

Abstract
Sunlight is captured and converted to chemical energy in illuminated environments. Although (bacterio)chlorophyll-based photosystems have been characterized in detail, retinal-based photosystems, rhodopsins, have only recently been identified as important mediators of light energy capture and conversion. Recent estimates suggest that up to 70% of cells in some environments harbor rhodopsins. However, because rhodopsin autofluorescence is low—comparable to that of carotenoids and significantly less than that of (bacterio)chlorophylls—these estimates are based on metagenomic sequence data, not direct observation. We report here the use of ultrasensitive total internal reflection fluorescence (TIRF) microscopy to distinguish between unpigmented, carotenoid-producing, and rhodopsin-expressing bacteria. Escherichia coli cells were engineered to produce lycopene, β-carotene, or retinal. A gene encoding an uncharacterized rhodopsin, actinorhodopsin, was cloned into retinal-producing E. coli. The production of correctly folded and membrane-incorporated actinorhodopsin was confirmed via development of pink color in E. coli and SDS-PAGE. Cells expressing carotenoids or actinorhodopsin were imaged by TIRF microscopy. The 561-nm excitation laser specifically illuminated rhodopsin-containing cells, allowing them to be differentiated from unpigmented and carotenoid-containing cells. Furthermore, water samples collected from the Delaware River were shown by PCR to have rhodopsin-containing organisms and were examined by TIRF microscopy. Individual microorganisms that fluoresced under illumination from the 561-nm laser were identified. These results verify the sensitivity of the TIRF microscopy method for visualizing and distinguishing between different molecules with low autofluorescence, making it useful for analyzing natural samples.
INTRODUCTION
Sunlight powers most carbon fixation, and the organic carbon produced via photosynthesis is the base of ecological metabolic interactions (1). Photoheterotrophs use sunlight for energy but consume organic carbon rather than produce it. The anoxygenic photoheterotrophs that harvest light with bacteriochlorophyll (BChl)-dependent photosystems have been characterized in detail, and their contributions to the global carbon cycle are well documented (2). Recent work suggests that a surprisingly large number of photoheterotrophic microbes may capture light energy with a retinal-based, single-polypeptide photosystem, rhodopsin (3), which uses light energy to generate an electrochemical gradient across the cytoplasmic membrane that can be used for motility (4), solute transport, or ATP synthesis (5, 6). In order to determine how light energy can be used by different organisms and to accurately quantify the organisms that use light, we must be able to identify not only chlorophyll (Chl)-containing organisms, but also rhodopsin-containing cells in environmental samples.
Rhodopsins are light-sensing membrane proteins with a retinal cofactor, which undergoes a conformational change in response to absorption of a photon (7). This conformational change drives either transfer of information, via protein-protein interactions and the regulation of the expression of other genes in response to light (8), or transport of an ion across the membrane (7). Photosensory rhodopsins, which sense light and transmit a signal to other proteins, are the basis for vision in vertebrates and many invertebrates and are found in plants and fungi, as well as a variety of prokaryotic species (7). Most characterized microbial rhodopsins transport protons in response to light and are thus used to maintain the proton motive force (7), though some pump Na+ or Cl− (9–11). Proton-pumping microbial rhodopsins include the proteorhodopsins of marine bacteria (12), bacteriorhodopsins of archaea (13), the xanthorhodopsins of Salinibacter ruber (6), and the recently identified actinorhodopsins (ActR) (14). Actinorhodopsins are predicted to be light-activated proton pumps and are found in freshwater Actinobacteria (14–16). Proton-pumping rhodopsins are hypothesized to supplement the cellular energy budget under low-nutrient conditions (3, 17–19), and heterologous expression experiments have demonstrated elevated ATP production in starved, proteorhodopsin-expressing Escherichia coli cells exposed to light (20).
Given the diversity of function, it is perhaps unsurprising that rhodopsins are widespread in illuminated environments. However, detection of rhodopsins in environmental samples has been hampered by the low fluorescence yield of rhodopsin and its light-absorbing cofactor retinal. Neither can be detected using standard fluorescence-based assays, such as those developed for in vivo quantification of Chl a or BChl a in natural samples (21–23). Instead, rhodopsin abundance has been calculated based on metagenomic sequence data (3, 12, 24, 25), amplicon sequencing (15, 26, 27), quantitative PCR (QPCR) (27, 28), or cultivation (15, 29–31). These estimates show that in some marine environments, up to 70% of the cells may host a rhodopsin (3), while up to 30% are Chl a-containing cyanobacteria (32, 33) and an additional 1 to 30% contain BChl a (34). In nonmarine aquatic environments, 35 to 62% of genomes within metagenomic assemblies harbor a rhodopsin (14), while analysis of freshwater bacterioplankton metagenomic assemblies and single amplified genomes from the same locations suggested the presence of a rhodopsin in 37 to 56% and 8 to 20% of the samples, respectively (35). These studies have demonstrated that rhodopsins are both more abundant and more diverse than previously suspected (10, 36–38). However, metagenomic and metatranscriptomic data cannot demonstrate that a rhodopsin is functional, nor can they consistently identify the organism that hosts the rhodopsin. To confirm the hypothesis that in some environments, the majority of prokaryotes respond to sunlight, it must be possible to detect and quantify rhodopsin-producing cells in natural samples.
Rhodopsin fluorescence, though faint (39), has a characteristic absorption peak in the 480- to 560-nm range and fluoresces in the 600- to 900-nm range (40), with a very low fluorescence yield, approximately 10−5 to 10−4 (40–42). Because of the low fluorescence yield, detecting and monitoring rhodopsin fluorescence has been difficult without bulk measurement of large numbers of cells or instrumentation that includes signal amplification for single-cell analysis (42). Here, we report a method that uses through-the-objective total internal reflection fluorescence (TIRF) microscopy (43) to differentiate between rhodopsin-containing and pigmented cells. TIRF microscopy relies on the total internal reflection phenomenon that takes place when light encounters an interface between two different refractive indexes (i.e., the coverglass and liquid medium). The evanescent field extends only a few hundred nanometers above the coverglass but is able to excite fluorescent molecules at the coverglass-cell interface. Because a relatively small excitation volume is created with TIRF microscopy, background contributions from Raman scattering of the liquid medium are greatly reduced, enabling the detection of weakly fluorescent surface-bound molecules. This method is sensitive enough to detect fluorescence due to rhodopsins and carotenoid pigments, precursors to the retinal cofactor in rhodopsins, and can differentiate between them with the appropriate excitation wavelength. Using TIRF microscopy, direct detection of rhodopsin-containing cells in natural samples becomes possible.
MATERIALS AND METHODS
Strains and growth conditions.
The actinobacterial strain Rhodoluna lacicola MWH-Ta8 (15, 16, 29) was grown in 3 g liter−1 NSY medium (44) at room temperature with gentle shaking. E. coli strain epi300 (Epicentre Biotechnology; catalog number EC300105) was used for carotenoid biosynthesis and actinorhodopsin expression. The cells were grown in Luria-Bertani (LB) medium supplemented with 100 mg liter−1 ampicillin and/or 34 mg liter−1 chloramphenicol, as appropriate, for plasmid propagation and with antibiotics and 0.02 to 0.2% l-arabinose for carotenoid expression.
Carotenoid-synthesizing E. coli strains.
Plasmids with genes encoding the synthesis of lycopene, β-carotene, and retinal were constructed by PCR amplification of the relevant genes from the marine alphaproteobacterial fosmid HF10_19P19 (20). (Table 1 lists primer sequences; Fig. 1 shows amplicons.) The plasmid pLY02, encoding production of lycopene (Fig. 1), was constructed by amplification of the region containing crtE (geranylgeranyldiphosphate synthase), crtI (phytoene dehydrogenase), and crtB (phytoene synthase) from HF10_19P19, using primers 19P19_F1 and 19P19_R1, and insertion into the TA cloning site of the pBAD-TOPO vector (Life Technologies K4300-40). The plasmid pBC01, encoding β-carotene biosynthesis, was constructed by amplifying a slightly larger region that also included the lycopene cyclase gene, crtY, using primers 19P19_F1 and 19P19_R2 (Table 1 and Fig. 1). The reverse primer for this reaction includes an endogenous XbaI site. To make plasmid pRET04, encoding retinal biosynthesis, the region immediately downstream of the region included in plasmid pBC01 was amplified from HF10_19P19 using primers 19P19_F3 (the reverse/complement of primer 19P19_R2) and 19P19_R3 (Fig. 1). This region covers the blh gene, encoding a β-carotene cleavage dioxygenase, which produces retinal from β-carotene. The PCR product was digested with XbaI, and plasmid pBC01 was digested with XbaI and the blunt cutter PmeI. Both the linearized plasmid and the digested PCR product were gel purified and ligated, and the ligation product was transformed into E. coli.
TABLE 1.
Primers used for cloning carotenoid and actinorhodopsin expression constructs and for detection of rhodopsins in Delaware River water
| Primer name | Primer sequencea (5′→3′) | Gene(s) in productb | Template | Restriction site |
|---|---|---|---|---|
| 19P19_F1 | ATG ACA GAG AAC ATA GCC AGC C | Fosmid HF10_19P19 | ||
| 19P19_R1 | GCG TTG TCT TGA GAG CTCGGT CTG C | crtE, crtI, crtB, crtY (partial) | Fosmid HF10_19P19 | |
| 19P19_R2 | CG CCG TCT AGA GGC GTT TTG C | crtE, crtI, crtB, crtY | Fosmid HF10_19P19 | XbaI |
| 19P19_F3 | G CAA AAC GCCTCT AGA CGG CG | blh | Fosmid HF10_19P19 | XbaI |
| 19P19_R3 | GCT TGT TCG GGT CAT GGC TGT G | Fosmid HF10_19P19 | ||
| F-apa-ta8 | CCC GGG CCC ATG AAC ACA TTG TCT AAT G | actR | R. lacicola genomic DNA | ApaI |
| R-ta8-bam | CGC GGA TCC TTA GGC GTC TTT GAA C | actR | R. lacicola genomic DNA | BamHI |
| SARPR_125F | THG GWG GAT AYT TAG GWG AAG C | pR (partial) | Delaware River genomic DNA | |
| SARPR_203R | ACC TAC TGT AAC RAT CAT TCT YA | pR (partial) | Delaware River genomic DNA |
Restriction sites are underlined.
pR, proteorhodopsin.
FIG 1.
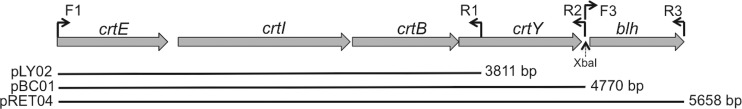
Carotenoid expression constructs. All genes were PCR amplified from fosmid HF10_19P19 (20). Plasmid pLY02 encodes lycopene biosynthesis, plasmid pBC01 encodes β-carotene biosynthesis, and pRET04 encodes retinal biosynthesis. Plasmids pLY02 and pBC01 were constructed by amplification of the regions of interest using primer pairs F1/R1 and F1/R2 (Table 1), respectively, and direct ligation of the product into the vector pBAD-TOPO. Plasmid pRET04 was constructed by amplification of the blh gene using primer pair F3/R3 and insertion of the product into the XbaI site of pBC01 (see Materials and Methods for more detail).
Cloning, expression, and partial purification of actinorhodopsin.
The gene actR (NCBI accession number FJ545221), encoding actinorhodopsin, was amplified from DNA extracted from R. lacicola strain MWH-Ta8 using primers F-apa-ta8 and R-ta8-bam (Table 1). PCRs were performed utilizing Phusion DNA polymerase (Thermo Scientific). The amplification steps were as follows: initial denaturation at 98°C for 30 s and then 35 cycles of 98°C for 10 s, 50°C for 30 s, and 72°C for 24 s, with a final elongation step of 72°C for 10 min. The ∼800-bp amplification product was inserted into plasmid pMCL200 (45) at the ApaI/BamHI restriction sites to produce plasmid pTAR, and the insert was sequenced. Plasmid pTAR was transformed into E. coli epi300/pRET04 to create a strain coexpressing actinorhodopsin and its cofactor, retinal. An empty-vector control strain was created by transforming pMCL200 into E. coli epi300/pRET04. Membranes from E. coli harboring pRET04, along with either pTAR or pMCL200, were partially purified by incubating the cells with an osmotic lysis buffer containing lysozyme (0.075 M Tris, pH 8.0, 2.0 mM MgSO4, 0.4 M sucrose, 10 mg ml−1 lysozyme) for 1 h at 37°C with shaking, followed by centrifugation at 4,500 × g for 20 min at 4°C. The supernatant was removed, and the cell pellet was resuspended in a high-salt buffer (50 mM Tris, pH 7.6, 10 mM MgSO4, 0.8 M NaCl) and briefly sonicated (46). After broken cells were centrifuged at 25,000 × g for 30 min at 4°C, a dull-colored cell debris pellet was obtained, covered by a brightly colored membrane film. The membrane film was removed and resuspended in 3% beta-octylglucopyranoside (Amresco) in 10 mM HEPES, pH 7.1, by vortexing overnight at 4°C in the dark. The detergent-solubilized membrane was centrifuged at 11,000 × g for 10 min at 4°C to remove insoluble material. Absorption spectra from 250 to 900 nm were recorded using a Thermo Scientific BioMate 3S UV-visible spectrophotometer. The membrane fraction of cells harboring pRET04 and pMCL200 was used as the blank. Membrane preparations to be analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) were incubated 1:1 in 2× loading buffer (250 mM Tris, 2% SDS, 30% glycerol, 10% 2-mercaptoethanol, 0.002% bromophenol blue) for 1 h at room temperature. Samples were loaded on a 10% Tris-buffered polyacrylamide resolving gel, topped with a 5% polyacrylamide stacking gel, and electrophoresed according to the method of Laemmli (47). The molecular mass standard was the PageRuler prestained protein ladder (10 to 170 kDa; Thermo Scientific).The gel was washed with deionized (DI) water, fixed with glacial acetic acid-methanol-water (10:25:65) for 15 min, and stained with LabSafe Gel Blue (G-Biosciences).
HPLC analysis.
For pigment analysis by high-performance liquid chromatography (HPLC), E. coli cells containing plasmids pLY02, pBC01, and pRET04 were grown in LB medium with 50 mg liter−1 ampicillin overnight at 30°C with shaking in the presence of either 0.2% glucose or 0.02% arabinose. Cells were harvested by centrifugation and washed once with TES buffer (200 mM Tris, pH 8.0, 20 mM EDTA, 200 mM NaCl). Pigments were extracted from the cells by sonication in acetone-methanol (7:2 [vol/vol]). Cell debris was removed by centrifugation, and the supernatants were filtered through 0.2-μm polytetrafluoroethylene syringe filters (Thermo Scientific) prior to injection into the HPLC system. The HPLC system was a Shimadzu Prominence system with a solvent degasser (DGU-20A5), a quaternary pump (LC-20AT), and a 996-element diode array detector (SPD-M20A) fitted with a Supelco Ascentis reverse-phase C18 column (100 by 3 mm; 3-μm beads; Sigma-Aldrich catalog number 581308-U). Solvent A was 62.5% water, 21% methanol, and 16.5% acetonitrile, buffered with 10 mM ammonium acetate, and solvent B was 50% methanol, 30% ethyl acetate, and 20% acetonitrile by volume (48). The gradient was as follows (minutes/percent solvent B): 0/20, 5/70, 12/100, and 25/100. The column was kept at a constant temperature of 35°C.
Delaware River water collection, genomic DNA isolation, and rhodopsin PCR.
Twenty liters of Delaware River water was collected on 28 October 2014 from Battery Park in New Castle, DE (39°39′27.1″N, 75°33′48.2″W). The water was filtered through 1-mm nylon into a washed and rinsed Nalgene bottle. At the time of collection, the water temperature was 18°C, and the salinity was ∼11 ppt. Genomic DNA was extracted from 100 ml of water that was filtered through a 1.0-μm cellulose nitrate filter (Whatman) and onto a 0.22-μm MoBio filter in triplicate. Genomic DNA was extracted using the MoBio Rapid Water kit (MoBio; catalog no. 14810-50-NF) according to the manufacturer's instructions. The genomic DNA was screened for the presence of rhodopsin genes using degenerate primers for actinorhodopsin (15) and proteorhodopsin (28) (Table 1; see Table S1 in the supplemental material). A positive result was obtained using the SARPR_125F and SARPR_203R primer pair with Taq polymerase (Sigma-Aldrich; catalog no. D4545-250UN) and thermocycling conditions of 94°C for 3 min, 40 cycles of 94°C for 1 min, 54°C for 1 min, and 72°C for 1 min, and then a final elongation step of 72°C for 5 min. The PCR products were cloned into the TOPO TA sequencing vector (Life Technologies; catalog no. K4575-01) and sequenced by the University of Delaware Sequencing and Genotyping Center. The sequences were trimmed of the vector sequence and aligned using the SeaView and Clustal programs.
Sample preparation for live-cell TIRF microscopy.
E. coli with plasmid(s) pLY02, pBC01, pRET04, pRET04/pMCL200, pRET04/pTAR, or pTAR was grown in LB medium with appropriate antibiotics overnight at 30°C with shaking. Expression of pigments and rhodopsin was induced with arabinose (0.2 g liter−1). One milliliter of cell culture was harvested by centrifugation at 3,500 × g for 5 min. The cells were washed with DI water twice and resuspended in 75 μl of water. Fifty microliters of cells was added to each chamber of a LabTekII chambered 1.5 German coverglass system (Nunc 155409) that had been previously treated with 100 μl of 0.5% (wt/vol) gelatin (Sigma; G6144) with 0.01% (wt/vol) chromium ammonium sulfate and then dried under vacuum (49). After 10 min, unattached cells were removed and 100 μl LB medium was added to the wells.
Sample preparation for fixed-cell TIRF microscopy.
Fisherbrand coverglasses (22 by 22 mm; no. 1.0) were cleaned with several washes of DI water, followed by sonication for 15 min in fresh DI water (2 times). The coverglasses were then placed in 0.1 N HCl for 1 h with shaking, washed 3 times with DI water, and soaked in 95% ethanol for 1 h with shaking. The coverglasses were rinsed 2 times with DI water and stored in 95% ethanol until use. The washed coverslips were removed from the ethanol, air dried, and sterilized with 15 min of exposure to UV light. The coverslips were then dipped in 0.5% (wt/vol) gelatin (Sigma; G6144) with 0.01% (wt/vol) chromium ammonium sulfate and air dried overnight at an angle. E. coli cells expressing retinal-containing actinorhodopsin were fixed with 4% paraformaldehyde for 15 min at 4°C and visualized by TIRF microscopy to confirm that fixing the cells did not affect the rhodopsin fluorescence (data not shown). Twenty milliliters of Delaware River water was filtered through a 1.0-μm cellulose nitrate filter (Whatman) and fixed with paraformaldehyde (Electron Microscopy Sciences; 4% final concentration) overnight at 4°C. The entire 20-ml volume was concentrated to ∼3 ml on a 25-mm-diameter, 0.2-μm-pore-size white Isopore polycarbonate filter (EMD Millipore) and stained with DAPI (4′,6-diamidino-2-phenylindole) (Life Technologies; catalog no. S33025) for 5 min (600 nM final concentration). The remaining 3 ml was filtered onto the polycarbonate filter. The filter was transferred to a gelatin-coated coverglass that had 1 μl of DI water on it to promote attachment between the filter and the gelatin. After 10 min, the filter was removed and the coverglass was sealed to a glass slide containing 10 μl DI water (50).
TIRF microscopy.
A laboratory-built laser microscopy system was used for TIRF, similar to the setup first described by Axelrod (43, 51). Briefly, images were acquired using a Zeiss Observer.A1 microscope with a 100×/1.46-numerical-aperture (NA) oil immersion lens, with an additional ×2 magnification after the tube lens. Laser light from 405-nm, 488-nm, 561-nm, and 641-nm sources (Coherent Cube [405-nm and 641-nm] and Coherent Sapphire [488-nm and 561-nm] lasers) was expanded to approximately 1-in. diameter and focused onto the back aperture of the objective using a 500-mm achromatic doublet lens. The laser beams were modulated using a computer-controlled acousto-optic modulator (model number AOTFnC-400.650; AA Opto-Electronic, Osray, France). Frames were acquired every 30 ms with a Peltier cooled (−75°C) Andor iXON DU897 electron-multiplying charge-coupled-device (eMCCD) (see Fig. 4 and 5; see Fig. S1 and S2 in the supplemental material) or Princeton Instruments Excelon ProEM512 charge-coupled-device (CCD) (see Fig. 6) camera using the software provided by the manufacturer with electron multiplier gain set to 300. The eMCCDs have similar sensitivities and pixel sizes, and the camera change did not affect detection capability. The laser intensity settings on the main laser module were 2.7 mW for 405 nm, 30 mW for 488 nm, 50 mW for 561 nm, and 75 mW for 641 nm.
FIG 4.
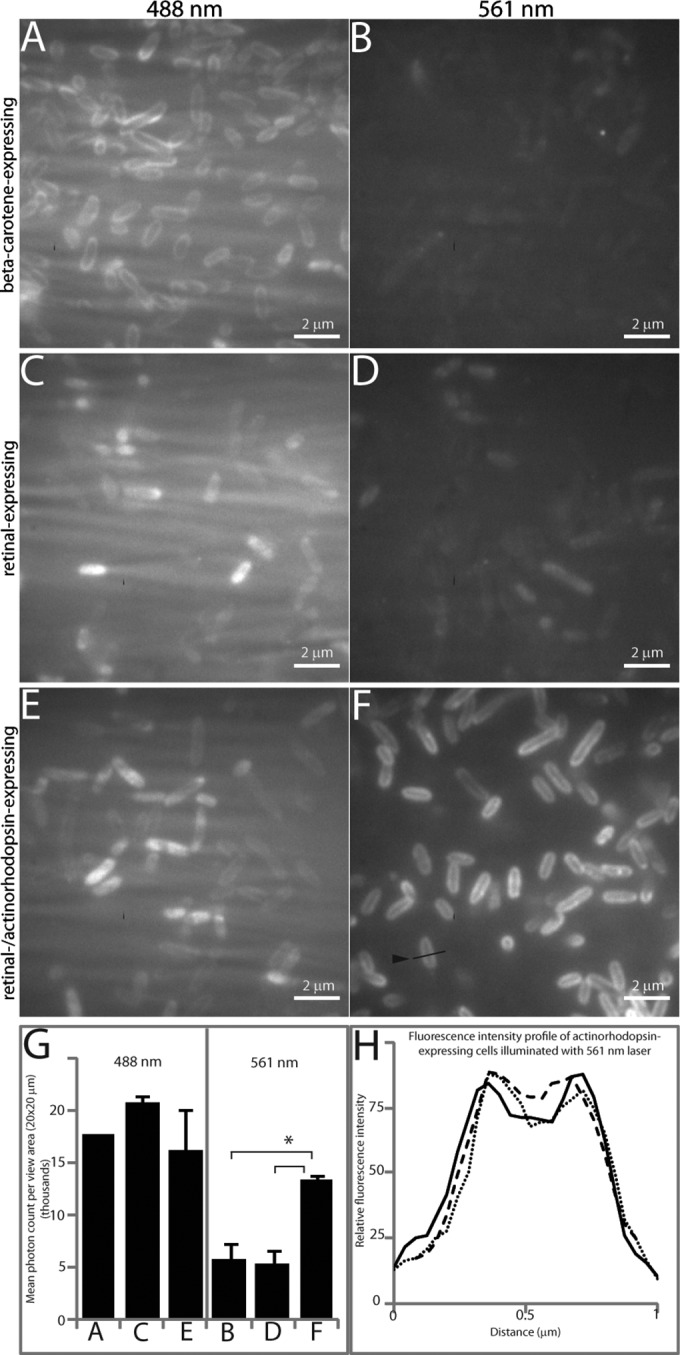
TIRF microscopy of engineered E. coli strains. Cells were affixed to a gelatin-coated chambered glass coverslip and viewed after being illuminated with a 488-nm laser (left column) or a 561-nm laser (right column). The 488-nm laser excites both carotenoids and rhodopsins, but the 561-nm laser excites rhodopsins exclusively. (A and B) E. coli with pBC01 (β-carotene expressing). (C and D) E. coli with pRET04/pMCL200 (retinal expressing). (E and F) E. coli with pRET04/pTAR (retinal and actinorhodopsin expressing). (G) Fluorescence intensities observed for each 20-μm2 view area when excited with the 488-nm or 561-nm laser. The asterisk indicates that significantly more fluorescence is emitted from actinorhodopsin-expressing cells than from any other cell type (P < 0.002). The error bars indicate standard deviations in observed fluorescence intensities. (H) Fluorescence intensity line profile across three individual actinorhodopsin-expressing cells excited with a 561-nm laser showing fluorescence localized to membranes. An example line profile is indicated by an arrowhead in panel F.
FIG 5.
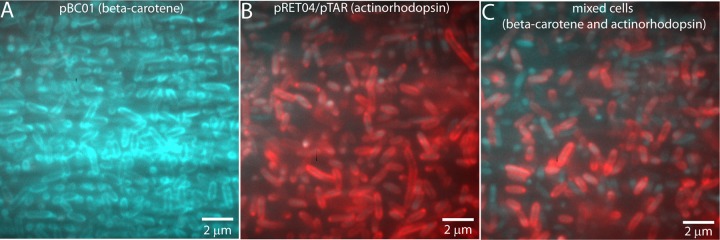
TIRF microscopy of pure cultures and mixed β-carotene- and actinorhodopsin-expressing E. coli cells. A glass coverslip was coated with 0.5% gelatin, and the cells were allowed to attach for 10 min. Unattached cells were washed away, and the chamber was flooded with medium before imaging. The same field of view was excited with the 488-nm laser and the 561-nm laser. Fluorescence observed from the 488-nm laser was colored cyan, while fluorescence from the 561-nm laser was colored red, and the images were merged. (A) A pure culture of E. coli with plasmid pBC01 (β-carotene-expressing cells) was imaged with both lasers sequentially. Most of the cells appear cyan because they were excited only with the 488-nm laser. (B) A pure culture of E. coli with plasmids pRET04 and pTAR (actinorhodopsin-expressing cells) was imaged with both lasers and appears red due to the fluorescence from the 561-nm laser excitation. (C) A mixed culture of β-carotene- and actinorhodopsin-expressing cells was imaged with both 488-nm and 561-nm lasers. Both populations of cells can be clearly viewed, as the β-carotene-expressing cells respond only to the 488-nm laser and thus are cyan, while the actinorhodopsin-expressing cells are illuminated by the 561-nm laser and are colored red.
FIG 6.
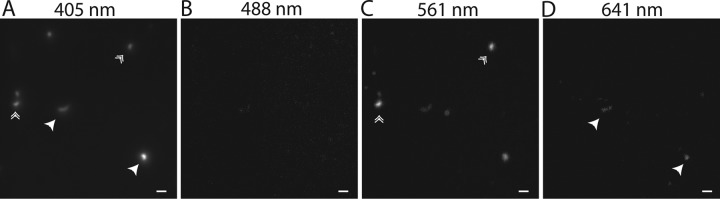
TIRF microscopy of natural water samples. Water samples were fixed, stained with DAPI, and filtered onto a 0.2-μm polycarbonate filter before being transferred to a gelatin-coated glass coverslip. Samples were viewed after being illuminated with a 405-nm laser (A), a 488-nm laser (B), a 561-nm laser (C), or a 641-nm laser (D). The 405-nm laser excited the DAPI stain, indicating the presence of DNA-containing cells. The 641-nm laser illuminated (bacterio)chlorophyll-containing cells (labeled with single arrowheads). Cells that fluoresce only when excited with the 561-nm laser (labeled with double arrowheads) are rhodopsin-containing cells. Each panel shows the same view of a single representative sample. Scale bars, 1 μm.
Image processing.
Images were processed using ImageJ version 1.47 (National Institutes of Health). Andor .sif files were read into ImageJ using the Read_SIF plug-in. Twenty-five sequential frames were summed to reduce random background noise. The minimum fluorescence level was normalized for all images acquired with the same laser. The average photon count over the entire view area was measured, and statistical significance was assessed using a t test. For the line profiles, a line was drawn across the midpoints of three individual bacteria, and the fluorescence intensity along that line was plotted as a function of distance. For the false-colored images, each laser (488 nm and 561 nm) was assigned a color, and the images were merged.
Nucleotide sequence accession numbers.
The nucleotide sequences from this study were deposited in GenBank with accession numbers KP343692, KP343693, KP343694, KP343695, KP343696, and KP343697.
RESULTS
Induction of carotenoid biosynthesis.
In previous work, a fosmid clone from the Hawaii Ocean Time Series (HOTS), HF10_19P19, was shown to encode retinal biosynthesis and a proteorhodopsin (20). In that study, the expression of these genes could be increased by increasing the copy number of the fosmid in E. coli but could not be controlled directly. Here, we cloned the genes for lycopene, β-carotene, and retinal biosynthesis (Fig. 1) into an expression vector downstream of the araBAD promoter so that the production of carotenoids in E. coli could be induced with addition of arabinose or inhibited with addition of glucose to the growth medium. In the presence of glucose, no lycopene, β-carotene, or retinal synthesis was observed (Fig. 2, dotted lines). In the presence of arabinose, E. coli harboring plasmid pLY02 synthesized lycopene (Fig. 2A, solid line), E. coli harboring plasmid pBC01 synthesized β-carotene (Fig. 2B, solid line), and E. coli harboring plasmid pRET04 synthesized retinal (Fig. 2C, solid line). The intermediate phytoene (not shown) was observed in all the samples, and some β-carotene accumulated in the retinal-producing strain.
FIG 2.

Products of carotenoid-producing E. coli strains. Pigments were extracted from E. coli grown with glucose, which represses expression due to the araC gene product (dotted lines), or with arabinose, which induces expression from the araBAD promoter (solid lines). (A) HPLC chromatogram of pigments extracted from E. coli harboring plasmid pLY02, monitored at 471 nm. (Right) These cells synthesize a single compound with absorption peaks at 471 and 502 nm, characteristic of lycopene. (B) HPLC chromatogram of pigments extracted from E. coli harboring plasmid pBC01, monitored at 452 nm. (Right) This strain produces a compound with absorption peaks at 452 and 478 nm, characteristic of β-carotene. (C) HPLC chromatogram of pigments extracted from E. coli harboring pRET04, monitored at 380 nm. (Right) The major pigment produced by these cells has an absorption peak at 380 nm, typical of retinal. Some β-carotene is also present in this strain (not shown).
Actinorhodopsin expression.
The actinorhodopsin gene (actR) from R. lacicola strain MWH-Ta8 (15, 16) was cloned into pMCL200 and expressed in retinal-producing E. coli (cells harboring plasmid pRET04). The retinal-producing cells were pale yellow but turned pink upon induction of actinorhodopsin expression (Fig. 3A), indicating that actinorhodopsin had folded correctly and incorporated the retinal cofactor (12). ActR was purified in E. coli membranes and was visible as a dark band with an apparent molecular mass of ∼22 kDa in denaturing gel electrophoresis (Fig. 3B). The absorption spectrum of the pink membrane fraction had a clear peak at 528 nm (Fig. 3C), which is in the typical range for microbial rhodopsins when retinal is bound.
FIG 3.
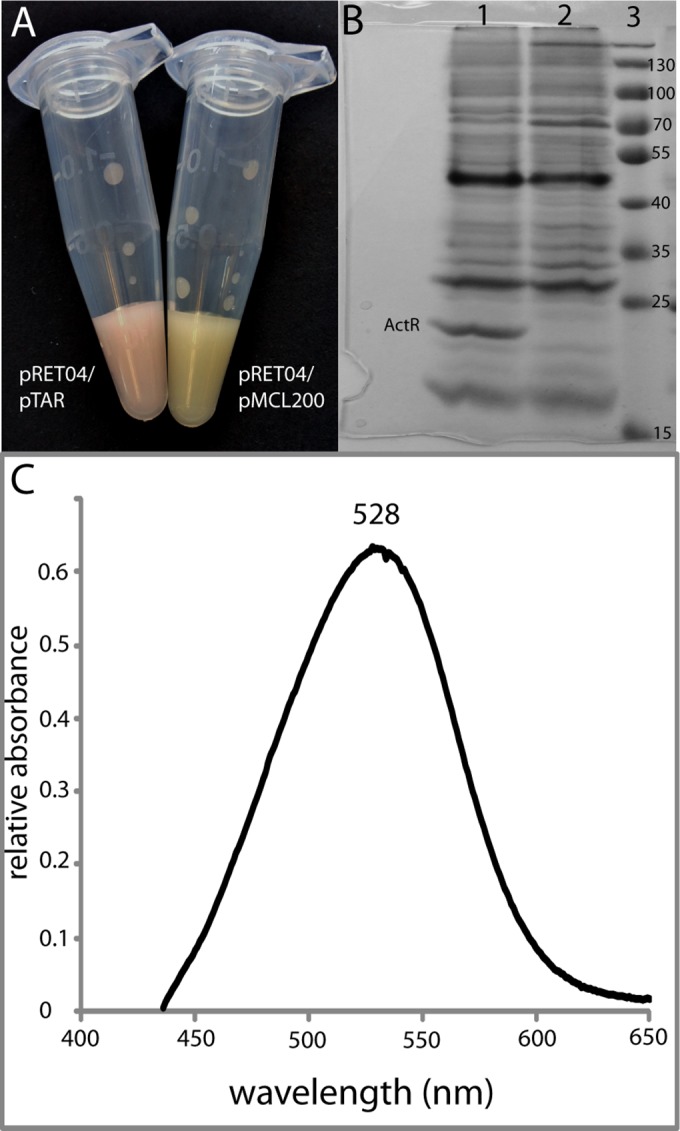
Expression of actinorhodopsin in E. coli. The actR gene was cloned into plasmid pMCL200 to produce plasmid pTAR and cotransformed into the epi300 strain, along with plasmid pRET04. (A) Concentrated cell solution of E. coli epi300 with plasmids pRET04 and pTAR (left) and pRET04 and pMCL200 (right). The pink color indicates that the actinorhodopsin has incorporated the retinal cofactor. (B) SDS-PAGE of partially purified membrane preparations from E. coli with plasmids pRET04 and pTAR (lane 1), partially purified membrane preparations from E. coli with plasmids pRET04 and pMCL200 (lane 2), and a protein standard (lane 3). The band corresponding to ActR at ∼22 kDa is labeled. (C) Absorption spectrum of partially purified membranes from E. coli with plasmids pRET04 and pTAR.
Visualization of actinorhodopsin using TIRF microscopy.
E. coli cells expressing lycopene, β-carotene, retinal, retinal and actinorhodopsin, or actinorhodopsin alone were imaged by TIRF microscopy (Fig. 4; see Fig. S1 in the supplemental material). Lasers with excitation wavelengths of 488 nm and 561 nm were utilized to view the cells. The 488-nm laser enabled visualization of the pigment-expressing cells (Fig. 4, left column; see Fig. S1, left column, in the supplemental material); however, light of this wavelength scattered through the gelatin, causing a streaking phenomenon. The 561-nm laser selectively excited the retinal-actinorhodopsin-expressing cells. The fluorescence emitted by these cells shows that the actinorhodopsin with its bound retinal cofactor is the chromophore (Fig. 4F), as cells expressing retinal alone (Fig. 4D) or actinorhodopsin alone (see Fig. S1F in the supplemental material) are not excited when illuminated with this laser. In addition, cells expressing the precursors to retinal, lycopene (see Fig. S1D in the supplemental material) and β-carotene (Fig. 4B), are also not excited at this wavelength. The average fluorescence intensities measured for each sample as a function of the view area size are summarized in Fig. 4G. The fluorescence observed from retinal- and actinorhodopsin-expressing cells excited with the 561-nm laser was significantly more than from any other sample type (P < 0.002). A fluorescence intensity profile across the midpoint of a cell indicated the fluorescence was highest in the membrane in the actinorhodopsin-expressing cells illuminated with the 561-nm laser (Fig. 4H).
TIRF microscopy has potential uses in identification of rhodopsin-expressing cells from environmental samples. However, environmental collections are mixed populations of cells, rather than pure cultures. To demonstrate that cells with and without rhodopsins in a mixed sample can be differentiated, equal volumes of cells expressing β-carotene and retinal-actinorhodopsin were mixed and then excited sequentially with the 561-nm and 488-nm lasers. The fluorescence emitted from cells excited with the 488-nm laser was falsely colored cyan, while the fluorescence from cells exposed to the 561-nm laser was colored red, and the images from both lasers were merged. Figure S2 in the supplemental material shows the images before the merge, clearly indicating the presence of discrete cell populations in the mixed culture. A pure culture of β-carotene-expressing cells was mostly cyan (Fig. 5A), while a pure culture of retinal- and actinorhodopsin-expressing cells was mostly red (Fig. 5B). A mixed culture (Fig. 5C) clearly showed two populations of cells, which could be differentiated using TIRF microscopy.
Detection of rhodopsin-containing cells in environmental samples.
To demonstrate that TIRF microscopy can detect rhodopsin-containing cells in natural samples, water was collected from the Delaware River and screened by PCR for the presence of rhodopsin-containing microorganisms. Rhodopsin genes were amplified using the degenerate primers SARPR_125F and SARPR_203R (28), and sequencing of the PCR products identified these rhodopsins as related to the rhodopsin found in the SAR11 clade (see Fig. S3 in the supplemental material).
Water samples were fixed, stained with DAPI, and concentrated onto polycarbonate filters. Cells were then transferred to gelatin-coated coverslips for imaging. Samples were excited using all four lasers sequentially on the same view area: 405 nm to detect DAPI, 488 nm to visualize carotenoid pigments, 561 nm to find potential rhodopsins, and 641 nm to image Chl-containing organisms (Fig. 6). A number of microorganisms exhibited fluorescence when excited with the 561-nm laser. Some fluoresced when excited with the 641-nm laser (single arrowheads), indicating the presence of Chl in these cells (Fig. 6D), but others were selectively excited with the 561-nm laser (double arrowheads) (Fig. 6C). These are rhodopsin-containing cells.
DISCUSSION
TIRF microscopy.
One of the primary challenges to detection of rhodopsins in natural samples has been the very low fluorescence yield of these pigment-protein complexes (40–42). As we demonstrate here, TIRF microscopy is capable of differentiating between unpigmented E. coli, E. coli producing weakly fluorescent, strongly absorbing carotenoid pigments, and E. coli producing weakly fluorescent, strongly absorbing rhodopsin proteins. In addition, in the relatively large E. coli cells, we can visualize the spatial localization of the pigments or proteins (Fig. 4F and H). The heterologously expressed actinorhodopsin is found in the E. coli membrane, as observed both in the membrane preparation (Fig. 3B) and in the fluorescence profiles of the cells (Fig. 4H).
Because of its sensitivity and low background fluorescence, TIRF microscopy has a variety of uses in the analysis of microbial rhodopsins. It has been used to characterize the mobility of the vertebrate photoreceptor rhodopsin within the cell membrane (52) and the subcellular localization of other bacterial proteins (53). Photocycle dynamics of both sensory and ion-pumping rhodopsins over very small temporal and spatial scales have been observed with TIRF microscopy using photochromic fluorescence resonance energy transfer (pcFRET) (54). Another high-resolution technique, confocal laser scanning microscopy (CLSM), also has the potential to detect fluorescence from rhodopsins if outfitted with a photomultiplier. CLSM can provide similar resolution and can be used on either fixed or live cells. However, TIRF microscopy is more powerful for imaging molecules located at or near the membrane (within 100 to 200 nm of the coverslip) and thus may be more useful for membrane-bound proteins, such as rhodopsins. (For a comparison of TIRF microscopy, CLSM, and other superresolution techniques, see the recent review by Schermelleh et al. [55].) In addition, the source of noise in single-molecule or single-cell imaging arises from Raman and Rayleigh scattering of the liquid (56). TIRF microscopy typically yields higher signal-to-noise ratios than CLSM because the imaging volume is much smaller and less background scattering is present. For example, the depth of a typical 40-μm by 40-μm image is around 100 nm for TIRF microscopy compared to 1 μm for CLSM; therefore, the volume imaged by TIRF is 10-fold smaller than that by CLSM.
We demonstrate here that, in addition to enabling analyses on the single-molecule and single-cell scales, TIRF microscopy can contribute to in situ analyses by differentiating between unpigmented, carotenoid-producing, Chl-producing and rhodopsin-producing microbes in environmental samples. TIRF microscopy is able to identify (B)Chl fluorescence as different from rhodopsin or carotenoid fluorescence. Chl a fluorescence emission is closer to 670 nm, while BChl a fluorescence emission is in the near-infrared (IR) (∼780 to 820 nm). In our system, (B)Chl-containing organisms fluoresce when excited with both the 561-nm and 641-nm lasers, while rhodopsin-containing organisms are selectively excited with the 561-nm laser.
Actinorhodopsin expression.
Actinorhodopsins (ActR) were identified first in a global analysis of metagenomic data (14) and subsequently in some cultivated freshwater Actinobacteria (15). They are predicted to be proton-pumping rhodopsins (15, 38). ActR distribution in Actinobacteria from freshwater environments suggests that they allow Actinobacteria to utilize one of the only resources universally available in those environments—sunlight—to supplement the cellular energy budget. Although their function is currently unconfirmed, we report here the expression of actR in a heterologous host. Expression of actR in a retinal-producing strain of E. coli results in E. coli cells with pink membranes, indicating a rhodopsin with bound retinal. Purification of ActR from the E. coli membrane and subsequent spectroscopy demonstrate that the retinal-bound form of actinorhodopsin has an absorption peak at 528 nm (Fig. 3), similar to the green-light-tuned forms of proteorhodopsin (57).
Production of carotenoid intermediates.
The plasmids described here encode the synthesis of lycopene, β-carotene, and retinal, under the control of the arabinose-inducible araBAD promoter (Fig. 1). This controllable expression construct allows the plasmids to be propagated without production of carotenoids, which tend to have a deleterious effect on the growth of E. coli. The coproduction of retinal and actinorhodopsin is clearly an effective way of supplying the actinorhodopsin with a cofactor; no additional proteins are necessary for insertion of the cofactor into actinorhodopsin, as evidenced by the color development (Fig. 3A). These plasmids were used here for proof-of-concept experiments demonstrating that TIRF microscopy can differentiate between unpigmented, carotenoid-producing, and rhodopsin-producing cells (Fig. 4 and 5).
Implications for rhodopsins in environmental samples.
Although rhodopsins are widespread among planktonic microbes, carotenoid pigments are even more common (25, 58–60). Even though the absorption spectrum of rhodopsin is red shifted relative to those of most carotenoids, the similarity in absorption spectra and fluorescence yields makes them difficult to differentiate in bulk samples using traditional methods. However, it is important to distinguish the two populations: bacteria with rhodopsins are able to utilize light, while carotenoids in the absence of a protein photosystem more likely protect the organism from light-induced damage (61, 62).
Whether a cell has a sensory rhodopsin or an ion-pumping rhodopsin, the rhodopsin mediates the ability to sense and use sunlight. The estimates of rhodopsin abundance based on metagenomic data indicate that an abundance of microbes in illuminated environments utilize sunlight. The discovery in 2001 that a large percentage of microbes in marine surface waters were aerobic anoxygenic phototrophs (AAPs) revolutionized our understanding of the contribution of sunlight to the biological oxidation of organic matter (2, 23). Current estimates indicate that rhodopsin-containing organisms are even more widespread than AAPs (3) and thus that sunlight may play an unexpectedly large role in organic carbon consumption. The TIRF microscopy method described here provides a way to directly identify rhodopsin-expressing cells, even in mixed cultures, and to detect rhodopsin-containing microorganisms in natural samples.
Supplementary Material
ACKNOWLEDGMENTS
We gratefully acknowledge Rovshan Mahmudov and Michael Davidson for technical assistance; the University of Delaware Sequencing and Genotyping Facility for sequence data; the Delaware Biotechnology Institute's Bioimaging Center for access to the TIRF microscope; and Bryan Ferlez (Penn State), Thomas Hanson, Clara Chan, and Holly Michael for useful discussions.
The research reported in this publication was supported by an Institutional Development Award (IDeA) from the National Institute of General Medical Sciences of the National Institutes of Health under grant number 5 P30 GM103519.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.00230-15.
REFERENCES
- 1.Geider RJ, Delucia EH, Falkowski PG, Finzi AC, Grime JP, Grace J, Kana TM, La Roche J, Long SP, Osborne BA, Platt T, Prentice IC, Raven JA, Schlesinger WH, Smetacek V, Stuart V, Sathyendranath S, Thomas RB, Vogelmann TC, Williams P, Woodward FI. 2001. Primary productivity of planet earth: biological determinants and physical constraints in terrestrial and aquatic habitats. Glob Chang Biol 7:849–882. doi: 10.1046/j.1365-2486.2001.00448.x. [DOI] [Google Scholar]
- 2.Moran MA, Miller WL. 2007. Resourceful heterotrophs make the most of light in the coastal ocean. Nat Rev Microbiol 5:792–800. doi: 10.1038/nrmicro1746. [DOI] [PubMed] [Google Scholar]
- 3.Kirchman DL, Hanson TE. 2013. Bioenergetics of photoheterotrophic bacteria in the oceans. Environ Microbiol Rep 5:188–199. doi: 10.1111/j.1758-2229.2012.00367.x. [DOI] [PubMed] [Google Scholar]
- 4.Hoff WD, Jung K-H, Spudich JL. 1997. Molecular mechanism of photosignaling by archaeal sensory rhodopsins. Annu Rev Biophys Biomol Struct 26:223–258. doi: 10.1146/annurev.biophys.26.1.223. [DOI] [PubMed] [Google Scholar]
- 5.Sharma AK, Spudich JL, Doolittle WF. 2006. Microbial rhodopsins: functional versatility and genetic mobility. Trends Microbiol 14:463–469. doi: 10.1016/j.tim.2006.09.006. [DOI] [PubMed] [Google Scholar]
- 6.Lanyi JK, Balashov SP. 2008. Xanthorhodopsin: a bacteriorhodopsin-like proton pump with a carotenoid antenna. Biochim Biophys Acta 1777:684–688. doi: 10.1016/j.bbabio.2008.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Spudich JL, Yang CS, Jung KH, Spudich EN. 2000. Retinylidene proteins: structures and functions from archaea to humans. Annu Rev Cell Dev Biol 16:365–392. doi: 10.1146/annurev.cellbio.16.1.365. [DOI] [PubMed] [Google Scholar]
- 8.Irieda H, Morita T, Maki K, Homma M, Aiba H, Sudo Y. 2012. Photo-induced regulation of the chromatic adaptive gene expression by Anabaena sensory rhodopsin. J Biol Chem 287:32485–32493. doi: 10.1074/jbc.M112.390864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schobert B, Lanyi JK. 1982. Halorhodopsin is a light-driven chloride pump. J Biol Chem 257:10306–10313. [PubMed] [Google Scholar]
- 10.Inoue K, Ono H, Abe-Yoshizumi R, Yoshizawa S, Ito H, Kogure K, Kandori H. 2013. A light-driven sodium ion pump in marine bacteria. Nat Commun 4:1678. doi: 10.1038/ncomms2689. [DOI] [PubMed] [Google Scholar]
- 11.Yoshizawa S, Kumagai Y, Kim H, Ogura Y, Hayashi T, Iwasaki W, DeLong EF, Kogure K. 2014. Functional characterization of flavobacteria rhodopsins reveals a unique class of light-driven chloride pump in bacteria. Proc Natl Acad Sci U S A 111:6732–6737. doi: 10.1073/pnas.1403051111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Beja O, Aravind L, Koonin EV, Suzuki MT, Hadd A, Nguyen LP, Jovanovich SB, Gates CM, Feldman RA, Spudich JL, Spudich EN, Delong EF. 2000. Bacterial rhodopsin: evidence for a new type of phototrophy in the sea. Science 289:1902–1906. doi: 10.1126/science.289.5486.1902. [DOI] [PubMed] [Google Scholar]
- 13.Oesterhelt D, Stockenius W. 1971. Rhodopsin-like protein from the purple membrane of Halobacterium halobium. Nat New Biol 233:149–152. [DOI] [PubMed] [Google Scholar]
- 14.Sharma AK, Zhaxybayeva O, Papke RT, Doolittle WF. 2008. Actinorhodopsins: proteorhodopsin-like gene sequences found predominantly in non-marine environments. Environ Microbiol 10:1039–1056. doi: 10.1111/j.1462-2920.2007.01525.x. [DOI] [PubMed] [Google Scholar]
- 15.Sharma AK, Sommerfeld K, Bullerjahn GS, Matteson AR, Wilhelm SW, Jezbera J, Brandt U, Doolittle WF, Hahn MW. 2009. Actinorhodopsin genes discovered in diverse freshwater habitats and among cultivated freshwater Actinobacteria. ISME J 3:726–737. doi: 10.1038/ismej.2009.13. [DOI] [PubMed] [Google Scholar]
- 16.Hahn M, Schmidt J, Taipale SJ, Doolittle WF, Koll U. 2014. Rhodoluna lacicola gen. nov., sp. nov., a planktonic freshwater bacterium with stream-lined genome. Int J Syst Evol Microbiol 64:3254–3263. doi: 10.1099/ijs.0.065292-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gómez-Consarnau L, Akram N, Lindell K, Pedersen A, Neutze R, Milton DL, González JM, Pinhassi J. 2010. Proteorhodopsin phototrophy promotes survival of marine bacteria during starvation. PLoS Biol 8:e1000358. doi: 10.1371/journal.pbio.1000358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gómez-Consarnau L, González JM, Coll-Lladó M, Gourdon P, Pascher T, Neutze R, Pedrós-Alió C, Pinhassi J. 2007. Light stimulates growth of proteorhodopsin-containing marine Flavobacteria. Nature 445:210–213. doi: 10.1038/nature05381. [DOI] [PubMed] [Google Scholar]
- 19.DeLong EF, Béjà O. 2010. The light-driven proton pump proteorhodopsin enhances bacterial survival during tough times. PLoS Biol 8:e1000359. doi: 10.1371/journal.pbio.1000359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Martinez A, Bradley AS, Waldbauer JR, Summons RE, Delong EF. 2007. Proteorhodopsin photosystem gene expression enables photophosphorylation in a heterologous host. Proc Natl Acad Sci U S A 104:5590–5595. doi: 10.1073/pnas.0611470104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li WKW, Wood AM. 1988. Vertical distribution of North Atlantic ultraphytoplankton: analysis by flow cytometry and epifluorescence microscopy. Deep Sea Res A Oceanogr Res Pap 35:1615–1638. doi: 10.1016/0198-0149(88)90106-9. [DOI] [Google Scholar]
- 22.Olson RJ, Zettler ER, Anderson OK. 1989. Discrimination of eukaryotic phytoplankton cell types from light scatter and autofluorescence properties measured by flow cytometry. Cytometry 10:636–643. doi: 10.1002/cyto.990100520. [DOI] [PubMed] [Google Scholar]
- 23.Kolber ZS, Van Dover CL, Niederman RA, Falkowski PG. 2000. Bacterial photosynthesis in surface waters of the open ocean. Nature 407:177–179. doi: 10.1038/35025044. [DOI] [PubMed] [Google Scholar]
- 24.Rusch DB, Halpern AL, Sutton G, Heidelberg KB, Williamson S, Yooseph S, Wu D, Eisen JA, Hoffman JM, Remington K, Beeson K, Tran B, Smith H, Baden-Tillson H, Stewart C, Thorpe J, Freeman J, Andrews-Pfannkoch C, Venter JE, Li K, Kravitz S, Heidelberg JF, Utterback T, Rogers Y-H, Falcón LI, Souza V, Bonilla-Rosso G, Eguiarte LE, Karl DM, Sathyendranath S, Platt T, Bermingham E, Gallardo V, Tamayo-Castillo G, Ferrari MR, Strausberg RL, Nealson K, Friedman R, Frazier M, Venter JC. 2007. The Sorcerer II global ocean sampling expedition: northwest Atlantic through eastern tropical Pacific. PLoS Biol 5:e77. doi: 10.1371/journal.pbio.0050077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McCarren J, DeLong EF. 2007. Proteorhodopsin photosystem gene clusters exhibit co-evolutionary trends and shared ancestry among diverse marine microbial phyla. Environ Microbiol 9:846–858. doi: 10.1111/j.1462-2920.2006.01203.x. [DOI] [PubMed] [Google Scholar]
- 26.Atamna-Ismaeel N, Sabehi G, Sharon I, Witzel KP, Labrenz M, Jurgens K, Barkay T, Stomp M, Huisman J, Beja O. 2008. Widespread distribution of proteorhodopsins in freshwater and brackish ecosystems. ISME J 2:656–662. doi: 10.1038/ismej.2008.27. [DOI] [PubMed] [Google Scholar]
- 27.Campbell BJ, Waidner LA, Cottrell MT, Kirchman DL. 2008. Abundant proteorhodopsin genes in the North Atlantic Ocean. Environ Microbiol 10:99–109. doi: 10.1111/j.1462-2920.2007.01436.x. [DOI] [PubMed] [Google Scholar]
- 28.Lami R, Cottrell MT, Campbell BJ, Kirchman DL. 2009. Light-dependent growth and proteorhodopsin expression by Flavobacteria and SAR11 in experiments with Delaware coastal waters. Environ Microbiol 11:3201–3209. doi: 10.1111/j.1462-2920.2009.02028.x. [DOI] [PubMed] [Google Scholar]
- 29.Hahn MW. 2009. Description of seven candidate species affiliated with the phylum Actinobacteria, representing planktonic freshwater bacteria. Int J Syst Evol Microbiol 59:112–117. doi: 10.1099/ijs.0.001743-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhao M, Chen F, Jiao N. 2009. Genetic diversity and abundance of flavobacterial proteorhodopsin in China seas. Appl Environ Microbiol 75:529–533. doi: 10.1128/AEM.01114-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yoshizawa S, Kawanabe A, Ito H, Kandori H, Kogure K. 2012. Diversity and functional analysis of proteorhodopsin in marine Flavobacteria. Environ Microbiol 14:1240–1248. doi: 10.1111/j.1462-2920.2012.02702.x. [DOI] [PubMed] [Google Scholar]
- 32.Allen LZ, Allen EE, Badger JH, McCrow JP, Paulsen IT. 2012. Influence of nutrients and currents on the genomic composition of microbes across an upwelling mosaic. ISME J 6:1403–1414. doi: 10.1038/ismej.2011.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yin Q, Fu B, Li B, Shi X, Inagaki F, Zhang X-H. 2013. Spatial variations in microbial community composition in surface seawater from the ultra-oligotrophic center to rim of the South Pacific Gyre. PLoS One 8:e55148. doi: 10.1371/journal.pone.0055148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Béjà O, Suzuki MT. 2008. Photoheterotrophic marine prokaryotes, p 131–157. In Kirchman DL. (ed), Microbial ecology of the oceans, 2nd ed John Wiley & Sons, New York, NY. [Google Scholar]
- 35.Martínez-García M, Swan BK, Poulton NJ, Gomez ML, Masland D, Sieracki ME, Stepanauskas R. 2012. High-throughput single-cell sequencing identifies photoheterotrophs and chemoautotrophs in freshwater bacterioplankton. ISME J 6:113–123. doi: 10.1038/ismej.2011.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ugalde JA, Podell S, Narasingarao P, Allen EE. 2011. Xenorhodopsins, an enigmatic new class of microbial rhodopsins horizontally transferred between archaea and bacteria. Biol Direct 6:52. doi: 10.1186/1745-6150-6-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kwon SK, Kim BK, Song JY, Kwak MJ, Lee CH, Yoon JH, Oh TK, Kim JF. 2013. Genomic makeup of the marine Flavobacterium Nonlabens (Donghaeana) dokdonensis and identification of a novel class of rhodopsins. Genome Biol Evol 5:187–199. doi: 10.1093/gbe/evs134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brown LS. 2014. Eubacterial rhodopsins—unique photosensors and diverse ion pumps. Biochim Biophys Acta 1837:553–561. doi: 10.1016/j.bbabio.2013.05.006. [DOI] [PubMed] [Google Scholar]
- 39.Alexiev U, Farrens DL. 2014. Fluorescence spectroscopy of rhodopsins: insights and approaches. Biochim Biophys Acta 1837:694–709. doi: 10.1016/j.bbabio.2013.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cheminal A, Léonard J, Kim SYY, Jung K-HH, Kandori H, Haacke S. 2013. Steady state emission of the fluorescent intermediate of Anabaena sensory rhodopsin as a function of light adaptation conditions. Chem Phys Lett 587:75–80. doi: 10.1016/j.cplett.2013.09.044. [DOI] [Google Scholar]
- 41.Kochendoerfer GG, Mathies RA. 1996. Spontaneous emission study of the femtosecond isomerization dynamics of rhodopsin. J Phys Chem 100:14526–14532. doi: 10.1021/jp960509+. [DOI] [Google Scholar]
- 42.Kralj JM, Douglass AD, Hochbaum DR, Maclaurin D, Cohen AE. 2012. Optical recording of action potentials in mammalian neurons using a microbial rhodopsin. Nat Methods 9:90–95. doi: 10.1038/nchembio.1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Axelrod D. 2008. Total internal reflection fluorescence microscopy, p 169–221. In Correia JJ, Detrich HW III (ed), Biophysical tools for biologists, vol 2: in vivo techniques Academic Press, London, United Kingdom. [Google Scholar]
- 44.Hahn MW, Stadler P, Wu QL, Pöckl M. 2004. The filtration-acclimatization method for isolation of an important fraction of the not readily cultivable bacteria. J Microbiol Methods 57:379–390. doi: 10.1016/j.mimet.2004.02.004. [DOI] [PubMed] [Google Scholar]
- 45.Nakano Y, Yoshida Y, Yamashita Y, Koga T. 1995. Construction of a series of pACYC-derived plasmid vectors. Gene 162:157–158. doi: 10.1016/0378-1119(95)00320-6. [DOI] [PubMed] [Google Scholar]
- 46.Bender GR, Sutton SV, Marquis RE. 1986. Acid tolerance, proton permeabilities, and membrane ATPases of oral streptococci. Infect Immun 53:331–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Laemmli UK. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 48.Maresca JA, Braff JC, DeLong EF. 2009. Characterization of canthaxanthin biosynthesis genes from an uncultured marine bacterium. Environ Microbiol Rep 1:524–534. doi: 10.1111/j.1758-2229.2009.00075.x. [DOI] [PubMed] [Google Scholar]
- 49.Doktycz MJ, Sullivan CJ, Hoyt PR, Pelletier DA, Wu S, Allison DP. 2003. AFM imaging of bacteria in liquid media immobilized on gelatin coated mica surfaces. Ultramicroscopy 97:209–216. doi: 10.1016/S0304-3991(03)00045-7. [DOI] [PubMed] [Google Scholar]
- 50.Ouverney CC, Fuhrman JA. 1999. Combined microautoradiography-16S rRNA probe technique for determination of radioisotope uptake by specific microbial cell types in situ. Appl Environ Microbiol 65:1746–1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Axelrod D. 1981. Cell-substrate contacts illuminated by total internal reflection fluorescence. J Cell Biol 89:141–145. doi: 10.1083/jcb.89.1.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kim T-Y, Ujii H, Möller M, Muls B, Hofkens J, Alexiev U. 2009. Monitoring the interaction of a single G-protein key binding site with rhodopsin disk membranes upon light activation. Biochemistry 48:3801–3803. doi: 10.1021/bi900308c. [DOI] [PubMed] [Google Scholar]
- 53.Garner EC, Bernard R, Wang W, Zhuang X, Rudner DZ, Mitchison T. 2011. Coupled, circumferential motions of the cell wall synthesis machinery and MreB filaments in B. subtilis. Science 333:222–225. doi: 10.1126/science.1203285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bayraktar H, Fields AP, Kralj JM, Spudich JL, Rothschild KJ, Cohen AE. 2012. Ultrasensitive measurements of microbial rhodopsin photocycles using photochromic FRET. Photochem Photobiol 88:90–97. doi: 10.1111/j.1751-1097.2011.01011.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schermelleh L, Heintzmann R, Leonhardt H. 2010. A guide to super-resolution fluorescence microscopy. J Cell Biol 190:165–175. doi: 10.1083/jcb.201002018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Funatsu T, Harada Y, Tokunaga M, Saito K, Yanagida T. 1995. Imaging of single fluorescent molecules and individual ATP turnovers by single myosin molecules in aqueous solution. Nature 374:555–559. doi: 10.1038/374555a0. [DOI] [PubMed] [Google Scholar]
- 57.Man D, Wang W, Sabehi G, Aravind L, Post AF, Massana R, Spudich EN, Spudich JL, Beja O. 2003. Diversification and spectral tuning in marine proteorhodopsins. EMBO J 22:1725–1731. doi: 10.1093/emboj/cdg183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Du H, Jiao N, Hu Y, Zeng Y. 2006. Diversity and distribution of pigmented heterotrophic bacteria in marine environments. FEMS Microbiol Ecol 57:92–105. doi: 10.1111/j.1574-6941.2006.00090.x. [DOI] [PubMed] [Google Scholar]
- 59.Asker D, Beppu T, Ueda K. 2007. Unique diversity of carotenoid-producing bacteria isolated from Misasa, a radioactive site in Japan. Appl Microbiol Biotechnol 77:383–392. doi: 10.1007/s00253-007-1157-8. [DOI] [PubMed] [Google Scholar]
- 60.Stafsnes M, Josefsen K, Kildahl-Andersen G, Valla S, Ellingsen T, Bruheim P. 2010. Isolation and characterization of marine pigmented bacteria from Norwegian coastal waters and screening for carotenoids with UVA-blue light absorbing properties. J Microbiol 48:16–23. doi: 10.1007/s12275-009-0118-6. [DOI] [PubMed] [Google Scholar]
- 61.Tuveson RW, Larson RA, Kagan J. 1988. Role of cloned carotenoid genes expressed in Escherichia coli in protecting against inactivation by near-UV light and specific phototoxic molecules. J Bacteriol 170:4675–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sandmann G, Kuhn S, Böger P. 1998. Evaluation of structurally different carotenoids in Escherichia coli transformants as protectants against UV-B radiation. Appl Environ Microbiol 64:1972–1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.