

Abstract
This study focused on identifying reproducible effects of dietary supplementation with a mannan oligosaccharide (MOS) on the broiler cecal bacterial community structure and function in a commercial production setting. Two separate trials, each with a control and a supplemented group, were carried out in the same commercial location and run concurrently. Approximately 10,000 birds from the same commercial hatchery were mirror imaged into each of four commercial broiler sheds and fed either a control or supplemented diet. Cecal contents were obtained on days 7, 21, and 35 posthatch from 12 randomly caught broilers from each group. Bacterial pyrosequencing was performed on all samples, with approximately 250,000 sequences obtained per treatment per time point. The predominant phyla identified at all three time points in both trials were Firmicutes, Bacteroidetes, Proteobacteria, Actinobacteria, and Tenericutes, representing >99% of all sequences. MOS supplementation altered the bacterial community composition from 7 days supplementation through 35 days supplementation. Bacteroidetes appeared to be replacing Firmicutes as a result of supplementation, with the most noticeable effects after 35 days. The effects of supplementation were reproducible across both trials. PICRUSt was used to identify differences between the functional potentials of the bacterial communities as a result of MOS supplementation. Using level 3 KEGG ortholog function predictions, differences between control and supplemented groups were observed, with very strong segregation noted on day 35 posthatch in both trials. This indicated that alterations of bacterial communities as a result of MOS are likely to alter the functional capability of the cecum.
INTRODUCTION
The gastrointestinal microbiota plays a vital role in nutritional, physiological, and protective functions in animals (1). An understanding and a description of the intestinal microbial communities in broilers are important for the development of new feed additives and the appropriate manipulation of diets to improve broiler performance, health, and welfare (2). The intestinal microbiota has a major impact on the bioavailability and bioactivity of dietary components by consuming, storing, and circulating nutrients effectively, while also impacting the host's ability to resist infection, thereby making an essential contribution to host health and performance (3). Poor intestinal health in poultry is associated with increased susceptibility to infectious disease and colonization by pathogens (4). Bacterial-disease outbreaks impose significant constraints on poultry production, adversely impacting the poultry industry by reducing animal welfare and productivity through disease, poor digestion, and poor nutrient absorption. This, in turn, can lead to significant losses for the farmer and can increase the potential for the contamination of poultry products marketed for human consumption (5).
Traditionally, antibiotics have been used in poultry feed at subtherapeutic levels to prevent clinical and subclinical levels of disease, leading to improved growth rates and feed efficiencies (6). An increase in food safety concerns resulting from extensive antibiotic use has seen the poultry industry challenged in recent years as safer meat, free from antibiotics and disease, has become a requirement within the European Union (7). Consumer demand for antibiotic-free meat has also increased within the United States and other antibiotic-using countries as a result of concerns about the spread of antibiotic resistance, making it necessary for poultry producers to find suitable replacements for antibiotic growth promoters (8). There are many feed supplements that are focused on stabilizing the gut microflora to aid intestinal health and decrease the animals' susceptibility to disease, e.g., prebiotics, probiotics, and organic acids (9). Prebiotics, such as mannan oligosaccharides (MOS), have been found to have beneficial effects on broilers. The effects of MOS supplementation on bird health and performance have been studied comprehensively, and MOS have proven effective at improving weight gain and feed conversion efficiencies while also protecting against infection through pathogen binding (10, 11). However, little is known about the effect of MOS supplementation on the unculturable bacterial community of birds. To understand and exploit the gut microbiota and the impact of its manipulation on health and performance, it is necessary to decipher the content, diversity, and particularly the function of the microbial gut community. Therefore, high-throughput sequencing of the V4 to V6 variable region of the 16S rRNA gene was used in this study to assess the cecal microbial diversity across two broiler trials in response to MOS supplementation in a standard production setting. Profiling phylogenetic marker genes, such as the 16S rRNA gene, is a key tool for studies trying to understand these microbial communities but does not provide direct evidence of a community's functional capabilities. This study also focused on using a computational approach, PICRUSt (http://picrust.github.io/picrust/), to predict the effect of altering the bacterial community structure with MOS material (Actigen; Alltech, Nicholasville, KY) on the overall function of the broiler cecal microbiota (12). The phylogenetic and functional capacities of the intestinal microbiota are of great interest to help understand its roles in health and physiological outcomes. More information is needed to help with designing and understanding the roles of specific nutritional intervention strategies. This knowledge will help the poultry industry in developing future nutritional strategies and preventing disease.
MATERIALS AND METHODS
Experimental design, sample collection, and preservation.
Two broiler trials were performed concurrently at a commercial production site in Ireland. All animals were taken from a commercial hatchery and transported to the commercial sheds on the day of hatching. Approximately 10,000 birds were mirror imaged from the hatchery into each of four sheds, where they received either a control standard commercial corn-soy diet or a standard diet plus MOS (Actigen; Alltech Biotechnology, Nicholasville, KY) at the manufacturer's recommended inclusion rates (800 grams per ton [g t−1] [starter ration] and 400 g t−1 [grower ration]). The birds were raised and fed under typical commercial production conditions, receiving feed and water ad libitum. All other conditions were kept uniform for all sheds. At days 7, 21, and 35 posthatch, the intact cecal pouches of 12 randomly caught birds per shed were removed immediately after euthanization. Animals were euthanized in accordance with humane killing protocols as set forth in European Union Council Regulation (EC) 1099/2009. The cecal contents were placed into sterile tubes containing sterilized 20% (wt/vol) maltodextrin, which acts as a lyoprotectant in the lyophilization process. The tubes were kept frozen on dry ice, transported within 8 h, lyophilized, and stored at −80°C.
Total DNA extraction and purification.
Cecal DNA was extracted as described previously (13). Briefly, 0.05 g of cecal contents was added to tubes containing 0.5 g of 0.1-mm glass beads and 0.5 g of 0.5-mm zirconia beads to which hexadecyltrimethylammonium bromide (CTAB) extraction buffer (10% CTAB in 0.7-M NaCl mixed with equal volumes of 240 mM K2HPO4, pH 8.0; Sigma-Aldrich, St. Louis, MO, USA) was added. The resulting DNA was purified using a High Pure PCR product purification kit (Roche, Basel, Switzerland) according to the manufacturer's instructions and was eluted in a final volume of 50 μl. The DNA was further diluted 1:10 with sterile Milli-Q water for subsequent analysis.
16S rRNA amplification and full-length V4 to V6 region sequencing.
The full-length V4 to V6 region from the bacterial 16S rRNA operon was amplified from cecal DNA using a universal primer set, 16S-0515F (5′-TGYCAGCMGCCGCGGTA-3′) and 16S-1061R (5′-TCACGRCACGAGCTGACG-3′), tailed on each end with the Roche multiplex identifiers (MIDs). This barcode-based primer approach allowed sequencing of multiple samples in a single sequencing run without the need for physical partitioning. The PCR conditions and reagents were similar to those described previously, and a standard concentration of 50 ng of cecal DNA was used in each reaction (13). The PCR products were purified using a High Pure PCR product purification kit (Roche, Basel, Switzerland).
Sequencing of the full-length V4 to V6 16S rRNA PCR product using Y-adapter ligation libraries and MID-barcoded PCR products integrated through PCR tailing made two distinctly barcoded rapid libraries for pooling on a GS-FLX run (http://www.454.com/). The blunt-ligation protocol generated approximately equal amounts of 5′→3′ and 3′→5′ products. Following sequencing, all the barcodes were sorted and removed, and the reads were quality assessed. The MIRA v. 3.2 assembler (http://sourceforge.net/projects/mira-assembler/files/MIRA/Older%20releases/V3.2.0/) was used to assemble the resulting forward and reverse sequences, typically with overlaps over 90% of their lengths, into contigs and singletons, with a 98% sequence similarity requirement. The assembler has a 454 sequencer-specific error model and is able to correct for base call errors, and by combining forward and reverse sequences, it recovers the full-length high-quality V4 to V6 amplicon consensus sequence, even for low-abundance taxa.
OTU picking and phylogenetic-diversity analysis.
For taxonomic assignments, two distinct de novo operational taxonomic unit (OTU) picking methods have been used. QIIME v. 1.9 (http://qiime.org/) was used on the unassembled reads to assign OTUs after de novo clustering, from GreenGenes 13.8 taxonomy (Lawrence Berkeley National Laboratory) using 97% identity, to assess alpha and beta diversity. Furthermore, BLASTN analysis was used on the assembled reads against an in-house-curated version of the RDP database (Michigan State University, East Lansing, MI) release 10.29, containing only nonredundant sequence entries with sufficiently detailed phylogenetic assignments. The best 25 BLAST hits per contig or singleton were assigned to OTUs from the NCBI taxonomy with MEGAN v. 4 (http://ab.inf.uni-tuebingen.de/software/megan4/). OTU counts were corrected for sequence numbers per contig to obtain final OTU tables (14). The generalized UniFrac (GUniFrac) (R Bioconductor v. 2 [http://www.bioconductor.org/]) approach was used to estimate pairwise distances between samples and to establish beta diversity after alpha rarefaction (15). Principal component analysis (PCA) was performed to establish two-dimensional projections of samples, reflecting the time point and the control/treatment status.
Establishment of predictive functional profiles.
QIIME v. 1.7 was used to pick OTUs from the GreenGenes 13.5 taxonomy, in closed reference mode and using 97% similarity (16, 17). Single rarefaction was performed to normalize the resulting OTU tables to 1,000 taxa per sample. PICRUSt v. 1.0 was used to correct OTU tables for known 16S rRNA gene copy numbers for each taxon and subsequently to predict metagenomes using precalculated KEGG ortholog (KO) and cluster of orthologous genes (COG) tables (12, 18). These metagenomes were summarized at different taxonomic levels prior to further normalization by single rarefaction at a depth of 1,000 gene copies per sample. The resulting BIOM (http://biom-format.org/) files were loaded into R, and functional domains with less than 100,000 gene copies cumulated across all 214 samples were filtered out. KEGG pathway gene copy numbers obtained at the class level (3) were normalized to reflect the levels observed in day 7 samples across all 24 treated individuals and controls (19). Hierarchical clustering was performed on samples using all pathways, whereas k-means partitioning (k = 5) was performed on the pathways using the day 35 treated individuals only.
Data availability.
The pyrosequencing data are accessible in the European Nucleotide Archive (Short Read Archive) under accession number ERP009698.
RESULTS AND DISCUSSION
Several factors, such as ingredients, dietary supplementation, breed, genetics, age, and environment, affect the broiler gut microbiota composition (20–23). The experiment was designed to control all nongenetic factors and, to some extent, genetic factors by selecting 2 crops of newly hatched chicks from the same hatchery and splitting each crop into a control and a supplemented unit, resulting in two separate trials in one location. The study described here provides in-depth information on the broiler cecal microbiota, with 1,547,400 high-quality 16S rRNA pyrosequencing reads from 144 samples. The purpose of this study was to identify reproducible alterations in the cecal bacterial microbiome in commercial broiler flocks in response to dietary supplementation with MOS and to attempt to attribute functional capabilities to these alterations.
Cecal microbial community dynamics in response to supplementation.
The influence of MOS on microbial community development in broiler cecal bacterial communities was analyzed by 454 sequencing. A total of 1,547,400 high-quality 16S rRNA pyrosequencing reads were obtained from 144 samples collected from the cecal contents of 12 birds at each time point, days 7, 21, and 35 posthatch, across two trials. After removing low-quality sequences and chimeras, the average number of reads generated for analysis was 10,121 (±7,172 [standard deviation {SD}]) sequences from each broiler cecum, with a median read length of 526 bases in all samples. Phylogenetic-diversity analysis indicated that our sequencing depth was sufficient for coverage of the microbial communities in the cecal samples across all time points (Fig. 1; see Fig. S1 in the supplemental material).
FIG 1.

Phylogenetic diversity (PD) indexes averaged over 12 samples per time point and treatment. (a) Trial 1. (b) Trial 2. The error bars indicate standard deviation (SD). PD_whole_tree:Timepoint, phylogenetic diversity of all samples per time point; T1, trial 1; T2, trial 2; C, control; A, MOS (Actigen); D07, day 7; D21, day 21; D35, day 35.
The sequence reads were taxonomically assigned to OTUs at 3% distance sequence similarity. Alterations in bacterial community compositions between samples were visualized using GUniFrac, and the permutational multivariate analysis of variance (PMAOV) was used to test for significance. The UniFrac distance measure is widely used to visualize distances between taxonomic trees; this method has a good balance between abundant and rare taxa. The implementation of the method requires that all taxa be at identical levels, and thus, the species levels from the per-sample taxonomies were extracted. The cecal bacterial communities of supplemented broilers were statistically distinct from those of unsupplemented broilers in both trial 1 and trial 2. Trial 1 saw significantly altered bacterial community structures at all time points (Fig. 2a to c) (P < 0.005), while trial 2 saw significantly different alterations on days 21 and 35 posthatch (Fig. 3a to c) (P < 0.005). Samples tended to cluster more strongly by time point than by treatment, indicating that age was a significant contributor to bacterial community composition (Fig. 4a and b). A gradual divergence of the bacterial community compositions as a result of dietary supplementation with MOS was noted from day 7 to days 21 and 35 posthatch in both trials (Fig. 2 and 3; see Fig. S2 in the supplemental material). The 16S rRNA gene survey data revealed that the starkest alterations in the bacterial community composition were observed at day 35 posthatch for both trials, with the earliest significant alteration observed on day 7 posthatch.
FIG 2.
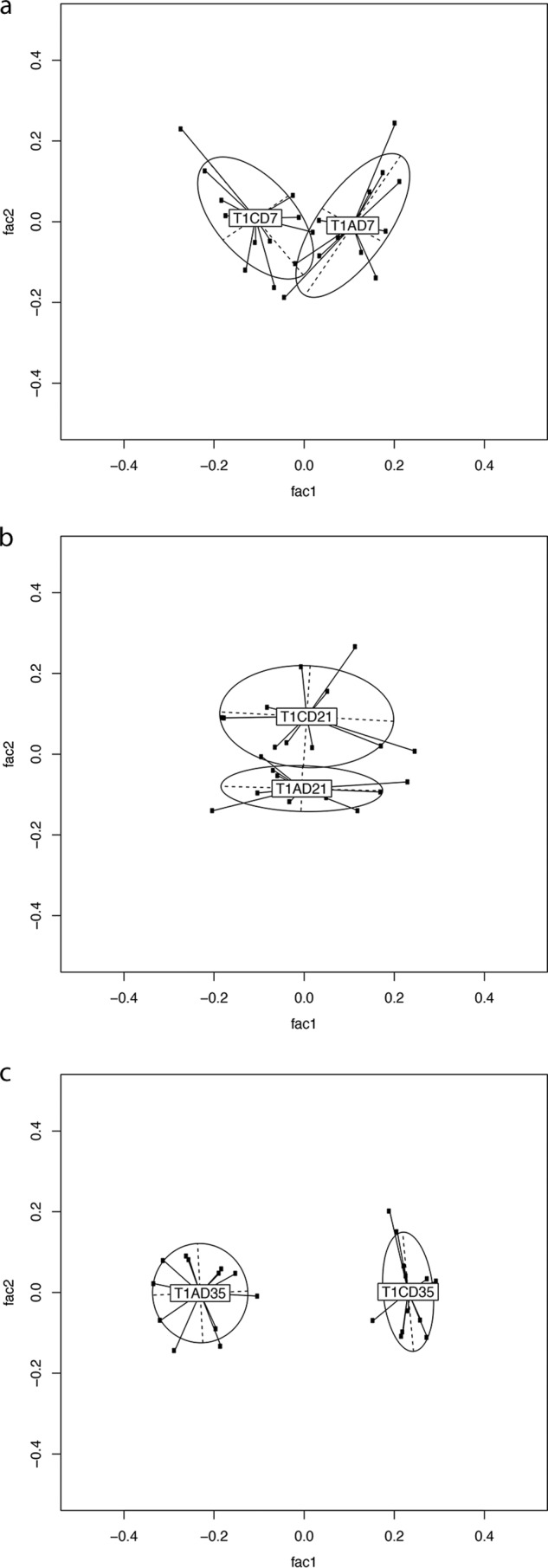
PCA was performed on pairwise distance estimates obtained from MEGAN 4 OTU picking, followed by generalized UniFrac analysis to assess for differences between control and supplemented groups at each time point from trial 1. (a) Seven days posthatch. (b) Twenty-one days posthatch. (c) Thirty-five days posthatch. n = 12 for each group. fac1 and fac2 are two principal components plotted using the s.class function from the R ade4 package. T1, trial 1; C, control; A, MOS (Actigen); D07, day 7; D21, day 21; D35, day 35.
FIG 3.
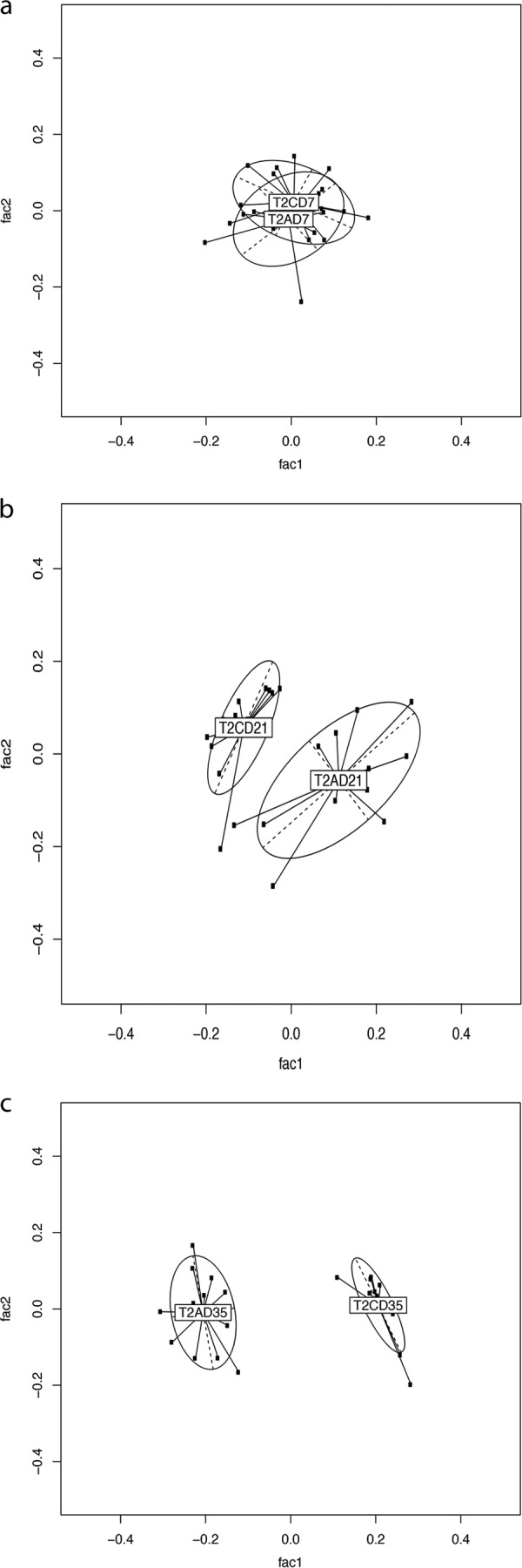
PCA was performed on pairwise distance estimates obtained from MEGAN 4 OTU picking, followed by generalized UniFrac analysis to assess for differences between control and supplemented groups from trial 2. (a) Seven days posthatch. (b) Twenty-one days posthatch. (c) Thirty-five days posthatch. n = 12 for each group. T1, trial 1; C, control; A, MOS (Actigen); D07, day 7; D21, day 21; D35, day 35.
FIG 4.
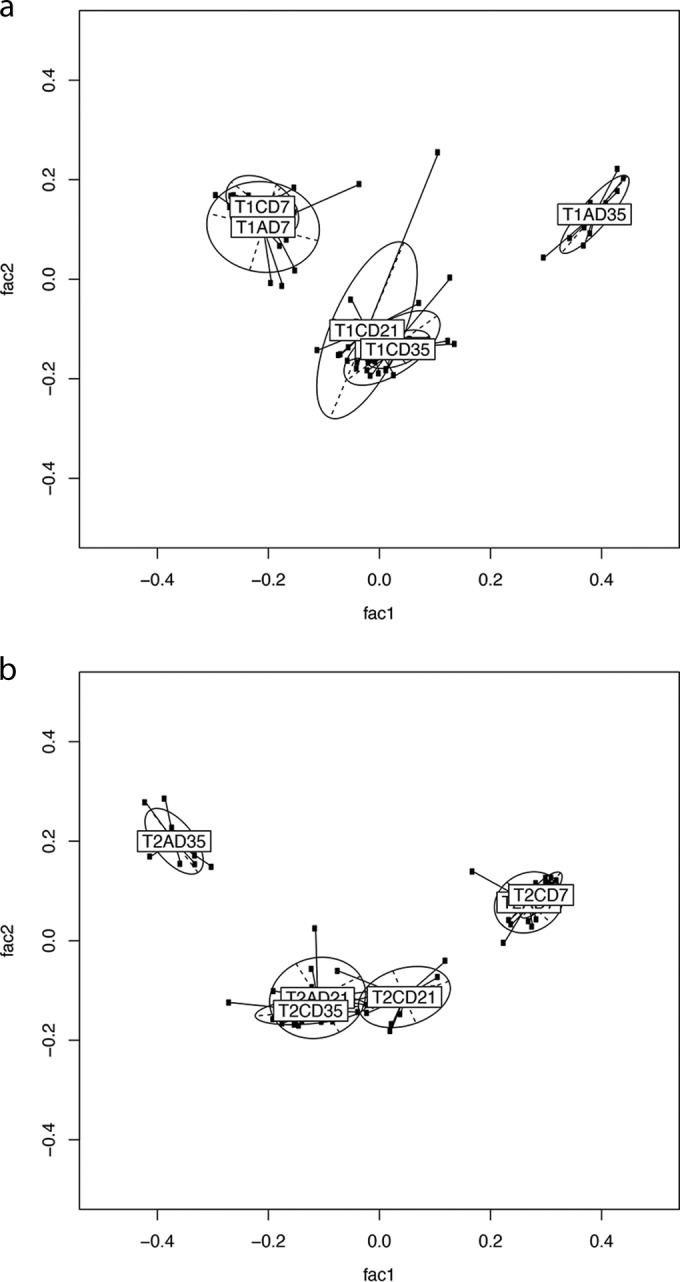
PCA was performed on pairwise distance estimates obtained from MEGAN 4 OTU picking, followed by generalized UniFrac analysis to assess for differences according to age rather than treatment. (a) Trial 1. (b) Trial 2. T1, trial 1; T2, trial 2; C, control; A, MOS (Actigen); D07, day 7; D21, day 21; D35, day 35.
The development and diversity of the bacterial intestinal microbiota in poultry have been well documented (24–28). In the present study, 454 sequencing was successfully used to compare the gut microbiota development during a total grow-out period in broilers fed either a control or MOS-supplemented diet. Sequencing analysis revealed that both diet and age had a significant impact on the composition of the gut microbiota. Relevant studies in broilers have confirmed increasing bacterial diversity and succession with age, and the current findings are in agreement with this (21, 24, 29). The increased diversification and succession of the bacterial community structure reflect the development of the immature cecal microflora toward a more mature and stable flora, a characteristic demonstrated in the juveniles of many species, including mice and humans (30, 31). Host physiology has been shown to have an impact on the development of the gastrointestinal microbiota (32). It is also likely that age-related differences arise due to a number of other factors, including resource competition between bacterial species, shifts in the host's diet, or age-related variation in the chemical and physiological state of the gastrointestinal tract (33). Broilers are harvested at a relatively young age, approximately 42 days posthatch. The results presented here support the idea that during this time, the gastrointestinal tract physiology is changing, as well as dietary content, and the cecal microflora must adapt accordingly, leaving the microbiota transient in nature.
Dietary supplementation has been shown to modify the cecal microbiota in broilers in many studies (2, 9). Previous studies involving MOS supplementation in poultry have been carried out in both research and clean facility settings and have demonstrated alterations of the bacterial community structure (13, 34). Due to the highly variable nature of the avian and broiler microbiotas across different flocks and even between birds in a flock (25, 35), there is concern that prebiotics and other supplements may be effective only under limited conditions and may never be usefully employed across the industry. As evidenced in the present study, MOS is capable of altering the microbial community structure of broiler cecal contents throughout the supplementation period in a typical production setting. Subsequently, the inclusion of MOS, despite the location, age, and growth setting, has been demonstrated, both here and in the relevant literature (13, 34), to have a direct influence on the bacterial community composition in broilers.
Taxonomic composition.
BLASTN analysis against the RDP database was used for taxonomic assignments. The best 25 BLAST hits per contig or singleton were assigned to OTUs from the NCBI taxonomy with MEGAN v. 4. On day 7 posthatch, the cecal bacterial communities were classified into five known bacterial phyla (Fig. 5). Another group of bacterial sequences, which could not be classified into any known phylum, were labeled “unassigned” in three of the four treatment groups. Six known bacterial phyla, along with an unassigned group of sequences, were found on day 21 posthatch, while on day 35 posthatch, eight known phyla and a group of unclassified sequences were identified (Fig. 5). At all three sampling time points and in both trials, the predominant phyla were Firmicutes, Bacteroidetes, Proteobacteria, Actinobacteria, and Tenericutes, representing >99% of all sequences, with the remaining percentage being made up of the phyla Cyanobacteria, Streptophyta, Verrucomicrobia, and Lentisphaerae. The predominant phylum in both trials on all sampling days for the control group was Firmicutes, followed by Bacteroidetes or Proteobacteria. This trend was also reflected in the supplemented group at days 7 and 21 posthatch, but not at day 35 posthatch. At that time point, phylum Bacteroidetes dominance increased to become the most dominant phylum in trial 1, and it comprised a significant proportion (≥41%) of all sequences in trial 2. Bacteroidetes appeared to be replacing Firmicutes as a result of supplementation with MOS, as the relative proportions of Firmicutes decreased to just 43% and 53% of all sequences, respectively, in trial 1 and trial 2 compared to 76% and 79% in the control groups at that time point.
FIG 5.

Bacterial phylum distributions (percentages) at each time point in control and MOS-supplemented groups for both trial 1 and trial 2. n = 12 for each treatment per time point.
The most dominant bacterial classes across all days and treatments were the Clostridia, Bacilli, and Bacteroidia, with the Negativicutes increasing to the third most dominant class on day 35 posthatch across all groups (Fig. 6). The class Clostridia dominated on days 7 and 21 posthatch, but at day 35, the shifts observed at the phylum level became more apparent, as the relative abundance of the class Bacteroidia significantly increased, with a corresponding decrease in the class Clostridia (Fig. 6). A consistent decrease across all bacterial families within the phylum Firmicutes was noted in the supplemented groups on day 35 in both trials and was most notable in the four most dominant families the Ruminococacceae, the Clostridiaceae, the Lachnospiraceae, and the Lactobacillaceae (see Fig. S3 in the supplemental material). Within the phylum Bacteroidetes, the family Bacteroidaceae was the only family seen to have its relative abundance increase as a result of supplementation compared with the control (see Fig. S4 in the supplemental material). No notable changes were observed for Proteobacteria families as a result of supplementation; however, the relative abundances of these families decreased on day 35 compared to day 21 (see Fig. S5 in the supplemental material).
FIG 6.

Bacterial class distributions (percentages) at each time point in control and MOS-supplemented groups for both trial 1 and trial 2. n = 12 for each treatment per time point.
During the present trials, three predominant phyla were identified across all time points in the broiler cecum—Firmicutes, Bacteroidetes, and Proteobacteria—which is consistent with what has been observed in the relevant literature for broilers and other avian species (13, 34–36). The relative abundances of the predominant phyla observed across both trials in this study were similar, and the bacterial communities were altered in the same manner as a result of supplementation. When comparing previous studies in broilers, the relative proportions of bacterial phyla, particularly the Firmicutes, Bacteroidetes, and Proteobacteria, have shown variation between broiler trials. Previous studies have attributed varying microbiota profiles across different trials to setting, location, and feed variation; in fact, the study by Waite and Taylor (35) showed that the biggest factor contributing to microbiota variation was the study itself. This variation was nullified in the case of the current trials, indicating the robustness of the experimental design. The consistency in the microbiota between trials observed here, therefore, may be due to the highly controlled nature of the experiment, with birds coming from the same hatchery and being fed the same diet in the same location. However, the eggs were from different flocks, which introduced a source of variation. The results here add to growing evidence that location is the biggest factor in determining the microbiota composition (35).
Previous work in both broilers and turkeys has noted increases in the phylum Firmicutes and decreases in Bacteroidetes and Proteobacteria as a result of supplementation with MOS (13, 34); however, members of the phylum Bacteroidetes were much more abundant in MOS-supplemented birds in both trials that were run for this experiment. This observation was repeated in both trial 1 and trial 2, and while large variation in broiler gut microbiota composition has been discussed, this appears to be a treatment effect, as all other traditional sources of variation have been eliminated. It is likely that the contrasting bacterial alterations between these trials in response to MOS supplementation (i.e., previously, an increase in Firmicutes, and currently, an increase in Bacteroidetes) are possibly due to two factors. The present trials were carried out in a commercial production environment, whereas the previous studies were carried out in a controlled clinical-type research setting, thus placing different selective pressures on the gut microbiota. Different approaches for phylogenetic identification were also used in the current study and previous studies. In-depth 454 sequencing was used for this study versus 16S rRNA clone library identification, with 454 sequencing giving much more depth of coverage than 16S rRNA clone library analysis. The effect of MOS supplementation on the Bacteroidetes has not been studied previously, perhaps because the phylum is not of major concern with regard to avian-pathogenic diseases and because of the difficulties associated with culturing these bacteria. Bacteroidetes species are common bacteria in the gut, involved in many important metabolic activities, including the fermentation of carbohydrates, the utilization of nitrogenous substances, the biotransformation of bile acids, and the prevention of pathogen colonization (25, 37, 38). Numerous efforts, especially dietary intervention and litter management, have been attempted to consistently modulate the microbiome to enhance feed conversion and gut health (9, 23, 26, 29). What we have observed here is a concordant effect of MOS supplementation on microbial modulation across two individual trials with birds raised in different sheds, using birds from the same hatchery but different crops.
Functional potential of bacterial community composition.
Using PICRUSt, we identified significant differences between the functional potentials of the bacterial community compositions using the level 3 KEGG ortholog function predictions (Fig. 7). Differences between control and supplemented groups using the top-level functional categories were observed, with very strong segregation noted on day 35 posthatch in both trials (Fig. 7). These functional categories included the notable enrichment of numerous pathways, including carbon fixation, chaperone and folding-catalyst, energy metabolism, homologous-recombination, oxidative-phosphorylation, and ribosome pathways in the supplemented groups, and are attributed strongly to the class Bacteroidia (Fig. 8). Energy metabolism-, chaperone- and folding-catalyst-, and carbon fixation-associated pathways and their associated relative gene copy numbers exhibited 35% to >50% increases in the day 35 supplemented group compared with the control group. Several functional categories, such as those associated with sporulation, transporters, transcription factors, and ABC transporters, diminished to 50% or more in the day 35 treated samples (Fig. 7). These pathways are attributed to the classes Clostridia and Bacilli (Fig. 8).
FIG 7.

Heat map reflecting hierarchical clustering of samples and k-means partitioning of normalized predicted gene copy number counts classified into KEGG pathways. The heat map color codes reflect changes. Dark blue represents −50% change, whereas dark red represents +50% change. T1, trial 1; T2, trial 2; C, control; A, MOS (Actigen); D07, day 7; D21, day 21; D35, day 35.
FIG 8.

Relative increases and decreases in abundance of OTUs and their associated functional potentials as a result of MOS supplementation. Actigen, MOS; ucrC=99, a sequence identity threshold of 99% using the UCLUST algorithm.
The hierarchical cluster analysis based on the metagenome functional potential associated with the KEGG database showed a pattern of clustering with respect to that obtained by taxonomic composition on day 35 posthatch in both trial 1 and trial 2. This partitioning of the predicted functional pathways indicated that alterations of bacterial communities as a result of MOS supplementation are also likely to alter the functional capability of the bacterial community in the cecum. From a functional standpoint, the enrichment of these pathways could have a number of implications. It must be noted that, although well validated, PICRUSt's predictive approach cannot confirm with absolute certainty the functional capabilities of the metagenome.
Microorganisms are able to gain energy from multiple metabolic pathways, such as carbon fixation, nitrogen metabolism, and sulfur metabolism (39). A relative increase in genes associated with energy metabolism pathways was observed in this study and may indicate a greater ability of the supplemented group to extract more energy from the same feed. Much research in both human and mouse models has shown that increases in the phylum Bacteroidetes are associated with lean phenotypes and increases in Firmicutes are related to the development of an obese phenotype (40, 41). It has been surmised that Firmicutes-dominant phenotypes have been related to an increased ability to harvest energy from the diet, with carbohydrate metabolism the important factor (42–44). It is unclear if these findings relate to poultry, due to the physiological differences and bacterial compositional differences. Singh et al. (28) examined the bacterial community composition changes in high-feed-conversion-ratio (FCR) and low-FCR broilers and found that those birds with a high FCR tended to have higher levels of Bacteroidetes than birds that had low FCRs, which contradicts what has been documented in humans and mice (28). The results predicted in this study using PICRUSt, however, align with what has been observed by Singh, i.e., that increases in Bacteroidetes may be related to increases in energy metabolism. It is unlikely that the findings based on human and mouse models can be compared to broilers with the expectation of similar results. Broilers have been reared specifically for rapid growth and weight gain, with very specific and precise diet formulations. This is in contrast to human studies, which are being examined for fat deposition as a result of overconsuming high-calorie energy-dense westernized diets. Verifying the results obtained by Singh et al. (28) was not possible, as monitoring individual broilers for FCR was beyond the scope of the study, but it warrants further investigation given the results obtained here following MOS supplementation.
Bacterial molecular chaperones and folding catalysts have been recognized as potent immunogens, active immunomodulators, and inducers of cross-reactive immunity and autoimmunity (45). Previous work carried out studying the effects of MOS on the immune system has shown improvements in immune response (46–48). These effects may be linked to alterations in the microbiota profile (2, 49). Prebiotics have been shown to enhance the immune response in chickens, resulting in enhanced ability to clear pathogens from invasion (50). The results in this study show an uptake in genes associated with molecular chaperones and folding catalysts, which potentially offers insight into why supplementation with MOS has been shown to have an immunomodulatory effect. The effects of dietary supplementation on the immune system have previously been documented by Janardhana et al. (51) and Baurhoo et al. (48), who demonstrated beneficial immune responses, such as attenuated inflammation with Salmonella lipopolysaccharide (LPS) and increased antibody titers as a result of supplementation with prebiotics. Improving our understanding of this could have major implications for the poultry industry, as improving performance and reducing disease are among the most important goals in poultry research.
The carbon fixation pathway in prokaryotes has also shown a strong uptake in relative gene copy numbers. Microbial fermentation of plant fiber polysaccharides within poultry feed is a major process in the cecum, and short-chain fatty acids (SCFA) are the major end products of this process (3, 52). In general, Bacteroidetes represent the primary fermenters that transform simple sugars derived from the breakdown of complex carbohydrates to organic acids, including SCFA and hydrogen. Secondary fermenters, such as Clostridium species and butyrate-producing bacteria, further utilize the organic acids to generate additional SCFA (53). The pathways of formation of the SCFA are not well elucidated, but propionate is known to be formed by a CO2 fixation pathway in the human colon (54, 55). It is possible that an uptake in genes associated with this pathway is related to the increased relative abundance of the Bacteroidetes microbiota, as shown in Fig. 8. Bacteroidetes are well-known plant polysaccharide degraders, as well as propionate producers, and so may have led to an increase in the SFCA concentration (38). SCFA concentrations were not measured as part of this study; however, future studies should take this into account to help better understand the functional implications of altering the bacterial community structure.
The significant depletion of the pathways related to sporulation, transporters, and transcription factors is possibly due to the associated decrease in the classes Clostridia and Bacilli. Sporulation is most commonly associated with the Bacilli and Clostridia (56). Induction of sporulation patterns in Bacillus spp. is usually attributed to nutrient starvation, and in many clostridial species, it is due to cessation of growth or exposure to oxygen in the presence of excess nutrients (57, 58). A reduction in the copy numbers of genes associated with these pathways is unlikely to be due to oxygen exposure, nutrient depletion, or increased growth but is most likely due to a reduction in the overall quantities of the bacteria associated with the genes. ABC transporters utilize the energy of ATP binding and hydrolysis to transport substrates across cellular membranes (59). They are essential to cell viability, virulence, and pathogenicity and vital for cell survival. Transcription factors bind to DNA, controlling the rate of transcription to RNA. It is possible that both of these pathways are reduced due to the decrease in Bacilli and Clostridia. Firmicutes play an important role in fermentation of dietary compounds, transporting many substrates across their cellular membranes, it is possible that a decrease in ABC transporter genes is related to a reduction in ATP-powered translocation of these substrates across membranes by a reduction in the number of Firmicutes, and this may be why we also see a reduction in transcription factors (60).
Conclusions.
A better understanding of the interactions of the intestinal microbiome with the host and with ingested feed is necessary to further develop poultry nutrition and gut health. A lack of adequate knowledge on the bacterial diversity, both phylogenetically and functionally, in poultry intestines is considered one of the major information gaps that limit our understanding of such interactions. Metagenomic analysis allows the relative abundances of most species to be determined and used to generate a data set for the assessment of the functional potential of each community. Intriguingly, recent metagenomic studies have suggested that a functional rather than a taxonomic core might be present within a given microbiome and that changes in these cores might lead to different physiological states (61, 62). The current study used a computational approach to project functions on the bacterial communities identified within the broiler cecum. It found that MOS supplementation consistently and reproducibly altered the cecal microbiota and increased the levels of Bacteroidetes in the broiler cecum.
The members of the genus Bacteroides generally have very high hydrolytic activity. They have been characterized as short-chain fatty-acid producers and are among the most effective degraders of indigestible carbohydrates, including resistant starch and cellulose. The prevalence of bacterial OTUs related to the degradation of cellulose and starch is unlikely to be coincidental, due to the composition of broiler feed. It is therefore to be expected that bacteria that play a major role in this digestion may be more abundant in those birds that are efficient at retaining and using energy. The results obtained here may help us to better understand the mode of action behind improved weight gain and FCR that are obtained as a result of MOS supplementation. Further research is warranted to see if these predicted functions translate into phenotypical changes in commercial broiler settings.
Supplementary Material
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.04194-14.
REFERENCES
- 1.Yeoman CJ, White BA. 2014. Gastrointestinal tract microbiota and probiotics in production animals. Annu Rev Anim Biosci 2:469–486. doi: 10.1146/annurev-animal-022513-114149. [DOI] [PubMed] [Google Scholar]
- 2.Sugiharto S. 12 June 2014. Role of nutraceuticals in gut health and growth performance of poultry. J Saudi Soc Agric Sci doi:http://dx.doi.org/10.1016/j.jssas.2014.06.001.
- 3.Rinttilä T, Apajalahti J. 2013. Intestinal microbiota and metabolites—implications for broiler chicken health and performance. J Appl Poult Res 22:647–658. doi: 10.3382/japr.2013-00742. [DOI] [Google Scholar]
- 4.Yegani M, Korver DR. 2008. Factors affecting intestinal health in poultry. Poult Sci 87:2052–2063. doi: 10.3382/ps.2008-00091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mead G, Lammerding AM, Cox N, Doyle MP, Humbert F, Kulikovskiy A, Panin A, do Nascimento VP, Wierup M. 2010. Scientific and technical factors affecting the setting of Salmonella criteria for raw poultry: a global perspective. J Food Prot 73:1566–1590. [DOI] [PubMed] [Google Scholar]
- 6.Marshall BM, Levy SB. 2011. Food animals and antimicrobials: impacts on human health. Clin Microbiol Rev 24:718–733. doi: 10.1128/CMR.00002-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Maron DF, Smith TJS, Nachman KE. 2013. Restrictions on antimicrobial use in food animal production: an international regulatory and economic survey. Globalization and Health 9:48–48. doi: 10.1186/1744-8603-9-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sofos JN. 2008. Challenges to meat safety in the 21st century. Meat Sci 78:3–13. doi: 10.1016/j.meatsci.2007.07.027. [DOI] [PubMed] [Google Scholar]
- 9.Yang Y, Iji PA, Choct M. 2009. Dietary modulation of gut microflora in broiler chickens: a review of the role of six kinds of alternatives to in-feed antibiotics. World Poult Sci J 65:97–114. doi: 10.1017/S0043933909000087. [DOI] [Google Scholar]
- 10.Spring P, Wenk C, Dawson KA, Newman KE. 2000. The effects of dietary mannaoligosaccharides on cecal parameters and the concentrations of enteric bacteria in the ceca of salmonella-challenged broiler chicks. Poult Sci 79:205–211. doi: 10.1093/ps/79.2.205. [DOI] [PubMed] [Google Scholar]
- 11.Connolly DM, Ha A. 2011. Meta-analysis summary of broiler chicken trials with dietary actigen (2009–2011). Int J Poul Sci 10:819–824. doi: 10.3923/ijps.2011.819.824. [DOI] [Google Scholar]
- 12.Langille MGI, Zaneveld J, Caporaso JG, McDonald D, Knights D, Reyes JA, Clemente JC, Burkepile DE, Vega Thurber RL, Knight R, Beiko RG, Huttenhower C. 2013. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat Biotechnol 31:814–821. doi: 10.1038/nbt.2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Corrigan A, Horgan K, Clipson N, Murphy RA. 2011. Effect of dietary supplementation with a Saccharomyces cerevisiae mannan oligosaccharide on the bacterial community structure of broiler cecal contents. Appl Environ Microbiol 77:6653–6662. doi: 10.1128/AEM.05028-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mitra S, Stark M, Huson DH. 2011. Analysis of 16S rRNA environmental sequences using MEGAN. BMC Genomics 12(Suppl 3):S17. doi: 10.1186/1471-2164-12-S3-S17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen J, Bittinger K, Charlson ES, Hoffmann C, Lewis J, Wu GD, Collman RG, Bushman FD, Li H. 2012. Associating microbiome composition with environmental covariates using generalized UniFrac distances. Bioinformatics 28:2106–2113. doi: 10.1093/bioinformatics/bts342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McDonald D, Price MN, Goodrich J, Nawrocki EP, DeSantis TZ, Probst A, Andersen GL, Knight R, Hugenholtz P. 2012. An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J 6:610–618. doi: 10.1038/ismej.2011.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Pena AG, Goodrich JK, Gordon JI, Huttley GA, Kelley ST, Knights D, Koenig JE, Ley RE, Lozupone CA, McDonald D, Muegge BD, Pirrung M, Reeder J, Sevinsky JR, Turnbaugh PJ, Walters WA, Widmann J, Yatsunenko T, Zaneveld J, Knight R. 2010. QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7:335–336. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kanehisa M, Goto S, Sato Y, Kawashima M, Furumichi M, Tanabe M. 2014. Data, information, knowledge and principle: back to metabolism in KEGG. Nucleic Acids Res 42:D199–D205. doi: 10.1093/nar/gkt1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kanehisa M, Goto S, Sato Y, Furumichi M, Tanabe M. 2012. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res 40:D109–D114. doi: 10.1093/nar/gkr988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Aggrey S, Karnuah A, Sebastian B, Anthony N. 2010. Genetic properties of feed efficiency parameters in meat-type chickens. Genet Sel Evol 42:25. doi: 10.1186/1297-9686-42-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lu J, Idris U, Harmon B, Hofacre C, Maurer JJ, Lee MD. 2003. Diversity and succession of the intestinal bacterial community of the maturing broiler chicken. Appl Environ Microbiol 69:6816–6824. doi: 10.1128/AEM.69.11.6816-6824.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Torok VA, Hughes RJ, Ophel-Keller K, Ali M, MacAlpine R. 2009. Influence of different litter materials on cecal microbiota colonization in broiler chickens. Poult Sci 88:2474–2481. doi: 10.3382/ps.2008-00381. [DOI] [PubMed] [Google Scholar]
- 23.Torok VA, Ophel-Keller K, Loo M, Hughes RJ. 2008. Application of methods for identifying broiler chicken gut bacterial species linked with increased energy metabolism. Appl Environ Microbiol 74:783–791. doi: 10.1128/AEM.01384-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Danzeisen JL, Kim HB, Isaacson RE, Tu ZJ, Johnson TJ. 2011. Modulations of the chicken cecal microbiome and metagenome in response to anticoccidial and growth promoter treatment. PLoS One 6:e27949. doi: 10.1371/journal.pone.0027949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sergeant MJ, Constantinidou C, Cogan TA, Bedford MR, Penn CW, Pallen MJ. 2014. Extensive microbial and functional diversity within the chicken cecal microbiome. PLoS One 9:e91941. doi: 10.1371/journal.pone.0091941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Singh KM, Shah T, Deshpande S, Jakhesara SJ, Koringa PG, Rank DN, Joshi CG. 2012. High throughput 16S rRNA gene-based pyrosequencing analysis of the fecal microbiota of high FCR and low FCR broiler growers. Mol Biol Rep 39:10595–10602. doi: 10.1007/s11033-012-1947-7. [DOI] [PubMed] [Google Scholar]
- 27.Stanley D, Geier MS, Hughes RJ, Denman SE, Moore RJ. 2013. Highly variable microbiota development in the chicken gastrointestinal tract. PLoS One 8:e84290. doi: 10.1371/journal.pone.0084290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Singh KM, Shah TM, Reddy B, Deshpande S, Rank DN, Joshi CG. 2014. Taxonomic and gene-centric metagenomics of the fecal microbiome of low and high feed conversion ratio (FCR) broilers. J Appl Genet 55:145–154. doi: 10.1007/s13353-013-0179-4. [DOI] [PubMed] [Google Scholar]
- 29.Torok VA, Allison GE, Percy NJ, Ophel-Keller K, Hughes RJ. 2011. Influence of antimicrobial feed additives on broiler commensal posthatch gut microbiota development and performance. Appl Environ Microbiol 77:3380–3390. doi: 10.1128/AEM.02300-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vaahtovuo J, Toivanen P, Eerola E. 2001. Study of murine faecal microflora by cellular fatty acid analysis; effect of age and mouse strain. Antonie Van Leeuwenhoek 80:35–42. doi: 10.1023/A:1012058107731. [DOI] [PubMed] [Google Scholar]
- 31.La Rosa PS, Warner BB, Zhou Y, Weinstock GM, Sodergren E, Hall-Moore CM, Stevens HJ, Bennett WE Jr, Shaikh N, Linneman LA, Hoffmann JA, Hamvas A, Deych E, Shands BA, Shannon WD, Tarr PI. 2014. Patterned progression of bacterial populations in the premature infant gut. Proc Natl Acad Sci U S A 111:12522–12527. doi: 10.1073/pnas.1409497111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Macfarlane GT, Macfarlane LE. 2009. Acquisition, evolution and maintenance of the normal gut microbiota. Dig Dis 27(Suppl 1):90–98. doi: 10.1159/000268127. [DOI] [PubMed] [Google Scholar]
- 33.van Dongen WF, White J, Brandl H, Moodley Y, Merkling T, Leclaire S, Blanchard P, Danchin E, Hatch S, Wagner R. 2013. Age-related differences in the cloacal microbiota of a wild bird species. BMC Ecol 13:11. doi: 10.1186/1472-6785-13-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Corrigan A, Horgan K, Clipson N, Murphy RA. 2012. Effect of dietary prebiotic (mannan oligosaccharide) supplementation on the caecal bacterial community structure of turkeys. Microb Ecol 64:826–836. doi: 10.1007/s00248-012-0046-6. [DOI] [PubMed] [Google Scholar]
- 35.Waite D, Taylor M. 2014. Characterising the avian gut microbiota: membership, driving influences, and potential function. Front Microbiol 5:223. doi: 10.3389/fmicb.2014.00223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wei S, Morrison M, Yu Z. 2013. Bacterial census of poultry intestinal microbiome. Poult Sci 92:671–683. doi: 10.3382/ps.2012-02822. [DOI] [PubMed] [Google Scholar]
- 37.Phillips ML. 2009. Gut reaction: environmental effects on the human microbiota. Environ Health Perspect 117:A198–A205. doi: 10.1289/ehp.117-a198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wexler HM. 2007. Bacteroides: the good, the bad, and the nitty-gritty. Clin Microbiol Rev 20:593–621. doi: 10.1128/CMR.00008-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tu Q, He Z, Li Y, Chen Y, Deng Y, Lin L, Hemme CL, Yuan T, Van Nostrand JD, Wu L, Zhou X, Shi W, Li L, Xu J, Zhou J. 2014. Development of HuMiChip for functional profiling of human microbiomes. PLoS One 9:e90546. doi: 10.1371/journal.pone.0090546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kotzampassi K, Giamarellos-Bourboulis EJ, Stavrou G. 2014. Obesity as a consequence of gut bacteria and diet interactions. ISRN Obes 2014:651895. doi: 10.1155/2014/651895. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 41.Ley RE, Bäckhed F, Turnbaugh P, Lozupone CA, Knight RD, Gordon JI. 2005. Obesity alters gut microbial ecology. Proc Natl Acad Sci U S A 102:11070–11075. doi: 10.1073/pnas.0504978102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Murphy EF, Cotter PD, Healy S, Marques TM, O'Sullivan O, Fouhy F, Clarke SF, O'Toole PW, Quigley EM, Stanton C, Ross PR, O'Doherty RM, Shanahan F. 2010. Composition and energy harvesting capacity of the gut microbiota: relationship to diet, obesity and time in mouse models. Gut 59:1635–1642. doi: 10.1136/gut.2010.215665. [DOI] [PubMed] [Google Scholar]
- 43.Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI. 2006. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 444:1027–1031. doi: 10.1038/nature05414. [DOI] [PubMed] [Google Scholar]
- 44.Tremaroli V, Backhed F. 2012. Functional interactions between the gut microbiota and host metabolism. Nature 489:242–249. doi: 10.1038/nature11552. [DOI] [PubMed] [Google Scholar]
- 45.Henderson B, Pockley AG. 2010. Molecular chaperones and protein-folding catalysts as intercellular signaling regulators in immunity and inflammation. J Leukoc Biol 88:445–462. doi: 10.1189/jlb.1209779. [DOI] [PubMed] [Google Scholar]
- 46.Xiao R, Power RF, Mallonee D, Routt K, Spangler L, Pescatore AJ, Cantor AH, Ao T, Pierce JL, Dawson KA. 2012. Effects of yeast cell wall-derived mannan-oligosaccharides on jejunal gene expression in young broiler chickens. Poult Sci 91:1660–1669. doi: 10.3382/ps.2011-02035. [DOI] [PubMed] [Google Scholar]
- 47.Che TM, Johnson RW, Kelley KW, Van Alstine WG, Dawson KA, Moran CA, Pettigrew JE. 2011. Mannan oligosaccharide modulates gene expression profile in pigs experimentally infected with porcine reproductive and respiratory syndrome virus. J Anim Sci 89:3016–3029. doi: 10.2527/jas.2010-3366. [DOI] [PubMed] [Google Scholar]
- 48.Baurhoo B, Ferket P, Ashwell CM, de Oliviera J, Zhao X. 2012. Cell walls of Saccharomyces cerevisiae differentially modulated innate immunity and glucose metabolism during late systemic inflammation. PLoS One 7:e30323. doi: 10.1371/journal.pone.0030323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kogut MH. 2013. The gut microbiota and host innate immunity: regulators of host metabolism and metabolic diseases in poultry? J Appl Poult Res 22:637–646. doi: 10.3382/japr.2013-00741. [DOI] [Google Scholar]
- 50.Lea H, Spring P, Taylor-Pickard J, Burton E. 2012. A natural carbohydrate fraction Actigen from Saccharomyces cerevisiae cell wall: effects on goblet cells, gut morphology and performance of broiler chickens. J Appl Anim Nutr 1:e9. doi: 10.1017/jan.2013.6. [DOI] [Google Scholar]
- 51.Janardhana V, Broadway MM, Bruce MP, Lowenthal JW, Geier MS, Hughes RJ, Bean AGD. 2009. Prebiotics modulate immune responses in the gut-associated lymphoid tissue of chickens. J Nutr 139:1404–1409. doi: 10.3945/jn.109.105007. [DOI] [PubMed] [Google Scholar]
- 52.Svihus B, Choct M, Classen HL. 2013. Function and nutritional roles of the avian caeca: a review. World Poult Sci J 69:249–264. doi: 10.1017/S0043933913000287. [DOI] [Google Scholar]
- 53.Sun Y, O'Riordan MXD. 2013. Regulation of bacterial pathogenesis by intestinal short-chain fatty acids. Adv Appl Microbiol 85:93–118. doi: 10.1016/B978-0-12-407672-3.00003-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Miller TL, Wolin MJ. 1996. Pathways of acetate, propionate, and butyrate formation by the human fecal microbial flora. Appl Environ Microbiol 62:1589–1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.den Besten G, van Eunen K, Groen AK, Venema K, Reijngoud D-J, Bakker BM. 2013. The role of short-chain fatty acids in the interplay between diet, gut microbiota and host energy metabolism. J Lipid Res 54:2325–2340. doi: 10.1194/jlr.R036012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Paredes CJ, Alsaker KV, Papoutsakis ET. 2005. A comparative genomic view of clostridial sporulation and physiology. Nat Rev Microbiol 3:969–978. doi: 10.1038/nrmicro1288. [DOI] [PubMed] [Google Scholar]
- 57.Veening J-W, Murray H, Errington J. 2009. A mechanism for cell cycle regulation of sporulation initiation in Bacillus subtilis. Genes Dev 23:1959–1970. doi: 10.1101/gad.528209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sauer U, Santangelo JD, Treuner A, Buchholz M, Dürre P. 1995. Sigma factor and sporulation genes in Clostridium. FEMS Microbiol Rev 17:331–340. doi: 10.1111/j.1574-6976.1995.tb00216.x. [DOI] [PubMed] [Google Scholar]
- 59.Davidson AL, Dassa E, Orelle C, Chen J. 2008. Structure, function, and evolution of bacterial ATP-binding cassette systems. Microbiol Mol Biol Rev 72:317–364. doi: 10.1128/MMBR.00031-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rees DC, Johnson E, Lewinson O. 2009. ABC transporters: the power to change. Nat Rev Mol Cell Biol 10:218–227. doi: 10.1038/nrm2646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Vallès Y, Artacho A, Pascual-García A, Ferrús ML, Gosalbes MJ, Abellán JJ, Francino MP. 2014. Microbial succession in the gut: directional trends of taxonomic and functional change in a birth cohort of Spanish infants. PLoS Genet 10:e1004406. doi: 10.1371/journal.pgen.1004406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Turnbaugh PJ, Hamady M, Yatsunenko T, Cantarel BL, Duncan A, Ley RE, Sogin ML, Jones WJ, Roe BA, Affourtit JP, Egholm M, Henrissat B, Heath AC, Knight R, Gordon JI. 2009. A core gut microbiome in obese and lean twins. Nature 457:480–484. doi: 10.1038/nature07540. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The pyrosequencing data are accessible in the European Nucleotide Archive (Short Read Archive) under accession number ERP009698.