

Abstract
The fluorinated d-glucose analog 18F-2-fluoro-2-deoxy-d-glucose (18F-FDG) is the most prevalent radiopharmaceutical for positron emission tomography (PET) imaging. P-Glycoprotein’s (P-gp, MDR1, and ABCB1) function in various cancer cell lines and tumors was shown to impact 18F-FDG incorporation, suggesting that P-gp function at the blood–brain barrier may also modulate 18F-FDG brain kinetics. We tested the influence of P-gp inhibition using the cyclosporine analog valspodar (PSC833; 5 μM) on the uptake of 18F-FDG in standardized human P-gp-overexpressing cells (MDCKII-MDR1). Consequences for 18F-FDG brain kinetics were then assessed using (i) 18F-FDG PET imaging and suitable kinetic modelling in baboons without or with P-gp inhibition by intravenous cyclosporine infusion (15 mg kg−1 h−1) and (ii) in situ brain perfusion in wild-type and P-gp/Bcrp (breast cancer resistance protein) knockout mice and controlled d-glucose exposure to the brain. In vitro, the time course of 18F-FDG uptake in MDR1 cells was influenced by the presence of valspodar in the absence of d-glucose but not in the presence of high d-glucose concentration. PET analysis revealed that P-gp inhibition had no significant impact on estimated brain kinetics parameters K1, k2, k3, VT, and CMRGlc. The lack of P-gp effect on in vivo18F-FDG brain distribution was confirmed in P-gp/Bcrp-deficient mice. P-gp inhibition indirectly modulates 18F-FDG uptake into P-gp-overexpressing cells, possibly through differences in the energetic cell level state. 18F-FDG is not a P-gp substrate at the BBB and 18F-FDG brain kinetics as well as estimated brain glucose metabolism are influenced by neither P-gp inhibition nor P-gp/Bcrp deficiencies in baboon and mice, respectively.
KEY WORDS: ABC transporters, blood–brain barrier, cyclosporine, glucose, multidrug resistance, nonhuman primate, positron emission tomography
INTRODUCTION
18F-2-Fluoro-2-deoxy-d-glucose (18F-FDG), a d-glucose analog, is the most widely prescribed radiopharmaceutical agent for positron emission tomography (PET) imaging in Nuclear Medicine. PET with 18F-FDG is a valuable diagnostic modality in various diseases. PET/CT (computed tomography) with 18F-FDG has become an important component of cancer imaging: 18F-FDG PET/CT is useful for the detection, staging and assessment of cancer recurrence (1). In some situation, PET/CT with 18F-FDG is also used to detect and/or predict response/resistance to therapy and thus impact treatment decision making and monitoring (2–4). PET with 18F-FDG is also an accurate method for the investigation of regional human brain metabolism in health and disease states (5). Alzheimer’s disease is characterized by a specific regional pattern of reduced 18F-FDG uptake, allowing early and preclinical diagnosis (6). Brain PET imaging using 18F-FDG is also performed routinely during presurgical evaluation in pharmacoresistant epilepsy. The locally reduced 18F-FDG uptake allows the noninvasive localization of the epileptic focus and is predictive of a seizure-free outcome after surgical resection (7).
From a molecular imaging perspective, it is important to understand the factors that govern 18F-FDG kinetics in tissues and assess the biological correlates of 18F-FDG uptake in both the tumor and the brain (8,9). Basically, 18F-FDG is transported through the cell membrane into the cytosol by glucose transporters (GLUT), phosphorylated by hexokinase (HK), and metabolically trapped in the cell as 18F-FDG-6-phosphate. Thus, 18F-FDG accumulation depends on its rate of transport through the cell membrane (influx and potential efflux), the HK activity, and the (low) rate of dephosphorylation in the cell (9). Tumors generally exhibit increased transport of glucose by overexpression and/or expression of avid GLUT isoform and increased HK activity (10,11). In the brain, GLUT1 (SLC2A1), a member of solute carrier (SLC) transporter family, is the predominant glucose transporter at the blood–brain barrier (BBB) and is also responsible for the transfer of 18F-FDG from the blood to the brain (12).
The P-glycoprotein (P-gp; ABCB1) is a member of the ATP-binding cassette (ABC) transporter family, also known as multidrug-resistant protein 1 (MDR1). P-gp mediates the unidirectional efflux of many structurally and mechanistically unrelated drugs across the plasma membrane. P-gp overexpression in cancer cells is a key component of the multidrug resistance (MDR) phenotype, which is associated with chemotherapeutic agent treatments failure (13). Numerous in vitro studies, performed using different cancer cell lines, have revealed that MDR or P-gp-overexpressing cells incorporate 18F-FDG at lower levels (14,15). This inverse relationship between P-gp function and 18F-FDG uptake was also reported in vivo with selected cancer cells implanted as xenografts: P-gp-overexpressing tumors showed reduced 18F-FDG uptake (16,17), which was restored using pharmacological P-gp inhibition (17). In patients, an inverse relationship was found between 18F-FDG uptake and the level of P-gp expression examined in tumor resection (lung cancer, hepatocellular carcinoma, and intrahepatic cholangiocarcinoma) (18–22). Therefore, it was hypothesized that 18F-FDG may be a substrate of P-gp (15,22). Conversely, it was also reported an enhanced 18F-FDG uptake in various P-gp-expressing cancer cells compared to P-gp negative in vitro (23–25). No explanation was given so far for this discrepancy. Nevertheless, these controversial results suggest that P-gp function may be an additional modulatory factor for 18F-FDG accumulation in tissues (8).
P-gp is also physiologically expressed at the BBB and limits the distribution of many drugs into the central nervous system (CNS) (26). The level of expression and/or functionality of P-gp can be modulated by inhibition, up- or down-regulation. This may affect the brain distribution and subsequent efficacy or toxicity of many P-gp substrates (27). Recent PET studies have revealed that P-gp function is decreased in patients with Alzheimer’s disease (28). P-gp overexpression is also proposed as a possible explanation for pharmacoresistance in epilepsy: Up-regulation of P-gp and other efflux transporters in capillaries, as well as aberrant expression in glial and neuronal cells, has been reproducibly demonstrated in surgically resected brain specimens (29).
Both the P-gp and GLUT1 are physiologically coexpressed at the BBB (26). Few studies have addressed the relationship between P-gp function at the BBB and 18F-FDG brain kinetics in a physiological condition. Tariquidar is a potent inhibitor of both the P-gp and the breast cancer resistance protein (BCRP and ABCG2), two major efflux transporters expressed at the BBB (26). It was reported that tariquidar induces a 45% increase in the brain uptake of 18F-FDG in rats, suggesting that ABC-transporter function at the BBB may also modulate 18F-FDG uptake by the brain (30). This also questions whether P-gp deficiency observed at the BBB in some physiopathological states may also impact 18F-FDG brain kinetics.
In the present study, we used standardized Madin–Darby canine kidney II (MDCKII) cells transfected with the human MDR1 (multidrug resistance 1) gene, instead of cancer cell lines, to test the influence of human P-gp function on 18F-FDG uptake. The overall influence of P-gp inhibition on 18F-FDG brain kinetics was then assessed using PET imaging and kinetic modeling in nonhuman primates. Finally, we used in situ brain perfusion in mice to test the influence of combined P-gp/Bcrp deficiency on 18F-FDG brain uptake under controlled d-glucose exposure to the brain.
MATERIAL AND METHODS
Animals
All animal use procedures were in accordance with the recommendations of the European Community (86/809/CEE) and the French National Committees (law 87/848) for the care and use of laboratory animals. The experimental protocol was evaluated by a local ethic committee for animal use (CETA reference no. 10-004). PET studies were carried out in adult male Papio anubis baboons (3–4 years old, 12–15 kg in weight during the study). Adult male Fvb mice (20–30 g, 7–11 weeks old) were obtained from Janvier (France). The dual P-gp/Bcrp-deficient mouse strain Mdr1a;Mdr1b;Bcrp(−/−;−/−;−/−) (derived from Fvb mice), also called P-gp/Bcrp(−/−), was bred in-house from progenitors obtained from the laboratory of Dr. Alfred H. Schinkel (The Netherlands Cancer Institute, The Netherlands).
Chemicals and Radiochemicals
Cyclosporine (CsA) was administered using the commercial drugs Sandimmun® (Novartis, Basel, Switzerland). Valspodar (PSC833), a nonimmunosuppressant CsA derivative, was a gift from Novartis. 18F-2-fluoro-2-deoxy-d-glucose (18F-FDG, Glucotep®) was purchased from Cyclopharma (Saint-Beauzire, France). 99mTc-sestamibi was prepared from the Cardiolite® kit (Iba Molecular, Gif-sur-Yvette, France).
Cell Culture and 18F-FDG Uptake Assay
The Stably Transfected Madin–Darby Canine Kidney Cell Lines
MDCKII-hMDR1 were a gift from Dr. Alfred H. Schinkel (The Netherlands Cancer Institute, The Netherlands). Culture and Western blot analysis of transporter expression was performed as previously described (31). Cells were seeded onto 24-well plates (3 × 105 cells/well) and allowed to grow for 3 days to reach confluence. On the day of the experiment, the culture medium was removed and cell monolayers washed twice with PBS. Monolayers were then incubated with 200 μL incubation buffer containing tracer dose 18 F-FDG (∼3.7 MBq/mL) and the P-gp substrate 99mTc-sestamibi (∼3.7 MBq/mL), as a positive control for P-gp function (32). Tracer uptake was measured in the presence of the P-gp inhibitor valspodar (5 μM) compared to vehicle [DMSO 0.5% (v/v) in incubation buffer]. The experiments were performed either in standard Dulbecco’s modified Eagle’s medium (DMEM) containing a 17.5-mM d-glucose concentration (“high d-glucose”) or in the absence of d-glucose. Plates were stored at 37°C under a humidified atmosphere. At selected times, ranging from 0 to 120 min, incubation medium was removed, the monolayers (n = 4–8 for each condition) were immediately washed twice with ice-cold PBS and cells lysed using 500 μL NaOH 1 N. Four hundred microliter samples from each well were counted using a calibrated γ-counter. The fluorine-18 energy channel was selected to estimate fluorine-18 decay corrected radioactivity expressed in Bq (half-life = 109.8 min). After 24-h decay, the samples were counted in the technetium-99m energy window to measure technetium-99m radioactivity (half-life = 6.02 h). Cell viability after 120-min incubation in glucose-free medium was assessed using trypan blue exclusion assay following the manufacturer instructions (Sigma-Aldrich, Saint-Quentin, France).
PET Imaging in Baboons
We studied the brain kinetics of 18F-FDG with or without P-gp inhibition using CsA in normoglycemic (12 h fasting) in three different adult baboons. Fasting animals were anesthetized, and PET data were acquired as previously described (33). Baboons were artificially ventilated, maintained, anesthetized with 66% N2O/1% isoflurane. Experiments were performed on a HR+ Tomograph (Siemens Healthcare, Knoxville, TN, USA) for brain PET studies. CsA (15 mg kg−1 h−1, i.v.) was infused 30 min before beginning and throughout PET scanning. This P-gp inhibition protocol, based on previous report (34), was validated in-house using the same baboon species and 11C-N-desmethyl-loperamide, a reliable PET probe for P-gp activity at the BBB (data not shown) (35). Heart rate, end-tidal pCO2, and rectal temperature were continuously monitored. After a transmission scan for the purpose of subsequent attenuation correction, animals were then injected i.v. with 100–200 MBq 18F-FDG. During PET acquisition (60 min), arterial plasma samples were obtained to estimate the arterial input function (at 0, 0.25, 0.5, 0.75, 1, 1.25, 1.5, 1.75, 2, 3, 5, 7.5, 10, 15, 20, 30, 40, 50, and 60 min) and blood glucose measurement (Cobas® 6000 analyzer, Roche Diagnostics, France) in each animal. Dynamic PET data were acquired over the brain in three-dimensional mode with the animal supine. Emission data were acquired for 60 min after intravenous injection of 18F-FDG. Each scan consisted of a sequence of 24 frames (1 × 15 s, 3 × 30 s, 1 × 45 min, 3 × 1 min, 1 × 1.5 min, 6 × 2 min, 1 × 3.5 min, and 8 × 5 min). Measured attenuation and scatter corrections were applied to the emission data. Data were reconstructed using the reprojection algorithm with an axial and transaxial Hanning filter that had a 0.5 cutoff frequency. The resulting images had a spatial resolution of approximately 5–7 mm in full width at half maximum.
Control and CsA PET images were coregistered (rigid coregistration) for each animal using PMOD® software (version 3.5). Regions of interest (ROI; brain hemispheres and cerebellum) were drawn manually on control conditions PET images. ROIs were then applied to coregistered dynamic PET data obtained with CsA. Time–activity curves (TACs) were generated by calculating the mean radioactivity in the brain hemispheres and cerebellum. TACs were generated by correcting the radioactivity in each region for fluorine-18 decay, injected dose, and animal weight. TACs are expressed as standard uptake value (SUV; i.e., mean radioactivity/injected dose/weight) versus time. Kinetic modeling was then performed using a two-tissue compartment model considering the arterial plasma input function with PMOD® software. Outcome parameters K1 (mL cm−3 min−1), k2 (L min−1), k3 (L min−1), k4 (L min−1) and VT (mL cm−3) were estimated in each condition from time–activity data in the brain and plasma (36). Cerebral metabolic rate of glucose utilization (CMRGlc ; μmol/min/100 g) was estimated in each region from measured plasma glucose and a lumped constant set at 0.89 (37), assuming that P-gp inhibition has no influence on the lumped constant.
CMRGlc parametric maps were then generated to allow visual comparison between tested conditions, taking arterial input function and plasma glucose into account (PMOD® software).
In Situ Brain Perfusion in Mice
The intrinsic initial transport rate (or brain uptake clearance) at the luminal BBB membrane for 18F-FDG was measured in wild-type mice and in P-gp/Bcrp(−/−) mice. Animals perfused with protein-free Krebs-carbonate buffer physiological saline containing a controlled and physiological concentration of d-glucose (9 mM), 18F-FDG (3.7 MBq/mL), and 99mTcO4− (3.7MBq/mL) used as a vascular integrity marker. The solution was gassed with 95% O2/5% CO2 for pH control (7.4) and warmed to 37°C. Once anesthetized with ketamine (140 mg/kg, i.p.) + xylazine (8 mg/kg, i.p.), the right common carotid artery was exposed, and the external carotid artery was ligated at the level of the bifurcation of the common and internal carotid arteries. The right common carotid artery was catheterized and the catheter connected to a syringe containing the perfusion fluid. At this time, the perfusion fluid replaces all the blood in the right brain vasculature. The thorax was then opened, the heart was cut, and perfusion started immediately at a flow rate of 2.5 mL/min (38). Each mouse was perfused for 120 s. The intrinsic transport rate (Kin; μL g−1 s−1, n = 5) for 18F-FDG was calculated as previously reported (38). The brain intravascular volume was taken into account using the distribution of 99mTcO4−, which does not measurably cross the BBB (39). The distribution volume of 18F-FDG (Vbrain; μL/g) and its brain uptake clearance (Kin = Vbrain/120 s ; μL g−1 s−1) were calculated from decay corrected 18F radioactive count in the right brain hemisphere.
Statistical Analysis
Results are shown as mean ± standard deviation. In vitro data were log transformed and compared using a two-way ANOVA with “valspodar” and “d-glucose presence” as two main factors followed by Tukey’s post hoc test. In vivo data were compared using the Student’s t test. Statistical significance was set at p < 0.05 (R software version 3.1.2 (40)). The size of the different groups was set from a previously reported study using similar methods in both the mice and baboons (33).
RESULTS
In Vitro18F-FDG Uptake Assay
In the absence of d-glucose, 18F-FDG uptake by MDCKII-MDR1 cells appeared linear in the first 60 min. Then, in a second phase, 18F-FDG uptake increased more slowly. In the presence of the P-gp inhibitor valspodar, 18F-FDG uptake increased linearly from 0- to 120-min incubation. Valspodar significantly reduced 18 F-FDG uptake from 0 to 60 min. Conversely, 18F-FDG uptake measured after 120-min incubation was ∼14% higher in the presence of valspodar (Fig. 1).
Fig. 1.
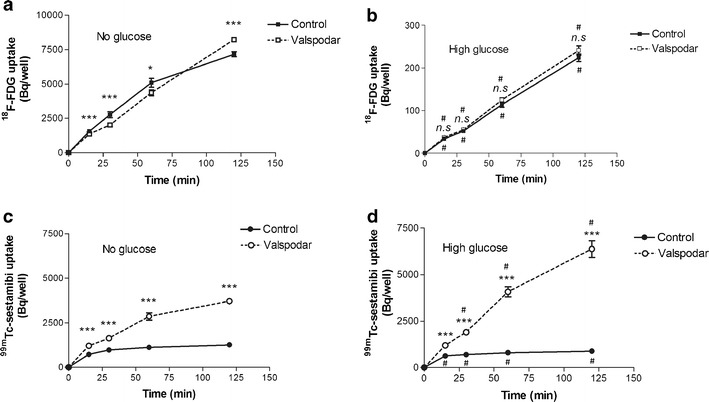
Time course of 18F-FDG and 99mTc-sestamibi accumulation in MDCKII-MDR1 cells. The uptake of 18F-FDG (a, b) and 99mTc-sestamibi (c, d) in MDCKII-MDR1 monolayers was measured in the presence and the absence of the P-gp inhibitor valspodar (mean ± SD; n = 4−8 vial for each condition). 99mTc-sestamibi, a P-gp substrate, is used as a positive control for P-gp activity. Data obtained in the absence of d-glucose in the incubation medium (a, c) were compared to those obtained in the presence of high d-glucose concentration (17.5 mM; b, d). ANOVA followed by followed by Tukey’s post hoc test was performed to assess statistical difference between groups: (i) absence compared to presence of valspodar, ***p < 0.001; *p < 0.05; n.s = nonsignificant and (ii) absence compared to presence of high d-glucose concentration # p < 0.001 when significant
The presence of a high d-glucose concentration in the incubation medium resulted in a significant ∼95% decrease in 18F-FDG uptake by MDR1 cells. ANOVA unveiled an interaction between the “d-glucose” and “valspodar” factors. P-gp inhibition using valspodar had no significant impact on 18F-FDG uptake in the presence of high d-glucose concentration in incubation medium over 120 min.
Valspodar significantly increased the accumulation of the known P-gp substrate 99mTc-sestamibi (p < 0.001) in the presence and the absence of d-glucose in the medium. When P-gp is inhibited, the presence of d-glucose significantly increased 99mTc-sestamibi cellular accumulation, while it was decreased in the presence of functionnal P-gp (p < 0.001) (Fig. 1c, d).
PET Study in Baboon
PET images revealed a similar distribution of 18F-FDG in brain regions with or without P-gp inhibition (Fig. 2). Representative TACs obtained in a monkey in the presence and the absence of CsA are shown in Fig. 3. PET quantification and kinetic analysis performed in several brain regions in baboon allowed the estimation of outcome parameters that were shown to reflect 18F-FDG kinetics and metabolism (36,41). Kinetic analysis revealed that P-gp inhibition with CsA does not influence 18F-FDG the transport rate from the blood to the brain (K1). Neither was the brain to blood efflux rate (k2) significantly affected by CsA. We found no difference in the k3 value that was shown to mainly reflect brain HK activity. We obtained much better fits to the data when k4 was included in the compartment model (i.e., k4 not set to zero). Nevertheless, the existence of significant dephosphorylation of 18F-FDG-6-P to free 18F-FDG is still a source of much debate (42). TACs unveiled a surprisingly high 18F-FDG uptake in the cerebellum (Fig. 3). CMRGlc was also higher in the cerebellum than in the brain hemispheres (Table I, Fig. 3).
Fig. 2.
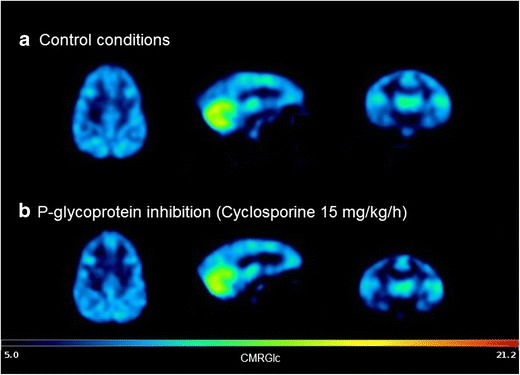
Representative CMRGlc parametric maps obtained in the brain of control (a) and cyclosporine-treated (b) baboons anesthetized using isoflurane. CMRGlc reflects the local glucose metabolism (μmol/min/100 g). Parametric map was generated using PMOD® software (version 3.5) starting from 18F-FDG dynamic PET images, arterial input function, plasma glucose, and a lumped constant set at 0.89
Fig. 3.
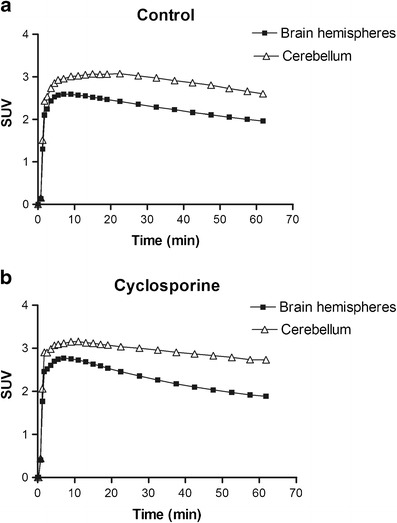
Representative time–activity curves obtained in brain hemispheres and cerebellum of control (a) and cyclosporine-treated (b) baboons. Data are expressed as standard uptake value (SUV) versus time
Table I.
18F-FDG Kinetic Parameters Measured in Brain Regions of the Baboon Using the 18F-FDG 2-tissue Compartment Model in Control Conditions (Mean ± S.D; n = 3) and in the Presence of the P-gp Inhibitor Cyclosporine 15 mg kg−1 h−1 (CsA, Mean ± SD; n = 3). Student’s t Test Analysis Revealed No Difference Between Control and CsA
| K 1 (mL cm−3 min−1) | k 2 (L min−1) | k 3 (L min−1) | k 4 (L min−1) | CMRGlc (μmol/min/100 g) | V T (mL cm−3) | ||
|---|---|---|---|---|---|---|---|
| Brain hemispheres | Control | 0.137 ± 0.031 | 0.263 ± 0.078 | 0.0485 ± 0.0037 | 0.0217 ± 0.0038 | 11.4 ± 0.9 | 1.73 ± 0.18 |
| CsA | 0.113 ± 0.016 | 0.202 ± 0.048 | 0.0367 ± 0.0139 | 0.0174 ± 0.0108 | 10.8 ± 2.7 | 2.06 ± 0.86 | |
| Cerebellum | Control | 0.164 ± 0.050 | 0.295 ± 0.103 | 0.0594 ± 0.0068 | 0.0233 ± 0.0053 | 14.9 ± 3.5 | 2.03 ± 0.17 |
| CsA | 0.136 ± 0.026 | 0.225 ± 0.053 | 0.0446 ± 0.0102 | 0.0190 ± 0.0035 | 14.4 ± 3.7 | 2.04 ± 0.24 |
18F-FDG in situ Brain Perfusion
We used the in situ brain perfusion technique and a perfusion fluid containing a controlled and physiological concentration of unlabeled d-glucose, so that 18F-FDG brain distribution is not likely to be influenced by peripheral blood glucose. 18F-FDG initial transport BBB rate measured in wild-type mice was 1.76 ± 0.24 μL g−1 s−1 (n = 5). The brain uptake clearance of d-glucose measured under similar conditions was Kin = 1.5 μL g−1 s−1 (38). This confirms previous data showing that 18F-FDG accumulates at a higher rate than d-glucose itself (42). We found no difference with the Kin of 18F-FDG measured in P-gp/Bcrp-deficient mice (1.71 ± 0.15 μL g−1 s−1; n = 5).
DISCUSSION
The relationship between 18F-FDG uptake and P-gp function was initially studied in cancer cells. MDR phenotype was mostly obtained by culturing drug-sensitive parental cells in the presence of cytotoxic drugs to promote selection. These in vitro studies concluded to a reduced (14,16) or enhanced (23–25) uptake of 18F-FDG in MDR cells as compared to MDR negative. First, it may be hypothesized that chemotherapeutic agents used for the selection of MDR clones may differentially affect glucose metabolism and affect 18F-FDG uptake. In addition to the variety of cell lines and culture condition (25), we show that these controversies may result from different incubation duration used in the different studies that ranged from 10 to 120 min. Our results show that, in the absence of d-glucose, P-gp inhibition using valspodar, a CsA derivative, enhanced 18F-FDG uptake after 120-min incubation, while it was reduced at earlier incubation times (Fig. 1). This time-dependent shift was also found in cancer cells (17,25). Interestingly, CsA was previously shown to abolish the difference in 18F-FDG uptake between MDR-positive and MDR-negative cell lines (23). In both the MDR-positive and MDR-negative cells, neither ATP consumption nor lactate production was influenced by CsA (43). This suggests that the effect of CsA, and probably valspodar, on 18F-FDG uptake is related to P-gp function and does not reflect a potential effect of these compounds on cellular glucose metabolism.
It was recently shown that high concentration FDG does not impact ATP hydrolysis in P-gp-overexpressing cells (25). However, inconsistencies for P-gp substrates to impact the P-gp ATPase activity have been reported, and ATPase assay appeared insufficient to conclude to a lack of P-gp mediated transport (44). Standardized transfected epithelial cell line such as MDCKII-MDR1 including a positive control are recommended to test the transport function of chemical entities by P-gp and bidirectional transport assay is usually preferred (44). In this model, the fast and extensive metabolism and trapping of 18F-FDG into cells would undoubtedly limit 18F-FDG recovery from the donor to the receiver chamber. We used uptake assay instead, also allowing comparison to previous studies performed using different cancer cell lines.
In the absence of d-glucose and in contrast to 99mTc-sestamibi, 18F-FDG uptake was found lower during the first 60-min incubation, when P-gp was inhibited. At 120 min, the effect of P-gp inhibition on 18F-FDG uptake in MDCKII-MDR1 cells (14% increase) was found significant but much lower than that previously obtained in breast carcinoma Bcap37-MDR1 cells (50% increase) (17). We may nonetheless hypothesize that this result may be biased by cell metabolism shift due to prolonged incubation in a d-glucose-free medium, although cell viability was >95% after 120-min incubation. MDCKII cells express GLUT1 and GLUT8 (45). We used a high concentration of d-glucose to unveil the potential transport of 18F-FDG by P-gp, apart from the GLUT activity. Unsurprisingly, 18F-FDG uptake was dramatically decreased in the presence of high d-glucose (Fig. 1). 18F-FDG cellular accumulation increased with time, suggesting significant 18F-FDG uptake, even in the presence of high d-glucose. In parallel, high d-glucose medium abolished the effect of P-gp inhibition on 18F-FDG uptake without impairing the ability of P-gp to mediate the efflux of 99mTc-sestamibi.
It was noticed that d-glucose deprivation decreased the cellular/mitochondrial retention of 99mTc-sestamibi in MDCKII-MDR1 cells when P-gp was inhibited (Fig 1d). 99mTc-sestamibi uptake is known to depend on the mitochondrial metabolic activity and cell viability (46). In the present study, trypan blue exclusion assay did not detect any additional cell death in the absence of d-glucose after 120-min incubation. This suggests that cellular metabolic shift induced by d-glucose deprivation may be detected in vitro using 99mTc-sestamibi.
Altogether, these results confirm that 18F-FDG is not transported by P-gp (25). Phosphorylation of 18F-FDG by HK, as well as P-gp-mediated transport, may competitively modulate d-glucose and intracellular ATP concentration. 99mTc-sestamibi is a hydrophilic molecule (log P = 0.7) and was incubated at same tracer concentration (approximately pM) in each vial (47). Therefore, the presence of 99mTc-sestamibi is not likely to influence ATPase activity (48). Despite differences in 18F-FDG uptake, it was reported that intracellular ATP content (14,49), HK activity (14), and GLUT transporter expression (25) are similar in MDR-positive and MDR-negative cells. Therefore, further experiments are needed to explain, at a molecular level, how P-gp function indirectly modulates the GLUT/HK-dependant 18F-FDG cellular distribution.
High level of P-gp and GLUT-1 expression were found in the endothelial cells of brain microvessels forming the BBB (26). In baboon, CsA did not impact K1, suggesting that 18F-FDG uptake by the brain is not influenced by this drug even at high dosing used for P-gp inhibition in primate (34). We found no influence on estimated k3 values, suggesting that CsA has no consequence on HK activity in the brain. CsA pretreatment did not either impact CMRGlu in the cerebellum and other regions of the brain (Table I). Altogether, these results, obtained in primates, suggest that 18F-FDG kinetics and glucose metabolism are influenced by neither P-gp inhibition nor CsA administration in the healthy brain. It was observed a surprisingly high 18F-FDG uptake in the cerebellum compared to the brain hemispheres. This phenomenon was observed in all tested animals, regardless of CsA exposure. It was reported that anesthetics may differentially influence 18F-FDG brain kinetics (50,51). In the present study, baboons received the same anesthetic protocol (ketamine followed by isoflurane). Further investigations are needed to test the potential influence of this anesthetic protocol on regional 18F-FDG brain kinetics.
In rat, it was shown that administration of tariquidar, a combined P-gp/Bcrp inhibitor, resulted in a ∼45% increase in CMRGlc (30). P-gp-unrelated effects of this molecule on peripheral and brain glucose metabolism was proposed as an explanation for its unexpected effect on the brain uptake of 18F-FDG (30). Therefore, we used transporter-deficient mice instead of pharmacological inhibition and performed in situ brain perfusion, so that 18F-FDG uptake by the brain is not influenced by peripheral blood glucose. This invasive method is an accurate approach to measure the influence of carrier-mediated transport on the initial distribution (e.g., brain uptake) of compounds at the BBB. In P-gp/Bcrp-deficient mice, the brain clearance parameter (Kin) of the P-gp substrate 99mTc-sestamibi was shown to be 2.7-fold greater than in wild-type mice (52). As P-gp and Bcrp seem to work together in a synergistic fashion, dual P-gp/Bcrp inhibition or deficiencies could sometimes be a better strategy to reveal the carrier-mediated transport of their common substrates at the BBB (53,54). Our results show that combined P-gp/Bcrp deficiency had no influence on 18F-FDG distribution by the healthy brain and that 18F-FDG is not either likely to be a substrate of the rodent P-gp or Bcrp.
CONCLUSION
In vitro assay confirmed the modulatory effect of valspodar on 18F-FDG uptake in P-gp-overexpressing cells, although 18F-FDG is not a P-gp substrate. In vivo, 18F-FDG brain kinetics was not measurably influenced by P-gp inhibition and/or depletion, starting from a physiological P-gp expression at the BBB. Clinical 18F-FDG PET provides useful information regarding the brain metabolism in several pathophysiological states in human. This study suggests that P-gp decline at the BBB is not a major factor of 18F-FDG brain kinetics and subsequent estimation of brain glucose metabolism.
ACKNOWLEDGMENTS
Salvatore Cisternino received a financial support from the Commissariat à l’énergie atomique et aux énergies alternatives (CEA) and l’Assistance publique–Hôpitaux de Paris (AP-HP) : “Postes d’accueil CEA/AP-HP”. Nicholas Bernards kindly reviewed the English text.
Disclosure/Conflict of Interest
The authors declare they have no conflict of interest.
REFERENCES
- 1.Delbeke D, Coleman RE, Guiberteau MJ, Brown ML, Royal HD, Siegel BA, et al. Procedure guideline for tumor imaging with 18F-FDG PET/CT 1.0. J Nucl Med. 2006;47:885–95. [PubMed] [Google Scholar]
- 2.De Geus-Oei L-F, Vriens D, van Laarhoven HWM, van der Graaf WTA, Oyen WJG. Monitoring and predicting response to therapy with 18F-FDG PET in colorectal cancer: a systematic review. J Nucl Med. 2009;50(Suppl 1):43S–54. doi: 10.2967/jnumed.108.057224. [DOI] [PubMed] [Google Scholar]
- 3.Ben-Haim S, Ell P. 18F-FDG PET and PET/CT in the Evaluation of Cancer Treatment Response. J Nucl Med. 2009;50:88–99. doi: 10.2967/jnumed.108.054205. [DOI] [PubMed] [Google Scholar]
- 4.Cook GJR, Yip C, Siddique M, Goh V, Chicklore S, Roy A, et al. Are pretreatment 18F-FDG PET tumor textural features in non-small cell lung cancer associated with response and survival after chemoradiotherapy? J Nucl Med. 2013;54:19–26. doi: 10.2967/jnumed.112.107375. [DOI] [PubMed] [Google Scholar]
- 5.Varrone A, Asenbaum S, Vander Borght T, Booij J, Nobili F, Någren K, et al. EANM procedure guidelines for PET brain imaging using [18F]FDG, version 2. Eur J Nucl Med Mol Imaging. 2009;36:2103–10. doi: 10.1007/s00259-009-1264-0. [DOI] [PubMed] [Google Scholar]
- 6.Frisoni GB, Bocchetta M, Chételat G, Rabinovici GD, de Leon MJ, Kaye J, et al. Imaging markers for Alzheimer disease: which vs how. Neurology. 2013;81:487–500. doi: 10.1212/WNL.0b013e31829d86e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kumar A, Juhász C, Asano E, Sood S, Muzik O, Chugani HT. Objective detection of epileptic foci by 18F-FDG PET in children undergoing epilepsy surgery. J Nucl Med. 2010;51:1901–7. doi: 10.2967/jnumed.110.075390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jadvar H, Alavi A, Gambhir SS. 18F-FDG uptake in lung, breast, and colon cancers: molecular biology correlates and disease characterization. J Nucl Med. 2009;50:1820–7. doi: 10.2967/jnumed.108.054098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhao S, Kuge Y, Mochizuki T, Takahashi T, Nakada K, Sato M, et al. Biologic correlates of intratumoral heterogeneity in 18F-FDG distribution with regional expression of glucose transporters and hexokinase-II in experimental tumor. J Nucl Med. 2005;46:675–82. [PubMed] [Google Scholar]
- 10.Avril N. GLUT1 expression in tissue and (18)F-FDG uptake. J Nucl Med. 2004;45:930–2. [PubMed] [Google Scholar]
- 11.Smith TA. The rate-limiting step for tumor [18F]fluoro-2-deoxy-D-glucose (FDG) incorporation. Nucl Med Biol. 2001;28:1–4. doi: 10.1016/S0969-8051(00)00177-3. [DOI] [PubMed] [Google Scholar]
- 12.Devraj K, Klinger ME, Myers RL, Mokashi A, Hawkins RA, Simpson IA. GLUT-1 glucose transporters in the blood–brain barrier: differential phosphorylation. J Neurosci Res. 2011;89:1913–25. doi: 10.1002/jnr.22738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Holohan C, Van Schaeybroeck S, Longley DB, Johnston PG. Cancer drug resistance: an evolving paradigm. Nat Rev Cancer. 2013;13:714–26. doi: 10.1038/nrc3599. [DOI] [PubMed] [Google Scholar]
- 14.Smith TAD, Sharma RI, Wang WG, Welch AE, Schweiger LF, Collie-Duguid ESR. Decreased [18F]fluoro-2-deoxy-d-glucose incorporation and increased glucose transport are associated with resistance to 5FU in MCF7 cells in vitro. Nucl Med Biol. 2007;34:955–60. doi: 10.1016/j.nucmedbio.2007.07.007. [DOI] [PubMed] [Google Scholar]
- 15.Yamada K, Brink I, Engelhardt R. Factors influencing [F-18] 2-fluoro-2-deoxy-D-glucose (F-18 FDG) accumulation in melanoma cells: is FDG a substrate of multidrug resistance (MDR)? J Dermatol. 2005;32:335–45. doi: 10.1111/j.1346-8138.2005.tb00904.x. [DOI] [PubMed] [Google Scholar]
- 16.Lorke DE, Krüger M, Buchert R, Bohuslavizki KH, Clausen M, Schumacher U. In vitro and in vivo tracer characteristics of an established multidrug-resistant human colon cancer cell line. J Nucl Med. 2001;42:646–54. [PubMed] [Google Scholar]
- 17.Yu C, Wan W, Zhang B, Deng S, Yen T-C, Wu Y. Evaluation of the relationship between [18F]FDG and P-glycoprotein expression: an experimental study. Nucl Med Biol. 2012;39:671–8. doi: 10.1016/j.nucmedbio.2011.12.007. [DOI] [PubMed] [Google Scholar]
- 18.Seo S, Hatano E, Higashi T, Nakajima A, Nakamoto Y, Tada M, et al. P-glycoprotein expression affects 18F-fluorodeoxyglucose accumulation in hepatocellular carcinoma in vivo and in vitro. Int J Oncol. 2009;34:1303–12. [PubMed] [Google Scholar]
- 19.Seo S, Hatano E, Higashi T, Hara T, Tada M, Tamaki N, et al. Fluorine-18 fluorodeoxyglucose positron emission tomography predicts tumor differentiation, P-glycoprotein expression, and outcome after resection in hepatocellular carcinoma. Clin Cancer Res. 2007;13:427–33. doi: 10.1158/1078-0432.CCR-06-1357. [DOI] [PubMed] [Google Scholar]
- 20.Seo S, Hatano E, Higashi T, Nakajima A, Nakamoto Y, Tada M, et al. Fluorine-18 fluorodeoxyglucose positron emission tomography predicts lymph node metastasis, P-glycoprotein expression, and recurrence after resection in mass-forming intrahepatic cholangiocarcinoma. Surgery. 2008;143:769–77. doi: 10.1016/j.surg.2008.01.010. [DOI] [PubMed] [Google Scholar]
- 21.Higashi K, Ueda Y, Ikeda R, Kodama Y, Guo J, Matsunari I, et al. P-glycoprotein expression is associated with FDG uptake and cell differentiation in patients with untreated lung cancer. Nucl Med Commun. 2004;25:19–27. doi: 10.1097/00006231-200401000-00004. [DOI] [PubMed] [Google Scholar]
- 22.Brito AF, Mendes M, Abrantes AM, Tralhão JG, Botelho MF. Positron emission tomography diagnostic imaging in multidrug-resistant hepatocellular carcinoma: focus on 2-deoxy-2-(18F)fluoro-D-glucose. Mol Diagn Ther. 2014;18:495–504. [DOI] [PubMed]
- 23.Márián T, Szabó G, Goda K, Nagy H, Szincsák N, Juhász I, et al. In vivo and in vitro multitracer analyses of P-glycoprotein expression-related multidrug resistance. Eur J Nucl Med Mol Imaging. 2003;30:1147–54. doi: 10.1007/s00259-003-1204-3. [DOI] [PubMed] [Google Scholar]
- 24.Krasznai ZT, Péli-Szabó J, Németh E, Balkay L, Szabó G, Goda K, et al. Paclitaxel modifies the accumulation of tumor-diagnostic tracers in different ways in P-glycoprotein-positive and negative cancer cells. Eur J Pharm Sci. 2006;28:249–56. doi: 10.1016/j.ejps.2006.02.006. [DOI] [PubMed] [Google Scholar]
- 25.Krasznai ZT, Trencsényi G, Krasznai Z, Mikecz P, Nizsalóczki E, Szalóki G, et al. 18FDG a PET tumor diagnostic tracer is not a substrate of the ABC transporter P-glycoprotein. Eur J Pharm Sci. 2014;64:1–8. doi: 10.1016/j.ejps.2014.08.002. [DOI] [PubMed] [Google Scholar]
- 26.Abbott NJ, Patabendige AAK, Dolman DEM, Yusof SR, Begley DJ. Structure and function of the blood–brain barrier. Neurobiol Dis. 2010;37:13–25. doi: 10.1016/j.nbd.2009.07.030. [DOI] [PubMed] [Google Scholar]
- 27.König J, Müller F, Fromm MF. Transporters and drug–drug interactions: important determinants of drug disposition and effects. Pharmacol Rev. 2013;65:944–66. doi: 10.1124/pr.113.007518. [DOI] [PubMed] [Google Scholar]
- 28.Van Assema DME, Lubberink M, Bauer M, van der Flier WM, Schuit RC, Windhorst AD, et al. Blood–brain barrier P-glycoprotein function in Alzheimer’s disease. Brain. 2012;135:181–9. doi: 10.1093/brain/awr298. [DOI] [PubMed] [Google Scholar]
- 29.Kwan P, Schachter SC, Brodie MJ. Drug-resistant epilepsy. N Engl J Med. 2011;365:919–26. doi: 10.1056/NEJMra1004418. [DOI] [PubMed] [Google Scholar]
- 30.La Fougère C, Böning G, Bartmann H, Wängler B, Nowak S, Just T, et al. Uptake and binding of the serotonin 5-HT1A antagonist [18F]-MPPF in brain of rats: effects of the novel P-glycoprotein inhibitor tariquidar. NeuroImage. 2010;49:1406–15. doi: 10.1016/j.neuroimage.2009.09.048. [DOI] [PubMed] [Google Scholar]
- 31.Tournier N, Chevillard L, Megarbane B, Pirnay S, Scherrmann J-M, Declèves X. Interaction of drugs of abuse and maintenance treatments with human P-glycoprotein (ABCB1) and breast cancer resistance protein (ABCG2) Int J Neuropsychopharmacol. 2010;13:905–15. doi: 10.1017/S1461145709990848. [DOI] [PubMed] [Google Scholar]
- 32.Tournier N, André P, Blondeel S, Rizzo-Padoin N, du Moulinet d’Hardemarre A, Declèves X, et al. Ibogaine labeling with 99mTc-tricarbonyl: synthesis and transport at the mouse blood–brain barrier. J Pharm Sci. 2009;98:4650–60. doi: 10.1002/jps.21771. [DOI] [PubMed] [Google Scholar]
- 33.Tournier N, Cisternino S, Peyronneau M-A, Goutal S, Dolle F, Scherrmann J-M, et al. Discrepancies in the P-glycoprotein-mediated transport of (18)F-MPPF: a pharmacokinetic study in mice and non-human primates. Pharm Res. 2012;29:2468–76. doi: 10.1007/s11095-012-0776-7. [DOI] [PubMed] [Google Scholar]
- 34.Ke AB, Eyal S, Chung FS, Link JM, Mankoff DA, Muzi M, et al. Modeling cyclosporine A inhibition of the distribution of a P-glycoprotein PET ligand, 11C-verapamil, into the maternal brain and fetal liver of the pregnant nonhuman primate: impact of tissue blood flow and site of inhibition. J Nucl Med. 2013;54:437–46. doi: 10.2967/jnumed.112.111732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kannan P, John C, Zoghbi SS, Halldin C, Gottesman MM, Innis RB, et al. Imaging the function of P-glycoprotein with radiotracers: pharmacokinetics and in vivo applications. Clin Pharmacol Ther. 2009;86:368–77. doi: 10.1038/clpt.2009.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Innis RB, Cunningham VJ, Delforge J, Fujita M, Gjedde A, Gunn RN, et al. Consensus nomenclature for in vivo imaging of reversibly binding radioligands. J Cereb Blood Flow Metab. 2007;27:1533–9. doi: 10.1038/sj.jcbfm.9600493. [DOI] [PubMed] [Google Scholar]
- 37.Graham MM, Muzi M, Spence AM, O’Sullivan F, Lewellen TK, Link JM, et al. The FDG lumped constant in normal human brain. J Nucl Med. 2002;43:1157–66. [PubMed] [Google Scholar]
- 38.Cattelotte J, André P, Ouellet M, Bourasset F, Scherrmann J-M, Cisternino S. In situ mouse carotid perfusion model: glucose and cholesterol transport in the eye and brain. J Cereb Blood Flow Metab. 2008;28:1449–59. doi: 10.1038/jcbfm.2008.34. [DOI] [PubMed] [Google Scholar]
- 39.Keyeux AJ, Ochrymowicz-Bemelmans DA, Charlier AA. Technetium-99m-pertechnetate as a whole blood marker for brain perfusion studies. J Nucl Med. 1994;35:479–83. [PubMed] [Google Scholar]
- 40.R Core Team (2014) R: A Language and Environment for Statistical Computing. http://www.R-project.org/.
- 41.Hong YT, Fryer TD. Kinetic modelling using basis functions derived from two-tissue compartmental models with a plasma input function: general principle and application to [18F]fluorodeoxyglucose positron emission tomography. NeuroImage. 2010;51:164–72. doi: 10.1016/j.neuroimage.2010.02.013. [DOI] [PubMed] [Google Scholar]
- 42.Hasselbalch SG, Knudsen GM, Holm S, Hageman LP, Capaldo B, Paulson OB. Transport of D-glucose and 2-fluorodeoxyglucose across the blood–brain barrier in humans. J Cereb Blood Flow Metab. 1996;16:659–66. doi: 10.1097/00004647-199607000-00017. [DOI] [PubMed] [Google Scholar]
- 43.Broxterman HJ, Pinedo HM, Schuurhuis GJ, Lankelma J. Cyclosporin A and verapamil have different effects on energy metabolism in multidrug-resistant tumour cells. Br J Cancer. 1990;62:85–8. [DOI] [PMC free article] [PubMed]
- 44.Giacomini KM, Huang S-M, Tweedie DJ, Benet LZ, Brouwer KLR, Chu X, et al. Membrane transporters in drug development. Nat Rev Drug Discov. 2010;9:215–36. [DOI] [PMC free article] [PubMed]
- 45.Quan Y, Jin Y, Faria TN, Tilford CA, He A, Wall DA, et al. Expression profile of drug and nutrient absorption related genes in Madin–Darby canine kidney (MDCK) cells grown under differentiation conditions. Pharmaceutics. 2012;4:314–33. [DOI] [PMC free article] [PubMed]
- 46.Furuta M, Nozaki M, Kawashima M, Iimuro M, Okayama A, Fukushima M, et al. Monitoring mitochondrial metabolisms in irradiated human cancer cells with (99m)Tc-MIBI. Cancer Lett. 2004;212:105–11. [DOI] [PubMed]
- 47.Cattelotte J, Tournier N, Rizzo-Padoin N, Schinkel AH, Scherrmann J-M, Cisternino S. Changes in dipole membrane potential at the mouse blood–brain barrier enhance the transport of 99mTechnetium Sestamibi more thaninhibiting Abcb1, Abcc1, or Abcg2. J Neurochem. 2009;108:767–75. [DOI] [PubMed]
- 48.Litman T, Nielsen D, Skovsgaard T, Zeuthen T, Stein WD. ATPase activity of P-glycoprotein related to emergence of drug resistance in Ehrlich ascites tumor cell lines. Biochim Biophys Acta. 1997;1361:147–58. [DOI] [PubMed]
- 49.Martell RL, Slapak CA, Levy SB. Effect of glucose transport inhibitors on vincristine efflux in multidrug-resistant murine erythroleukaemia cells overexpressing the multidrug resistance-associated protein (MRP) and two glucose transport proteins, GLUT1 and GLUT3. Br J Cancer. 1997;75:161–8. [DOI] [PMC free article] [PubMed]
- 50.Noda A, Takamatsu H, Minoshima S, Tsukada H, Nishimura S. Determination of kinetic rate constants for 2-[18F]fluoro-2-deoxy-d-glucose and partition coefficient of water in conscious macaques and alterations in aging or anesthesia examined on parametric images with an anatomic standardization technique. J Cereb Blood Flow Metab. 2003;23:1441–7. [DOI] [PubMed]
- 51.Vollenweider FX, Leenders KL, Scharfetter C, Antonini A, Maguire P, Missimer J, et al. Metabolic hyperfrontality and psychopathology in the ketamine model of psychosis using positron emission tomography (PET) and [18F]fluorodeoxyglucose (FDG). Eur Neuropsychopharmacol. 1997;7:9–24. [DOI] [PubMed]
- 52.Cattelotte J, Tournier N, Rizzo-Padoin N, Schinkel AH, Scherrmann J-M, Cisternino S. Changes in dipole membrane potential at the mouse blood–brain barrier enhance the transport of 99mTechnetium Sestamibi more than inhibiting Abcb1, Abcc1, or Abcg2. J Neurochem. 2009;108:767–75. [DOI] [PubMed]
- 53.Cisternino S, Mercier C, Bourasset F, Roux F, Scherrmann J-M. Expression, up-regulation, and transport activity of the multidrug-resistance protein Abcg2 at the mouse blood–brain barrier. Cancer Res. 2004;64:3296–301. [DOI] [PubMed]
- 54.Hosten B, Boisgard R, Jacob A, Goutal S, Saubaméa B, Dollé F, et al. [11C]befloxatone brain kinetics is not influenced by Bcrp function at the blood–brain barrier: a PET study using Bcrp TGEM knockout rats. Eur J Pharm Sci. 2013;50:520–5. [DOI] [PubMed]