, mGluR2-analog of BINA, mGluR3-RO-4491533, mGluR4-VU0364770, mGluR4-ADX88178, mGluR5-CDPPB, mGluR7-AMN082, and mGluR8-AZ12216052 (pose 1). The alignments of the conformations between before-MD and after-MD were shown in Figure S11. Our results showed that almost all the conformations of liangds and proteins were stable during the MD simulation. It is noted that the structure of CDPPB differs from other modulators of mGluR5 that discussed in the present work. The stable interactions between CDPPB and 7TMD of mGluR5 indicated that the amide group in CDPPB played a similar role as alkyne. Moreover, for mGluR8-AZ12216052 (pose 1), our results showed that this binding pose was very stable during the MD, as well as the corresponding interactions between AZ12216052 and mGluR8. We suggest that the first binding pose (pose 1) between AZ12216052 and mGluR8 is more reasonable than the second binding pose (pose 2). All these results indicate that the binding poses discussed in the present work are valid.
CONCLUSION
We completed six mGluRs by using homology modeling and systematically reported the allosteric binding site/pocket, the important residues, and the selective residues. Val6643.36, Ser6683.40, Thr8157.32, and Ala8187.35 in mGluR1 played important roles for its selectivity. Gly6242.45, Ile6252.46, Gly6282.49, Pro6553.40, and Ser8097.39 in mGluR5 may play key roles in the recognition of ligands. Our results of mGluR1 and mGluR5 were congruent with the findings in their crystal structures (27,28). Based on our studies, Phe643/Phe6523.40 was important for both of mGluR2 and mGluR3 in group II. Furthermore, Asp7445.47 and Val7485.51 played roles for the selectivity in mGluR3. For the other four mGluRs in Group III, Leu7565.43 in mGluR4, Thr6613.40, Cys7545.43, and Leu8247.36 in mGluR6, Ile7565.40 in mGluR7 were of importance for the recognitions of ligands. Two different binding poses of AZ12216052 (selective PAM for mGluR8) can be found in mGluR8. The 100 ns MD simulations of mGluR5 (residue 26-832) bound with antagonist/NAM or agonist/PAM were carried out to explore the activation mechanism of class C GPCRs. Most of the findings in mGluR5 of class C GPCRs were congruent with the findings in class A of GPCRs. The binding poses presented in our work were validated using the MD simulations.
Additionally, the mGluR6 plays a key role in visual signal transduction, with certain mutations in mGluR6 cause congenital stationary night blindness. In spite of the importance of mGluR6, the knowledge of the molecular basis of its function and the selective modulator and antagonist is lacking. It is imperative use allosteric pocket in homology model of mGluR6 to screen small chemical molecules, so that potential selective allosteric modulator can be discovered.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Figure S1 shows the sequence alignments among mGluRs family. Figure S2 shows the comparisons of the crystal structure between mGluR1 and mGluR5. Figure S3 shows the Ramachandran plots of other six mGluRs, including mGluR2, mGluR3, mGluR4, mGluR6, mGluR7, and mGluR8. Figure S4 shows the common 3D structure of mGluRs family. Figure S5 shows the comparisons of the NAM between mGluR1 and mGluR5. Figure S6 shows the superposition and comparison of mGluR5 (class C) with D3R (class A), CRF1R (class B), and SMO receptor (class F). Figure S7 shows the structural information of compounds discussed in the present work. Figure S8 shows the comparisons of binding mode of MTEP and MPEP in mGluR5. Figure S9 shows the comparisons of mGluR5 bound with different compounds during 100 ns MD simulation. Figure S10 shows conformational changes (view from extracellular side) of 7TMD bound with different compounds after 100 ns MD simulation. Figure S11 shows the alignments of other nine complexes between before-MD and after-MD. Table S1 shows sequence identities of six subtypes with two group I mGluRs (mGluR1 and mGluR5). This material is available free of charge via the Internet at http://www.aaps.org. (DOCX 13644 kb)
Acknowledgments
The project is supported by funding from the NIDA P30DA035778A1 and NIH R01DA025612.
References
- 1.Dingledine R, Borges K, Bowie D, Traynelis SF. The glutamate receptor ion channels. Pharmacol Rev. 1999;51:7–62. [PubMed] [Google Scholar]
- 2.Sugiyama H, Ito I, Hirono C. A new type of glutamate receptor linked to inositol phospholipid metabolism. Nature. 1987;325:531–3. doi: 10.1038/325531a0. [DOI] [PubMed] [Google Scholar]
- 3.Mølck C, Harpsøe K, Gloriam DE, Mathiesen JM, Nielsen SM, Bräuner-Osborne H. mGluR5: exploration of orthosteric and allosteric ligand binding pockets and their applications to drug discovery. Neurochem Res. 2014;39:1–14. doi: 10.1007/s11064-014-1248-8. [DOI] [PubMed] [Google Scholar]
- 4.Gregory KJ, Dong EN, Meiler J, Conn PJ. Allosteric modulation of metabotropic glutamate receptors: structural insights and therapeutic potential. Neuropharmacology. 2011;60:66–81. doi: 10.1016/j.neuropharm.2010.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Niswender CM, Conn PJ. Metabotropic glutamate receptors: physiology, pharmacology, and disease. Annu Rev Pharmacol Toxicol. 2010;50:295. doi: 10.1146/annurev.pharmtox.011008.145533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nicoletti F, Bockaert J, Collingridge G, Conn P, Ferraguti F, Schoepp D, et al. Metabotropic glutamate receptors: from the workbench to the bedside. Neuropharmacology. 2011;60:1017–41. doi: 10.1016/j.neuropharm.2010.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ferraguti F, Shigemoto R. Metabotropic glutamate receptors. Cell Tissue Res. 2006;326:483–504. doi: 10.1007/s00441-006-0266-5. [DOI] [PubMed] [Google Scholar]
- 8.Conn PJ, Lindsley CW, Meiler J, Niswender CM. Opportunities and challenges in the discovery of allosteric modulators of GPCRs for treating CNS disorders. Nat Rev Drug Discov. 2014;13:692–708. doi: 10.1038/nrd4308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brauner-Osborne H, Wellendorph P, Jensen AA. Structure, pharmacology and therapeutic prospects of family C G-protein coupled receptors. Curr Drug Targets. 2007;8:169–84. doi: 10.2174/138945007779315614. [DOI] [PubMed] [Google Scholar]
- 10.Flor PJ, Acher FC. Orthosteric versus allosteric GPCR activation: the great challenge of group-III mGluRs. Biochem Pharmacol. 2012;84:414–24. doi: 10.1016/j.bcp.2012.04.013. [DOI] [PubMed] [Google Scholar]
- 11.Pin J-P, Galvez T, Prézeau L. Evolution, structure, and activation mechanism of family 3/C G-protein-coupled receptors. Pharmacol Ther. 2003;98:325–54. doi: 10.1016/S0163-7258(03)00038-X. [DOI] [PubMed] [Google Scholar]
- 12.Dalton J, Gómez-Santacana X, Llebaria A, Giraldo J. A computational analysis of negative and positive allosteric modulator binding and function in metabotropic glutamate receptor 5 (in) activation. J Chem Inf Model. 2014;54:1476–87. doi: 10.1021/ci500127c. [DOI] [PubMed] [Google Scholar]
- 13.Kunishima N, Shimada Y, Tsuji Y, Sato T, Yamamoto M, Kumasaka T, et al. Structural basis of glutamate recognition by a dimeric metabotropic glutamate receptor. Nature. 2000;407:971–7. doi: 10.1038/35039564. [DOI] [PubMed] [Google Scholar]
- 14.Muto T, Tsuchiya D, Morikawa K, Jingami H. Structures of the extracellular regions of the group II/III metabotropic glutamate receptors. Proc Natl Acad Sci U S A. 2007;104:3759–64. doi: 10.1073/pnas.0611577104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kniazeff J, Prézeau L, Rondard P, Pin J-P, Goudet C. Dimers and beyond: the functional puzzles of class C GPCRs. Pharmacol Ther. 2011;130:9–25. doi: 10.1016/j.pharmthera.2011.01.006. [DOI] [PubMed] [Google Scholar]
- 16.Wellendorph P, Bräuner‐Osborne H. Molecular basis for amino acid sensing by family CG‐protein‐coupled receptors. Br J Pharmacol. 2009;156:869–84. doi: 10.1111/j.1476-5381.2008.00078.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Feng Z, Alqarni MH, Yang P, Tong Q, Chowdhury A, Wang L, et al. Modeling, molecular dynamics simulation and mutation validation for structure of cannabinoid receptor 2 based on known crystal structures of GPCRs. J Chem Inf Model. 2014;54:2483–2499. doi: 10.1021/ci5002718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Goudet C, Vilar B, Courtiol T, Deltheil T, Bessiron T, Brabet I, et al. A novel selective metabotropic glutamate receptor 4 agonist reveals new possibilities for developing subtype selective ligands with therapeutic potential. FASEB J. 2012;26:1682–93. doi: 10.1096/fj.11-195941. [DOI] [PubMed] [Google Scholar]
- 19.Burford NT, Watson J, Bertekap R, Alt A. Strategies for the identification of allosteric modulators of G-protein-coupled receptors. Biochem Pharmacol. 2011;81:691–702. doi: 10.1016/j.bcp.2010.12.012. [DOI] [PubMed] [Google Scholar]
- 20.Conn PJ, Christopoulos A, Lindsley CW. Allosteric modulators of GPCRs: a novel approach for the treatment of CNS disorders. Nat Rev Drug Discov. 2009;8:41–54. doi: 10.1038/nrd2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Emmitte KA. mGlu5 negative allosteric modulators: a patent review (2010–2012) Expert Opin Ther Pat. 2013;23:393–408. doi: 10.1517/13543776.2013.760544. [DOI] [PubMed] [Google Scholar]
- 22.Rocher J-P, Bonnet B, Bolea C, Lutjens R, Le Poul E, Poli S, et al. mGluR5 negative allosteric modulators overview: a medicinal chemistry approach towards a series of novel therapeutic agents. Curr Top Med Chem. 2011;11:680–95. doi: 10.2174/1568026611109060680. [DOI] [PubMed] [Google Scholar]
- 23.Lavreysen H, Langlois X, Ahnaou A, Drinkenburg W, te Riele P, Biesmans I, et al. Pharmacological characterization of JNJ-40068782, a new potent, selective, and systemically active positive allosteric modulator of the mGlu2 receptor and its radioligand [3H] JNJ-40068782. J Pharmacol Exp Ther. 2013;346:514–27. doi: 10.1124/jpet.113.204990. [DOI] [PubMed] [Google Scholar]
- 24.Hopkins CR. Is there a path forward for mGlu2 positive allosteric modulators for the treatment of schizophrenia? ACS Chem Neurosci. 2013;4:211–3. doi: 10.1021/cn400023y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Macdonald GJ, Lindsley CW. A unique industrial–academic collaboration towards the next generation of schizophrenia therapeutics. Curr Top Med Chem. 2014;14:304–12. doi: 10.2174/1568026613666131127154443. [DOI] [PubMed] [Google Scholar]
- 26.Lindsley CW, Hopkins CR. Metabotropic glutamate receptor 4 (mGlu4)-positive allosteric modulators for the treatment of Parkinson’s disease: historical perspective and review of the patent literature. Expert Opin Ther Pat. 2012;22:461–81. doi: 10.1517/13543776.2012.679437. [DOI] [PubMed] [Google Scholar]
- 27.Wu H, Wang C, Gregory KJ, Han GW, Cho HP, Xia Y, et al. Structure of a class C GPCR metabotropic glutamate receptor 1 bound to an allosteric modulator. Science. 2014;344:58–64. doi: 10.1126/science.1249489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Doré AS, Okrasa K, Patel JC, Serrano-Vega M, Bennett K, Cooke RM, et al. Structure of class C GPCR metabotropic glutamate receptor 5 transmembrane domain. Nature. 2014;511:557–62. doi: 10.1038/nature13396. [DOI] [PubMed] [Google Scholar]
- 29.SYBYL-X 1.3, Tripos International, 1699 South Hanley Rd., St. Louis, MO, 63144, USA. 2010.
- 30.Martí-Renom MA, Stuart AC, Fiser A, Sánchez R, Melo F, Šali A. Comparative protein structure modeling of genes and genomes. Annu Rev Biophys Biomol. 2000;29:291–325. doi: 10.1146/annurev.biophys.29.1.291. [DOI] [PubMed] [Google Scholar]
- 31.Xie XQ, Chen JZ, Billings EM. 3D structural model of the G‐protein‐coupled cannabinoid CB2 receptor. Proteins Struct Funct Bioinf. 2003;53:307–19. doi: 10.1002/prot.10511. [DOI] [PubMed] [Google Scholar]
- 32.Wiederstein M, Sippl MJ. ProSA-web: interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007;35:W407–10. doi: 10.1093/nar/gkm290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Crystallogr. 1993;26:283–91. doi: 10.1107/S0021889892009944. [DOI] [Google Scholar]
- 34.Lovell SC, Davis IW, Arendall WB, de Bakker PI, Word JM, Prisant MG, et al. Structure validation by Cα geometry: ϕ, ψ and Cβ deviation. Proteins. 2003;50:437–50. doi: 10.1002/prot.10286. [DOI] [PubMed] [Google Scholar]
- 35.Jain AN. Scoring noncovalent protein-ligand interactions: a continuous differentiable function tuned to compute binding affinities. J Comput Aided Mol Des. 1996;10:427–40. doi: 10.1007/BF00124474. [DOI] [PubMed] [Google Scholar]
- 36.Chen J-Z, Wang J, Xie X-Q. GPCR structure-based virtual screening approach for CB2 antagonist search. J Chem Inf Model. 2007;47:1626–37. doi: 10.1021/ci7000814. [DOI] [PubMed] [Google Scholar]
- 37.Søndergaard CR, Olsson MH, Rostkowski M, Jensen JH. Improved treatment of ligands and coupling effects in empirical calculation and rationalization of p K a values. J Chem Theory Comput. 2011;7:2284–95. doi: 10.1021/ct200133y. [DOI] [PubMed] [Google Scholar]
- 38.Pedretti A, Villa L, Vistoli G. VEGA: a versatile program to convert, handle and visualize molecular structure on Windows-based PCs. J Mol Graph Model. 2002;21:47–9. doi: 10.1016/S1093-3263(02)00123-7. [DOI] [PubMed] [Google Scholar]
- 39.Hsin J, Arkhipov A, Yin Y, Stone JE, Schulten K. Using VMD: an introductory tutorial. Curr Protoc Bioinformatics. 2008; 5.7. 1-5.7. 48. [DOI] [PMC free article] [PubMed]
- 40.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of simple potential functions for simulating liquid water. J Chem Phys. 1983;79:926. doi: 10.1063/1.445869. [DOI] [Google Scholar]
- 41.Kalé L, Skeel R, Bhandarkar M, Brunner R, Gursoy A, Krawetz N, et al. NAMD2: greater scalability for parallel molecular dynamics. J Comput Phys. 1999;151:283–312. doi: 10.1006/jcph.1999.6201. [DOI] [Google Scholar]
- 42.Brooks BR, Bruccoleri RE, Olafson BD. CHARMM: a program for macromolecular energy, minimization, and dynamics calculations. J Comput Chem. 1983;4:187–217. doi: 10.1002/jcc.540040211. [DOI] [Google Scholar]
- 43.MacKerell AD, Bashford D, Bellott M, Dunbrack RL, Evanseck JD, Field MJ, et al. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J Phys Chem B. 1998;102:3586–616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 44.Essmann U, Perera L, Berkowitz ML, Darden T, Lee H, Pedersen LG. A smooth particle mesh Ewald method. J Chem Phys. 1995;103:8577–93. doi: 10.1063/1.470117. [DOI] [Google Scholar]
- 45.Feng Z, Hou T, Li Y. Studies on the interactions between β2 adrenergic receptor and Gs protein by molecular dynamics simulations. J Chem Inf Model. 2012;52:1005–14. doi: 10.1021/ci200594d. [DOI] [PubMed] [Google Scholar]
- 46.Palczewski K, Kumasaka T, Hori T, Behnke CA, Motoshima H, Fox BA, et al. Crystal structure of rhodopsin: AG protein-coupled receptor. Science. 2000;289:739–45. doi: 10.1126/science.289.5480.739. [DOI] [PubMed] [Google Scholar]
- 47.Chien EY, Liu W, Zhao Q, Katritch V, Han GW, Hanson MA, et al. Structure of the human dopamine D3 receptor in complex with a D2/D3 selective antagonist. Science. 2010;330:1091–5. doi: 10.1126/science.1197410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hollenstein K, Kean J, Bortolato A, Cheng RK, Doré AS, Jazayeri A, et al. Structure of class B GPCR corticotropin-releasing factor receptor 1. Nature. 2013;499:438–43. doi: 10.1038/nature12357. [DOI] [PubMed] [Google Scholar]
- 49.Wang C, Wu H, Katritch V, Han GW, Huang X-P, Liu W, et al. Structure of the human smoothened receptor bound to an antitumour agent. Nature. 2013;497:338–43. doi: 10.1038/nature12167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kohara A, Takahashi M, Yatsugi S-i, Tamura S, Shitaka Y, Hayashibe S, et al. Neuroprotective effects of the selective type 1 metabotropic glutamate receptor antagonist YM-202074 in rat stroke models. Brain Res. 2008;1191:168–79. doi: 10.1016/j.brainres.2007.11.035. [DOI] [PubMed] [Google Scholar]
- 51.Kohara A, Nagakura Y, Kiso T, Toya T, Watabiki T, Tamura S, et al. Antinociceptive profile of a selective metabotropic glutamate receptor 1 antagonist YM-230888 in chronic pain rodent models. Eur J Pharmacol. 2007;571:8–16. doi: 10.1016/j.ejphar.2007.05.030. [DOI] [PubMed] [Google Scholar]
- 52.Pałucha A, Brański P, Szewczyk B, Wierońska JM, Kłak K, Pilc A. Potential antidepressant-like effect of MTEP, a potent and highly selective mGluR5 antagonist. Pharmacol Biochem Behav. 2005;81:901–6. doi: 10.1016/j.pbb.2005.06.015. [DOI] [PubMed] [Google Scholar]
- 53.Turlington M, Noetzel MJ, Chun A, Zhou Y, Gogliotti RD, Nguyen ED, et al. Exploration of allosteric agonism structure–activity relationships within an acetylene series of metabotropic glutamate receptor 5 (mGlu5) positive allosteric modulators (PAMs): discovery of 5-((3-fluorophenyl) ethynyl)-N-(3-methyloxetan-3-yl) picolinamide (ML254) J Med Chem. 2013;56:7976–96. doi: 10.1021/jm401028t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lindsley CW, Wisnoski DD, Leister WH, O’Brien JA, Lemaire W, Williams DL, et al. Discovery of positive allosteric modulators for the metabotropic glutamate receptor subtype 5 from a series of N-(1, 3-diphenyl-1 H-pyrazol-5-yl) benzamides that potentiate receptor function in vivo. J Med Chem. 2004;47:5825–8. doi: 10.1021/jm049400d. [DOI] [PubMed] [Google Scholar]
- 55.Bridges TM, Rook JM, Noetzel MJ, Morrison RD, Zhou Y, Gogliotti RD, et al. Biotransformation of a novel positive allosteric modulator of metabotropic glutamate receptor subtype 5 contributes to seizure-like adverse events in rats involving a receptor agonism-dependent mechanism. Drug Metab Dispos. 2013;41:1703–14. doi: 10.1124/dmd.113.052084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pomierny-Chamioło L, Rup K, Pomierny B, Niedzielska E, Kalivas PW, Filip M. Metabotropic glutamatergic receptors and their ligands in drug addiction. Pharmacol Ther. 2014;142:281–305. doi: 10.1016/j.pharmthera.2013.12.012. [DOI] [PubMed] [Google Scholar]
- 57.Dhanya R-P, Sidique S, Sheffler DJ, Nickols HH, Herath A, Yang L, et al. Design and synthesis of an orally active metabotropic glutamate receptor subtype-2 (mGluR2) positive allosteric modulator (PAM) that decreases cocaine self-administration in rats. J Med Chem. 2010;54:342–53. doi: 10.1021/jm1012165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Woltering TJ, Wichmann J, Goetschi E, Knoflach F, Ballard TM, Huwyler J, et al. Synthesis and characterization of 1, 3-dihydro-benzo[1, 4] diazepin-2-one derivatives: Part 4. In vivo active potent and selective non-competitive metabotropic glutamate receptor 2/3 antagonists. Bioorg Med Chem Lett. 2010;20:6969–74. doi: 10.1016/j.bmcl.2010.09.125. [DOI] [PubMed] [Google Scholar]
- 59.Mølck C, Harpsøe K, Gloriam DE, Clausen RP, Madsen U, Pedersen LØ, et al. Pharmacological characterization and modeling of the binding sites of novel 1, 3-bis (pyridinylethynyl) benzenes as metabotropic glutamate receptor 5-selective negative allosteric modulators. Mol Pharmacol. 2012;82:929–37. doi: 10.1124/mol.112.078808. [DOI] [PubMed] [Google Scholar]
- 60.Gregory KJ, Nguyen ED, Reiff SD, Squire EF, Stauffer SR, Lindsley CW, et al. Probing the metabotropic glutamate receptor 5 (mGlu5) positive allosteric modulator (PAM) binding pocket: discovery of point mutations that engender a “molecular switch” in PAM pharmacology. Mol Pharmacol. 2013;83:991–1006. doi: 10.1124/mol.112.083949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Vinson PN, Conn PJ. Metabotropic glutamate receptors as therapeutic targets for schizophrenia. Neuropharmacology. 2012;62:1461–72. doi: 10.1016/j.neuropharm.2011.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Matosin N, Fernandez-Enright F, Frank E, Deng C, Wong J, Huang X-F, et al. Metabotropic glutamate receptor mGluR2/3 and mGluR5 binding in the anterior cingulate cortex in psychotic and nonpsychotic depression, bipolar disorder and schizophrenia: implications for novel mGluR-based therapeutics. J Psychiatry Neurosci. 2014;39:407. doi: 10.1503/jpn.130242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lundström L, Bissantz C, Beck J, Wettstein J, Woltering T, Wichmann J, et al. Structural determinants of allosteric antagonism at metabotropic glutamate receptor 2: mechanistic studies with new potent negative allosteric modulators. Br J Pharmacol. 2011;164:521–37. doi: 10.1111/j.1476-5381.2011.01409.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rovira X, Malhaire F, Scholler P, Rodrigo J, Gonzalez-Bulnes P, Llebaria A, Pin J-P, Giraldo J, Goudet C. Overlapping binding sites drive allosteric agonism and positive cooperativity in type 4 metabotropic glutamate receptors. FASEB J. 2014;29:116–130. [DOI] [PubMed]
- 65.Doherty A, Palmer M, Bortolotto Z, Hargreaves A, Kingston A, Ornstein P, et al. A novel, competitive mGlu5 receptor antagonist (LY344545) blocks DHPG‐induced potentiation of NMDA responses but not the induction of LTP in rat hippocampal slices. Br J Pharmacol. 2000;131:239–44. doi: 10.1038/sj.bjp.0703574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Feng Z, Hou T, Li Y. Selectivity and activation of dopamine D3R from molecular dynamics. J Mol Model. 2012;18:5051–63. doi: 10.1007/s00894-012-1509-x. [DOI] [PubMed] [Google Scholar]
- 67.Feng Z, Hou T, Li Y. Docking and MD study of histamine H4R based on the crystal structure of H1R. J Mol Graph Model. 2013;39:1–12. doi: 10.1016/j.jmgm.2012.10.003. [DOI] [PubMed] [Google Scholar]
- 68.Rasmussen SG, DeVree BT, Zou Y, Kruse AC, Chung KY, Kobilka TS, et al. Crystal structure of the β2 adrenergic receptor-Gs protein complex. Nature. 2011;477:549–55. doi: 10.1038/nature10361. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 shows the sequence alignments among mGluRs family. Figure S2 shows the comparisons of the crystal structure between mGluR1 and mGluR5. Figure S3 shows the Ramachandran plots of other six mGluRs, including mGluR2, mGluR3, mGluR4, mGluR6, mGluR7, and mGluR8. Figure S4 shows the common 3D structure of mGluRs family. Figure S5 shows the comparisons of the NAM between mGluR1 and mGluR5. Figure S6 shows the superposition and comparison of mGluR5 (class C) with D3R (class A), CRF1R (class B), and SMO receptor (class F). Figure S7 shows the structural information of compounds discussed in the present work. Figure S8 shows the comparisons of binding mode of MTEP and MPEP in mGluR5. Figure S9 shows the comparisons of mGluR5 bound with different compounds during 100 ns MD simulation. Figure S10 shows conformational changes (view from extracellular side) of 7TMD bound with different compounds after 100 ns MD simulation. Figure S11 shows the alignments of other nine complexes between before-MD and after-MD. Table S1 shows sequence identities of six subtypes with two group I mGluRs (mGluR1 and mGluR5). This material is available free of charge via the Internet at http://www.aaps.org. (DOCX 13644 kb)