

Abstract
Core structures of current drugs have been assembled and their structural relationships and activity profiles have been explored. Drug scaffolds were frequently involved in different types of structural relationships. In addition, a variety of activity profile relationships between structurally related drug scaffolds were detected, ranging from closely overlapping to distinct profiles. Furthermore, when structural and activity profile relationships of scaffolds from drugs and bioactive compounds were compared, systematic differences were detected. Consensus activity profiles were introduced as a new approach for the qualitative and quantitative assessment of activity similarity of structurally related drugs represented by the same scaffold. On the basis of consensus activity profiles, scaffolds representing drugs active against distinct targets can be distinguished from drugs having similar target profiles and target hypotheses can be derived for individual drugs. Given the results of our analysis, drug scaffolds have been systematically organized according to structural and activity profile criteria. Our scaffold sets and the associated information are made freely available.
Electronic supplementary material
The online version of this article (doi:10.1208/s12248-015-9737-5) contains supplementary material, which is available to authorized users.
KEY WORDS: activity profiles, bioactive compounds, approved drugs, scaffolds, structural relationships
INTRODUCTION
Building blocks of drug-like molecules and drugs are of high interest in pharmaceutical research. For the analysis of molecular building blocks in active compounds, the scaffold concept is usually adapted (1–3). According to the definition that is most widely applied in medicinal chemistry, originally introduced by Bemis and Murcko (3), scaffolds are extracted from compounds by removing all substituents (R-groups), while retaining aliphatic linkers between ring systems (3). One of the primary goals of scaffold analysis is the association of molecular building blocks or core structures with specific biological activities (4–7). Knowledge of such preferred core structures is thought to be of high relevance for target- or target family-directed compound design (2–4). Scaffolds have also been monitored in compounds at different pharmaceutical development stages including leads, clinical trials compounds, experimental drugs, and marketed drugs (8). In addition, the scaffold concept can also be applied to establish structural relationships among bioactive compounds or drugs (9), i.e., compounds or drugs with structurally or topologically related core structures can be identified (9). Especially for drugs, scaffold analyses have predominantly been carried out to identify core structures that most frequently occur in drugs (3,10,11). The seminal work of Bemis and Murcko (3) identified the 41 most frequently occurring drug scaffolds (frameworks) in the Comprehensive Medicinal Chemistry database (12), Wang and Hou (10) isolated the 50 most frequently occurring ring systems from FDA-approved drugs in DrugBank (13,14) and the World Drug Index (15), and Taylor et al. (11) identified the 100 most frequent rings in drugs from the FDA Orange Book (16). Because these studies have applied different core structure or ring system definitions and used different drug sources, the results are not directly comparable (given different definitions, they are also not comparable to scaffold analysis). From these studies, it has become evident, however, that scaffolds and ring structures of drugs occur with different frequencies and that many building blocks are recurrent in different drugs. As an extension of core structure and frequency-of-occurrence analysis, it has also been investigated if drugs might contain unique scaffolds that were not found in other compounds (17). Therefore, scaffolds systematically isolated from drugs and bioactive compounds were compared. In this study, 221 drug scaffolds were identified that were not contained in currently available bioactive compounds; a rather unexpected finding. When structural relationships between these drug-unique scaffolds and the universe of bioactive scaffolds were explored, a variety of relationships were detected. However, many drug-unique scaffolds were structurally unrelated or only very distantly related to scaffolds from bioactive compounds (17). Reasons for the existence of drug-unique scaffolds are currently unclear. It is difficult to rationalize why there are no structural analogs for more than 200 drugs among currently available bioactive compounds. However, these findings indicate that current drug space is chemically at least in part only little explored, which raises another relevant question: If structural relationships between drug scaffolds and bioactive scaffolds are frequently limited, how related are drug scaffolds to each other? Herein, we have addressed this question and systematically explored different types of structural relationships between drug scaffolds. In addition, to complement the study of structural relationships, activity profiles of drug scaffolds were systematically generated and compared. Consensus activity profiles were introduced as a new measure to comprehensively capture target information associated with structurally related drugs at the level of scaffolds.
MATERIALS AND METHODS
Scaffolds from Drugs and Bioactive Compounds
ChEMBL was used as a source for bioactive compounds (18). This database represents the primary repository of compounds from medicinal chemistry sources including the scientific literature and patents. In addition, ChEMBL contains large numbers of molecules from public domain screening campaigns (18). As other repositories of bioactive compounds, ChEMBL collects activity records of compounds but does not contain information about the number of assays compounds were tested in (and found to be active or inactive). This is relevant for promiscuity analysis, one of the aspects analyzed and discussed below. From ChEMBL release 18, compounds with direct interactions (i.e., assay relationship type “D”) against human targets at the highest confidence level (i.e., assay confidence score 9) were assembled. Two types of activity measurements were considered, including (assay-independent) equilibrium constants (Ki values) and (assay-dependent) IC50 values. Only compounds with explicitly defined Ki and/or IC50 values were taken into consideration. In addition, compounds with multiple Ki and/or IC50 measurements for the same target were retained if all values fell within the same order of magnitude. This set of compound selection criteria has been shown to ensure high data confidence and provides a reliable basis for large-scale data analysis (19). From DrugBank 4.1 (13,14), all approved small molecule drugs with available structures and activity information were collected. For each qualifying drug, all reported “drug action” targets, metabolizing enzymes, transporters, and carriers were assembled and corresponding UniProt (20) accession IDs were collected.
From all bioactive compounds and approved drugs, scaffolds based upon the conventional definition by Bemis and Murcko (3) were extracted by removal of all substituents and retaining linkers between rings, as described above. This scaffold definition has also been applied in our previous studies on pharmaceutically relevant compounds (9,21). An intrinsic aspect of the scaffold concept is that compounds containing the same scaffold are consistently assigned to this scaffold, regardless of the degree to which substituent patterns differ. Tautomeric scaffolds are listed separately. We also note that no hybridization states are assigned to scaffolds following the applied definition. Hence, scaffolds must be viewed as algorithmically generated core structures, not as actual compounds, consistent with medicinal chemistry conventions.
In the following, scaffolds isolated from drugs and bioactive compounds are designated drug scaffolds and bioactive scaffolds, respectively. Furthermore, scaffolds were also transformed into cyclic skeletons (CSKs) (22) by converting all heteroatoms to carbon and setting all bond orders to one. Hence, CSKs represent a further abstraction from scaffold structure and each CSK covers a set of topologically equivalent scaffolds.
Structural Relationships
Four different types of structural relationships were systematically and separately determined among all drug scaffolds and bioactive scaffolds.
Matched molecular pair (MMP) relationship: an MMP is defined as a pair of compounds that only differ by a structural change at a single site (23). The exchange of a pair of substructures that transforms compounds into each other is termed a chemical transformation (24). Three size restrictions were applied to limit structural differences between compounds to small replacements (typically involving R-groups) (25). First, the invariant core fragment was required to have at least twice the size of each exchanged fragment. Second, the maximal size of an exchanged fragment was limited to 13 non-hydrogen atoms. Third, the size difference between two exchanged fragments was set to maximally eight non-hydrogen atoms. Accordingly, the generation of these transformation size-restricted MMPs provides a conservative measure of structural similarity. All scaffold-based MMPs were calculated using an in-house version of the algorithm by Hussain and Rea (24) that utilizes the OpenEye toolkit (26).
Synthetic relationship: as an alternative to standard fragmentation of exocyclic single bonds for MMP generation, a set of 13 published rules from the retrosynthetic combinatorial analysis procedure (RECAP) (27) was applied to generate RECAP-MMPs (28). Following retrosynthetic rules, bonds were only fragmented according to reaction information. Thus, compounds forming RECAP-MMPs are synthetically related. Transformation size restrictions detailed above were also applied to scaffold-based RECAP-MMPs. All RECAP-MMPs were calculated using in-house Java code and the OpenEye toolkit (26).
Substructure relationship: a scaffold is a substructure of another larger scaffold if its structure is entirely contained in the larger one. Thus, scaffolds forming substructure relationships have different sizes and contain different numbers of rings. To limit the assessment of substructure relationships to scaffolds having moderate size differences (similar to the structural differences permitted for MMPs) and avoid the detection of very distant relationships, substructure relationships were only considered if the participating scaffolds differed by one or at most two rings (17). In addition, the most generic scaffold, benzene, was excluded from the assessment of substructure relationships.
CSK equivalence: two scaffolds yielding the same CSK are topologically equivalent. Such pairs of scaffolds only differ by one or more heteroatoms and/or bond orders and hence have the same size. The most generic CSK, cyclohexane, was excluded from topological analysis.
These four types of structural relationships are illustrated in Fig. 1a. A pair of scaffolds might be involved in different types of structural relationships. Only substructure relationships and CSK equivalences are mutually exclusive. In Fig. 1b, c, two exemplary pairs of scaffolds are shown that are each involved in three different types of structural relationships (the largest possible number).
Fig. 1.

Structural relationships. In a, four different types of structural relationships are illustrated including scaffold-based MMP and RECAP-MMP relationships, substructure relationship (Sub), and CSK equivalence. In b and c, two exemplary pairs of scaffolds representing three types of structural relationships are shown. For each pair of scaffolds, structural differences are highlighted using bold lines and letters
Activity Profiles
For each scaffold, an activity profile was generated by combining the target annotations of all compounds represented by the scaffold. We note that activity profiles for large-scale promiscuity analysis are generated from positive activity data (in the absence of negative data, as further discussed below). The activity profile definition as reported herein has been consistently applied in previous studies to assess the biological activity and the degree of promiscuity associated with scaffolds or chemotypes (7,21,29,30). In our current analysis, activity profiles were used for drug scaffold promiscuity assessment. Promiscuity refers to the ability of a compound to specifically interact with multiple targets (31). From activity profiles, the degree of scaffold-based promiscuity was calculated as the total number of target annotations comprising the profile. It should be noted that two scaffolds would yield the same degree of promiscuity if they were active against the same number of identical, overlapping, or distinct targets. Therefore, in addition to numerical analysis, the activity profiles of structurally related scaffolds were further categorized as identical, overlapping, or distinct.
Furthermore, consensus activity profiles were generated for drug scaffolds by monitoring drug annotations on a per target basis. Figure 2 shows a consensus activity profile for hypothetical drug scaffold S that represents four drugs active against a total of seven targets. For each target associated with the drug scaffold, the relative drug frequency (RDF) is calculated with respect to all drugs. For example, only one of four drugs is active against T1. Therefore, the RDF value of scaffold S for T1 is 0.25. For scaffold S, the mean RDF value is then calculated by considering all associated targets. This value can be used as a measure to assess the activity similarity of drugs represented by a given drug scaffold.
Fig. 2.
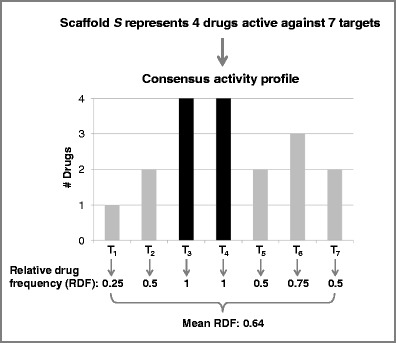
Consensus activity profile. Shown is the consensus activity profile of a hypothetical drug scaffold S. This scaffold represents four drugs that are active against a total of seven targets (T1—T7). The distribution of drugs over individual targets is represented in a bar plot. Bars corresponding to targets T3 and T4 that all four drugs are active against are shown in black
RESULTS AND DISCUSSION
Drug Scaffolds and Bioactive Scaffolds
From ChEMBL release 18, a total of 143,424 bioactive compounds with high-confidence Ki and/or IC50 measurements for 1376 human targets were obtained. From these compounds, 52,007 unique bioactive scaffolds were extracted, 35,076 of which (~67.4%) only represented a single compound. On average, each bioactive scaffold was associated with ~2.8 compounds. In addition, 20,399 unique CSKs were obtained from the pool of bioactive scaffolds. Scaffold distributions in bioactive compounds and the corresponding activity profiles were determined for comparison with drug scaffolds.
From DrugBank 4.1, 1429 approved drugs annotated with 1657 target proteins were collected. These drugs yielded 779 unique drug scaffolds and 479 unique CSKs. A subset of 620 of these scaffolds (~79.6%) represented a single approved drug. Hence, compared to bioactive scaffolds, many drug scaffolds were under-represented. On average, each drug scaffold represented ~1.8 drugs. We note that the bioactive compound-to-scaffold or the drug-to-scaffold ratio is not directly related to the frequency of occurrence of scaffolds. However, drugs must represent structural novelty as a key intellectual property criterion. Therefore, there is an intrinsically low tendency for drugs to share the same scaffold.
Structural Relationships
For the exploration of structural relationships, all drug scaffolds and bioactive scaffolds were compared in a pairwise manner, respectively. For 779 drug scaffolds, a total of 303,031 possible scaffold pairs were generated. For ~52,000 bioactive scaffolds, more than 1,300,000,000 possible pairs were obtained. For each scaffold pair, four different types of structural relationships illustrated in Fig. 1a were evaluated. As reported in Table I, a total of 627, 70, 671, and 1337 drug scaffold pairs formed MMP, RECAP-MMP, substructure, and CSK relationships, respectively. Hence, CSK equivalence was the most frequently observed structural relationship between drug scaffolds, but involved less than 50% of all drug scaffolds, while 68% of drug scaffolds were involved in substructure relationships (Table I). Taken together, these findings indicated that drug scaffolds frequently shared the same topology but were chemically distinct, due to heteroatom and/or bond order variation and that specific substructure relationships also often occurred. By contrast, only 70 scaffold pairs were found to form RECAP-MMPs indicating that the majority of drug scaffolds could not be transformed into each other on the basis of standard retrosynthetic rules.
Table I.
Structural Relationship Statistics
| Drug scaffold-based structural relationships | ||||
| Number of | MMP | RECAP | Sub | CSK |
| Scaffold pairs | 627 | 70 | 671 | 1337 |
| Scaffolds in pairs | 252 | 98 | 530 | 379 |
| % Scaffolds in pairs | 32.3% | 12.6% | 68.0% | 48.7% |
| Bioactive scaffold-based structural relationships | ||||
| Number of | MMP | RECAP | Sub | CSK |
| Scaffold pairs | 594,495 | 72,137 | 107,684 | 511,794 |
| Scaffolds in pairs | 43,821 | 29,476 | 46,969 | 38,454 |
| % Scaffolds in pairs | 84.3% | 56.7% | 90.3% | 73.9% |
For drug scaffolds and bioactive scaffolds, the number of scaffold pairs forming different types of structural relationships is reported. In addition, the number of scaffolds forming pairs and the corresponding percentage of scaffolds involved in each type of relationships are given
MMP matched molecular pair, RECAP retrosynthetic combinatorial analysis procedure, Sub substructure, CSK cyclic skeleton
The large number of bioactive scaffolds yielded many structural relationships, as also reported in Table I. Most bioactive scaffolds were found to be involved in structural relationships, ranging from ~56.7% of scaffolds forming RECAP-MMPs to more than 90% (substructure relationships). Table I shows that the proportion of drug scaffolds involved in structural relationships was consistently much lower than the proportion of bioactive scaffolds. Thus, compared to bioactive scaffolds, drug scaffolds were overall much less related to each other structurally and also much less related from a retrosynthetic chemical perspective.
Figure 3a reports the proportion of drug scaffolds involved in increasing numbers of structural relationships and further corroborates the findings discussed above. If drug scaffolds were involved in structural relationships, they often formed only one or two relationships. By contrast, bioactive scaffolds were typically found to be involved in many more relationships (taking into consideration that the much larger sample size yields an increased statistical likelihood of relationship formation), as shown in Figure S1 of the Supporting Information. For instance, more than 54% of all bioactive scaffolds formed MMPs with at least five other scaffolds. By contrast, only ~6.5% of the drug scaffolds were involved in the formation of five or more MMPs.
Fig. 3.
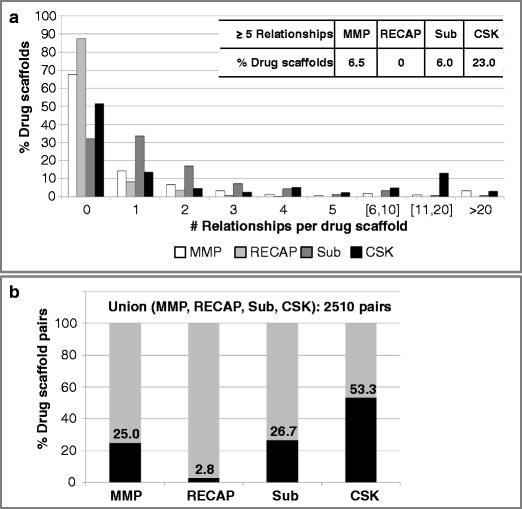
Distribution of structural relationships. a For each type of structural relationships, the percentage of drug scaffolds forming increasing numbers of relationships is reported. In addition, for each type of structural relationships, the proportion of drug scaffolds involved in at least five relationships is given in a table insert. b Different types of structural relationships were combined by determining the union of pairs involved in these relationships (given at the top). For each type, the proportion (black bar) is shown for drug scaffolds
Furthermore, scaffold pairs displayed only limited overlap in structural relationships. Therefore, the union of scaffold pairs involved in different structural relationships was determined, as reported in Fig. 3b and Figure S2 of the Supporting Information for drug scaffolds and bioactive scaffolds, respectively. A total of 2510 and 1,150,457 unique pairs were found for drug scaffolds and bioactive scaffolds, respectively. Different trends were observed. For drug scaffolds, ~53.3% of pairs (or structural relationships) established CSK equivalences and approximately one fourth of pairs represented MMP or substructure relationships. By contrast, for bioactive scaffolds, the largest number of structural relationships resulted from MMP relationships, followed by CSK equivalences, but only less than 10% of pairs formed substructure relationships.
The observation that many bioactive scaffolds display more structural relationships than drug scaffolds can be rationalized in light of the point made above concerning the intellectual property-related issue of structural novelty that generally applies to drugs. At the same time, drug candidates and drugs typically represent end points of an intense chemical optimization efforts and it is not unusual that there are many structural analogs of approved synthetic small molecular drugs available, at least some of which might ultimately find their way into the pool of bioactive compounds and form scaffold relationships, which further supports the presence of differences in the degree of structural relatedness between bioactive compounds and drugs.
Promiscuity of Structurally Related Scaffolds
For scaffolds forming structural relationships, we also compared the degree of scaffold-based promiscuity. Fig. 4a and Figure S3 of the Supporting Information give the percentage of pairs of scaffolds forming different types of structural relationships that displayed increasingly large differences in promiscuity for drug scaffolds and bioactive scaffolds, respectively. Depending on the types of structural relationships, scaffolds having the same degree of promiscuity were found in ~3 to ~6% of drug scaffold pairs and ~24 to ~66% of bioactive scaffold pairs. Differences in the degree of promiscuity of more than 10 targets were observed for drug scaffolds forming ~36 to ~60% of structural relationships but only for bioactive scaffolds forming ~2 to ~36% of the relationships. Therefore, most of the structurally related drug scaffolds displayed large differences in promiscuity, whereas the majority of structurally related bioactive scaffolds had the same or a similar degree of promiscuity. These significant differences can be rationalized by considering the global distribution of promiscuity across these scaffold sets. Of all 779 drug scaffolds, 87 scaffolds (~11.2%) were only annotated with a single target. On average, a drug scaffold was associated with 9.4 targets. However, for bioactive scaffolds, 33,886 (~65.2%) scaffolds represented compounds known to be active against a single target. In this case, the average degree of promiscuity was only 1.8 targets per bioactive scaffold. Therefore, pairs of bioactive scaffolds were much more likely to display the same or a similar degree of promiscuity than pairs of drug scaffolds. Drug candidates or drugs are often more intensely profiled than many bioactive compounds, which is likely to influence detectable promiscuity rates. However, drugs have consistently been shown to have on average higher intrinsic promiscuity than compounds at earlier stages of the drug discovery pathway (32), which cannot only be attributed to differences in assay or profiling frequency (32). Low promiscuity rates have also been established for screening compounds tested in many different assays (33).
Fig. 4.

Promiscuity and activity profile comparison of structurally related drug scaffolds. For drug scaffolds, the number of scaffold pairs forming different types of structural relationships and a having increasing differences in the degree of scaffold-based promiscuity (∆Promiscuity), and b having identical, distinct, or overlapping activity profiles is reported
It should also be noted that ChEMBL and other major compound repositories do not contain any records of inactivity or information concerning the number of assays compounds have been tested in. This information is usually not provided in scientific literature sources. This sets promiscuity analysis across different target families apart from profiling experiments focusing on a limited number of related targets. Clearly, the absence of an activity annotation for a bioactive compound from ChEMBL does not imply that the compound has been tested and found to be inactive. The absence of negative data gives rise to the well-appreciated issue of data sparseness (31,34). This also means that assay frequency cannot be directly accounted for in compound or scaffold promiscuity analysis. One limited exception is provided by the analysis of screening libraries used for publicly available screening campaigns in PubChem (33) that does not, however, contain explicit drug information. From PubChem data, assay frequency of screening compounds has previously been extracted and related to activity records, confirming generally low promiscuity rates for frequently tested screening compounds (33), as referred to above.
Activity Profiles of Structurally Related Scaffolds
Next, we compared the activity profiles of structurally related scaffolds. The distribution of scaffold pairs displaying identical, distinct, or overlapping activity profiles is reported in Fig. 4b and Figure S4 of the Supporting Information for drug scaffolds and bioactive scaffolds, respectively. For drug scaffolds, 0 to ~0.2% of scaffold pairs had identical, ~20 to ~60% distinct, and ~40 to ~80% overlapping activity profiles. Figure 5 shows an exemplary drug scaffold that formed different types of structural relationships with others. These scaffolds had overlapping or distinct activity profiles. Compared to drug scaffolds, a larger proportion of bioactive scaffold pairs shared the same activity profile (~7 to ~57%) or distinct profiles (~23 to ~88%), depending on the types of structural relationships (Figure S4). Therefore, although drug scaffolds often displayed significant differences in promiscuity, the majority of these scaffolds had overlapping activity profiles. By contrast, bioactive scaffolds having the same or a similar degree of promiscuity were often associated with distinct sets of targets (Figure S4).
Fig. 5.
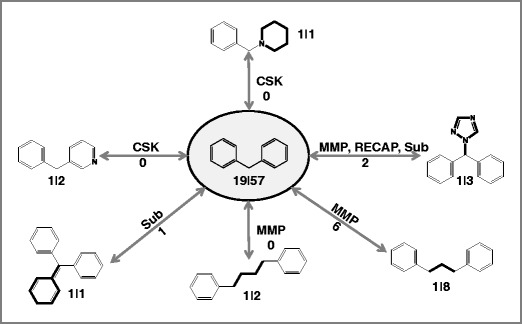
Structurally related drug scaffolds. An exemplary drug scaffold (center) and six related scaffolds are shown that form different structural relationships. For each scaffold, the number of approved drugs and targets is reported. For example, “19|57” indicates that the scaffold represents 19 approved drugs active against a total of 57 targets. Furthermore, for each scaffold pair, the type(s) of their structural relationship(s) and the number of shared targets (number next to arrows) are provided
Drug Scaffolds with Most Structural Relationships
For each type of structural relationships, drug scaffolds forming the largest number of relationships were determined (Fig. 6). In Fig. 6a, 19 drug scaffolds are shown that formed MMPs with 25 or more other drug scaffolds. Interestingly, most of these scaffolds contained two terminal benzene rings that were connected by linkers of different length and/or heteroatom content. Hence, linker fragments mostly distinguished these scaffolds chemically from others.
Fig. 6.
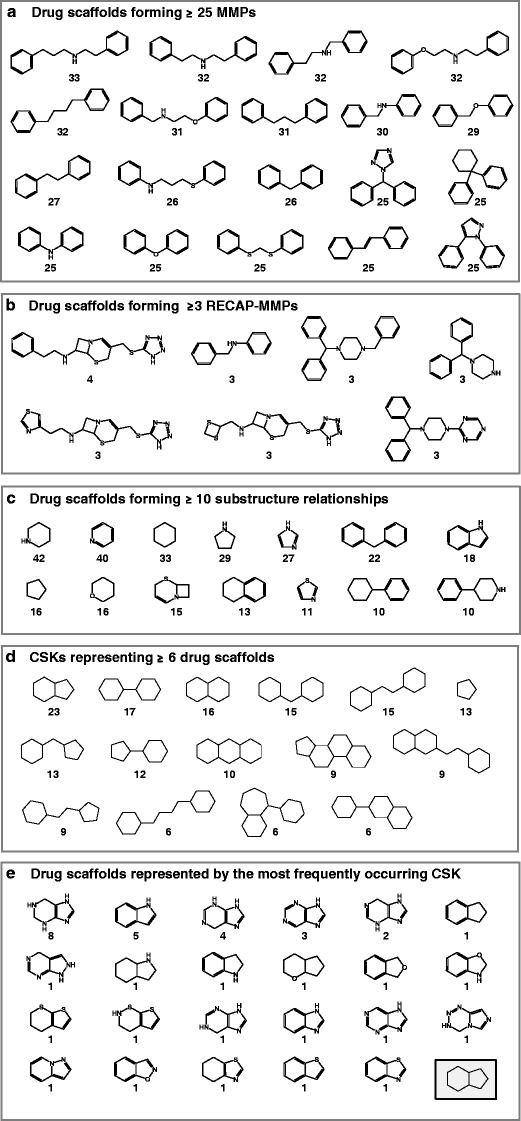
Drug scaffolds with largest numbers of structural relationships. Shown are drug scaffolds that formed at least a 25 MMPs, b three RECAP-MMPs, and c 10 substructure relationships. For each drug scaffold, the number of corresponding structural relationships is provided. d 15 CSKs are shown that represented more than five drug scaffolds. For each CSK, the number of drug scaffolds is reported. e all 23 scaffolds are shown that were represented by hydrindane (bottom right on a gray background), the CSK covering most drug scaffolds. For each of these scaffolds, the number of approved drugs it represented is given
Compared to standard MMPs, only a relatively small number of drug scaffold pairs were identified that formed retrosynthetic MMPs. In fact, most retrosynthetically related drug scaffolds only formed a single RECAP-MMP. Figure 6b shows those seven drug scaffolds that were involved in three or maximally four RECAP-MMPs. These scaffolds contained several amines and/or thioethers and were in part structurally very similar (forming two subsets of analogous scaffolds).
In Fig. 6c, 14 drug scaffolds are shown that formed most (10 or more) substructure relationships. These scaffolds were structurally generic and often consisted of only one five-membered and/or one six-membered ring.
Figure 6d shows 15 CSKs each representing more than five drug scaffolds. Hence, these CSKs covered subsets of topologically equivalent drug scaffolds with largest heteroatom and/or bond order variation. The CSKs displayed varying degrees of structural complexity, ranging from one five-membered ring to structures comprising four fused ring systems. The CSK representing most drug scaffolds was hydrindane. All 23 drug scaffolds covered by hydrindane are shown in Fig. 6e. These topologically equivalent scaffolds represented varying numbers of approved drugs. For example, indole was detected in five drugs, whereas benzimidazole (with only one more nitrogen atom) only represented one drug. Only one scaffold was contained in more than five drugs. Eighteen of these 23 scaffolds only represented a single drug. Hence, many drugs contained unique scaffolds, rendering them chemically distinct from others.
Preferred Chemical Transformations
In order to further investigate the origins of structural modifications between scaffolds forming MMPs, as exemplified in Fig. 6a, all 517 unique chemical transformations derived from 627 drug scaffold MMPs were analyzed. A total of 21 transformations were found in three or more MMPs, as shown in Fig. 7. This small subset of transformations represented the most frequently occurring chemical changes between drug scaffolds. Surprisingly, only four transformations involved the addition of a ring, while most transformations resulted from structural modifications of linker fragments including, for example, the extension of the linker by one or more (carbon, nitrogen, or oxygen) atoms or heteroatom replacements. Therefore, many drug scaffolds consisted of the same ring(s) connected by different linkers, a straightforward approach to render scaffolds chemically distinct.
Fig. 7.
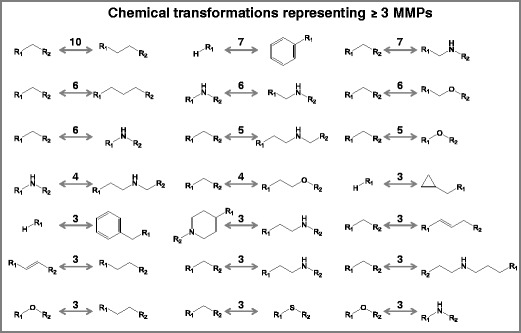
Most frequently occurring chemical transformations. Shown are 21 transformations that represented three or more drug scaffold-based MMPs. For each transformation, the number of corresponding MMPs is provided
Consensus Activity Profiles
In order to explore whether drugs represented by the same scaffold might display similar biological activity, we also generated consensus activity profiles for individual drug scaffolds, as described above and illustrated in Fig. 2. From consensus activity profiles, mean RDF values were calculated for all 159 drug scaffolds that represented multiple drugs. Scaffolds representing drugs with differential activity against multiple targets yield low mean RDF values (close to 0). By contrast, scaffolds representing drugs having little target variation yield high mean RDF values (close to 1). Therefore, mean RDF values serve as a measure to quantitatively evaluate the activity similarity among drugs represented by the same scaffold.
The distribution of mean RDF values is reported in Fig. 8a. The values covered a wide range from 0.02 to 1. The majority of mean RDF values ranged from 0.2 to 0.7. A total of 28 drug scaffolds yielded mean RDF values greater than 0.7. An exemplary scaffold with a value of 0.80 is shown in Fig. 8b together with its consensus activity profile. The scaffold represented four drugs with a total of 26 targets. The target profile is provided in Table S1 of the Supporting Information. All four drugs were active against 14 of these targets, and three drugs were active against six targets. Only three targets were associated with a single drug. In Fig. 8c, a scaffold with a low mean RDF value of 0.33 is shown, which was contained in six drugs active against 19 targets, as listed in Table S2 of the Supporting Information. Only one target was found that all six drugs were active against (target 7). The majority of targets (14 of 19) were only associated with a single drug represented by the given scaffold.
Fig. 8.
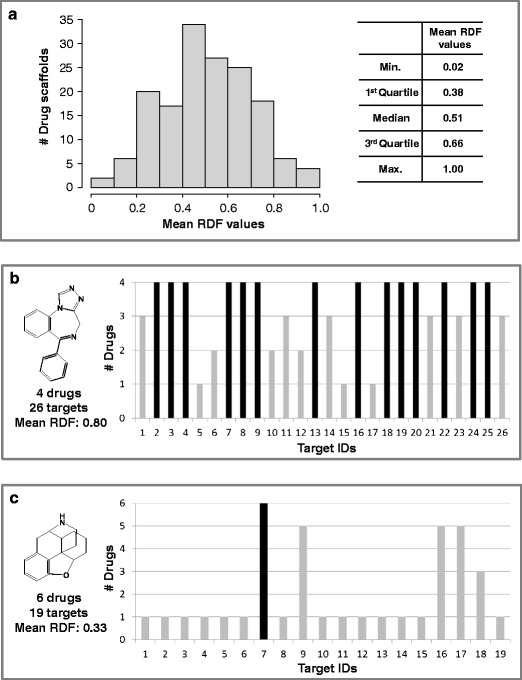
Mean RDF values for drug scaffolds. a Shown is the distribution of mean RDF values for 159 drug scaffolds that represented multiple drugs. The minimum, 1st quartile, median, 3rd quartile, and maximum mean RDF values are reported in a table insert. In addition, two representative drug scaffolds with b high and c low mean RDF values and the corresponding consensus activity profiles (represented according to Fig. 2) are shown. For each scaffold, the number of drugs it represented, the number of associated targets, and the mean RDF value are reported. The target profiles of these two scaffolds are provided in Table S1 and S2 of the Supporting Information
The selection of drug scaffold consensus activity profiles on the basis of associated mean RDF values and their comparison makes it possible to explore activity similarity among structurally related drugs represented by a given scaffold. For example, scaffolds representing drugs with high activity similarity can be readily identified. Furthermore, given sparsely populated consensus activity profiles, potential targets can be suggested for associated drugs.
The systematic exploration of compound-scaffold-CSK and MMP relationships in combination with activity profile analysis is comprehensive and yields all possible structure-activity relationships for further consideration. As such, this analysis goes far beyond substructure or similarity searching that is hypothesis driven (i.e., structural similarity, however assessed, is used as a hypothetical indicator of activity similarity) and limited to individual compound series and their sequential assessment.
CONCLUSIONS
The analysis reported herein has focused on a systematic exploration of structural relationships between drug scaffolds, target annotations of corresponding drugs, activity profiles of drug scaffolds, and activity similarity of structurally related drugs. For the latter purpose, consensus activity profiles and the relative drug frequency measure were introduced. Results obtained for drug scaffolds were compared to scaffolds from bioactive compounds (used as a general reference for drug scaffold properties). In addition to the scaffold concept and the generation of activity profiles, the use of MMP analysis (including standard and retrosynthetic MMPs) for the systematic assessment of structural relationships has been a central aspect of our study. Scaffold mapping and MMP analysis have made frequent contributions to drug discovery, for example, through the identification of novel bioactive scaffolds along structural decomposition pathways (6,7) or the generation of additional target hypotheses for drugs through systematic exploration of MMP relationships with bioactive analogs (35).
In our study, a total of 779 scaffolds were extracted from approved drugs and four different structural relationships were systematically investigated. It was observed that drug scaffolds frequently shared the same topology but varied in heteroatom and/or bond order composition. In addition, specific substructure relationships were frequently detected. By contrast, only a small number of drug scaffolds were found to be involved in retrosynthetic relationships. Moreover, the majority of structurally related drug scaffolds displayed notable differences in promiscuity but had often overlapping sets of targets.
From the analysis of structural relationships, promiscuity, and activity profiles, three major differences between drug scaffolds and bioactive scaffolds emerged. Firstly, structural relationships were formed at higher rates between bioactive scaffolds than drug scaffolds. Secondly, drug scaffolds and bioactive scaffolds displayed different preferences for types of structural relationships. For example, the majority of drug scaffold pairs were formed by topologically equivalent scaffolds. Thirdly, structurally related bioactive scaffolds displayed much lower differences in the degree of promiscuity than drug scaffolds.
Drug scaffolds most frequently involved in different types of structural relationships were identified and characteristic structural features became apparent. For example, many drug scaffolds involved in matched molecular pair relationships were found to contain the same set of ring systems and only differed by the composition of linker fragments between rings. Recurrent chemical transformations producing these features were identified. Furthermore, CSKs covering more than five drug scaffolds represented the most frequent drug topologies.
To assess the activity similarity of structurally related drugs represented by the same scaffold, consensus activity profiles were generated and mean RDF values calculated. On the basis of this analysis, scaffolds representing drugs active against distinct targets were systematically distinguished from drugs having similar target profiles. On the basis of our analysis, consensus activity profiles are indicated to be useful tools to explore and numerically quantify the activity patterns of structurally related drugs and their differences. From consensus activity profiles, additional drug targets can be predicted.
The collection of drug scaffolds, their structural organization and associated target information, activity profiles, and subsets of drug scaffolds with distinct activity profiles is made freely available as an open access ZENODO deposition (36).
Electronic Supplementary Material
(DOC 168 kb)
REFERENCES
- 1.Hu Y, Stumpfe D, Bajorath J. Lessons learned from molecular scaffold analysis. J Chem Inf Model. 2011;51(8):1742–53. doi: 10.1021/ci200179y. [DOI] [PubMed] [Google Scholar]
- 2.Brown N, Jacoby E. On scaffolds and hopping in medicinal chemistry. Mini-Rev Med Chem. 2006;6(11):1217–29. doi: 10.2174/138955706778742768. [DOI] [PubMed] [Google Scholar]
- 3.Bemis GW, Murcko MA. The properties of known drugs. 1. Molecular frameworks. J Med Chem. 1996;39(15):2887–93. doi: 10.1021/jm9602928. [DOI] [PubMed] [Google Scholar]
- 4.Müller G. Medicinal chemistry of target family-directed masterkeys. Drug Discov Today. 2003;8(15):681–91. doi: 10.1016/S1359-6446(03)02781-8. [DOI] [PubMed] [Google Scholar]
- 5.Sutherland JJ, Higgs RE, Watson I, Vieth M. Chemical fragments as foundations for understanding target space and activity prediction. J Med Chem. 2008;51(9):2689–700. doi: 10.1021/jm701399f. [DOI] [PubMed] [Google Scholar]
- 6.Schuffenhauer A, Ertl P, Roggo S, Wetzel S, Koch MA, Waldmann H. The scaffold tree—visualization of the scaffold universe by hierarchical scaffold classification. J Chem Inf Model. 2007;47(1):47–58. doi: 10.1021/ci600338x. [DOI] [PubMed] [Google Scholar]
- 7.Renner S, Van Otterlo WAL, Seoane MD, Möcklinghoff S, Hoffmann B, Wetzel S, et al. Bioactivity-guided mapping and navigation of chemical space. Nat Chem Biol. 2009;5(8):585–92. doi: 10.1038/nchembio.188. [DOI] [PubMed] [Google Scholar]
- 8.Hu Y, Bajorath J. Scaffold distributions in bioactive molecules, clinical trials compounds, and drugs. ChemMedChem. 2010;5(2):187–90. doi: 10.1002/cmdc.200900419. [DOI] [PubMed] [Google Scholar]
- 9.Hu Y, Bajorath J. Rationalizing structure and target relationships between current drugs. AAPS J. 2012;14(4):764–71. doi: 10.1208/s12248-012-9392-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang J, Hou T. Drug and drug candidate building block analysis. J Chem Inf Model. 2010;50(1):55–67. doi: 10.1021/ci900398f. [DOI] [PubMed] [Google Scholar]
- 11.Taylor RD, MacCoss M, Lawson ADG. Rings in drugs. J Med Chem. 2014;57(14):5845–59. doi: 10.1021/jm4017625. [DOI] [PubMed] [Google Scholar]
- 12.Comprehensive Medicinal Chemistry (CMC) database: http://accelrys.com/products/databases/bioactivity/comprehensive-medicinal-chemistry.html.
- 13.Law V, Knox C, Djoumbou Y, Jewison T, Guo AC, Liu Y, et al. DrugBank 4.0: shedding new light on drug metabolism. Nucleic Acids Res. 2014;42(Database issue):D1091–7. doi: 10.1093/nar/gkt1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.DrugBank: http://www.drugbank.ca.
- 15.World Drug Index (WDI): http://thomsonreuters.com/world-drug-index/.
- 16.FDA Orange Book: http://www.fda.gov/Drugs/InformationOnDrugs/ucm129662.htm.
- 17.Hu Y, Bajorath J. Many drugs contain unique scaffolds with varying structural relationships to scaffolds of currently available bioactive compounds. Eur J Med Chem. 2014;76:427–34. doi: 10.1016/j.ejmech.2014.02.040. [DOI] [PubMed] [Google Scholar]
- 18.Bento AP, Gaulton A, Hersey A, Bellis LJ, Chambers J, Davies M, et al. The ChEMBL bioactivity database: an update. Nucleic Acids Res. 2014;42(Database issue):D1083–90. doi: 10.1093/nar/gkt1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hu Y, Bajorath J. Influence of search parameters and criteria on compound selection, promiscuity, and pan assay interference characteristics. J Chem Inf Model. 2014;54(11):3056–66. doi: 10.1021/ci5005509. [DOI] [PubMed] [Google Scholar]
- 20.UniProt Consortium The Universal Protein Resource (UniProt) in 2010. Nucleic Acids Res. 2010;38(Database issue):D142–8. doi: 10.1093/nar/gkp846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hu Y, Bajorath J. How promiscuous are pharmaceutically relevant compounds? A data-driven assessment. AAPS J. 2013;15(1):104–11. doi: 10.1208/s12248-012-9421-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xu YJ, Johnson M. Using molecular equivalence numbers to visually explore structural features that distinguish chemical libraries. J Med Chem. 2002;42(4):912–26. doi: 10.1021/ci025535l. [DOI] [PubMed] [Google Scholar]
- 23.Kenny PW, Sadowski J. Structure modification in chemical databases. In: Oprea TI, editor. Chemoinformatics in drug discovery. Weinheim: Wiley-VCH; 2004. pp. 271–85. [Google Scholar]
- 24.Hussain J, Rea C. Computationally efficient algorithm to identify matched molecular pairs (MMPs) in large data sets. J Chem Inf Model. 2010;50(3):339–48. doi: 10.1021/ci900450m. [DOI] [PubMed] [Google Scholar]
- 25.Hu X, Hu Y, Vogt M, Stumpfe D, Bajorath J. MMP-cliffs: systematic identification of activity cliffs on the basis of matched molecular pairs. J Chem Inf Model. 2012;52(5):1138–45. doi: 10.1021/ci3001138. [DOI] [PubMed] [Google Scholar]
- 26.OEChem, version 1.7.7, OpenEye Scientific Software, Inc., Santa Fe, NM, USA. 2012. http://www.eyesopen.com.
- 27.Lewell XQ, Judd DB, Watson SP, Hann MM. RECAP—retrosynthetic combinatorial analysis procedure: a powerful new technique for identifying privileged molecular fragments with useful applications in combinatorial chemistry. J Chem Inf Comput Sci. 1998;38(3):511–22. doi: 10.1021/ci970429i. [DOI] [PubMed] [Google Scholar]
- 28.de la Vega de León A, Bajorath J. Matched molecular pairs derived by retrosynthetic fragmentation. Med Chem Commun. 2014;5:64–7. doi: 10.1039/C3MD00259D. [DOI] [Google Scholar]
- 29.Cases M, Mestres J. A chemogenomic approach to drug discovery: focus on cardiovascular diseases. Drug Discov Today. 2009;14(9–10):479–85. doi: 10.1016/j.drudis.2009.02.010. [DOI] [PubMed] [Google Scholar]
- 30.Ertl P. Intuitive ordering of scaffolds and scaffold similarity searching using scaffold keys. J Chem Inf Model. 2014;54(6):1617–22. doi: 10.1021/ci5001983. [DOI] [PubMed] [Google Scholar]
- 31.Hu Y, Bajorath J. Compound promiscuity—what can we learn from current data. Drug Discov Today. 2013;18(13–14):644–50. doi: 10.1016/j.drudis.2013.03.002. [DOI] [PubMed] [Google Scholar]
- 32.Hu Y, Bajorath J. High-resolution view of compound promiscuity. F1000Res. 2013;2:144. doi: 10.12688/f1000research.2-144.v1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hu Y, Bajorath J. What is the likelihood of an active Compound to be promiscuous? Systematic assessment of compound promiscuity on the basis of PubChem confirmatory bioassay data. AAPS J. 2013;15(3):808–15. doi: 10.1208/s12248-013-9488-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mestres J, Gregori-Puigjane E, Valverde S, Sole RV. Data completeness—the achilles heel of drug-target networks. Nat Biotechnol. 2008;26(9):983–4. doi: 10.1038/nbt0908-983. [DOI] [PubMed] [Google Scholar]
- 35.Hu Y, Lounkine E, Bajorath J. Many approved drugs have bioactive analogs with different target annotations. AAPS J. 2014;16(4):847–59. doi: 10.1208/s12248-014-9621-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hu Y, Bajorath J. Drug scaffolds and their structural relationships. ZENODO. 2014; 10.5281/zenodo.14947.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(DOC 168 kb)