

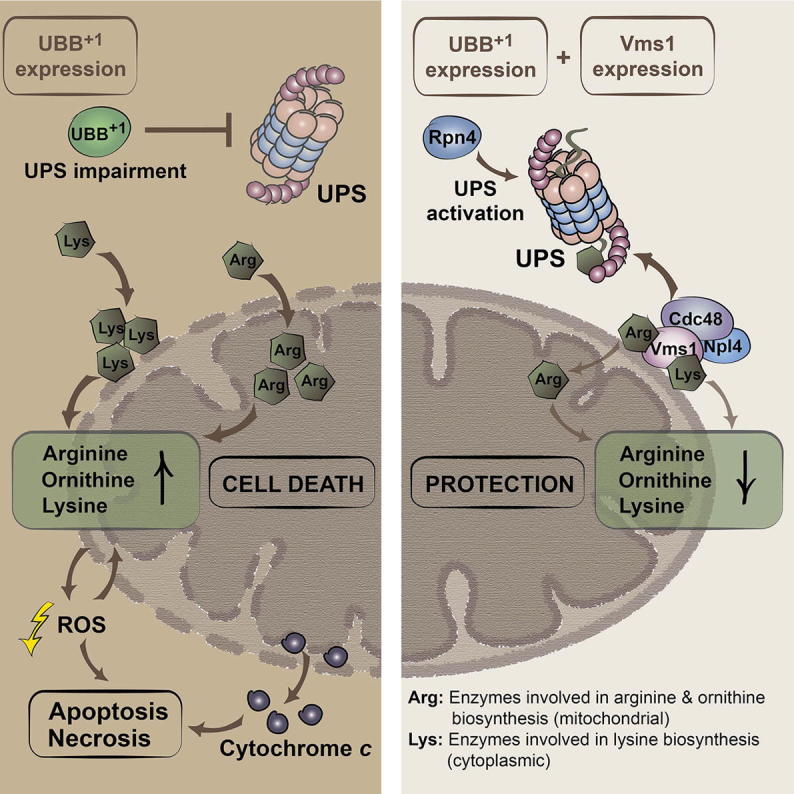
Summary
Neuronal accumulation of UBB+1, a frameshift variant of ubiquitin B, is a hallmark of Alzheimer’s disease (AD). How UBB+1 contributes to neuronal dysfunction remains elusive. Here, we show that in brain regions of AD patients with neurofibrillary tangles UBB+1 co-exists with VMS1, the mitochondrion-specific component of the ubiquitin-proteasome system (UPS). Expression of UBB+1 in yeast disturbs the UPS, leading to mitochondrial stress and apoptosis. Inhibiting UPS activity exacerbates while stimulating UPS by the transcription activator Rpn4 reduces UBB+1-triggered cytotoxicity. High levels of the Rpn4 target protein Cdc48 and its cofactor Vms1 are sufficient to relieve programmed cell death. We identified the UBB+1-induced enhancement of the basic amino acids arginine, ornithine, and lysine at mitochondria as a decisive toxic event, which can be reversed by Cdc48/Vms1-mediated proteolysis. The fact that AD-induced cellular dysfunctions can be avoided by UPS activity at mitochondria has potentially far-reaching pathophysiological implications.
Graphical Abstract
Highlights
-
•
UBB+1 co-exists with the UPS component VMS1 in neurofibrillary tangles
-
•
UBB+1 accumulation impairs the UPS and mitochondria, triggering cell death
-
•
UBB+1 causes accumulation of basic amino acids at mitochondria
-
•
Vms1 reverts UBB+1-triggered basic amino acid accumulation and cell death
Braun et al. demonstrate that basic amino acid accumulation at mitochondria is a decisive toxic event upon cellular accumulation of UBB+1, an Alzheimer’s-disease-associated ubiquitin variant. Triggering the mitochondrion-specific branch of the ubiquitin-proteasome system is sufficient to prevent UBB+1-triggered cytotoxicity, which has potentially far-reaching pathophysiological implications.
Introduction
UBB+1, a loss-of-function variant of ubiquitin B (UBB), accumulates in neurofibrillary tangles, a pathological hallmark in Alzheimer’s disease (AD) (van Leeuwen et al., 1998). UBB+1 is translated from an aberrant mRNA encoding a +1 frameshift protein in which the C-terminal glycine residue required for ubiquitylation is replaced by an extension of 20 amino acids (Dennissen et al., 2010). The detrimental impact of UBB+1 has been studied in neuronal cell cultures, transgenic mice, and yeast (De Vrij et al., 2001; Fischer et al., 2009; Tank and True, 2009). UBB+1 is a substrate for truncation, ubiquitylation, and proteasomal degradation (Dennissen et al., 2011; Lindsten et al., 2002; van Tijn et al., 2007, 2010). Whereas the ubiquitin-proteasome system (UPS) can assure the degradation of low levels of UBB+1, higher levels impair the UPS and subvert the homeostatic mechanisms allowing for its elimination (Fischer et al., 2009; Lindsten et al., 2002; van Tijn et al., 2007, 2010). At high levels, UBB+1 affects mitochondrial dynamics and triggers neuronal cell death (De Vrij et al., 2001; Tan et al., 2007) through as-yet elusive mechanisms.
Yeast is an established model for studying programmed cell death mechanisms that are often shared with animal cells, including the contribution of caspases and mitochondrion-associated cell death proteins, such as cytochrome c (Carmona-Gutierrez et al., 2010). Yeast models have been used to explore cell killing by neurotoxic proteins, such as Parkinson-disease-associated α-synuclein, and the outcome could be successfully translated to fly, worm, and murine disease models, as well as to human disease (Braun et al., 2010; Büttner et al., 2013).
Driven by these premises, we established a yeast cell death model for UBB+1-triggered neurotoxicity. Our findings revealed that UBB+1 interfered with the UPS and triggered the perturbation of the mitochondrion-associated basic amino acid synthesis executing cell death. The mitochondrion-associated UPS subroutine, depending on the AAA-ATPase Cdc48 and its co-factor Vms1, strongly antagonized UBB+1 cytotoxicity. Since VMS1, the human homolog of yeast Vms1, co-exists with UBB+1 in neurofibrillary tangles, these data imply a potential pivotal role of the UPS at mitochondria in AD.
Results
Expression of Human UBB+1 in Yeast Recapitulates Hallmarks of UBB+1 in Neurons
To investigate whether the introduction of UBB+1 into yeast recapitulates hallmarks of UBB+1 accumulation in neurons, we expressed monomeric ubiquitin B (UBB), UBB+1, as well as an UBB+1 variant lacking two lysine residues (K29,48R) that are important for its ubiquitylation. When expressing UBB, we detected a discrete immunoreactive band at the size of monomeric ubiquitin (9 kDa), and an immunoreactive smear across a wide range of the immunoblot that corresponds to ubiquitylated proteins (Figure 1A). This smear was not detectable upon transformation with UBB+1 or UBB+1-K29,48R, reflecting their loss of function. Instead, UBB+1 or UBB+1-K29,48R were detectable as 12 and 9 kDa protein species (full-length and truncated UBB+1; fl-UBB+1 and tUBB+1) that accumulated over time (Figures 1A, S1A, and S1B). In cells expressing UBB+1, a faint higher molecular weight species corresponding to the size of monoubiquitylated fl-UBB+1 (21 kDa) appeared (Figure 1A, FLAG long exposure, asterisks). Consistent with a role of lysines 29 and/or 48 in the ubiquitylation of UBB+1, this band was absent in cells expressing UBB+1-K29,48R. These results suggest that in yeast human UBB (but not UBB+1) can serve as a substrate for ubiquitin ligases and that, like in neurons, UBB+1 is ubiquitylated and truncated.
Figure 1.

Expression of UBB+1 in Yeast and Its Effect on UPS Activity
(A) Proteins were expressed for 1, 2, or 3 days and determined by immunoblotting of cell extracts using antibodies directed against the N-terminal FLAG-tag, or the specific C terminus of UBB+1. Hexokinase (Hxk) was used as loading control. #, unspecific protein band; fl-UBB+1, full-length UBB+1; tUBB+1, truncated UBB+1.
(B) The level of polyubiquitylated proteins and of UBB+1 in cell extracts was determined by immunoblotting using an antibody directed against ubiquitin. ∗Uncharacterized ubiquitin variant.
(C) Quantification of (B). The levels of polyubiquitylated proteins of cells transformed with vector controls were set to 100% in every experiment.
(D) Cellular level of ubiquitin-G76V-GFP upon UBB+1 expression. GFP fluorescence (relative fluorescence units, RFUs) was normalized to optical densities (OD600).
Data: percentage change values (C) and mean values (D), respectively. Error bars: SE. p values: ∗p < 0.05, ∗∗p < 0.01. See Table S1 and Figure S1.
Next, we investigated whether UBB+1 expression results in UPS impairment by means of three complementary assays: (1) the measurement of polyubiquitylated endogenous proteins by immunoblot; (2) the assessment of the abundance of transgenic ubiquitin-G76V-GFP, which is a substrate of the ubiquitin-fusion degradation pathway; and (3) an enzymatic assay designed to quantify the chymotrypsin-like proteasomal activity. Cells expressing UBB+1 or UBB+1-K29,48R contained a higher level of polyubiquitylated proteins than cells transformed with vector controls (Figures 1B and 1C), suggesting decreased UPS-dependent protein turnover. The steady-state levels of ubiquitin-G76V-GFP were significantly increased upon expression of UBB+1 or UBB+1-K29,48R (Figure 1D). In contrast, UBB+1 or UBB+1-K29,48R expression did not reduce chymotrypsin-like proteasomal activities (Figure S1C). These data suggest that, in yeast like in neurons, UBB+1 expression impairs the UPS. However, in yeast UBB+1 does neither directly affect the enzymatic activity of proteasomes, nor is its ubiquitylation essential for UPS dysfunction.
UBB+1 Triggers Oxidative Stress and Programmed Cell Death upon Protracted Expression
To assess its effects on the fitness of proliferating cells, we performed growth assays on agar plates and in liquid cultures. As a positive control of cytotoxicity, TDP-43, a causal factor for motor neuron degeneration, was expressed. In sharp contrast with TDP-43, UBB+1 and UBB+1-K29,48R failed to compromise the growth of cells on agar plates (Figure 2A), and in liquid cultures (Figure 2B), suggesting that UBB+1 is unable to kill proliferating cells.
Figure 2.

Cytotoxicity and Cell Death upon UBB+1 Expression
(A) Growth on solid media. Cultures were spotted in serial dilutions onto solid media inducing or repressing expression.
(B) Growth in liquid media. Left, growth curves. Right, cell densities during stationary phase.
(C) Yeast cells were evaluated for clonogenicity (colony forming units [CFUs]) at the indicated time points after inducing expression.
(D) Oxidative stress levels (DHE staining) were measured using a fluorescence plate reader at the indicated time points after inducing expression.
(E and F) Apoptosis and necrosis. (E) 2 days after inducing expression, yeast cells were measured for (early) apoptosis (Annexin V+/PI−), necrosis (Annexin V−/PI+), and (late) apoptosis/secondary necrosis (Annexin V+/PI+). (F) TUNEL-positive cells are referred to be apoptotic.
Data: mean values (B–F). Error bars: SD (B), and SE (C–F), respectively. p values: ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001. See Table S1 and Figure S2.
Next, we studied the effects of UBB+1 or UBB+1-K29,48R on chronologically aged cultures. For this, the proportion of viable cells capable of forming a colony (clonogenicity) on nutrient-containing solid medium was studied at different time points following UBB+1 or UBB+1-K29,48R expression. Consistent with the growth assays, 16 hr (day 1) after UBB+1 or UBB+1-K29,48R expression cells exhibited a similar clonogenic potential as did cells expressing vector controls (Figure 2C). In contrast, we observed a 10% and 25% decrease in clonogenic cell survival when expressing UBB+1 for 2 and 3 days, respectively. Exogenously applied stressors, including acetate and hydrogen peroxide, further enhanced the cytotoxicity of prolonged UBB+1 expression (Figures S2A and S2B, left). Upon both chronological aging and stress experiments, UBB+1-K29,48R turned out to be slightly less cytotoxic as compared to UBB+1 (Figures 2C, S2A, and S2B, left), suggesting that ubiquitylated UBB+1 is slightly more cytotoxic than UBB+1.
We next examined whether the UBB+1-induced loss of clonogenicity correlated with the manifestation of oxidative stress, which can be detected by the intracellular conversion of the reactive oxygen species (ROS)-sensitive stain dihydroethidium (DHE) to fluorescent ethidium. We observed indistinguishable low levels of oxidative stress after expressing UBB+1 or UBB+1-K29,48R for 16 hr (day 1) (Figure 2D). At later time points, the levels of oxidative stress progressively increased in all cultures with chronological aging, and UBB+1-expressing cells exhibited a mild but significant increase in oxidative stress as compared to vector controls. Thus, upon UBB+1 expression increased markers of oxidative stress coincided with decreased clonogenic cell survival (cf. Figures 2C, 2D, and S2C). When combined with protracted UBB+1 expression, acetate or hydrogen peroxide exacerbated the signs of oxidative stress (Figures S2A and S2B, right). As shown for clonogenic survival, UBB+1-K29,48R demonstrated slightly decreased levels of oxidative stress upon chronological aging or exogenously applied stress as compared to UBB+1 (Figures 2D, S2A, and S2B).
To determine the mode of cell death triggered by the expression of UBB+1 or UBB+1-K29,48R, we performed double staining with Annexin V-FITC and propidium iodide (PI). Annexin V-FITC labels externalized phosphatidylserine that appears on the surface of apoptotic cells, whereas PI is a vital dye that stains cells that have lost plasma membrane integrity during necrosis. Two days after UBB+1 expression the frequencies of early apoptotic (Annexin V-FITC+ PI−), late apoptotic or secondary necrotic (Annexin V-FITC+ PI+), and necrotic cells (Annexin V-FITC− PI+) were increased, as compared with vector controls (Figures 2E and S2D). Apoptosis induction by UBB+1 could be confirmed by the terminal deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL) that detects fragmentation of nuclear DNA (Figures 2F and S2E). Consistent with the results obtained from the clonogenic survival and oxidative stress experiments, UBB+1-K29,48R triggered cell death in a lower number of cells as compared to UBB+1 (Figures 2E and 2F). Altogether, these results indicate that the protracted expression of UBB+1 can induce apoptotic and necrotic killing of yeast cells, and that ubiquitylated UBB+1 is a slightly better killer than UBB+1.
The UPS Capacity and the Ratio of Mutant to Wild-Type Ubiquitin Determine UBB+1-Triggered Cytotoxicity
To investigate the putative contribution of dysfunctional UPS to UBB+1-triggered cytotoxicity, we measured the cytotoxic potential of UBB+1 in the context of enhanced or suppressed UPS. Since full knockout of genes coding for proteasomal subunits is lethal, yeast strains bearing point mutations in one or two proteasomal genes were employed (Heinemeyer et al., 1993). The chymotrypsin-like proteasomal activity was reduced in strains carrying mutant alleles in the proteasomal subunits Pre1 and Pre2 by >88% (Figure 3A). In these conditions of close-to-complete proteasomal inactivation, significantly reduced clonogenic cell survival was only observed in the pre1-1 and the pre1-1/pre2-2 strains upon UBB+1 expression for day 1, and in the pre1-1/pre2-2 strain upon UBB+1 expression for day 2 (Figures 3B and S3A), as compared to wild-type strain. One explanation for the increased UBB+1-triggered cytotoxicity would be that UBB+1 accumulates in these strains due to impaired UBB+1 degradation. However, we could not observe increased steady-state levels of UBB+1 in these strains (neither fl-UBB+1, nor tUBB+1, nor ubiquitylated fl-UBB+1) (Figures S3D–S3F; data not shown). Thus, although severe proteasomal inactivation can increase UBB+1-triggered cell death, there is no strict correlation between the loss of proteasomal capacity on the one hand, and the increase in UBB+1-triggered cytotoxicity or the increase in the steady-state levels of UBB+1 on the other hand.
Figure 3.

UBB+1-Triggered Cytotoxicity in Yeast Strains with Various UPS Capacities
(A) Cultures were grown in logarithmic phase in YPD at 30°C and chymotrypsin-like activities were determined in proteasomal mutant strains. The relative luminescence units (RLUs) obtained using wild-type cells were set to 100% in every experiment.
(B) Clonogenicity in proteasomal mutant strains 1 day after inducing expression. The CFUs obtained using cells expressing vector controls were set to 100% in every experiment.
(C) Cultures were grown in logarithmic phase in YPD at 30°C and chymotrypsin-like activities were determined in UPS knockout strains. The RLUs obtained using wild-type cells were set to 100% in every experiment.
(D) Clonogenicity in UPS knockout strains 2 days after inducing expression following acetate treatment. The CFUs obtained using cells expressing vector controls were set to 100% in every experiment.
(E) Cultures were grown in logarithmic phase in defined medium inducing expression of the transcription activator RPN4, and chymotrypsin-like proteasomal activities were determined. The RLUs obtained using cells carrying vector controls were set to 100% in every experiment.
(F) Clonogenicity of UBB+1-expressing cultures in strains with endogenous (vector control) and elevated levels of Rpn4 (Rpn4), respectively. Clonogenicity was determined 1 day after inducing expression followed by acetate treatment. The CFUs obtained using cells with endogenous and elevated levels of Rpn4, respectively, but lacking UBB+1, were set to 100% in every experiment (not shown).
Data: percentage change values. Error bars: SE. p values: xp < 0.1, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001. See Table S1 and Figure S3.
Next, we measured UBB+1-induced cytotoxicity in knockout strains lacking selective UPS genes, including (1) UBI4 encoding ubiquitin (Finley et al., 1987), (2) RPN4 encoding a major transcriptional UPS activator (Mannhaupt et al., 1999), (3) UBR2 encoding the E3 ligase responsible for Rpn4 degradation (Kruegel et al., 2011), (4) YUH1 encoding the ubiquitin protease that cleaves fl-UBB+1 into tUBB+1 (Dennissen et al., 2011), and (5) UBP6 encoding a deubiquitinase, which can be inhibited by extended ubiquitin proteins (Krutauz et al., 2014). Only RPN4 deletion significantly impaired the chymotrypsin-like proteasomal capacity of the cells (Figure 3C). Notably, upon comparable fl-UBB+1 steady-state levels (Figures S3G and S3I), UBB+1-triggered cytotoxicity was significantly increased in Δubi4 as compared to Δrpn4 upon both stressed und unstressed conditions (Figures 3D and S3B), although the proteasomal capacity was lower in Δrpn4 as compared to Δubi4 (Figure 3C). These data propose that the ratio of mutant ubiquitin (UBB+1) to wild-type ubiquitin (encoded by UBI4) is more relevant for determining UBB+1-triggered cytotoxicity than the proteasomal capacity.
Upon stressed conditions, UBB+1-triggered cytotoxicity was markedly increased in Δyuh1 as compared to wild-type cells upon comparable fl-UBB+1 steady-state levels (Figures 3D, S3B, S3H, and S3I). These data suggest that UBB+1 truncation is a putative protective event, for instance, as part of a mechanism to degrade excessive UBB+1. In Δubp6 as compared to wild-type cells UBB+1-triggered cytotoxicity was unaltered upon comparable fl-UBB+1 steady-state levels (Figures 3D, S3B, S3G, and S3I), suggesting that Ubp6 activity is not protective against the accumulation of the extended ubiquitin UBB+1.
UBB+1-triggered cytotoxicity was significantly relieved in Δubr2 cells upon stressed conditions (Figures 3D and S3B), in which Rpn4 is stabilized and consequently the UPS activity is increased (Kruegel et al., 2011). Consistently, Rpn4 expression, which also leads to increased UPS activities (Figure 3E), was protective for UBB+1-expressing wild-type cells (Figures 3F and S3C) but not for cells lacking UBI4 (Figure S3C). In both cases, the protective effect cannot be explained by decreased steady-state levels of UBB+1 (Figures S3G and S3I–S3K). These data show that increasing UPS capacity is protective for UBB+1-expressing cells, but not by affecting the turnover of UBB+1 itself but rather by interrupting the lethal signaling cascade triggered by UBB+1.
UBB+1 Causes Lethal Mitochondrial Dysfunction
Oxidative stress and mitochondrial impairment are hallmarks of neurotoxin-elicited death in yeast and neurons (Braun, 2012; Debattisti and Scorrano, 2013). Therefore, we analyzed whether oxidative stress, which occurred starting by day 2 of UBB+1 expression (Figure 4A), correlated with mitochondrial impairment. Two days after inducing UBB+1 expression, the mitochondrial network was fragmented in both UBB+1-expressing cells, as well as in cells carrying vector controls (data not shown), which is typical for stationary phase cultures. However, after shifting these cultures to fresh growth medium (which represses UBB+1 expression) the recovery of the mitochondrial network was significantly compromised in cultures transformed with UBB+1-encoding constructs as compared with vector controls (Figures 4B and S4A). These data suggest that mitochondrial and oxidative stresses coincide in cells expressing UBB+1.
Figure 4.

Pivotal Mitochondrial Impairment upon UBB+1 Expression
(A) Oxidative stress levels were measured by flow cytometry 2 days after inducing expression.
(B) Mitochondrial fragmentation. UBB+1 and RFP fused with a mitochondrial targeting sequence were expressed. 2 days after induction, cultures were shifted to fresh media repressing expression, and after 3 hr the proportion of cells with fragmented mitochondria was quantified.
(C–E) Cellular oxygen consumption (C), mitochondrial membrane potential (D), and cellular ATP levels (E) were determined 2 and 3 days after inducing UBB+1 expression. The oxygen consumption (C), mitochondrial membrane potential (D), and ATP levels (E) measured using cells carrying vector controls were set to 100% in every experiment.
(F and G) Protein alterations in crude mitochondria. UBB+1 was expressed for 24 hr and crude mitochondria were isolated by differential centrifugation. (F) Immunoblot demonstrating the steady-state levels of Rip1, cytochrome c (Cyt. c), and the mitochondrial outer membrane protein Por1 as loading control. (G) Quantification of (F). The immunoreactive signals obtained using cells carrying vector controls were set to 100% in every strain and experiment.
(H and I) UBB+1-triggered cytotoxicity in strains deleted from genes encoding mitochondrial cell death (H), and ER-associated proteins (I), respectively. Clonogenicity was determined 2 days after inducing expression followed by acetate treatment. The CFUs obtained using cells carrying vector controls were set to 100% in every experiment.
Data: mean values (A and B), and percentage change values (C–E, G–I), respectively. Error bars: SE. p values: ∗p ≤ 0.05, ∗∗p < 0.01. See Table S1 and Figure S4.
We further tested for mitochondrial impairment by measuring the cellular oxygen consumption, the mitochondrial membrane potential, and the ATP levels in cells expressing UBB+1 for days 2 and 3. Whereas the cellular oxygen consumption and mitochondrial membrane potential were significantly increased by day 3 in (surviving) cells expressing UBB+1 (Figures 4C and 4D), the cellular ATP levels were significantly decreased by days 2 and 3 (Figure 4E). These data hint at hyperactive mitochondria, which are incapable to prevent a metabolic crisis in UBB+1-expressing cells.
In yeast, alterations in the cytochrome bc1 complex of the mitochondrial respiratory chain may contribute to the loss of respiratory capacity and the production of lethal ROS (Diaz et al., 2012; Eisenberg et al., 2007). For instance, loss of the Rieske iron-sulfur protein Rip1, a key component of the cytochrome bc1 complex, results in increased ROS generation and mitochondrial dysfunction (Diaz et al., 2012). Indeed, the cellular level of Rip1 was markedly decreased by days 2 and 3 upon UBB+1 expression as compared with vector controls (Figures S4B and S4C). Consistently, Rip1 and cytochrome c were depleted in the mitochondrial fraction of UBB+1-expressing cells (Figures 4F and 4G). These data further hint at a major UBB+1-induced mitochondrial dysfunction, in which the respiratory chain is impaired (depletion of Rip1 and cytochrome c), leading to the production of ROS (for which cellular oxygen is needed), and the decline of cellular ATP levels.
Hyperpolarization of mitochondria may precede mitochondrion-dependent yeast death (Eisenberg et al., 2007); therefore, we expressed UBB+1 in strains deleted for genes encoding a range of mitochondrial cell death proteins, including the yeast BH3-only protein (Ybh3) that translocates to mitochondria to mediate their permeabilization, and several potentially cytotoxic proteins that can be released from mitochondria such as apoptosis-inducing factor 1 (Aif1), endonuclease G (Nuc1), and the two cytochrome c isoforms (Cyc1, Cyc7). Deletion of NUC1 resulted in a paradoxical increase in UBB+1-triggered cytotoxicity, and loss of Ybh3 did not have any effect upon both stressed and unstressed conditions (Figures 4H and S4D). In contrast, UBB+1-mediated cytotoxicity was significantly decreased in strains depleted from isoform 2 of cytochrome c (Δcyc7) upon stressed conditions (Figures 4H and S4D). The steady-state levels of UBB+1 were not decreased in the Δcyc7 as compared to wild-type strain (Figure S4F), and this strain maintained a normal state of respiratory competence (presumably due to the presence of the cytochrome c isoform 1 Cyc1) (Figure S4I), excluding trivial explanations for the cytoprotective action of Δcyc7. Thus, our data suggest the implication of mitochondria in UBB+1-triggered cell death.
Next, we tested for a possible role of the unfolded protein response (UPR) and the ER in UBB+1-triggered cytotoxicity and expressed UBB+1 for 2 days in cells lacking the UPR kinase Ire1 and its downstream target Hac1, as well as in cells lacking the ER cell death protease Kex1 (which executes cell death in which mitochondria play a pivotal role [Hauptmann and Lehle, 2008]). Upon stress, UBB+1-triggered cytotoxicity was relieved in Δire1 and Δkex1 but not in Δhac1 cells (Figures 4I and S4E), under conditions where the steady-state levels of UBB+1 were comparable (Figures S4G and S4H). These data suggest for an implication of the ER in UBB+1-triggered cytotoxicity, but, due to the lack of rescue in the Δhac1 cells, a critical involvement of the UPR is unlikely.
Perturbation of Basic Amino Acid Synthesis at Mitochondria Is a Decisive Toxic Event upon UBB+1 Accumulation
Next, we performed quantitative proteomic analyses of crude mitochondria after “stable isotope labeling by amino acids in cell culture” (SILAC). This approach led to the identification of 16 proteins whose abundance was significantly altered (increased for ten or decreased for six proteins) upon UBB+1 expression (Figure 5A; Table S2). Among the proteins with established mitochondrial localization, three were enzymes participating in amino acid metabolism, namely, Put1 (involved in proline degradation), Arg5,6, and Arg8 (involved in arginine and ornithine biosynthesis). In addition, UBB+1 induced the accumulation of the cytosolic enzyme Lys1 (involved in lysine biosynthesis), an increase in the motor protein Myo3 and the (putative) peroxisomal proteins Gpd1 and Str3, in crude mitochondria. Upon acetate stress, deletion of the ARG5,6, ARG8, and the LYS1 genes restored the clonogenic potential of UBB+1-expressing cells, whereas the deletion of all other genes had no effect (Figures 5B and S5A). These data point to a hitherto unexpected involvement of the biosynthesis of basic amino acids (arginine, ornithine, and lysine) in UBB+1-triggered cytotoxicity.
Figure 5.

Perturbation of Basic Amino Acid Synthesis upon UBB+1 Expression
(A) Protein alterations in crude mitochondria were quantified by SILAC in two independent experiments. Changes are shown that were significant in both experiments.
(B) UBB+1-triggered cytotoxicity in strains deleted from genes encoding proteins accumulating in crude mitochondria upon UBB+1 expression. Clonogenicity was determined 2 days after inducing expression followed by acetate treatment. The CFUs obtained using cells carrying vector controls were set to 100% in every experiment.
(C) Basic amino acids were isolated from cultures expressing UBB+1 or vector controls, respectively. The mean values of amino acids from cells carrying vector controls were set to 1.0 for every amino acid.
(D) Arginine and ornithine biosynthetic pathway in S. cerevisiae. Green: deletion of genes encoding these enzymes does significantly prevent (green) and does not prevent (red) from UBB+1-triggered oxidative stress, respectively (see E and Figure S5B); Black: no data.
(E) Oxidative stress in strains with disrupted arginine/ornithine biosynthesis was measured 2 days after inducing UBB+1 expression followed by acetate treatment. The oxidative stress levels obtained using cells carrying vector controls were set to 100% in every experiment.
(F) Cell death in strains with disrupted arginine/ornithine biosynthesis and increased levels of arginine in the growth media was measured 2 days after inducing UBB+1 expression. The proportion of dead cells carrying vector controls was set to 100% in every experiment.
(G) Cell death in strains with disrupted lysine biosynthesis and increased levels of lysine in the growth media was measured 2 days after inducing UBB+1 expression. The proportion of dead cells carrying vector controls was set to 100% in every experiment.
Data: percentage change values (B and E–G) and mean values (C), respectively. Error bars: SE. p values: xp < 0.1, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001. See Tables S1, S2, and S4 and Figure S5.
To challenge this hypothesis, we measured the cellular steady-state levels of arginine, ornithine, and lysine in cultures expressing UBB+1 (Figure 5C). Indeed, we observed a marked increase in the cellular levels of all three basic amino acids, in particular, ornithine, upon UBB+1 accumulation. To weigh the contribution of arginine and ornithine (as opposed to their metabolic intermediates) to UBB+1 cytotoxicity, we measured ROS production upon UBB+1 expression in strains depleted from the arginine and ornithine biosynthetic enzymes (Figure 5D). Depletion of all enzymes operating upstream of cytosolic ornithine (Arg2, Arg5,6, Arg7, and Ort1) significantly relieved UBB+1-triggered cytotoxicity both in unstressed and acetate-stressed conditions (Figures 5E and S5B). In contrast, none of the enzymes downstream of cytosolic ornithine (Arg3, Arg1, and Arg4, which are needed for the conversion of ornithine into arginine) were required for the cytotoxic action of UBB+1. Notably, all tested enzymes operating upstream of cytosolic ornithine are mitochondrion-associated (Ljungdahl and Daignan-Fornier, 2012). Therefore, we concluded that UBB+1 triggers the mitochondrion-associated biosynthesis of ornithine, leading to increased cytosolic levels of ornithine (and its product arginine). This plays a decisive role in executing UBB+1-triggered cell death.
If this model is true, increasing cytosolic levels of either ornithine or arginine (which can easily be interconverted into each other) should recover the cytotoxic effect of UBB+1 in strains with interrupted mitochondrion-associated biosynthesis of ornithine. Therefore, we measured UBB+1-triggered cell death in the strain depleted for the mitochondrial protein Ort1 in growth media with increasing concentrations of arginine and ornithine, respectively. It turned out that Δort1 cells were not able to efficiently uptake ornithine from the growth media, because the severe growth deficit of the Δort1 strain in growth media lacking arginine could not be relieved by increasing concentrations of ornithine in the growth media (data not shown). In contrast, Δort1 cells grew well in the presence of arginine in growth media lacking ornithine (data not shown), demonstrating the efficient cellular uptake of arginine. As expected, yeast cells lacking Ort1 were protected from UBB+1-triggered cell death upon moderate concentrations of arginine (30 and 50 mg/l) in the growth media (Figure 5F). In contrast, elevated concentrations of arginine in the growth media (150 and 300 mg/l) recovered the cytotoxic effect of UBB+1 (Figure 5F), substantiating the decisive role of increased cellular levels of arginine (and cytosolic ornithine) in executing UBB+1-triggered cell death.
In order to address the role of cellular levels of lysine, we measured UBB+1-triggered cell death in the strain depleted from Lys1 in growth media with increasing concentrations of lysine. Whereas deletion of LYS1 relieved UBB+1-triggered cytotoxicity as compared to wild-type strain (Figure 5B), increasing the lysine concentrations did not promote cytotoxicity in the Δlys1 strain (Figure 5G). Thus, in contrast to arginine/ornithine the cellular lysine level appears to be negligible in accelerating UBB+1-triggered cell death.
Cdc48/Vms1-Stimulated Mitochondrial UPS Protects from UBB+1-Triggered Cytotoxicity
The aforementioned data incriminate mitochondria and the UPS in the execution of UBB+1-triggered cytotoxicity, notably because of the protective impact of the removal of mitochondrial enzymes involved in basic amino acid synthesis and the overexpression of the transcriptional UPS activator Rpn4. Among the known Rpn4 targets are the conserved AAA-ATPase Cdc48 and its cofactor Npl4 (Bosis et al., 2010). Cdc48 and Npl4 are involved in the UPS, and determined by their cofactor Vms1, regulate mitochondrion-associated protein degradation (Heo et al., 2010). Driven by these premises, we evaluated the involvement of the Cdc48/Vms1/Npl4-dependent UPS pathway to UBB+1-triggered cytotoxicity. For this, we measured UBB+1-triggered cytotoxicity in normal and acetate-stressed conditions in strains expressing increased levels of wild-type Cdc48 or the pro-apoptotic Cdc48-S565G variant (Madeo et al., 1997), which is characterized by decreased Vms1 binding and mitochondrion-associated degradation (Heo et al., 2010). We also determined the cytotoxicity of UBB+1 in strains depleted from the Cdc48 cofactors Npl4 and Vms1, or overexpressing Vms1. UBB+1-triggered cytotoxicity was markedly attenuated in cultures expressing increased levels of wild-type Cdc48, as compared to cells expressing Cdc48-S565G or controls with endogenous Cdc48 only (Figures 6A and S6A). UBB+1-triggered cytotoxicity was significantly increased in cultures depleted from Npl4 under non-stressed conditions (Figures 6B and S6B). Depletion of Vms1 resulted in a marked elevation in cytotoxicity upon stress (Figures 6C and S6C), while overexpression of Vms1 significantly protected against UBB+1 upon acetate stress, as measured by the clonogenic approach (Figures 6D and S6D). High levels of Vms1 also protected from cell death and oxidative stress induced by UBB+1 expression (Figures 6E and 6F). Notably, high amounts of Cdc48 and Cdc48-S565G resulted in markedly decreased steady-state levels of UBB+1 (Figures S6E and S6F), whereas neither the deletion of VMS1, nor its overexpression had an effect on the cellular UBB+1 amounts (Figures S6G–S6J). These data point to a protective role of Vms1, which is independent from UBB+1 degradation, potentially by improving the quality control at mitochondria. In contrast, the beneficial role of high amounts of Cdc48 could be due to both increased Vms1-independent UBB+1 degradation and improved Vms1-dependent mitochondrial quality control.
Figure 6.

Role of Cdc48/Npl4/Vms1 Complex in UBB+1-Triggered Cytotoxicity
(A–C) UBB+1 was expressed in strains with elevated levels of Cdc48 or Cdc48-S565G (A) and strains deleted for NPL4 (B) and VMS1 (C). Clonogenicity was determined 2 days after inducing expression before (B) and after acetate stress (A and C). The CFUs obtained using cells carrying vector controls were set to 100% in every experiment.
(D) Clonogenicity of UBB+1-expressing cultures in strains with endogenous (vector control) and elevated levels of Vms1 (Vms1), respectively. Clonogenicity was determined 2 days after inducing expression followed by acetate treatment. The CFUs obtained using cells with endogenous and elevated levels of Vms1, respectively, but lacking UBB+1, were set to 100% in every experiment (not shown).
(E and F) Cell death and oxidative stress was measured 1, 2, and 3 days after inducing expression of UBB+1 and/or Vms1.
(G–I) UBB+1 was expressed in wild-type strain with endogenous (vector ctrl) and increased levels of Vms1 (Vms1). Cellular oxygen consumption (G), mitochondrial membrane potential (H), and cellular ATP levels (I) were determined 2 and 3 days after inducing expression. The oxygen consumption (G), mitochondrial membrane potential (H), and ATP levels (I) measured using cells with endogenous Vms1 were set to 100% in every experiment.
(J) Protein alterations in crude mitochondria upon Vms1 expression. Mitochondria were isolated from cultures expressing UBB+1 in cells with increased or endogenous levels of Vms1, respectively. Protein alterations were quantified by SILAC in two independent experiments. Changes were shown that were significant in both experiments. Blue-labeled proteins are inversely regulated as compared to Figure 5A.
(K) Cellular levels of basic amino acids upon Vms1 expression. Basic amino acids were isolated from cultures expressing UBB+1 in cells with endogenous (vector control) or increased levels of Vms1 (Vms1), respectively. The mean values of amino acids from cells with endogenous Vms1 levels were set to 1.0 for every amino acid.
Data: percentage change values (A–D, G–I) and mean values (E, F, K), respectively. Error bars: SE. p values: xp < 0.1, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001. See Tables S1, S3, and S4 and Figure S6.
In order to address whether elevated Vms1 levels prevent from UBB+1-triggered mitochondrial impairment, we measured the cellular oxygen consumption, the mitochondrial membrane potential, and the cellular ATP levels in cells expressing UBB+1 upon endogenous or elevated amounts of Vms1 (Figures 6G–6I). Whereas the cellular oxygen consumption and the mitochondrial membrane potential were significantly decreased by day 3 and days 2 and 3, respectively, cellular ATP levels were significantly increased by day 2 upon high amounts of Vms1. In other words, high amounts of Vms1 reverted the mitochondrial damage induced by high levels of UBB+1 (see Figures 4C–4E).
In a next step, we used SILAC technology to comparatively assess alterations of the mitochondrial proteome between UBB+1-expressing cells with endogenous and high levels of Vms1 (Figure 6J; Table S3). We observed that among the 16 proteins whose abundance levels were altered by UBB+1 as compared with the vector control (Figure 5A; Table S2), ten were no more altered upon expression of both UBB+1 and Vms1 (Figure 6J, blue-labeled proteins). Among these ten proteins, which were particularly stringently associated with the cytopathic activity of UBB+1, the basic amino acid synthesis enzymes Arg5,6, Arg8, and Lys1 were significantly decreased in UBB+1-expressing cells upon high levels of Vms1, as compared to endogenous Vms1 levels. Consistently, Vms1 overexpression blunted the UBB+1-mediated increase in the steady-state levels of arginine, ornithine, and lysine (Figure 6K, see Figure 5C). These data point to a pivotal role of the Vms1-dependent mitochondrial UPS activity in avoiding the UBB+1-triggered lethal overproduction of basic amino acids.
VMS1 Co-exists with tau and UBB+1 in Hippocampal Neurons from AD Patients
The hippocampus is severely affected during AD progression. Pathological hallmarks include intracellular neurofibrillary tangles comprising aberrant forms of the microtubule-associated protein tau, UBB+1, and the mitochondrial outer membrane voltage-dependent anion channel 1 (VDAC1) (Reddy, 2013; van Leeuwen et al., 1998). Immunohistochemistry revealed expression of VMS1, the human homolog of yeast Vms1, in pyramidal cells within the hippocampi from AD patients and aged non-demented controls (Figure 7A, all arrows; Tables S5 and S6). VMS1 stained structures reminiscent of tau pathology, including tangle-like (yellow arrows) and neuropil thread-like structures (blue arrows), as well as other cellular staining patterns (green arrows), were observed in samples from AD patients, and aged non-demented controls with tau pathology. We also observed these tangle-like and thread-like staining patterns when analyzing the sections for aberrant tau, UBB+1, and VDAC1 (Figure S7). Immunohistochemistry of consecutive paraffin sections from the hippocampi of AD patients (Figure 7B) confirmed the identification of pyramidal cells with intracellular tangle-like structures, which co-stained for aberrant tau and VMS1 (Figure 7C, violet arrows), and for UBB+1, VMS1, and VDAC1 (Figure 7D, orange arrows). These data suggest that VMS1 is a component of neurofibrillary tangles comprising aberrant tau, UBB+1, and VDAC1, underscoring a potential role of the Cdc48/VMS1 complex in UBB+1-mediated AD progression.
Figure 7.

VMS1 Co-existence with Aberrant tau, UBB+1, or VDAC1 in Hippocampi of AD Patients
(A) VMS1 staining in AD patient and non-demented control.
(B) Principle of the staining of consecutive sections from the hippocampus of an AD patient shown in (C) and (D).
(C) Co-existence of aberrant tau (MC1) and VMS1.
(D) Co-existence of UBB+1 (Ubi2a), VMS1, and VDAC1.
Discussion
We established a yeast model for dissecting cell death mechanisms triggered by UBB+1, in which mitochondria play a pivotal role in the execution of cell death (see the Supplemental Discussion). UBB+1 triggers neuronal apoptosis accompanied by reduced mitochondrial movement (Tan et al., 2007), and mitochondrial impairment likely contributes to AD (Rodolfo et al., 2010). Thus, our yeast model corroborates features of cell-death-relevant mitochondrion dysfunctions found in AD neurons.
Yeast strains expressing UBB+1 accumulated the basic amino acids arginine, ornithine, and lysine. Deletion of mitochondrion-associated enzymes involved in their synthesis abolished UBB+1-triggered cell killing, which could be recovered by increasing concentrations of arginine in the growth media. The accumulation of basic amino acids may trigger mitochondrial damage and cell death in mammalian cells and in yeast (Almeida et al., 2007; Biczó et al., 2011). For instance, increased production of nitric oxide from arginine executes yeast apoptosis (Almeida et al., 2007) and increased levels of polyamines, which are produced from ornithine, may impair neuronal ion channel activities (Inoue et al., 2013). The levels of arginine, ornithine, and/or their polyamine products were altered in aged human brains and in brains from AD patients (Inoue et al., 2013; Liu et al., 2014; Rushaidhi et al., 2012). The results of these studies are controversial, and it remains not yet clear whether the observed alterations are cause or consequence of AD. Despite that, our data suggest that perturbed basic amino acid synthesis is a decisive event triggering mitochondrion-dependent cell death upon UBB+1 accumulation in yeast. Further studies aiming at analyzing the role of arginine/ornithine metabolism during aging or AD progression should consider a potential pivotal contribution of UPS and mitochondrial dysfunctions.
We observed that UBB+1 accumulation impaired the UPS, and that the UPS activity, in turn, determined UBB+1 cytotoxicity. Yeast cultures that were depleted from ubiquitin (Δubi4) were highly vulnerable to UBB+1. In contrast, yeast cultures in which the UPS was stimulated by the transcriptional activator Rpn4 were insensitive to UBB+1, but not in cells lacking the ubiquitin gene UBI4. Extended ubiquitin variants have been proposed to be specific inhibitors of the deubiquitinase Ubp6 in yeast (Krutauz et al., 2014). Since the UBB+1-triggered cytotoxicity was unaltered in a strain deleted for UBP6 as compared to wild-type strain, our data suggest that the lethal effect of the extended ubiquitin UBB+1 does not essentially depend on Ubp6. It is tempting to speculate that the ratio of mutant (UBB+1) to wild-type ubiquitin determines UBB+1-triggered cytotoxicity with UBB+1 as a competitive inhibitor of wild-type ubiquitin, affecting numerous ubiquitin-regulated cellular processes.
We established that elevated amounts of Cdc48 or its cofactor Vms1 conferred tolerance against UBB+1 expression. More specifically, Vms1 overexpression relieved the UBB+1-triggered mitochondrial damage and accumulation of the basic amino acids arginine, ornithine, and lysine. The Cdc48/Vms1 complex enables the degradation of mitochondrion-associated proteins (Heo et al., 2010). Whereas under normal conditions, this complex is predominantly cytosolic, Vms1 recruits Cdc48 to the mitochondrial outer membrane upon stress, presumably with the scope of improving the local quality of proteins. Our data suggest that Cdc48/Vms1-mediated processes can prevent the UBB+1-triggered lethal derangement of mitochondria. In one possible scenario, Cdc48/Vms1 might remove protein junk from the mitochondrial outer membrane. Alternatively, Cdc48/Vms1 might specifically prevent the accumulation of arginine, ornithine, and lysine, through regulation of the turnover of the enzymes Arg5,6, Arg8, and Lys1, which are pivotal for their synthesis. Whereas the activity of the cytoplasmic enzyme Lys1 could be regulated by its degradation, the activities of the mitochondrion-associated Arg5,6 and Arg8 could be controlled by preventing their import into mitochondria via ubiquitylation and proteasomal degradation. Lys1, Arg5,6, and Arg8 are known targets for ubiquitylation (Xu et al., 2009), and the UPS regulates the import of mitochondrial intermembrane space proteins (Bragoszewski et al., 2013; Harbauer et al., 2014). It is tempting to speculate for a UPS-dependent regulation of the import of the mitochondrial matrix proteins Arg5,6 and Arg8. Further studies are needed to address the influence of UPS (dys)function on the turnover of these and other mitochondrial proteins. This is important because recent studies demonstrated that UPS dysfunction can lead to mitochondrial dysfunction and vice versa (Livnat-Levanon et al., 2014; Maharjan et al., 2014; Segref et al., 2014), and our data revealed the unexpected link between UBB+1-triggered UPS dysfunction and the accumulation of functional enzymes in the mitochondrial matrix leading to potentially cytotoxic accumulation of basic amino acids.
Human VMS1 and mitochondrial VDAC1 co-existed with UBB+1 in neurofibrillary tangles of AD patients and aged non-demented controls with tau pathology. UBB+1 accumulates and the number of neurofibrillary tangles and damaged mitochondria markedly increase during AD progression (Dennissen et al., 2010; Rodolfo et al., 2010). We propose that VMS1-dependent mitochondrial quality control might retard the AD-associated neuronal dysfunction, which is elicited by the accumulation of both aberrant tau and UBB+1.
Experimental Procedures
Yeast Strains and Growth Conditions
Yeast expression constructs, strains, and growth conditions were described in the Supplemental Experimental Procedures. Gene expression was under the control of galactose-regulated promoters. For stressing cells, cultures were treated for 4 hr with acetate. For stable isotope labeling (SILAC), cells expressing vector controls, UBB+1, or UBB+1 and Vms1 were grown in media supplemented either with Lys0 and Arg0 (normal isotopes), or with Lys4 and Arg6, or with Lys8 and Arg10 (heavy isotopes, Silantes).
Measuring Cytotoxicity Based on Growth and Clonogenicity
Assays were performed as described in the Supplemental Experimental Procedures. Briefly, growth deficits upon expression of proteins of interest on solid or liquid media, as compared to vector controls, suggest for cytotoxic effects of these proteins on (growing) yeast cells. For clonogenic assays, 500 cells from liquid yeast cultures expressing proteins of interest or vector controls, respectively, were plated on agar plates, on which expression is repressed. The number of colonies (colony forming units [CFUs]) formed after 2 days of incubation correlates with the fitness of the culture.
Measurement of Oxidative Stress, Cell Death, Apoptosis, and Necrosis
Oxidative stress was determined by measuring the conversion of dihydroethidium (DHE, Sigma-Aldrich) to the red fluorescent ethidium applying a fluorescence plate reader or a flow cytometer. Cell death was measured by the incorporation of the “vital dye” propidium iodide (PI, Sigma-Aldrich) in cells that have lost their plasma membrane integrity using a flow cytometer. Annexin V/PI co-staining (Annexin V-FLUOS Staining Kit, Roche Applied Science) for discriminating early and late apoptosis, as well as necrosis, and terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) for measuring apoptosis (In Situ Cell Death Detection Kit, Roche Applied Science) were performed by flow cytometry. See the Supplemental Experimental Procedures for details.
Measurement of Cellular Oxygen Consumption, Mitochondrial Membrane Potential, and Cellular ATP Levels
Oxygen consumption of stationary yeast cultures was analyzed using the FireSting optical oxygen sensor system (Pyro Science). The decrease of the oxygen concentration over time in yeast cultures was determined. Mitochondrial membrane potential was assessed with flow cytometry after staining cells with tetramethylrhodamine methyl ester (TMRM, Molecular Probes, Life Technologies), a fluorescent dye that accumulates within mitochondria dependent on their membrane potential. To determine the ATP level of yeast cultures, intracellular metabolites were obtained using hot ethanol extraction. ATP was measured using the ATP Determination Kit (Molecular Probes, Life Technologies). This assay is based on an ATP-dependent reaction of recombinant firefly luciferase, which induces bioluminescence of its substrate D-luciferin and is directly correlated with the ATP content. All data were normalized to the number of living cells within the samples. See the Supplemental Experimental Procedures for details.
Measurement of UPS Activities
For determining the level of polyubiquitylated proteins in cellular extracts, immunoblots of cellular extracts were incubated with an ubiquitin-specific antibody and immunosignals were quantified with ImageJ 1.47 m. For measuring the turnover of UPS substrates, the ubiquitin-fusion protein ubiquitin-G76V-GFP was co-expressed with UBB+1 or vector controls. GFP fluorescence (relative fluorescence units [RFUs]) and optical densities (OD600) were determined using the FLUOstar Omega plate reader. RFU was normalized to OD600, in order to determine the level of ubiquitin-GFP fusion proteins per culture. Measurement of chymotrypsin-like proteasomal activities were performed using the FLUOstar Omega plate reader, applying the luminescence-based Proteasome-Glo Cell-Based Assay (Promega). See the Supplemental Experimental Procedures for details.
Generation of Cell Extracts, SDS-PAGE, and Immunoblot Analyses
Yeast cultures were incubated in expression media (SCGal) for the indicated time points. Cell extracts were generated by pre-treating yeast pellets in NaOH followed by heating in SDS lysis buffer. Protein extracts were separated on Tricine-SDS polyacrylamide gels, transferred on PVDF membranes, and incubated with primary and secondary antibodies coupled to horseradish peroxidase. Immunodetection was done using luminol. Membranes were digitized in an ImageQuant LAS 4000 (GE Healthcare). Images were processed with Adobe Photoshop CS6. Immunoblot quantification was done with the gel analysis method in ImageJ 1.47 m. See the Supplemental Experimental Procedures for details.
Mass Spectrometry
Crude mitochondrial extracts were taken up in SDS lysis buffer, thawed, reduced with DTT, and alkylated using iodoacetamide (Sigma-Aldrich). Protein mixtures were separated by SDS-PAGE using Bis-Tris gels (NuPAGE, Invitrogen). The gel lanes were cut into slices, which were in-gel digested with trypsin (Promega), and the resulting peptide mixtures were processed on STAGE tips. Mass spectrometry was performed on a LTQ Orbitrap XL mass spectrometer (Thermo Fisher Scientific) coupled to an Eksigent NanoLC-ultra. See the Supplemental Experimental Procedures for details.
Metabolomics
For extraction of metabolites cultures were harvested by filtration, washed with ddH2O, and quenched in liquid nitrogen. Metabolites were extracted by acid extraction using trichloroacetic acid and by hot ethanol extraction. Extracts obtained from uniformly 13C-labeled yeast cells served as internal standard. Metabolites were determined using ion pair reversed-phase liquid chromatography coupled to negative electro spray high-resolution mass spectrometry (IP-RP-LC/HRMS). LC/MS measurements were normalized to the total number of cells of each sample. See the Supplemental Experimental Procedures for details.
Immunohistochemistry
Experiments with human materials were in accordance with the local ethical committees at the Universities of Bayreuth (Germany) and Maastricht (the Netherlands). Postmortem tissues of hippocampi from AD patients and non-demented controls were obtained from the Netherlands Brain Bank (Table S6) as paraffin sections. For immunohistochemistry, sections were deparaffinated, incubated with primary antibodies against the indicated proteins, and with biotin-coupled secondary antibodies followed by the avidin-biotin-peroxidase complex. Immunodetection was performed by the colorimetric reaction of 3,3′-diaminobenzidine. Sections were dehydrated and coverslipped. See the Supplemental Experimental Procedures for details.
Author Contributions
R.J.B. and F.M. initiated the project; R.J.B., C.S., F.M., T.E., C.M., J.D., and F.W.v.L. designed the experiments; R.J.B., C.S., C.L., R.J.G.G., V.I.D., K.P., T.E., L.H., and G.T. performed the experiments; R.J.B., C.S., C.L., V.I.D., T.E., R.J.G.G., F.W.v.L., K.P., and G.T. analyzed the data; R.J.B., C.S., F.M., and F.W.v.L. prepared figures and tables; R.J.B., F.M., and G.K. wrote the manuscript. See detailed author contributions in the Supplemental Information.
Acknowledgments
We would like to thank Benedikt Westermann for critical reading of the manuscript and Jasmin Großer, Adil Günal, and Daniel Lux for technical support. We are grateful to the Deutsche Forschungsgemeinschaft (DFG) for grant BR 3706/3-1 to R.J.B., to the Federation of European Biochemistry Societies (FEBS) for short-term fellowship to R.J.B., to the Fonds zur Förderung der wissenschaftlichen Forschung (FWF) for grant DKplus Metabolic and Cardiovascular Disease to C.S., L.H., and F.M., for grants LIPOTOX, I1000, P23490-B12, and P24381-B20 to F.M., and to the Internationale Stichting Alzheimer Onderzoek (ISAO) for project 09-514 to F.W.v.L. T.E. is a recipient of an APART fellowship of the Austrian Academy of Sciences at the Institute of Molecular Biosciences, University of Graz. V.I.D. and J.D. are supported by the Excellence Initiative of the German Federal and State Governments through FRIAS and the excellence cluster BIOSS. G.T., C.M., F.S., and T.P. are grateful to the Austrian Federal Ministry for Transport, Innovation and Technology (bmvit) for project Met2Net. G.K. is financed by the Ligue contre le Cancer (équipe labelisée); Agence National de la Recherche (ANR); Association pour la recherche sur le cancer (ARC); Cancéropôle Ile-de-France; Institut National du Cancer (INCa); Fondation Bettencourt-Schueller; Fondation de France; Fondation pour la Recherche Médicale (FRM); the European Commission (ArtForce); the European Research Council (ERC); the LabEx Immuno-Oncology; the SIRIC Stratified Oncology Cell DNA Repair and Tumor Immune Elimination (SOCRATE); the SIRIC Cancer Research and Personalized Medicine (CARPEM); and the Paris Alliance of Cancer Research Institutes (PACRI). This publication was funded by the University of Bayreuth in the funding program Open Access Publishing.
Footnotes
This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/3.0/).
Contributor Information
Ralf J. Braun, Email: ralf.braun@uni-bayreuth.de.
Frank Madeo, Email: frank.madeo@uni-graz.at.
Supplemental Information
References
- Almeida B., Büttner S., Ohlmeier S., Silva A., Mesquita A., Sampaio-Marques B., Osório N.S., Kollau A., Mayer B., Leão C. NO-mediated apoptosis in yeast. J. Cell Sci. 2007;120:3279–3288. doi: 10.1242/jcs.010926. [DOI] [PubMed] [Google Scholar]
- Biczó G., Hegyi P., Dósa S., Shalbuyeva N., Berczi S., Sinervirta R., Hracskó Z., Siska A., Kukor Z., Jármay K. The crucial role of early mitochondrial injury in L-lysine-induced acute pancreatitis. Antioxid. Redox Signal. 2011;15:2669–2681. doi: 10.1089/ars.2011.4065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosis E., Salomon D., Ohayon O., Sivan G., Bar-Nun S., Rabinovich E. Ssz1 restores endoplasmic reticulum-associated protein degradation in cells expressing defective cdc48-ufd1-npl4 complex by upregulating cdc48. Genetics. 2010;184:695–706. doi: 10.1534/genetics.109.111419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bragoszewski P., Gornicka A., Sztolsztener M.E., Chacinska A. The ubiquitin-proteasome system regulates mitochondrial intermembrane space proteins. Mol. Cell. Biol. 2013;33:2136–2148. doi: 10.1128/MCB.01579-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun R.J. Mitochondrion-mediated cell death: dissecting yeast apoptosis for a better understanding of neurodegeneration. Front. Oncol. 2012;2:182. doi: 10.3389/fonc.2012.00182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun R.J., Büttner S., Ring J., Kroemer G., Madeo F. Nervous yeast: modeling neurotoxic cell death. Trends Biochem. Sci. 2010;35:135–144. doi: 10.1016/j.tibs.2009.10.005. [DOI] [PubMed] [Google Scholar]
- Büttner S., Habernig L., Broeskamp F., Ruli D., Vögtle F.N., Vlachos M., Macchi F., Küttner V., Carmona-Gutierrez D., Eisenberg T. Endonuclease G mediates α-synuclein cytotoxicity during Parkinson’s disease. EMBO J. 2013;32:3041–3054. doi: 10.1038/emboj.2013.228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmona-Gutierrez D., Ruckenstuhl C., Bauer M.A., Eisenberg T., Büttner S., Madeo F. Cell death in yeast: growing applications of a dying buddy. Cell Death Differ. 2010;17:733–734. doi: 10.1038/cdd.2010.10. [DOI] [PubMed] [Google Scholar]
- De Vrij F.M., Sluijs J.A., Gregori L., Fischer D.F., Hermens W.T., Goldgaber D., Verhaagen J., Van Leeuwen F.W., Hol E.M. Mutant ubiquitin expressed in Alzheimer’s disease causes neuronal death. FASEB J. 2001;15:2680–2688. doi: 10.1096/fj.01-0438com. [DOI] [PubMed] [Google Scholar]
- Debattisti V., Scorrano L. D. melanogaster, mitochondria and neurodegeneration: small model organism, big discoveries. Mol. Cell. Neurosci. 2013;55:77–86. doi: 10.1016/j.mcn.2012.08.007. [DOI] [PubMed] [Google Scholar]
- Dennissen F.J., Kholod N., Steinbusch H.W., Van Leeuwen F.W. Misframed proteins and neurodegeneration: a novel view on Alzheimer’s and Parkinson’s diseases. Neurodegener. Dis. 2010;7:76–79. doi: 10.1159/000285510. [DOI] [PubMed] [Google Scholar]
- Dennissen F.J., Kholod N., Hermes D.J., Kemmerling N., Steinbusch H.W., Dantuma N.P., van Leeuwen F.W. Mutant ubiquitin (UBB+1) associated with neurodegenerative disorders is hydrolyzed by ubiquitin C-terminal hydrolase L3 (UCH-L3) FEBS Lett. 2011;585:2568–2574. doi: 10.1016/j.febslet.2011.06.037. [DOI] [PubMed] [Google Scholar]
- Diaz F., Enríquez J.A., Moraes C.T. Cells lacking Rieske iron-sulfur protein have a reactive oxygen species-associated decrease in respiratory complexes I and IV. Mol. Cell. Biol. 2012;32:415–429. doi: 10.1128/MCB.06051-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenberg T., Büttner S., Kroemer G., Madeo F. The mitochondrial pathway in yeast apoptosis. Apoptosis. 2007;12:1011–1023. doi: 10.1007/s10495-007-0758-0. [DOI] [PubMed] [Google Scholar]
- Finley D., Ozkaynak E., Varshavsky A. The yeast polyubiquitin gene is essential for resistance to high temperatures, starvation, and other stresses. Cell. 1987;48:1035–1046. doi: 10.1016/0092-8674(87)90711-2. [DOI] [PubMed] [Google Scholar]
- Fischer D.F., van Dijk R., van Tijn P., Hobo B., Verhage M.C., van der Schors R.C., Li K.W., van Minnen J., Hol E.M., van Leeuwen F.W. Long-term proteasome dysfunction in the mouse brain by expression of aberrant ubiquitin. Neurobiol. Aging. 2009;30:847–863. doi: 10.1016/j.neurobiolaging.2008.06.009. [DOI] [PubMed] [Google Scholar]
- Harbauer A.B., Zahedi R.P., Sickmann A., Pfanner N., Meisinger C. The protein import machinery of mitochondria-a regulatory hub in metabolism, stress, and disease. Cell Metab. 2014;19:357–372. doi: 10.1016/j.cmet.2014.01.010. [DOI] [PubMed] [Google Scholar]
- Hauptmann P., Lehle L. Kex1 protease is involved in yeast cell death induced by defective N-glycosylation, acetic acid, and chronological aging. J. Biol. Chem. 2008;283:19151–19163. doi: 10.1074/jbc.M801303200. [DOI] [PubMed] [Google Scholar]
- Heinemeyer W., Gruhler A., Möhrle V., Mahé Y., Wolf D.H. PRE2, highly homologous to the human major histocompatibility complex-linked RING10 gene, codes for a yeast proteasome subunit necessary for chrymotryptic activity and degradation of ubiquitinated proteins. J. Biol. Chem. 1993;268:5115–5120. [PubMed] [Google Scholar]
- Heo J.M., Livnat-Levanon N., Taylor E.B., Jones K.T., Dephoure N., Ring J., Xie J., Brodsky J.L., Madeo F., Gygi S.P. A stress-responsive system for mitochondrial protein degradation. Mol. Cell. 2010;40:465–480. doi: 10.1016/j.molcel.2010.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue K., Tsutsui H., Akatsu H., Hashizume Y., Matsukawa N., Yamamoto T., Toyo’oka T. Metabolic profiling of Alzheimer’s disease brains. Sci. Rep. 2013;3:2364. doi: 10.1038/srep02364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruegel U., Robison B., Dange T., Kahlert G., Delaney J.R., Kotireddy S., Tsuchiya M., Tsuchiyama S., Murakami C.J., Schleit J. Elevated proteasome capacity extends replicative lifespan in Saccharomyces cerevisiae. PLoS Genet. 2011;7:e1002253. doi: 10.1371/journal.pgen.1002253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krutauz D., Reis N., Nakasone M.A., Siman P., Zhang D., Kirkpatrick D.S., Gygi S.P., Brik A., Fushman D., Glickman M.H. Extended ubiquitin species are protein-based DUB inhibitors. Nat. Chem. Biol. 2014;10:664–670. doi: 10.1038/nchembio.1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsten K., de Vrij F.M., Verhoef L.G., Fischer D.F., van Leeuwen F.W., Hol E.M., Masucci M.G., Dantuma N.P. Mutant ubiquitin found in neurodegenerative disorders is a ubiquitin fusion degradation substrate that blocks proteasomal degradation. J. Cell Biol. 2002;157:417–427. doi: 10.1083/jcb.200111034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P., Fleete M.S., Jing Y., Collie N.D., Curtis M.A., Waldvogel H.J., Faull R.L., Abraham W.C., Zhang H. Altered arginine metabolism in Alzheimer’s disease brains. Neurobiol. Aging. 2014;35:1992–2003. doi: 10.1016/j.neurobiolaging.2014.03.013. [DOI] [PubMed] [Google Scholar]
- Livnat-Levanon N., Kevei É., Kleifeld O., Krutauz D., Segref A., Rinaldi T., Erpapazoglou Z., Cohen M., Reis N., Hoppe T., Glickman M.H. Reversible 26S proteasome disassembly upon mitochondrial stress. Cell Rep. 2014;7:1371–1380. doi: 10.1016/j.celrep.2014.04.030. [DOI] [PubMed] [Google Scholar]
- Ljungdahl P.O., Daignan-Fornier B. Regulation of amino acid, nucleotide, and phosphate metabolism in Saccharomyces cerevisiae. Genetics. 2012;190:885–929. doi: 10.1534/genetics.111.133306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madeo F., Fröhlich E., Fröhlich K.U. A yeast mutant showing diagnostic markers of early and late apoptosis. J. Cell Biol. 1997;139:729–734. doi: 10.1083/jcb.139.3.729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maharjan S., Oku M., Tsuda M., Hoseki J., Sakai Y. Mitochondrial impairment triggers cytosolic oxidative stress and cell death following proteasome inhibition. Sci. Rep. 2014;4:5896. doi: 10.1038/srep05896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mannhaupt G., Schnall R., Karpov V., Vetter I., Feldmann H. Rpn4p acts as a transcription factor by binding to PACE, a nonamer box found upstream of 26S proteasomal and other genes in yeast. FEBS Lett. 1999;450:27–34. doi: 10.1016/s0014-5793(99)00467-6. [DOI] [PubMed] [Google Scholar]
- Reddy P.H. Is the mitochondrial outermembrane protein VDAC1 therapeutic target for Alzheimer’s disease? Biochim. Biophys. Acta. 2013;1832:67–75. doi: 10.1016/j.bbadis.2012.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodolfo C., Ciccosanti F., Giacomo G.D., Piacentini M., Fimia G.M. Proteomic analysis of mitochondrial dysfunction in neurodegenerative diseases. Expert Rev. Proteomics. 2010;7:519–542. doi: 10.1586/epr.10.43. [DOI] [PubMed] [Google Scholar]
- Rushaidhi M., Jing Y., Kennard J.T., Collie N.D., Williams J.M., Zhang H., Liu P. Aging affects L-arginine and its metabolites in memory-associated brain structures at the tissue and synaptoneurosome levels. Neuroscience. 2012;209:21–31. doi: 10.1016/j.neuroscience.2012.02.021. [DOI] [PubMed] [Google Scholar]
- Segref A., Kevei É., Pokrzywa W., Schmeisser K., Mansfeld J., Livnat-Levanon N., Ensenauer R., Glickman M.H., Ristow M., Hoppe T. Pathogenesis of human mitochondrial diseases is modulated by reduced activity of the ubiquitin/proteasome system. Cell Metab. 2014;19:642–652. doi: 10.1016/j.cmet.2014.01.016. [DOI] [PubMed] [Google Scholar]
- Tan Z., Sun X., Hou F.S., Oh H.W., Hilgenberg L.G., Hol E.M., van Leeuwen F.W., Smith M.A., O’Dowd D.K., Schreiber S.S. Mutant ubiquitin found in Alzheimer’s disease causes neuritic beading of mitochondria in association with neuronal degeneration. Cell Death Differ. 2007;14:1721–1732. doi: 10.1038/sj.cdd.4402180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tank E.M., True H.L. Disease-associated mutant ubiquitin causes proteasomal impairment and enhances the toxicity of protein aggregates. PLoS Genet. 2009;5:e1000382. doi: 10.1371/journal.pgen.1000382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Leeuwen F.W., de Kleijn D.P., van den Hurk H.H., Neubauer A., Sonnemans M.A., Sluijs J.A., Köycü S., Ramdjielal R.D., Salehi A., Martens G.J. Frameshift mutants of beta amyloid precursor protein and ubiquitin-B in Alzheimer’s and Down patients. Science. 1998;279:242–247. doi: 10.1126/science.279.5348.242. [DOI] [PubMed] [Google Scholar]
- van Tijn P., de Vrij F.M., Schuurman K.G., Dantuma N.P., Fischer D.F., van Leeuwen F.W., Hol E.M. Dose-dependent inhibition of proteasome activity by a mutant ubiquitin associated with neurodegenerative disease. J. Cell Sci. 2007;120:1615–1623. doi: 10.1242/jcs.03438. [DOI] [PubMed] [Google Scholar]
- van Tijn P., Verhage M.C., Hobo B., van Leeuwen F.W., Fischer D.F. Low levels of mutant ubiquitin are degraded by the proteasome in vivo. J. Neurosci. Res. 2010;88:2325–2337. doi: 10.1002/jnr.22396. [DOI] [PubMed] [Google Scholar]
- Xu P., Duong D.M., Seyfried N.T., Cheng D., Xie Y., Robert J., Rush J., Hochstrasser M., Finley D., Peng J. Quantitative proteomics reveals the function of unconventional ubiquitin chains in proteasomal degradation. Cell. 2009;137:133–145. doi: 10.1016/j.cell.2009.01.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.