

Murray et al. examine the correspondence between Thal amyloid phase, tau pathology and clinical characteristics in a large Alzheimer’s disease autopsy series. They extrapolate their findings to an autopsy cohort for which Pittsburgh compound-B imaging data are available, and evaluate the neuropathological significance of a quantitative amyloid-β imaging cut-off point.
Keywords: Alzheimer’s disease, neuropathology, Thal amyloid phase, Pittsburgh compound B, Braak tangle stage
Murray et al. examine the correspondence between Thal amyloid phase, tau pathology and clinical characteristics in a large Alzheimer’s disease autopsy series. They extrapolate their findings to an autopsy cohort for which Pittsburgh compound-B imaging data are available, and evaluate the neuropathological significance of a quantitative amyloid-β imaging cut-off point.
Abstract
Thal amyloid phase, which describes the pattern of progressive amyloid-β plaque deposition in Alzheimer’s disease, was incorporated into the latest National Institute of Ageing – Alzheimer’s Association neuropathologic assessment guidelines. Amyloid biomarkers (positron emission tomography and cerebrospinal fluid) were included in clinical diagnostic guidelines for Alzheimer’s disease dementia published by the National Institute of Ageing – Alzheimer’s Association and the International Work group. Our first goal was to evaluate the correspondence of Thal amyloid phase to Braak tangle stage and ante-mortem clinical characteristics in a large autopsy cohort. Second, we examined the relevance of Thal amyloid phase in a prospectively-followed autopsied cohort who underwent ante-mortem 11C-Pittsburgh compound B imaging; using the large autopsy cohort to broaden our perspective of 11C-Pittsburgh compound B results. The Mayo Clinic Jacksonville Brain Bank case series (n = 3618) was selected regardless of ante-mortem clinical diagnosis and neuropathologic co-morbidities, and all assigned Thal amyloid phase and Braak tangle stage using thioflavin-S fluorescent microscopy. 11C-Pittsburgh compound B studies from Mayo Clinic Rochester were available for 35 participants scanned within 2 years of death. Cortical 11C-Pittsburgh compound B values were calculated as a standard uptake value ratio normalized to cerebellum grey/white matter. In the high likelihood Alzheimer’s disease brain bank cohort (n = 1375), cases with lower Thal amyloid phases were older at death, had a lower Braak tangle stage, and were less frequently APOE-ε4 positive. Regression modelling in these Alzheimer’s disease cases, showed that Braak tangle stage, but not Thal amyloid phase predicted age at onset, disease duration, and final Mini-Mental State Examination score. In contrast, Thal amyloid phase, but not Braak tangle stage or cerebral amyloid angiopathy predicted 11C-Pittsburgh compound B standard uptake value ratio. In the 35 cases with ante-mortem amyloid imaging, a transition between Thal amyloid phases 1 to 2 seemed to correspond to 11C-Pittsburgh compound B standard uptake value ratio of 1.4, which when using our pipeline is the cut-off point for detection of clear amyloid-positivity regardless of clinical diagnosis. Alzheimer’s disease cases who were older and were APOE-ε4 negative tended to have lower amyloid phases. Although Thal amyloid phase predicted clinical characteristics of Alzheimer’s disease patients, the pre-mortem clinical status was driven by Braak tangle stage. Thal amyloid phase correlated best with 11C-Pittsburgh compound B values, but not Braak tangle stage or cerebral amyloid angiopathy. The 11C-Pittsburgh compound B cut-off point value of 1.4 was approximately equivalent to a Thal amyloid phase of 1–2.
Introduction
Alzheimer’s disease is a multi-proteinopathy, involving both extracellular amyloid-β deposits (i.e. amyloid-β plaques) and intracellular accumulation of post-translationally modified tau proteins [i.e. neurofibrillary tangles (NFT)], recently reviewed by Murray et al. (2014). Both proteins are considered to develop in a topographic pattern that can be stereotypically assessed using Thal amyloid phase for amyloid-β plaques (Thal et al., 2002) and Braak NFT staging (Braak and Braak, 1991). Braak NFT staging was incorporated in the 1997 National Institute on Ageing (NIA) consensus recommendations for post-mortem diagnosis of Alzheimer’s disease, and is widely validated by clinicopathological studies that demonstrate strong associations with greater severity of cognitive decline (Duyckaerts et al., 1997; Bennett et al., 2004; Sabbagh et al., 2010; Nelson et al., 2012). Thal amyloid phase was recently incorporated into the latest NIA-Alzheimer’s Association’s (NIA-AA) neuropathologic assessment guidelines (Hyman et al., 2012; Montine et al., 2012), which is of particular importance as amyloid biomarkers (e.g. PET and amyloid-β CSF) have been included in NIA-AA clinical diagnostic guidelines for Alzheimer’s disease dementia (Albert et al., 2011; Jack et al., 2011; McKhann et al., 2011).
Ante-mortem imaging with amyloid labelling ligands, such as 11C-Pittsburgh compound B (PiB), is a direct measure of amyloid-β plaque load (Klunk et al., 2004; Bacskai et al., 2007; Johnson et al., 2007; Lockhart et al., 2007; Sojkova et al., 2011; Driscoll et al., 2012; Kantarci et al., 2012b). Since its introduction a decade ago (Klunk et al., 2004), investigative work with PiB-PET imaging continues to rapidly evolve. Amyloid imaging is now incorporated into research diagnostic criteria for Alzheimer’s disease as well as preclinical stages of the disease (Jack et al., 2011; McKhann et al., 2011; Sperling et al., 2011). Given the limits of a decade or less of experience with the technique, however, there are still many unanswered questions about what it is measuring. Cut-off points denoting PiB-positivity are based on quantitative measures of PiB retention (Rowe et al., 2007; Jack et al., 2008). The relationship between the Consortium to Establish a Registry for Alzheimer’s Disease (CERAD) ‘neuritic’ plaque score and ante-mortem PiB-PET imaging shows a range between moderate to high agreement (Sojkova et al., 2011; Driscoll et al., 2012). The CERAD score is a semiquantitative assessment of neuritic and cored plaques (Mirra et al., 1991), both of which have fibrillar forms of amyloid that bind the fluorescent analogue of PiB or radiolabelled PiB (Lockhart et al., 2007; Ikonomovic et al., 2008). CERAD does not capture the severity of diffuse plaques (non-fibrillar amyloid), which are commonly found at autopsy in ageing brains (Nelson et al., 2012). There is evidence, however, that PiB binds to diffuse plaques, as well as cerebral amyloid angiopathy in the vessels and parenchyma (Johnson et al., 2007; Lockhart et al., 2007; Ikonomovic et al., 2008; Niedowicz et al., 2012). Thal amyloid phase does not discriminate among plaque types, but neither does it consider cerebral amyloid angiopathy. Thus, prompting us to investigate neuropathologic measures that drive the association with cortical PiB-PET standard uptake value (SUV) ratios.
Our first goal was to retrospectively classify a large brain bank cohort using recommended criteria for Thal amyloid phase (Thal et al., 2002) to examine the range of quantitative senile plaque pathology and Braak NFT stage (Braak and Braak, 1991). Our second goal was to examine demographic [e.g. gender, education, apolipoprotein E (APOE) genotype] and clinicopathological associations (e.g. age of onset, disease duration) with Thal amyloid phase in a subset of Alzheimer’s disease cases from the brain bank. Our third goal was to examine which Thal amyloid phase corresponds to a PiB-PET SUV ratio cut-off point score of 1.4 in a separate prospectively-followed autopsied cohort with ante-mortem PiB-PET; and lastly to investigate the relevance of Braak NFT stage and severity of cerebral amyloid angiopathy.
Materials and methods
Study samples
The brain bank for neurodegenerative disorders at Mayo Clinic Jacksonville was queried for all cases with cortical amyloid-β plaques that were received on or before 14 November 2013. Of the 4493 available autopsies, 3618 had sufficient information to retrospectively assign a Thal amyloid phase (Thal et al., 2002) (described below). Cases were not excluded for co-existing pathology. The final study cohort included 3618 autopsied individuals (1940 males and 1678 females; age at death 29–105 years) who had a Thal amyloid phase 0 through phase 5. Note that case selection was performed based on neuropathological diagnosis and not ante-mortem clinical diagnosis.
Of the 63 participants with an ante-mortem PiB-PET scan who came to autopsy between May 2008 and June 2014, 35 cases were available for this study (24 males and 11 females; aged 56–95). They were prospectively followed in the Mayo Clinic Alzheimer’s Disease Research Centre or Mayo Clinic Study of Ageing in Rochester and had an ante-mortem PET and MRI within 2 years of death. All patients or their informants signed consent to disclosure of clinical information, neuroimaging, and brain donation with appropriate ethical approval from the Mayo Clinic Institutional Review Board.
Procedures
Ante-mortem clinical history for age at onset of cognitive symptoms, education, and Mini-Mental State Examination (MMSE) score (Folstein et al., 1975) was retrospectively abstracted from clinical reports for the Mayo Clinic Jacksonville series, with investigators blinded to Thal amyloid phase. Time elapsed from age of onset and last MMSE to death was calculated by subtracting the date of onset or MMSE test date, respectively. Time was converted to years by dividing by 365.25. The final MMSE score was recorded if MMSE test date was within 3 years of death.
Upon neuropathologic examination, Mayo Clinic Jacksonville brains were received for evaluation with the left hemibrain formalin-fixed and the right frozen at −80°. Brain weight represents the fixed specimen that was calculated based upon doubling the weight of the available (usually left) hemibrain. A standardized dissection and sampling method was used as described previously (Terry et al., 1987). Tissue samples were processed and embedded in paraffin blocks. Mayo Clinic Rochester brains were sampled and examined according to the CERAD protocol (Mirra et al., 1991). Comparable cortical and subcortical slides were sent to Mayo Clinic Jacksonville for staining and assessment. Senile plaques and NFT were assessed and severity of amyloid angiopathy scored with thioflavin-S fluorescent microscopy, as previously described (Murray et al., 2011). The thioflavin-S staining protocol we used for these studies is sensitive to all senile plaque types (e.g. diffuse, cored, and neuritic) (Dickson et al., 1992), which were each included with a truncated maximum of 50 plaques per 3 mm2 using a ×10 objective (Supplementary Fig. 1). At the time of diagnosis Mayo Clinic Jacksonville brains were assigned a Braak NFT stage using thioflavin-S (Braak and Braak, 1991), with retrospective assessment on Mayo Clinic Rochester brains performed subsequent to thioflavin-S staining. Thal amyloid phase was assigned for Mayo Clinic Jacksonville brains by retrospectively assessing senile plaque quantification of neocortex (i.e. mid-frontal, inferior parietal or superior temporal cortex) and hippocampus using thioflavin-S staining results as supported in the latest NIA-AA recommendations (Hyman et al., 2012; Montine et al., 2012). Neuropathologic reports were abstracted for non-database material on basal ganglia and cerebellum to complete Thal amyloid phase evaluation (Thal et al., 2002). We do not currently evaluate senile plaques in the superior colliculus or substantia nigra, but the CA4 of the hippocampus performs as well if not better (Thal et al., 2002). The maximum Thal amyloid phase was assigned if senile plaques were found in: Phase 1: neocortex; Phase 2: CA1/subiculum of the hippocampus; Phase 3: basal ganglia; Phase 4: CA4 of the hippocampus; and Phase 5: cerebellar molecular layer. Supplementary Fig. 1 illustrates regional assessment of senile plaques on thioflavin-S microscopy for Thal amyloid phase. TAR DNA binding protein 43 (TARDBP, previously known as TDP-43) positivity (Amador-Ortiz et al., 2007) and Lewy body disease type (Uchikado et al., 2006) was assessed using immunohistochemical methods, as previously described. The presence of significant vascular disease was based on Jellinger and Attems (2003) incidence study. Global cerebral amyloid angiopathy was assessed using a semiquantitative severity measure of 0 = none; 1 = mild; 2 = moderate; 3 = severe from the most severely affected region.
PET imaging was performed in Mayo Clinic Rochester using the 11C amyloid tracer PiB with four 5-min dynamic frames acquired 40–60 min after injection, as previously described (Jack et al., 2008; Kantarci et al., 2012a). Standard corrections, co-registrations, and normalization to internal references were applied (Jack et al., 2008; Kantarci et al., 2012a). Briefly, PiB-PET images were co-registered to the T1-weighted MRI scan of the subject with a custom modified anatomical labelling atlas. Atlas labels in the custom template space were warped to the native T1 MRI space of the subject, as previously described (Kantarci et al., 2012a). PiB-PET cortical regions of interest were partial volume corrected and included both grey matter and white matter without segmentation. PiB-PET uptake was normalized to cerebellar grey/white matter uptake to obtain SUV ratios for brain regions. The meta-region of interest used to measure cortical retention of PiB included an average of the bilateral prefrontal, orbitofrontal, temporal, parietal, anterior cingulate, posterior cingulate, and precuneus regions (Jack et al., 2008). Using our pipeline we defined the threshold for PiB-positivity at the cut-off point that corresponded to a 90% sensitivity of clinically diagnosed Alzheimer’s disease subjects with an abnormal PET scan (Jack et al., 2014), which corresponds to a PiB-PET SUV ratio ≥ 1.4.
For APOE genotyping, DNA was obtained from frozen brain tissue using standard protocols. Each sample was genotyped for APOE-ε2, -ε3, and -ε4 with TaqMan® chemistry (Applied Biosystems).
Statistical analysis
SigmaPlot v12 and SAS (version 9.3, SAS Institute Inc.) were used to perform all statistical analyses. The median and interquartile ranges are reported along with the unadjusted P-values from a two-sided, two-sample Wilcoxon rank sum test or Kruskal Wallis test for the continuous variables. For categorical variables, the number of subjects is reported as well as the per cent and P-values from a chi-squared test. On Mayo Clinic Jacksonville Alzheimer’s disease cases, multiple linear regression analyses are reported between clinical course (age of onset, disease duration, final MMSE score) and pathology variables—adjusted for age at death, brain weight, APOE-ε4, and presence of co-existing Lewy body disease. Regression analysis reported on the final score of MMSE was additionally adjusted by education. On Mayo Clinic Rochester autopsied PiB-PET participants, a multiple linear regression analysis of PiB-PET SUV ratio and pathology variables (Thal amyloid phase, Braak NFT stage, CERAD ‘neuritic’ plaque score, and severity of cerebral amyloid angiopathy) was adjusted for APOE-ε4 status, age at PET scan, and time between PET scan and death.
Results
Amyloid-β plaque burden and Braak NFT stage differs across Thal amyloid phase
The Mayo Clinic Jacksonville Brain Bank cases stratified by Thal amyloid phase 0–5 and thioflavin-S plaque counts are graphically displayed in Fig. 1. Primary cortices were not used to assign Thal amyloid phase, but were included in the graph to gain a perspective difference across association cortices, primary cortices, and allocortical (hippocampal) regions. What is perhaps most striking is the number of plaques in allocortical regions is far less compared to association cortices, which were already truncated to a maximum count of 50 plaques. Despite expected differences between cortical and hippocampal regions with each increment in Thal amyloid phase, the interquartile range included, as part of Supplementary Table 1 demonstrates, the range of amyloid-β plaque severity within a given Thal amyloid phase. Braak NFT stage differed across Thal amyloid phase (P < 0.001, chi-square), as shown by Fig. 2. A Braak NFT stage of <IV is consistent with NFTs restricted to limbic regions and not yet involving cortical regions (Braak and Braak, 1991). The proportion of cases with Braak <IV by Thal amyloid phase was: Phase 0 = 97%, Phase 1 = 92%, Phase 2 = 89%, Phase 3 = 76%, Phase 4 = 41%, and Phase 5 = 6%.
Figure 1.

Stereotypic progression of plaques in Thal amyloid phase. Regional distribution of thioflavin-S plaque counts by Thal amyloid phase from the Mayo Clinic Jacksonville brain bank series (n = 3618). From left to right, three association cortices, two primary cortices, and three allocortical (hippocampal) regions are displayed. The 3D distribution plots of median thioflavin-S plaque counts across Thal phase 0–5 demonstrate the incremental involvement of subsequent regions. Note the striking difference between involvement of association cortices and allocortices throughout each phase. Senile plaque counts were maximally counted to 50 per 3 mm2 using a ×10 objective.
Figure 2.

Proportion of Braak NFT stage by Thal amyloid phase in Mayo Clinic Jacksonville Brain Bank. The proportion of each Braak NFT stages from the total for each Thal amyloid phase (found above each bar), was plotted across Thal amyloid Phase 0–5. The proportion of cases with Braak ≥IV by Thal amyloid phase was: Phase 0 = 3%, Phase 1 = 8%, Phase 2 = 11%, Phase 3 = 24%, Phase 4 = 59%, and Phase 5 = 94%.
Clinicopathological associations with Thal amyloid phase
Given the higher proportion of Braak NFT stage ≥IV in the latter Thal amyloid phases, clinicopathological group-wise comparisons of Mayo Clinic Jacksonville Alzheimer’s disease cases were analysed across Thal amyloid Phases 3 to 5. Alzheimer’s disease cases in Thal amyloid Phase 3 were older at death, but did not differ with respect to gender or education (Table 1). The presence of an APOE-ε4 allele did not differ across Alzheimer’s disease cases in Thal amyloid Phase 3–5; however, when genotype frequency was considered, cases with Thal amyloid Phase 3 had a lower frequency of APOE-ε3ε4 and APOE-ε4ε4. Brain weight was higher and Braak NFT stage was lower in Thal amyloid Phase 3. The presence of TDP-43 and vascular disease did not differ across Alzheimer’s disease cases in Thal amyloid Phases 3–5. The presence of co-existing Lewy body disease pathology was lower in Phase 3, amygdala predominant Lewy bodies was more common with increasing phase, and diffuse Lewy body disease was more frequent in Thal amyloid Phase 4. The three Alzheimer’s disease subtypes classified by NFT distribution (i.e. hippocampal sparing, typical, and limbic predominant) (Murray et al., 2011) did not show differences in Thal amyloid phase (all: median Thal = 5, P = 0.120).
Table 1.
Demographic and neuropathologic characteristics of Mayo Clinic Jacksonville Alzheimer’s disease brain bank cases by Thal amyloid phase
| Characteristic (n = 1375) | Phase 3 | Phase 4 | Phase 5 | |
|---|---|---|---|---|
| Number (%) | 106 (8%) | 143 (10%) | 1126 (82%) | |
| Demographic characteristics | ||||
| Age, years | 83 (77,88) | 80 (75,85) | 80 (73,86) | <0.001 |
| Females (%) | 52 (49%) | 75 (52%) | 623 (55%) | 0.402 |
| Education, years | 16 (12,16) | 13 (12,16) | 4 (12,16) | 0.145 |
| APOE-ε4 presence (%) | 40/79 (51%) | 63/101 (64%) | 501/834 (60%) | 0.219 |
| APOE-ε4 frequency (%) | 0.004 | |||
| 0 alleles (%) | 39/78 (49%) | 38/101 (38%) | 333 (40%) | |
| 1 allele (%) | 31/78 (39%) | 37/101 (36%) | 393 (47%) | |
| 2 alleles (%) | 9/78 (11%) | 25/101 (26%) | 108 (13%) | |
| Neuropathologic characteristics | ||||
| Brain weight, g | 1120 (1020,1220) | 1080 (1000,1180) | 1030 (920, 1140) | <0.001 |
| Braak NFT stage | IV-V (IV,V) | V (V,VI) | VI (V,VI) | <0.001 |
| TARDBP+/total (%) | 17/68 (25%) | 25/82 (30%) | 212/723 (29%) | 0.723 |
| Vascular disease/total (%) | 32/106 (30%) | 38/143 (27%) | 300/1126 (27%) | 0.730 |
| LBD (%) | 0.015 | |||
| None (%) | 68 (64%) | 85 (59%) | 689 (61%) | |
| ALB (%) | 8 (8%) | 16 (11%) | 167 (15%) | |
| BLBD or TLBD (%) | 9 (8%) | 12 (8%) | 129 (11%) | |
| DLBD (%) | 21 (20%) | 30 (21%) | 141 (13%) | |
| Clinical characteristics | ||||
| Age of onset, years | 76 (70,81) | 73 (66,79) | 70 (62,77) | <0.001b |
| Disease duration, years | 7 (6,9) | 8 (6,10) | 9 (6,12) | <0.001b |
| MMSE final score, pointsa | 17 (12,20) | 16 (8,21) | 12 (6,19) | 0.018b |
LBD = Lewy body disease; ALB = amygdala predominant Lewy bodies; BLBD = brainstem LBD; TLBD = transitional LBD; DLBD = diffuse LBD.
Data are median (25th, 75th), n (% of phase), or n/N (% of phase). Group-wise comparisons were done with the Kruskal-Wallis for continuous values and chi-square test for categorical values.
aScore assessed within 3 years of death (final).
bMultiple linear regression modelling when adjusted for Braak NFT stage showed no significant differences across Thal amyloid phase (Table 2).
Table 1 displays the clinical characteristics of Mayo Clinic Jacksonville Alzheimer’s disease cases with Thal amyloid Phases 3–5. Cases with Thal amyloid Phase 5 Alzheimer’s disease were younger at onset, had a longer disease duration and lower final score on the MMSE. Three linear regression models were constructed to account for the ability of Thal amyloid phase and Braak NFT stage to predict age of onset of cognitive symptoms, disease duration, and final MMSE score (Table 2). Significant variables from the univariate analyses of demographic and neuropathologic characteristics were used as covariates in each model: age at death, brain weight, number of APOE-ε4 alleles, and the presence of Lewy body disease. Education was additionally included as a covariate when predicting MMSE final score (O'Connor et al., 1989). After controlling for each covariate, Braak NFT stage remained the significant predictor and not Thal amyloid phase. Impressively, Model 1 accounted for 84% of the variability in age of onset in this autopsied Alzheimer’s disease cohort, with Braak NFT stage showing a 1.2 year younger age of onset for each increasing Braak NFT stage (adjusted P < 0.001). Model 2 accounted for 18% of the variance in disease duration, with Braak NFT stage showing a 1.3 year longer disease duration for each increasing Braak NFT stage (adjusted P < 0.001). Model 3 accounted for 26% of the variance in MMSE, with each increasing Braak NFT stage showing nearly five points lower on the MMSE for each increasing Braak NFT stage (adjusted P < 0.001).
Table 2.
Clinicopathologic characteristics and multiple linear regression modelling of Alzheimer’s disease brain bank cases by Thal amyloid phase
| Characteristic | Model 1: Age of onset | Model 2: Disease duration | Model 3: MMSE final scorea | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Independent variables | 0.26 | ||||||||
| Thal amyloid phase | 0.088 | 0.75 | 0.056 | 0.84 | −3.82 | 0.59 | |||
| Braak NFT stage | −1.2 | <0.001 | 1.2 | <0.001 | −4.7 | <0.001 | |||
| Confounding variables | |||||||||
| Age at death | 0.94 | <0.001 | 0.055 | 0.001 | 0.032 | 0.60 | |||
| Brain weight | 0.0082 | <0.001 | −0.0083 | <0.001 | 0.011 | 0.010 | |||
| APOE-ε4 | −0.33 | 0.14 | 0.39 | 0.070 | 1.2 | 0.15 | |||
| LBD presence | −0.67 | 0.031 | 0.79 | 0.009 | −0.95 | 0.42 | |||
LBD = Lewy body disease.
aMMSE final score was additionally adjusted for education.
Association of PiB-PET retention and neuropathological measures
Mayo Clinic Rochester participants who had an ante-mortem PiB-PET within 2 years of death (median 1.3, range 0.2–2.0) were assessed for Thal amyloid phase. Subject characteristics are described in Table 3 and PiB-PET images from a representative participant are shown in Fig. 3 for each Thal phase. Of the 35 participants who came to autopsy, 20% were considered PiB-negative as they were below the SUV ratio cut-off point of 1.4 when cerebellar grey and white matter was used as the normalization reference region (Rowe et al., 2007). There was no difference in age at death, gender, or education between PiB-negative and PiB-positive cases. PiB-positive cases were more frequently APOE-ε4 positive. Brain weight and coexisting vascular disease, or Lewy body disease did not differ between the PiB-negative and PiB-positive autopsied participants. Thal amyloid phase, Braak NFT stage, CERAD ‘neuritic’ plaque score, and cerebral amyloid angiopathy, however, were higher in PiB-positive cases.
Table 3.
Demographic and clinicopathologic characteristics of autopsied participants with ante-mortem PiB-PET within 2 years of death
| Characteristic (n = 35) | PiB SUV ratio < 1.4 | |
|---|---|---|
| Number (% of phase) | 7 (20%) | 28 (80%) |
| Demographic characteristics | ||
| Age at death, years | 77 (74, 86) | 81 (77, 89) |
| Females (% of phase) | 3 (43%) | 8 (28%) |
| Education, years | 18 (13, 19) | 16 (13, 18) |
| APOE-ε4 presence (%)* | 0 (0%) | 15 (56%) |
| Neuropathological characteristics | ||
| Brain weight, g | 1269 (1188, 1355) | 1363 (1213, 1445) |
| Thal amyloid phase | 0 (0, 0) | 4 (3, 4) |
| Braak NFT stage | II (I, III) | IV (II, VI) |
| CERAD ‘neuritic’ plaque score | 0 (0, 0) | 2 (1.5, 2.5) |
| Cerebral amyloid angiopathy score | 0 (0, 0) | 1.5 (1, 2) |
| Vascular disease/total (%) | 2 (28%) | 14 (50%) |
| LBD (% of phase) | 1 (14%) | 8 (28%) |
| Clinicoradiologic characteristics | ||
| PiB-PET, SUV ratio | 1.30 (1.28, 1.41) | 2.09 (1.55, 2.42) |
| Age at PET scan, years | 75 (73, 84) | 81 (75, 88) |
| Time to death from PET scan, years | 1.68 (140, 1.86) | 1.20 (0.82, 1.62) |
| CDR global score | 0 (0, 3) | 0.5 (0, 3) |
| CDR sum of boxes score | 0 (0, 14) | 3.5 (0, 10) |
| MMSE final score, points | 27 (24, 28) | 23 (16, 27) |
CDR = Clinical Dementia Rating scale; LBD = Lewy body disease.
Data are median (25th, 75th), n (% of phase), or n/N (%).
Pairwise comparisons were done with the Wilcoxon rank sum test for median values and chi-square test for categorical values.
Figure 3.

Comparison of 11C-PiB SUV ratio images across each Thal amyloid phase. The PiB-PET SUV ratio values can be found below each Thal amyloid phase. Representative axial, sagittal, and coronal slices from PiB-PET of six Mayo Clinic Rochester participants’ shows increasing PiB-positivity with each subsequent Thal amyloid phase. All example images are spatially and intensity normalized. The heat map index (left) shows the start of green at a SUV ratio level of 1.4, which is the cut-off point used to assess PiB-positivity. Of note, cerebellar PiB-PET uptake in Thal amyloid Phase 5 is not visible as this is the region used to normalize the scan. SUVR = SUV ratio.
Correspondence of ante-mortem PiB-PET SUV ratio cut-off point with neuropathologic measures
PiB-positivity using the 1.4 cut-off point demonstrated that this threshold occurs between Thal Phase 1 and Thal Phase 2—with the only Thal 0 outlier found right at the cut-off point with a PiB-PET SUV ratio of 1.40 exactly (Supplementary Fig. 2). Given the range of plaque density within each Thal amyloid phase (Supplementary Table 1), findings from the large Mayo Clinic Jacksonville Brain Bank were used to gain a broader perspective of Thal amyloid Phase 1–2 cases. Consideration of amyloid-β plaque density regarding Alzheimer’s disease pathologic diagnosis will be 30 plaques per ×10 field—following recommendations from Khachaturian (1985) criteria. Of the 600 Thal amyloid Phase 1–2 cases, 416 (69%) had insufficient plaque pathology for Alzheimer’s disease diagnosis (i.e. <30 plaques/field), with only 22/416 having a Braak NFT stage ≥IV (i.e. tangle predominant dementia cases) (Janocko et al., 2012). Of the 184 (31%) Thal amyloid Phase 1–2 cases with sufficient plaques for Alzheimer’s disease diagnosis, 155 are Braak NFT stage <IV (i.e. pathological ageing cases) (Murray and Dickson, 2014) and only 29 are Braak ≥IV (i.e. advanced Alzheimer’s disease cases). The autopsied PiB-PET participants at incremental ranges of PiB SUV ratio values and by distribution of neuropathologic severity are shown in Table 4 using the collapsed NIA-AA recommended neuropathologic criteria for Alzheimer’s disease, where ‘A’ represents Thal amyloid phase, ‘B’ Braak NFT stage, and ‘C’ CERAD neuritic plaque score.
Table 4.
Distribution of autopsied PiB-PET participants at each given range of SUV ratio values
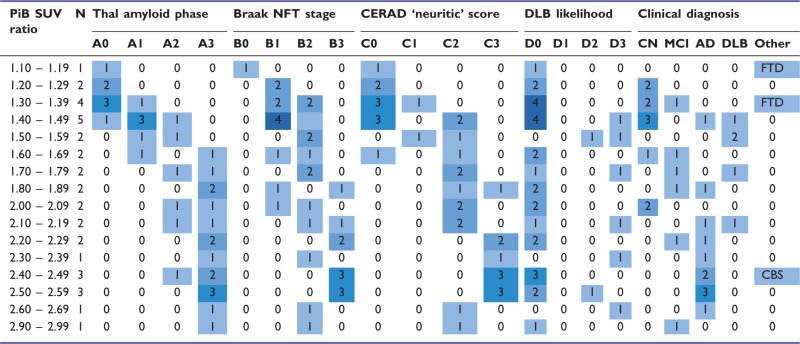 |
The 1.4 PiB-PET cut-off point is used to compare differences in NIA-AA ‘ABC’ neuropathologic criteria, dementia with Lewy bodies neuropathologic classification, and clinical diagnosis at the time of PET scanning between PiB-positive and PiB-negative patients.
CERAD = Consortium to Establish a Registry for Alzheimer’s Disease; DLB = dementia with Lewy bodies; CN = cognitively normal; MCI = mild cognitive impairment; AD = Alzheimer’s disease; CBD = corticobasal syndrome; FTD = frontotemporal dementia.
Coloured boxed numbers correspond to number of cases per range of PiB SUV ratio and clinicopathological information column. There are no autopsied cases with an ante-mortem PiB SUV ratio 1.9 – 1.99 and 2.70 – 2.89.
According to NIA-AA criteria (Hyman et al., 2012) Thal amyloid phase: A0 = Phase 0, A1 = Phase 1 or 2, A2 = Phase 3, A3 = Phase 4 or 5; Braak NFT stage: B0 = Stage 0, B1 = Stage 1 or 2, B2 = Stage 3 or 4, B3 = Stage 5 or 6, CERAD neuritic score: C0 = absent, C1 = sparse, C2 = moderate, C3 = frequent. Dementia with Lewy bodies likelihood was patterned after the NIA-AA scheme and presented based on recommendations from the DLB consortium (McKeith et al., 2005).
PiB-PET SUV ratio values differed across Thal amyloid Phases 0–5 and cerebral amyloid angiopathy severity scores (Supplementary Table 2). A multiple linear regression model of cortical PiB-PET SUV ratio (dependent variable) examined whether Thal amyloid phase, Braak NFT stage, CERAD ‘neuritic’ plaque score, or severity of cerebral amyloid angiopathy was predictive of higher PiB binding. After adjusting for APOE-ε4 status, time between PET scan to death, and age at PET scan the model accounted for 68% of the variability in PiB-PET SUV ratio. For every increase in Thal amyloid phase, the model predicted an increase of 0.16 SUV ratio units (Table 5). None of the other neuropathological variables predicted PiB-PET SUV ratio. Modelled after the NIA-AA designations (Hyman et al., 2012), Table 4 shows ‘ABCD’ criteria—incorporating both recommended Alzheimer’s disease neuropathologic measures and likelihood of dementia with Lewy bodies neuropathologic classification (McKeith et al., 2005). Of the n = 7 cases who have a PiB-PET SUV ratio < 1.4, all n = 7 were Thal Phase 0 (A0) or Thal Phase 1 and 2 (A1) and CERAD none (C0) or CERAD sparse (C1)—with a sensitivity of 86% for predicting PiB-negativity for both measures. Of the n = 28 that have a PiB ≥1.4, only n = 1 was a Thal Phase 0 (A0), whereas n = 4 were CERAD none (C0). Thus, the sensitivity of predicting PiB-positivity measured by Thal amyloid phase was 96% and by CERAD was 86%. Of the n = 8 cases with coexisting Lewy body disease pathology, all were classified as intermediate (D2) and high likelihood of dementia with Lewy bodies (D3); and all were found to be PiB-positive. Five of the cases with Lewy body disease were clinically diagnosed as dementia with Lewy bodies; and three were clinically diagnosed as Alzheimer’s disease and showed the highest PiB-PET uptake.
Table 5.
Multiple linear regression modelling of PiB-PET SUV ratio values and Alzheimer’s disease neuropathologic change for Mayo Clinic Rochester study participants autopsied within 2 years of death
| Characteristic | β-coefficient | P-value | Adjusted-R2 |
|---|---|---|---|
| Independent variables | 0.675 | ||
| Thal amyloid phase | 0.16 | 0.007 | |
| Braak NFT stage | 0.066 | 0.224 | |
| CERAD ‘neuritic’ plaque score | 0.078 | 0.502 | |
| Cerebral amyloid angiopathy score | −0.053 | 0.412 | |
| Confounding variables | |||
| APOE-ε4 presence | 0.11 | 0.403 | |
| Time to death from PET scan | 0.15 | 0.163 | |
| Age at PET scan | 0.000054 | 0.992 |
Discussion
Using quantitative data from more than 3500 autopsied Mayo Clinic Jacksonville brains, our findings demonstrate each progressive Thal amyloid phase shows a steady increase in the proportion of cases with a Braak NFT stage ≥IV (i.e. cortical tau involvement). This is of particular clinical importance given the high degree of association between NFT accumulation and cognitive decline (Duyckaerts et al., 1997; Bennett et al., 2004; Sabbagh et al., 2010; Nelson et al., 2012). In a subset of ∼1500 autopsy-confirmed Alzheimer’s disease brains, we show that although Thal amyloid phase highly correlates with age of onset, disease duration, and MMSE final score; these associations were driven by Braak NFT stage. Using a separate prospectively-followed Mayo Clinic Rochester cohort, this study confirms previous findings that ante-mortem PiB binding is associated with accumulation of underlying amyloid-β neuropathology (Lockhart et al., 2007; Ikonomovic et al., 2008; Driscoll et al., 2012). More importantly, to the best of our knowledge it demonstrates for the first time that cortical PiB SUV ratio values significantly associate with Thal amyloid phase (Thal et al., 2002), but not with Braak NFT stage (Braak and Braak, 1991), CERAD ‘neuritic’ plaque score (Mirra et al., 1991), or severity of cerebral amyloid angiopathy using multiple linear regression analysis. In this series of 35 autopsied participants with ante-mortem PiB-PET within 2 years of death, a cut-off point value of 1.4 SUV ratio units corresponds to Thal amyloid Phase 1–2.
Our observations reaffirm the concept that amyloid-β burden itself is not the proximate causal pathology for cognitive decline. There have now been many studies of the association between cognition and amyloid-β accumulation at different points along the Alzheimer’s disease spectrum (Morris et al., 1996; Schmitt et al., 2000; Knopman et al., 2003; Petersen et al., 2006; Price et al., 2009; Maarouf et al., 2011; Mathis et al., 2013). The generally modest relationship can be understood by the imaging-pathological associations described in this study. Abnormal accumulation of amyloid-β and NFT pathology likely occurs in parallel with one another, eventually resulting in incompletely understood crosstalk between the pathologies. The initial sites of neuropathologic insults greatly differ between the two proteins with amyloid-β found in the cortex 10–15 years before cognitive decline (Price and Morris, 1999) and NFTs found in brainstem structures of individuals in their 20’s and 30’s (Braak et al., 2011). Although cortical accumulation of amyloid-β was considered sufficient to influence the risk of clinical Alzheimer’s disease, we and others demonstrate that NFT accumulation mediates the contribution of amyloid-β pathology to cognitive decline (Bennett et al., 2004). To the extent that rising levels of cerebral amyloid-β are associated with higher Braak NFT stages, and that Braak NFT stage is more strongly linked to cognition—the relationship between amyloid-β accumulation and cognition can be seen as rather noisy.
Thal amyloid phase describes a progressive stereotypic topographic distribution of amyloid-β plaque pathology (Thal et al., 2002; Alafuzoff et al., 2009). Neuropathologically diagnosed Alzheimer’s disease cases are typically found in Thal amyloid Phase 3–5 (Thal et al., 2002; Hyman et al., 2012; Montine et al., 2012). Thal amyloid Phase 3 Alzheimer’s disease cases are older at death, have a lower genotype frequency of APOE-ε4, larger brain weights, lower Braak NFT stage, and infrequent co-existing Lewy body disease. Atypical distributions of NFT pathology are known to disproportionately affect neocortex relative to limbic regions and vice versa in ∼25% of Alzheimer’s disease cases (Galton et al., 2000; Alladi et al., 2007; Murray et al., 2011; Whitwell et al., 2012). There were no differences, however, in Thal amyloid phase across atypical and typical Alzheimer’s disease subtypes. This finding supports ante-mortem PiB-PET studies that demonstrate amyloid-β binding is widely distributed in sporadic Alzheimer’s disease without focal involvement (Lehmann et al., 2013; Laforce et al., 2014)—with the exception of cases with posterior cortical atrophy (Crutch et al., 2012; Ossenkoppele et al., 2014).
Previous studies showed evidence that PiB may bind to vessels laden with amyloid, which was attributed to retention of the PiB compound in cerebral amyloid angiopathy (Johnson et al., 2007; Lockhart et al., 2007; Ikonomovic et al., 2008). Thus, in addition to Thal amyloid phase, we examined the contribution of cerebral amyloid angiopathy severity. Significant group-wise differences are found across Thal amyloid phases and cerebral amyloid angiopathy severity scores. Unlike the linear association of PiB SUV ratio values and Thal amyloid phase, PiB SUV ratio values first increase dramatically from none to mild cerebral amyloid angiopathy and then decrease in cases with moderate and severe amyloid angiopathy. We show for the first time that Thal amyloid phase is the sole predictor of PiB SUV ratio values, but not Braak NFT stage, CERAD ‘neuritic’ plaque score, or cerebral amyloid angiopathy using linear regression modelling. This is an important finding given the previous suggestions of PiB binding to vascular amyloid-β deposits and weak binding to a subset of extracellular ghost NFTs (Johnson et al., 2007; Lockhart et al., 2007; Ikonomovic et al., 2008). Our cortical PiB-PET SUV ratio values, however, are derived from a meta-region of interest that does not include the occipital lobe. Given the predominance of cerebral amyloid angiopathy in the occipital lobe, visual grading of PiB-PET scans should be performed with the concept that greater occipital-to-global PiB ratio may occur in patients with Alzheimer’s disease dementia with suspected cerebral amyloid angiopathy (Johnson et al., 2007; Kantarci et al., 2013). Our study supports findings of variable agreement between CERAD ‘neuritic’ plaque score and in vivo PiB binding (Sojkova et al., 2011). Moreover, our data show that Thal amyloid phase remains the significant predictor of PiB SUV ratio adding new insight to the contribution of diffuse plaque pathology to increasing PiB retention in the presence of other forms of amyloid-β pathology. Progressive accumulation of PiB-PET binding with each increasing Thal amyloid phase is evident in Fig. 3 with the caveat of cerebellar PiB-PET retention. Cerebellar amyloid is characterized as diffuse amyloid-β plaque pathology in the molecular layer that only weakly binds PiB-PET ligands (Ikonomovic et al., 2008; Niedowicz et al., 2012) with less frequent deposition of cored plaques in the Purkinje cell layer (Joachim et al., 1989). Weak binding is compounded by the fact that the cerebellum is routinely used as the PiB-PET reference region.
A PiB SUV ratio cut-off point of 1.4 is consistent with a 90% sensitivity of clinically diagnosed Alzheimer’s disease subjects with an abnormal PET scan using our pipeline (Jack et al., 2014). We demonstrate that the 1.4 PiB cut-off point corresponds to a Thal amyloid Phase 1–2 or A1 in the Mayo Clinic Rochester series of autopsied PiB-PET participants. Based on our brain bank findings, the PiB-PET cut-off point corresponding to Thal amyloid Phase 1–2 will likely comprise patients with insufficient NFT pathology for neuropathologic diagnosis of Alzheimer’s disease (i.e. lacking cortical NFTs at Braak NFT stage <IV); but all will have accumulation of cortical amyloid-β plaques transitioning from insufficient to sufficient for neuropathological diagnosis of Alzheimer’s disease. Amyloid-β pathology is known to modulate cholinergic activity in the cortex by altering neurotrophic signalling in Alzheimer’s disease (Yaar et al., 1997; Bulbarelli et al., 2009). Increasing evidence, however, suggests that neurotrophic signalling is not affected by amyloid-β pathology in the prodromal stages of Alzheimer’s disease dementia (Perez et al., 2015). A longitudinal PiB study in individuals without dementia shows that higher PiB retention associates with cognitive decline in verbal memory, thus tracking well with disease progression (Resnick et al., 2010). Taking these studies into account, our data provide neuropathologic support for therapeutic intervention prior to crossing the threshold to PiB-positivity—when patients are still in Thal amyloid Phases 0 and early Phase 1, but lack significant cortical NFT pathology. Future studies should consider topographic differences with relation to stereotyped progression in Thal amyloid phase in order to elucidate subtle differences that may distinguish individuals who meet criteria for Thal amyloid Phase 1–2, but lack significant neurodegeneration (e.g. cortical NFTs) and perhaps use lower thresholds for PiB SUV ratio if the goal is to identify individuals at early stages of the Alzheimer’s disease for preventive interventions.
The strengths of our study include the large number of subjects with pathologically confirmed Alzheimer’s type pathology from our brain bank series, which we use to inform our interpretations of 35 prospectively-followed autopsied patients with ante-mortem PiB-PET scans. No previous study that investigated pathologic correlates of PiB binding attempted to address the relevance of PiB cut-off point values with respect to Thal amyloid phase. We also accounted for other potential pathologic and demographic confounders, including APOE-ε4 status, age at PET scan, time between PET scan to death, and brain weight—accounting for 68% of the variability in PiB-PET SUV ratio values in this cohort.
Limitations of our study include the fact that our regression analysis of PiB-PET did not allow us to take into account non-linearity between time of scan and death. PiB-PET binding in patients with Alzheimer’s disease dementia, however, does not increase appreciably over 2 years; (Engler et al., 2006) and annual change within 1 year does not differ among cognitively normal, mild cognitive impairment and Alzheimer’s disease dementia patients (Jack et al., 2009). Regardless, we included time of scan-to-death as a covariate, and limited scan-to-death interval to 2 years in an attempt to control for any higher PiB SUV ratio that occurred prior to death. Another limitation is that our large series with Alzheimer’s type pathology is a brain bank sample of convenience with retrospective clinical data abstraction. Our finding that Braak NFT stage and not Thal amyloid phase drives differences in clinical course should be confirmed in a prospectively followed cohort. Although our PiB-PET cohort is one of the largest to be autopsied within 2 years of scanning, we recognize the limitation of generalizability from a sample size of 35. Moreover, replication in other cohorts and with other amyloid imaging compounds is warranted.
This study shows that cortical PiB-PET values are a good biomarker of underlying accumulation of amyloid-β stereotyped by Thal amyloid phase, and hence support their use for prescreening patients for inclusion in clinical trials. Thal amyloid phase significantly associates with variables of clinical course (i.e. age of onset, disease duration, and peri-mortem MMSE score); however, association with Thal amyloid phase did not remain significant after accounting for Braak NFT stage. Thus, the findings from this study suggest that therapeutic trials targeting clinical course may additionally benefit from using a multimodal approach that combines PiB-PET with a neurodegeneration biomarker (e.g. structural MRI, CSF).
Funding
R01-AG040042 (PI Kantarci), R01-AG011378 (PI Jack), R01-AG041851 (coPI Jack), P50-AG016574 (PI Petersen)/P1, U01-AG006786 (PI Petersen), P50-NS072187 (PI Dickson); Mangurian Foundation; Robert H. and Clarice Smith and Abigail vanBuren Alzheimer’s disease Research Program; the Elsie and Marvin Dekelboum Family Foundation; Donors Cure Foundation New Vision Award (PI Murray); The Alexander Family.
Supplementary material
Supplementary material is available at Brain online.
Acknowledgements
We thank the patients and their families who have participated in these prospective clinical and imaging studies, and especially for the generous donation of their brain tissue to help further our knowledge in Alzheimer’s disease. The authors would like to acknowledge the continuous commitment and teamwork offered by Linda G. Rousseau, Virginia R. Phillips, and Monica Castanedes-Casey. We would like to also thank Kris Johnson for assistance in collection of pathologic material.
Glossary
Abbreviations
- CERAD
Consortium to Establish a Registry for Alzheimer's Disease
- MMSE
Mini-Mental State Examination
- NIA-AA
National Institute on Ageing–Alzheimer’s Association
- NFT
neurofibrillary tangle
- PiB
11C-Pittsburgh compound B
- SUV
standard uptake value
References
- Amador-Ortiz C, Lin WL, Ahmed Z, Personett D, Davies P, Duara R, et al. TDP-43 immunoreactivity in hippocampal sclerosis and Alzheimer's disease. Ann Neurol. 2007;61:435–45. doi: 10.1002/ana.21154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alafuzoff I, Thal DR, Arzberger T, Bogdanovic N, Al-Sarraj S, Bodi I, et al. Assessment of beta-amyloid deposits in human brain: a study of the BrainNet Europe Consortium. Acta Neuropathol. 2009;117:309–20. doi: 10.1007/s00401-009-0485-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albert MS, DeKosky ST, Dickson D, Dubois B, Feldman HH, Fox NC, et al. The diagnosis of mild cognitive impairment due to Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 2011;7:270–9. doi: 10.1016/j.jalz.2011.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alladi S, Xuereb J, Bak T, Nestor P, Knibb J, Patterson K, et al. Focal cortical presentations of Alzheimer's disease. Brain. 2007;130(Pt 10):2636–45. doi: 10.1093/brain/awm213. [DOI] [PubMed] [Google Scholar]
- Bacskai BJ, Frosch MP, Freeman SH, Raymond SB, Augustinack JC, Johnson KA, et al. Molecular imaging with Pittsburgh Compound B confirmed at autopsy: a case report. Arch Neurol. 2007;64:431–4. doi: 10.1001/archneur.64.3.431. [DOI] [PubMed] [Google Scholar]
- Bennett DA, Schneider JA, Wilson RS, Bienias JL, Arnold SE. Neurofibrillary tangles mediate the association of amyloid load with clinical Alzheimer disease and level of cognitive function. Arch Neurol. 2004;61:378–84. doi: 10.1001/archneur.61.3.378. [DOI] [PubMed] [Google Scholar]
- Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991;82:239–59. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- Braak H, Thal DR, Ghebremedhin E, Del Tredici K. Stages of the pathologic process in Alzheimer disease: age categories from 1 to 100 years. J Neuropathol Exp Neurol. 2011;70:960–9. doi: 10.1097/NEN.0b013e318232a379. [DOI] [PubMed] [Google Scholar]
- Bulbarelli A, Lonati E, Cazzaniga E, Re F, Sesana S, Barisani D, et al. TrkA pathway activation induced by amyloid-beta (Abeta) Mol Cell Neurosci. 2009;40:365–73. doi: 10.1016/j.mcn.2008.12.006. [DOI] [PubMed] [Google Scholar]
- Crutch SJ, Lehmann M, Schott JM, Rabinovici GD, Rossor MN, Fox NC. Posterior cortical atrophy. Lancet Neurol. 2012;11:170–8. doi: 10.1016/S1474-4422(11)70289-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickson DW, Crystal HA, Mattiace LA, Masur DM, Blau AD, Davies P, et al. Identification of normal and pathological aging in prospectively studied nondemented elderly humans. Neurobiol Aging. 1992;13:179–89. doi: 10.1016/0197-4580(92)90027-u. [DOI] [PubMed] [Google Scholar]
- Driscoll I, Troncoso JC, Rudow G, Sojkova J, Pletnikova O, Zhou Y, et al. Correspondence between in vivo (11)C-PiB-PET amyloid imaging and postmortem, region-matched assessment of plaques. Acta Neuropathol. 2012;124:823–31. doi: 10.1007/s00401-012-1025-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duyckaerts C, Bennecib M, Grignon Y, Uchihara T, He Y, Piette F, et al. Modeling the relation between neurofibrillary tangles and intellectual status. Neurobiol Aging. 1997;18:267–73. doi: 10.1016/s0197-4580(97)80306-5. [DOI] [PubMed] [Google Scholar]
- Engler H, Forsberg A, Almkvist O, Blomquist G, Larsson E, Savitcheva I, et al. Two-year follow-up of amyloid deposition in patients with Alzheimer's disease. Brain. 2006;129(Pt 11):2856–66. doi: 10.1093/brain/awl178. [DOI] [PubMed] [Google Scholar]
- Folstein MF, Folstein SE, McHugh PR. “Mini-mental state”. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res. 1975;12:189–98. doi: 10.1016/0022-3956(75)90026-6. [DOI] [PubMed] [Google Scholar]
- Galton CJ, Patterson K, Xuereb JH, Hodges JR. Atypical and typical presentations of Alzheimer's disease: a clinical, neuropsychological, neuroimaging and pathological study of 13 cases. Brain. 2000;123(Pt 3):484–98. doi: 10.1093/brain/123.3.484. [DOI] [PubMed] [Google Scholar]
- Hyman BT, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Carrillo MC, et al. National Institute on Aging-Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease. Alzheimers Dement. 2012;8:1–13. doi: 10.1016/j.jalz.2011.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikonomovic MD, Klunk WE, Abrahamson EE, Mathis CA, Price JC, Tsopelas ND, et al. Post-mortem correlates of in vivo PiB-PET amyloid imaging in a typical case of Alzheimer's disease. Brain. 2008;131(Pt 6):1630–45. doi: 10.1093/brain/awn016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR, Jr, Albert MS, Knopman DS, McKhann GM, Sperling RA, Carrillo MC, et al. Introduction to the recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 2011;7:257–62. doi: 10.1016/j.jalz.2011.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR, Jr, Lowe VJ, Senjem ML, Weigand SD, Kemp BJ, Shiung MM, et al. 11C PiB and structural MRI provide complementary information in imaging of Alzheimer's disease and amnestic mild cognitive impairment. Brain. 2008;131(Pt 3):665–80. doi: 10.1093/brain/awm336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR, Jr, Lowe VJ, Weigand SD, Wiste HJ, Senjem ML, Knopman DS, et al. Serial PIB and MRI in normal, mild cognitive impairment and Alzheimer's disease: implications for sequence of pathological events in Alzheimer's disease. Brain. 2009;132(Pt 5):1355–65. doi: 10.1093/brain/awp062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR, Jr, Wiste HJ, Weigand SD, Rocca WA, Knopman DS, Mielke MM, et al. Age-specific population frequencies of cerebral beta-amyloidosis and neurodegeneration among people with normal cognitive function aged 50-89 years: a cross-sectional study. Lancet Neurol. 2014;13:997–1005. doi: 10.1016/S1474-4422(14)70194-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janocko NJ, Brodersen KA, Soto-Ortolaza AI, Ross OA, Liesinger AM, Duara R, et al. Neuropathologically defined subtypes of Alzheimer's disease differ significantly from neurofibrillary tangle-predominant dementia. Acta Neuropathol. 2012;124:681–92. doi: 10.1007/s00401-012-1044-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jellinger KA, Attems J. Incidence of cerebrovascular lesions in Alzheimer's disease: a postmortem study. Acta Neuropathol. 2003;105:14–7. doi: 10.1007/s00401-002-0634-5. [DOI] [PubMed] [Google Scholar]
- Joachim CL, Morris JH, Selkoe DJ. Diffuse senile plaques occur commonly in the cerebellum in Alzheimer's disease. Am J Pathol. 1989;135:309–19. [PMC free article] [PubMed] [Google Scholar]
- Johnson KA, Gregas M, Becker JA, Kinnecom C, Salat DH, Moran EK, et al. Imaging of amyloid burden and distribution in cerebral amyloid angiopathy. Ann Neurol. 2007;62:229–34. doi: 10.1002/ana.21164. [DOI] [PubMed] [Google Scholar]
- Kantarci K, Gunter JL, Tosakulwong N, Weigand SD, Senjem MS, Petersen RC, et al. Focal hemosiderin deposits and beta-amyloid load in the ADNI cohort. Alzheimers Dement. 2013;9(5 Suppl):S116–23. doi: 10.1016/j.jalz.2012.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kantarci K, Lowe VJ, Boeve BF, Weigand SD, Senjem ML, Przybelski SA, et al. Multimodality imaging characteristics of dementia with Lewy bodies. Neurobiol Aging. 2012a;33:2091–105. doi: 10.1016/j.neurobiolaging.2011.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kantarci K, Yang C, Schneider JA, Senjem ML, Reyes DA, Lowe VJ, et al. Ante mortem amyloid imaging and β-amyloid pathology in a case with dementia with Lewy bodies. Neurobiol Aging. 2012b;33:878–85. doi: 10.1016/j.neurobiolaging.2010.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khachaturian ZS. Diagnosis of Alzheimer's disease. Arch Neurol. 1985;42:1097–105. doi: 10.1001/archneur.1985.04060100083029. [DOI] [PubMed] [Google Scholar]
- Klunk WE, Engler H, Nordberg A, Wang Y, Blomqvist G, Holt DP, et al. Imaging brain amyloid in Alzheimer's disease with Pittsburgh Compound-B. Ann Neurol. 2004;55:306–19. doi: 10.1002/ana.20009. [DOI] [PubMed] [Google Scholar]
- Knopman DS, Parisi JE, Salviati A, Floriach-Robert M, Boeve BF, Ivnik RJ, et al. Neuropathology of cognitively normal elderly. J Neuropathol Exp Neurol. 2003;62:1087–95. doi: 10.1093/jnen/62.11.1087. [DOI] [PubMed] [Google Scholar]
- Laforce R, Jr, Tosun D, Ghosh P, Lehmann M, Madison CM, Weiner MW, et al. Parallel ICA of FDG-PET and PiB-PET in three conditions with underlying Alzheimer's pathology. Neuroimage Clin. 2014;4:508–16. doi: 10.1016/j.nicl.2014.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann M, Ghosh PM, Madison C, Laforce R, Jr, Corbetta-Rastelli C, Weiner MW, et al. Diverging patterns of amyloid deposition and hypometabolism in clinical variants of probable Alzheimer's disease. Brain. 2013;136(Pt 3):844–58. doi: 10.1093/brain/aws327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lockhart A, Lamb JR, Osredkar T, Sue LI, Joyce JN, Ye L, et al. PIB is a non-specific imaging marker of amyloid-beta (Abeta) peptide-related cerebral amyloidosis. Brain. 2007;130(Pt 10):2607–15. doi: 10.1093/brain/awm191. [DOI] [PubMed] [Google Scholar]
- Maarouf CL, Daugs ID, Kokjohn TA, Walker DG, Hunter JM, Kruchowsky JC, et al. Alzheimer's disease and non-demented high pathology control nonagenarians: comparing and contrasting the biochemistry of cognitively successful aging. PLoS One. 2011;6:e27291. doi: 10.1371/journal.pone.0027291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathis CA, Kuller LH, Klunk WE, Snitz BE, Price JC, Weissfeld LA, et al. In vivo assessment of amyloid-beta deposition in nondemented very elderly subjects. Ann Neurol. 2013;73:751–61. doi: 10.1002/ana.23797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKeith IG, Dickson DW, Lowe J, Emre M, O'Brien JT, Feldman H, et al. Diagnosis and management of dementia with Lewy bodies: third report of the DLB consortium. Neurology. 2005;65:1863–72. doi: 10.1212/01.wnl.0000187889.17253.b1. [DOI] [PubMed] [Google Scholar]
- McKhann GM, Knopman DS, Chertkow H, Hyman BT, Jack CR, Jr, Kawas CH, et al. The diagnosis of dementia due to Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer's association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 2011;7:263–9. doi: 10.1016/j.jalz.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirra SS, Heyman A, McKeel D, Sumi SM, Crain BJ, Brownlee LM, et al. The Consortium to Establish a Registry for Alzheimer's Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer's disease. Neurology. 1991;41:479–86. doi: 10.1212/wnl.41.4.479. [DOI] [PubMed] [Google Scholar]
- Montine TJ, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Dickson DW, et al. National Institute on Aging-Alzheimer's association guidelines for the neuropathologic assessment of Alzheimer's disease: a practical approach. Acta Neuropathol. 2012;123:1–11. doi: 10.1007/s00401-011-0910-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris JC, Storandt M, McKeel DW, Jr, Rubin EH, Price JL, Grant EA, et al. Cerebral amyloid deposition and diffuse plaques in “normal” aging: Evidence for presymptomatic and very mild Alzheimer's disease. Neurology. 1996;46:707–19. doi: 10.1212/wnl.46.3.707. [DOI] [PubMed] [Google Scholar]
- Murray ME, Dickson DW. Is pathological aging a successful resistance against amyloid-beta or preclinical Alzheimer's disease? Alzheimers Res Ther. 2014;6:24. doi: 10.1186/alzrt254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray ME, Graff-Radford NR, Ross OA, Petersen RC, Duara R, Dickson DW. Neuropathologically defined subtypes of Alzheimer's disease with distinct clinical characteristics: a retrospective study. Lancet Neurol. 2011;10:785–96. doi: 10.1016/S1474-4422(11)70156-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray ME, Kouri N, Lin WL, Jack CR, Jr, Dickson DW, Vemuri P. Clinicopathologic assessment and imaging of tauopathies in neurodegenerative dementias. Alzheimers Res Ther. 2014;6:1. doi: 10.1186/alzrt231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niedowicz DM, Beckett TL, Matveev S, Weidner AM, Baig I, Kryscio RJ, et al. Pittsburgh compound B and the postmortem diagnosis of Alzheimer disease. Ann Neurol. 2012;72:564–70. doi: 10.1002/ana.23633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson PT, Alafuzoff I, Bigio EH, Bouras C, Braak H, Cairns NJ, et al. Correlation of Alzheimer disease neuropathologic changes with cognitive status: a review of the literature. J Neuropathol Exp Neurol. 2012;71:362–81. doi: 10.1097/NEN.0b013e31825018f7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ossenkoppele R, Schonhaut DR, Baker SL, O'Neil JP, Janabi M, Ghosh PM, et al. Tau, amyloid, and hypometabolism in a patient with posterior cortical atrophy. Ann Neurol. 2015;77:338–42. doi: 10.1002/ana.24321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connor DW, Pollitt PA, Treasure FP, Brook CP, Reiss BB. The influence of education, social class and sex on Mini-Mental State scores. Psychol Med. 1989;19:771–6. doi: 10.1017/s0033291700024375. [DOI] [PubMed] [Google Scholar]
- Perez SE, He B, Nadeem M, Wuu J, Scheff SW, Abrahamson EE, et al. doi: 10.1016/j.biopsych.2013.12.016. Resilience of precuneus neurotrophic signaling pathways despite amyloid pathology in prodromal Alzheimer's disease. Biol Psychiatry 2015; 77: 693–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen RC, Parisi JE, Dickson DW, Johnson KA, Knopman DS, Boeve BF, et al. Neuropathologic features of amnestic mild cognitive impairment. Arch Neurol. 2006;63:665–72. doi: 10.1001/archneur.63.5.665. [DOI] [PubMed] [Google Scholar]
- Price JL, McKeel DW, Jr, Buckles VD, Roe CM, Xiong C, Grundman M, et al. Neuropathology of nondemented aging: presumptive evidence for preclinical Alzheimer disease. Neurobiol Aging. 2009;30:1026–36. doi: 10.1016/j.neurobiolaging.2009.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price JL, Morris JC. Tangles and plaques in nondemented aging and “preclinical” Alzheimer's disease. Annals of neurology. 1999;45:358–68. doi: 10.1002/1531-8249(199903)45:3<358::aid-ana12>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- Resnick SM, Sojkova J, Zhou Y, An Y, Ye W, Holt DP, et al. Longitudinal cognitive decline is associated with fibrillar amyloid-beta measured by [11C]PiB. Neurology. 2010;74:807–15. doi: 10.1212/WNL.0b013e3181d3e3e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowe CC, Ng S, Ackermann U, Gong SJ, Pike K, Savage G, et al. Imaging beta-amyloid burden in aging and dementia. Neurology. 2007;68:1718–25. doi: 10.1212/01.wnl.0000261919.22630.ea. [DOI] [PubMed] [Google Scholar]
- Sabbagh MN, Cooper K, DeLange J, Stoehr JD, Thind K, Lahti T, et al. Functional, global and cognitive decline correlates to accumulation of Alzheimer's pathology in MCI and AD. Curr Alzheimer Res. 2010;7:280–6. doi: 10.2174/156720510791162340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt FA, Davis DG, Wekstein DR, Smith CD, Ashford JW, Markesbery WR. “Preclinical” AD revisited: neuropathology of cognitively normal older adults. Neurology. 2000;55:370–6. doi: 10.1212/wnl.55.3.370. [DOI] [PubMed] [Google Scholar]
- Sojkova J, Driscoll I, Iacono D, Zhou Y, Codispoti KE, Kraut MA, et al. In vivo fibrillar beta-amyloid detected using [11C]PiB positron emission tomography and neuropathologic assessment in older adults. Arch Neurol. 2011;68:232–40. doi: 10.1001/archneurol.2010.357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperling RA, Aisen PS, Beckett LA, Bennett DA, Craft S, Fagan AM, et al. Toward defining the preclinical stages of Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 2011;7:280–92. doi: 10.1016/j.jalz.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terry RD, Hansen LA, DeTeresa R, Davies P, Tobias H, Katzman R. Senile dementia of the Alzheimer type without neocortical neurofibrillary tangles. Journal of neuropathology and experimental neurology. 1987;46:262–8. doi: 10.1097/00005072-198705000-00003. [DOI] [PubMed] [Google Scholar]
- Thal DR, Rub U, Orantes M, Braak H. Phases of A beta-deposition in the human brain and its relevance for the development of AD. Neurology. 2002;58:1791–800. doi: 10.1212/wnl.58.12.1791. [DOI] [PubMed] [Google Scholar]
- Uchikado H, Lin WL, DeLucia MW, Dickson DW. Alzheimer disease with amygdala Lewy bodies: a distinct form of alpha-synucleinopathy. J Neuropathol Exp Neurol. 2006;65:685–97. doi: 10.1097/01.jnen.0000225908.90052.07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitwell JL, Dickson DW, Murray ME, Weigand SD, Tosakulwong N, Senjem ML, et al. Neuroimaging correlates of pathologically defined subtypes of Alzheimer's disease: a case-control study. Lancet Neurol. 2012;11:868–77. doi: 10.1016/S1474-4422(12)70200-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaar M, Zhai S, Pilch PF, Doyle SM, Eisenhauer PB, Fine RE, et al. Binding of beta-amyloid to the p75 neurotrophin receptor induces apoptosis. A possible mechanism for Alzheimer's disease. J Clin Invest. 1997;100:2333–40. doi: 10.1172/JCI119772. [DOI] [PMC free article] [PubMed] [Google Scholar]