

Abstract
The traditional markerless gene deletion technique based on overlap extension PCR has been used for generating gene deletions in multidrug-resistant Acinetobacter baumannii. However, the method is time-consuming because it requires restriction digestion of the PCR products in DNA cloning and the construction of new vectors containing a suitable antibiotic resistance cassette for the selection of A. baumannii merodiploids. Moreover, the availability of restriction sites and the selection of recombinant bacteria harboring the desired chimeric plasmid are limited, making the construction of a chimeric plasmid more difficult. We describe a rapid and easy cloning method for markerless gene deletion in A. baumannii, which has no limitation in the availability of restriction sites and allows for easy selection of the clones carrying the desired chimeric plasmid. Notably, it is not necessary to construct new vectors in our method. This method utilizes direct cloning of blunt-end DNA fragments, in which upstream and downstream regions of the target gene are fused with an antibiotic resistance cassette via overlap extension PCR and are inserted into a blunt-end suicide vector developed for blunt-end cloning. Importantly, the antibiotic resistance cassette is placed outside the downstream region in order to enable easy selection of the recombinants carrying the desired plasmid, to eliminate the antibiotic resistance cassette via homologous recombination, and to avoid the necessity of constructing new vectors. This strategy was successfully applied to functional analysis of the genes associated with iron acquisition by A. baumannii ATCC 19606 and to ompA gene deletion in other A. baumannii strains. Consequently, the proposed method is invaluable for markerless gene deletion in multidrug-resistant A. baumannii.
INTRODUCTION
Acinetobacter baumannii is a major opportunistic pathogen causing nosocomial infections in both community and hospital settings (1, 2). A. baumannii infections cause serious diseases in immunocompromised human hosts, including pneumonia, bacteremia, sepsis, and meningitis (3). The increase in the resistance of the bacterium to major antimicrobial drugs has become a major worldwide concern (4). However, the pathogenesis of this bacterium has been poorly characterized despite its clinical significance.
The increasing availability of bacterial genome sequences and genomewide association tools, such as transcriptome and comparative genome analysis, have led to the identification of various genes that play important roles in the pathogenesis of A. baumannii (5–7). This circumstance has given rise to the need for fast and efficient gene disruption systems, which are critical tools for the functional analysis of genes. The insertion of antibiotic resistance markers into targeted genes using suicide vectors, such as pEX100T and pJQ200, has been widely used for the disruption of A. baumannii genes (8–10). Gene replacement using a linear PCR fragment carrying an antibiotic resistance cassette was developed for A. baumannii (11). However, these methods result in serious problems with respect to complementation of the mutant and the construction of double gene knockout mutants, especially in multidrug-resistant A. baumannii strains. Hence, a method for markerless gene deletion is absolutely essential for the construction of A. baumannii mutants.
To date, a cloning method based on overlap extension PCR has been widely used to introduce gene deletions into A. baumannii (12, 13). This system involves a procedure in which the downstream and upstream regions of the target gene to be deleted are combined by using overlap extension PCR with locus-specific primers containing the recognition sites of restriction enzymes. The PCR product digested with the restriction enzymes is cloned into a suicide vector digested with the same restriction enzymes. The chimeric plasmid is integrated within the target sequence via homologous recombination and is then excised by a second single-crossover homologous-recombination event, resulting in allelic exchange. However, this method can be limited by the availability of restriction sites in the cloning steps. Moreover, the insertion of an unmarked DNA fragment into the plasmid results in problems such as a high background of false-positive colonies (without the desired chimeric plasmid) caused by the self-ligation of digested plasmids. Notably, the construction of new vectors containing a suitable antibiotic resistance cassette is required for the construction of mutants of clinical isolates that exhibit different profiles of resistance to a variety of antibiotics. Therefore, this method is time-consuming and inefficient for the construction of mutants of multidrug-resistant A. baumannii.
Recently, Tucker et al. (14) developed a method based on RecET recombinase and flippase (FLP)/FLP recognition target (FRT) sites for markerless gene deletion in A. baumannii. This method generates PCR products by using primers with 100- to 125-nucleotide extensions that are homologous to regions adjacent to the gene to be disrupted and a template plasmid carrying an antibiotic resistance cassette that is flanked by FRT sites. The method utilizes RecET recombinase and FLP/FRT systems for the recombination and elimination of the antibiotic resistance cassette, respectively. Consequently, two different antibiotic resistance markers, one for the template plasmid and one for the RecET/FLP system, are absolutely necessary for the method. Hence, its application is inevitably restricted for multidrug-resistant clinical isolates of A. baumannii, since available antibiotic resistance markers are extremely limited.
In this study, we describe a new method for markerless gene deletion that is based on a rapid and simple cloning strategy using direct insertion of a blunt-end PCR product into a blunt-end suicide vector. Briefly, the blunt-end DNA fragments containing upstream and downstream regions of the target gene and an antibiotic resistance cassette are sequentially assembled via overlap extension PCR and are then directly cloned into the blunt-end suicide vector developed in this study. The recombinant bacteria carrying the desired chimeric plasmid are easily selected on a medium containing the antibiotic that corresponds to the antibiotic resistance cassette inserted outside the downstream region of the target gene. The plasmid-borne deletion allele with the antibiotic resistance cassette is integrated into the targeted sequence by using a conjugation-based gene transfer system and homologous recombination. A. baumannii merodiploids are selected on the medium supplemented with the antibiotic corresponding to the antibiotic resistance cassette inserted outside the downstream region of the target gene. Importantly, the plasmid and antibiotic resistance cassette are simultaneously eliminated via a second single-crossover homologous-recombination event by sucrose counterselection, resulting in allelic exchange. In the present study, the effectiveness of the proposed method was demonstrated by deletion of basD and bauA, which are essential for the survival of A. baumannii ATCC 19606 under iron-depleted conditions (15, 16). The proposed method was also applied to the functional analysis of genes in siderophore gene cluster 1 of A. baumannii ATCC 19606. Finally, we chose two different clinical isolates of A. baumannii, ATCC 17978 and 1656-2, with which to investigate the applicability of the proposed method. In order to choose a suitable antibiotic resistance cassette in mutant construction, the antibiotic susceptibilities of A. baumannii ATCC 17978 and 1656-2 were examined. Each of the ompA genes in these A. baumannii strains was successfully deleted.
MATERIALS AND METHODS
Bacterial strains, plasmids, and culture conditions.
The bacterial strains and plasmids used in this study are listed in Table 1. Escherichia coli and A. baumannii were grown in Luria-Bertani (LB) broth alone or in LB broth containing 1.5% (wt/vol) agar at 37°C. For plasmid maintenance in E. coli, chloramphenicol (20 μg/ml), erythromycin (400 μg/ml), or kanamycin (50 μg/ml) was added to the growth medium. After conjugation, A. baumannii merodiploids were selected on the medium supplemented with ampicillin (100 μg/ml) and kanamycin (30 μg/ml) or erythromycin (30 μg/ml). All the medium components were purchased from Difco (Detroit, MI, USA), and the chemicals were purchased from Sigma-Aldrich (St. Louis, MO, USA).
TABLE 1.
Bacterial strains and plasmids used in this study
| Strain or plasmid | Relevant characteristic(s)a | Reference or source |
|---|---|---|
| Bacterial strains | ||
| A. baumannii | ||
| ATCC 19606 | Prototype strain | 26 |
| ATCC 17978 | Prototype strain | 27 |
| 1656-2 | Clinical isolate | 18 |
| HKD01 | ATCC 19606 with ΔbasD | This study |
| HKD02 | ATCC 19606 with ΔbauA | This study |
| HKD03 | ATCC 19606 with ΔrimL | This study |
| HKD04 | ATCC 19606 with ΔpiuB | This study |
| HKD05 | ATCC 19606 with ΔcirA | This study |
| HKD06 | ATCC 19606 with ΔmenG | This study |
| HKD07 | ATCC 19606 with ΔfhuF | This study |
| HKD08 | ATCC 19606 with ΔrhbC1 | This study |
| HKD09 | ATCC 19606 with ΔrhbC2 | This study |
| HKD10 | ATCC 19606 with ΔaraJ | This study |
| HKD11 | ATCC 19606 with ΔiucD | This study |
| HKD12 | ATCC 19606 with ΔrhbC3 | This study |
| HKD13 | HKD02 with ΔcirA | This study |
| HKD14 | ATCC 17978 with ΔompA | This study |
| HKD15 | 1656-2 with ΔompA | This study |
| E. coli | ||
| DH5α λpir | supE44 ΔlacU169 (ϕ80 lacZΔM15) hsdR17 recA1 endA1 gyrA96 thi-1 relA1λpir (phage lysogen); plasmid replication | Laboratory collection |
| S17-l λpir | λpir lysogen; thi pro hsdR hsdM+ recA RP4-2 Tc::Mu-Km::Tn7; Tpr Smr; host for π-requiring plasmids; conjugal donor | 28 |
| Plasmids | ||
| pDS132 | Suicide vector; oriR6K sacB; Cmr | 19 |
| pUC4K | pUC4 with nptI; Apr Kmr | Pharmacia |
| pIL252 | Emr; pAMβ1 ori | 29 |
| pHKD01 | pDS132, multicloning sites; oriR6K sacB; Cmr | This study |
| pHKD02 | pHKD01 with ΔbasD::nptI; Cmr Kmr | This study |
| pHKD03 | pHKD01 with ΔbauA::nptI; Cmr Kmr | This study |
| pHKD04 | pHKD01 with ΔrimL::nptI; Cmr Kmr | This study |
| pHKD05 | pHKD01 with ΔpiuB::nptI; Cmr Kmr | This study |
| pHKD06 | pHKD01 with ΔcirA::nptI; Cmr Kmr | This study |
| pHKD07 | pHKD01 with ΔmenG::nptI; Cmr Kmr | This study |
| pHKD08 | pHKD01 with ΔfhuF::nptI; Cmr Kmr | This study |
| pHKD09 | pHKD01 with ΔrhbC1::nptI; Cmr Kmr | This study |
| pHKD10 | pHKD01 with ΔrhbC2::nptI; Cmr Kmr | This study |
| pHKD11 | pHKD01 with ΔaraJ::nptI; Cmr Kmr | This study |
| pHKD12 | pHKD01 with ΔiucD::nptI; Cmr Kmr | This study |
| pHKD13 | pHKD01 with ΔrhbC3::nptI; Cmr Kmr | This study |
| pHKD14 | pHKD01 with ATCC 17978 ΔompA::nptI; Cmr Emr | This study |
| pHKD15 | pHKD01 with 1656-2 ΔompA::nptI; Cmr Emr | This study |
Apr, ampicillin resistant; Cmr, chloramphenicol resistant; Emr, erythromycin resistant; Kmr, kanamycin resistant.
DNA manipulations.
Genomic and plasmid DNAs were purified from bacteria using a SolGent Genomic DNA prep kit (SolGent, Daejeon, Republic of Korea) and an AccuPrep plasmid extraction kit (Bioneer, Daejeon, Republic of Korea), respectively. DNA fragments were purified with an AccuPrep gel purification kit (Bioneer). Restriction and DNA-modifying enzymes were purchased from New England BioLabs (Ipswich, MA, USA). Routine DNA manipulations were performed as described previously (17) or according to the manufacturer's recommendations.
Construction of the pHKD01 vector.
For direct cloning of blunt-end PCR products, the multicloning sites of pDS132 were replaced by annealed oligonucleotide cloning. To generate the annealed oligonucleotides, each complementary single-stranded oligonucleotide (Fig. 1) was resuspended in annealing buffer containing 10 mM Tris (pH 7.5), 50 mM NaCl, and 1 mM EDTA. The reaction was initiated by mixing equal amounts (2 μg) of each oligonucleotide in a total volume of 50 μl. Annealing conditions were set to 95°C for 5 min and 25°C for 60 min. The annealed oligonucleotides were ligated with pDS132 digested with PstI.
FIG 1.

Construction of pHKD01. Complementary oligonucleotides were designed to generate restriction enzyme sites for PstI, FspI, NotI, and BglII. Annealing between the oligonucleotides was performed as described in Materials and Methods. The annealed oligonucleotides were ligated with PstI-digested pDS132. The added restriction enzyme sites are indicated by different colors. sacB, levansucrase-encoding gene; R6K ori, replication origin; cat, chloramphenicol resistance gene; mob RP4, plasmid mobilization region.
Primers, PCR, and plasmid construction.
All oligonucleotides used for mutant construction are listed in Table 2. All primers were designed to yield melting temperatures of 54 to 56°C and PCR fragments of approximately 1,000 to 1,200 bp. The primers were designed to combine the upstream and downstream regions of the coding regions of the target genes with the kanamycin or erythromycin resistance cassette by overlap extension PCR. Reverse primers for amplification of the upstream and downstream sections contained 25 additional nucleotides at their 5′ ends that were homologous to the downstream region and the antibiotic resistance cassette, respectively. The genomic DNAs purified from A. baumannii strains and pUC4K (or pIL252) for amplification of the kanamycin resistance cassette (or erythromycin resistance cassette) were used as templates for the PCR. PrimeSTAR GXL Taq DNA polymerase (TaKaRa, Shiga, Japan), which exhibits 30 mismatched bases per 486,923 total bases and higher fidelity than Pfu DNA polymerase, was chosen to prevent errors in the PCR products generated by overlap extension PCR and to produce blunt-end PCR products. PCR was performed using 1.25 U PrimeSTAR GXL Taq DNA polymerase, 10 μl of 5× PrimeSTAR GXL buffer, 0.2 mM deoxynucleoside triphosphate (dNTP) mixture, 10 pM of each primer, and template DNA (100 ng). Each mutated fragment was amplified by two-step PCR. For the first step (PCR-1), PCR cycle conditions were as follows: 30 cycles of 98°C for 10 s, 54°C for 40 s, and 68°C for 2 min, with a final elongation at 68°C for 10 min. The products obtained from the first step of PCR were then mixed at equimolar concentrations and were subjected to overlap extension PCR with the forward primers corresponding to the upstream region and the reverse primers corresponding to the antibiotic resistance cassette. PCR cycle conditions were as follows: 30 cycles of 98°C for 10 s, 54°C for 40 s, and 68°C for 4 min, with a final elongation at 68°C for 10 min. The blunt-end PCR products containing each deleted target gene with the antibiotic resistance cassette were ligated with FspI-digested pHKD01 to generate chimeric plasmids (Table 1). The plasmids constructed were confirmed by DNA sequencing.
TABLE 2.
Oligonucleotides used for the construction of mutants
| Oligonucleotide function and designation | Oligonucleotide sequence (5′ → 3′)a | Target gene | Locus tag |
|---|---|---|---|
| Gene deletion in A. baumannii ATCC 19606 | |||
| BASD01F | GAAGCAATTGAGCGGTTCAGG | basD | HMPREF0010_02303 |
| BASD01R | AAATGATGAAAGTTCAAAATACGAATTGTTAATCATTTCCAATTTTGCTGT | ||
| BASD02F | TTCGTATTTTGAACTTTCATCATTTTGG | ||
| BASD02R | GCAACACCTTCTTCACGAGGCAGACTGTGCAAACAGGTTAAACGCAG | ||
| BAUA01F | GTCGATTTGGAGAAAACGTAAGC | bauA | HMPREF0010_02301 |
| BAUA01R | CATAAATTTTCACTATTTTGCTATGATAAAAAAACCCGCAATCGC | ||
| BAUA02F | CATAGCAAAATAGTGAAAATTTATGCTC | ||
| BAUA02R | GCAACACCTTCTTCACGAGGCAGACATCATAGCGGCAGCTGTCAC | ||
| RIML01F | ACTTTGCTTCTATCTGGATGCG | rimL | HMPREF0010_00723 |
| RIML01R | CTTGCAGGTCTACCCATACCAGAAACGGTCCTTGCTATGACATAACC | ||
| RIML02F | TTTCTGGTATGGGTAGACCTGC | ||
| RIML02R | GCAACACCTTCTTCACGAGGCAGACGGGATAAATTGATCAAGATCGG | ||
| PIUB01F | GGCTTAAATCTGGGATTGACTGG | piuB | HMPREF0010_00725 |
| PIUB01R | CCCAAAATAATGATGCAAAGCTCATTTAAGCCACTCCCCATTTAGCTA | ||
| PIUB02F | ATGAGCTTTGCATCATTATTTTGG | ||
| PIUB02R | GCAACACCTTCTTCACGAGGCAGACGCACATGTCTTCTACTACAGCCG | ||
| CIRA01F | AGTTAGCGAAAATAGGTCGCGTACC | cirA | HMPREF0010_00727 |
| CIRA01R | ACAGGCATCTCTTTCGTTAGTTGCATTCTCCCTAGCCCAAATGTTACTCAAC | ||
| CIRA02F | TGCAACTAACGAAAGAGATGCCTG | ||
| CIRA02R | GCAACACCTTCTTCACGAGGCAGACAACAGTCCAGTAAAGGCCATCACC | ||
| MENG01F | TGGTCTGTACTTGCTCATGCGT | menG | HMPREF0010_00728 |
| MENG01R | CGAACATTAATGAGAATGATTTTTACTTTAAATTTTCCGATTTCTTTTAAACGTAAG | ||
| MENG02F | TAAAAATCATTCTCATTAATGTTCGTATT | ||
| MENG02R | GCAACACCTTCTTCACGAGGCAGACGAGTAATCAGGTCCGTAGTCAGTATCC | ||
| FHUF01F | CCGATAGGAATTAAAGCTCTTGG | fhuF | HMPREF0010_00729 |
| FHUF01R | CTTTAGAAGTATCAATTTGATTCATCGTCAGATATCACTAACAATTTAAGGC | ||
| FHUF02F | ATGAATCAAATTGATACTTCTAAAGCACT | ||
| FHUF02R | GCAACACCTTCTTCACGAGGCAGACAGCAACTTTACGACCTGCCG | ||
| RHBC101F | GCCCAACAATCTCTACGCAC | rhbC1 | HMPREF0010_00730 |
| RHBC101R | AAGACATATTCATCGTCAGATATCAAGATAAATCTCACTTCACCTGCAAT | ||
| RHBC102F | TGATATCTGACGATGAATATGTCTTTTAA | ||
| RHBC102R | GCAACACCTTCTTCACGAGGCAGACGACTCAATCCGAACCGTACG | ||
| RHBC201F | CCATGATCATGCTAACCTTAGCAG | rhbC2 | HMPREF0010_00731 |
| RHBC201R | GCTGAAGTGCATTCATAGATAAATCTTTTATTAATCCTATTTCTTTAATTGGTCG | ||
| RHBC202F | GATTTATCTATGAATGCACTTCAGCC | ||
| RHBC202R | GCAACACCTTCTTCACGAGGCAGACCCAAGAGCTTTAATTCCTATCGG | ||
| ARAJ01F | GGGTACAACTCCACATTTACCAG | araJ | HMPREF0010_00732 |
| ARAJ01R | TGTAATTGTCCCATTTTTATTAATCATCCTCTGTTTAGCTTATTCAATATGC | ||
| ARAJ02F | GATTAATAAAAATGGGACAATTACATCC | ||
| ARAJ02R | GCAACACCTTCTTCACGAGGCAGACGCAACCAGTCTGTAATAATTGGC | ||
| IUCD01F | GATATTGGTTTACGTGGATGGC | iucD | HMPREF0010_00733 |
| IUCD01R | TCCATACACATATCCTCTGTTTAGCTCAAGCATAACTCACATCCTTCTGT | ||
| IUCD02F | GCTAAACAGAGGATATGTGTATGGAAC | ||
| IUCD02R | GCAACACCTTCTTCACGAGGCAGACTCATTAGCGCAAAACCAGTCC | ||
| RHBC301F | ATTGTTTTTTGTTCGGTGTTCG | rhbC3 | HMPREF0010_00734 |
| RHBC301R | GTCCAATTCCAATAAAATCAAGCATCTGACTACATCCTATTCAGAATCTTAAGC | ||
| RHBC302F | ATGCTTGATTTTATTGGAATTGGA | ||
| RHBC302R | GCAACACCTTCTTCACGAGGCAGACTGCTGCAATAACACAATCAGCC | ||
| Gene deletion in A. baumannii ATCC 17978 | |||
| OMPA101F | ATGTCAGTCATTGTTGTAATCTCCG | ompA | A1S_2840 |
| OMPA101R | CTTTTTTACTGTTCAAGAACTCAAAGTGCAATACGACTC AATTTCATGG | ||
| OMPA102F | TGAGTTCTTGAACAGTAAAAAAGCG | ||
| OMPA102R | ATGGTGCAAGTCAGCACGAACACGAGGTCTTGTCCGAATGCTTCG | ||
| Gene deletion in A. baumannii 1656-2 | |||
| OMPA201F | TGACTTCTTCGACATCTGTAGGC | ompA | ABK1_3141 |
| OMPA201R | TCGCTTTTTTACTGTTCAAGAACTCGGATATCCTCCAGAG ATAACAATTG | ||
| OMPA202F | GAGTTCTTGAACAGTAAAAAAGCGAC | ||
| OMPA202R | ATGGTGCAAGTCAGCACGAACACGACCATTGAACGGAAAGTGCC | ||
| Amplification of the kanamycin resistance cassette | |||
| U1 | GTCTGCCTCGTGAAGAAGGTG | nptI | |
| U2 | GATCCGTCGACCTGCAGG | ||
| Amplification of the erythromycin resistance cassette | |||
| U3 | TCGTGTTCGTGCTGACTTGC | ermAM | |
| U4 | GACCTCTTTAGCTCCTTGGAAGC |
The oligonucleotides were designed using the genome sequences of A. baumannii ATCC 19606 (GenBank accession number ACQB00000000), ATCC 17978 (GenBank accession number CP000521), and 1656-2 (GenBank accession number CP001921). Regions of oligonucleotides not complementary to the corresponding templates are underlined.
Conjugation and plasmid curing with an antibiotic resistance cassette.
The E. coli S17-1 λpir strains containing each constructed plasmid were used as conjugal donors for A. baumannii strains. The transconjugants were conjugated and isolated as follows. Donor and recipient strains were grown in LB broth until the late-log phase (optical density at 600 nm [OD600], 0.8). The bacterial cells were then mixed at an equal ratio and were spotted onto an LB plate. The bacterial cells were incubated for 12 h at 30°C. The bacteria were resuspended in LB broth and were then plated onto LB agar plates containing ampicillin and kanamycin or erythromycin in order to eliminate the donor strains and select the merodiploid strains. Plasmids with the antibiotic resistance cassette were excised from the bacterial chromosome on solid LB medium with 10% sucrose and without NaCl (13). Each mutant was confirmed by PCR analysis.
qRT-PCR and survival under iron-depleted conditions.
A. baumannii was grown in an iron-replete medium, LB medium. LB medium with 2,2-dipyridyl (DIP) added to a final concentration of 0.2 mM was used as an iron-depleted medium. Total RNAs from A. baumannii grown to an OD600 of 0.7 were isolated by using an RNeasy minikit (Qiagen, Valencia, CA, USA). For quantitative real-time PCR (qRT-PCR), cDNA was synthesized by using an iScript cDNA synthesis kit (Bio-Rad Laboratories, Hercules, CA, USA), and real-time PCR amplification of the cDNA was carried out in a Chromo4 real-time PCR detection system (Bio-Rad Laboratories) with a pair of specific primers, as listed in Table 3. The relative expression levels of the targeted genes were calculated by using the 16S rRNA expression level as the internal control for normalization.
TABLE 3.
Oligonucleotides used for quantitative real-time PCR
| Oligonucleotide | Oligonucleotide sequence (5′ → 3′) | Target gene |
|---|---|---|
| RIML03F | GCTACTCATTACGTCAGGTTCAG | rimL |
| RIML03R | CCACTGAGGAATAACGTGTGG | |
| PIUB03F | AAAGATGGCGAGTCACAGTAG | piuB |
| PIUB03R | CCCAATAGCTCTCCGGTATTAG | |
| CIRA03F | GTAGTAACTGCGACTCGTACAC | cirA |
| CIRA03R | GCCGTAGCTTGCTGGATAA | |
| MENG03F | CGTTGTGGTCTTAGGTGCTATT | menG |
| MENG03R | CCCTTCATTGCGAGTGGTTA | |
| FHUF03F | GTGCGTTTATGGAATGGATGG | fhuF |
| FHUF03R | AGGGACAATTACGGCATAAGG | |
| RHBC103F | CAAAGCTTGGCACATAGATTCC | rhbC1 |
| RHBC103R | CTTGCTGACTGAGACCTTCTT | |
| RHBC203F | GGCAAAGTCAGGATCGCTATT | rhbC2 |
| RHBC203R | ACGTGAGCTTGATGTGTTAGTT | |
| ARAJ03F | CATGGCTTAGGAATGGGACTT | araJ |
| ARAJ03R | GGCCGCTTGAGTAACTAGATG | |
| IUCD03F | TGATGTGGAGGTAGTGTGTAATG | iucD |
| IUCD03R | GCTTTCTGGTAAATGTGGAGTTG | |
| RHBC303F | GAGCAGCCAATTTCAGATCAAG | rhbC3 |
| RHBC303R | GAGCTTGCCAAGGATGTAGT |
The survival of A. baumannii under iron-depleted conditions was investigated by measuring the growth of the bacteria cultured in the iron-depleted LB medium. A. baumannii was grown in LB medium overnight at 37°C. The overnight cultures of A. baumannii were washed using phosphate-buffered saline (pH 7.4). One hundred microliters of the cultures was inoculated into 100 ml of the iron-depleted LB medium and was shaken at 37°C. After 10 h of culture, the bacterial growth was monitored by measuring the OD600 of the cultures.
Biofilm assay.
Biofilms formed by A. baumannii were measured using a gentian violet staining assay (18). Briefly, overnight cultures were adjusted to an OD600 of 2.0 and were diluted 200-fold in LB medium. Aliquots (1 ml) of the bacterial suspension were then transferred to 5-ml polystyrene tubes. Biofilms were formed by incubating these cultures for 14 h at 37°C without shaking. Planktonic cells were removed, and the tubes were then washed twice with 1 ml of phosphate-buffered saline (pH 7.4). The biofilm cells on the wall were stained with 0.1% (wt/vol) gentian violet solution for 15 min at room temperature. The biofilms were quantitated by measuring the amount of gentian violet eluted from the biofilms as the OD570 normalized to total bacterial growth (OD600).
Data analysis and statistics.
Averages and standard errors of the means (SEM) were calculated from at least three independent experiments. All data were analyzed by Student t tests with SAS software (SAS Institute Inc., Cary, NC, USA). Differences between experimental groups were considered significant at a P value of <0.005.
RESULTS
pHKD01 construction for direct cloning of blunt-end PCR products.
pDS132, a pCVD442 derivative in which the IS1 element was removed to prevent untargeted integration of the plasmid into bacterial chromosomes, has been widely used for gene allele exchange in bacteria (19). However, only four restriction enzyme sites for XbaI, SalI, PstI, and SphI exist in the multicloning sites of pDS132. The restricted availability of multicloning sites causes difficulties during the cloning step. Therefore, we constructed a pDS132 derivative carrying alternative cloning sites for direct cloning of blunt-end PCR products. The construction of pHKD01 is presented in Fig. 1. The multicloning sites of pDS132 were replaced via annealed oligonucleotide cloning. We designed complementary oligonucleotides (Fig. 1), in which the recognition sites for PstI, FspI, NotI, and BglII exist consecutively, to be cloned directly into the overhangs of the pDS132 plasmid generated by PstI digestion. Especially, the FspI restriction site was chosen to produce a blunt-end vector. The annealed oligonucleotides were ligated with the PstI-digested pDS132. Correct plasmid construction was confirmed by DNA sequencing. The new suicide plasmid was named pHKD01.
Markerless deletion of the basD and bauA genes in A. baumannii ATCC 19606.
The principle for the markerless gene deletion method is diagramed in Fig. 2. The amplification of mutated DNA fragments is accomplished in two PCR steps (PCR-1 and PCR-2) with six PCR primers. Four of these are unique to the upstream and downstream regions (approximately 1 kb each) of the coding region of the target gene to be deleted. Two of these are common primers for the amplification of nptI, conferring kanamycin resistance. For example, the upstream and downstream regions of target gene X are amplified from the genomic DNA of A. baumannii using primer pairs S1/S2 and S3/S4 (Fig. 2), respectively. The kanamycin resistance cassette is amplified using primer pair U1/U2 (Table 2) with pUC4K as the template. In particular, primers S2 and S4 contain 25 nucleotides at their 5′ ends that are homologous to the downstream region of the target gene and the kanamycin resistance cassette, respectively. The three PCR products obtained in the first step are mixed at equimolar concentrations and are subjected to overlap extension PCR with primers S1 and U2 (Fig. 2). The mutated DNA fragment generated by overlap extension PCR is ligated directly into the blunt-end plasmid pHKD01, which was created by digestion with FspI. The plasmid is integrated into the chromosome of A. baumannii by using conjugation-based gene transfer and homologous recombination. In the first single-crossover homologous-recombination event, merodiploids that possess an integrated copy of pHKD01 containing the mutated DNA fragment are readily obtained on LB agar plates containing kanamycin and ampicillin. The second single-crossover homologous-recombination event in the bacteria is achieved with sacB, conferring sucrose sensitivity, on LB agar plates containing sucrose. This step allows the bacterial cells to excise the plasmid with nptI. The bacteria sensitive to kanamycin are selected, and the target gene deletion is then confirmed by PCR analysis.
FIG 2.

Schematic representation of markerless gene deletion based on rapid and simple cloning. The first step consists of independently amplifying the upstream and downstream regions of target gene X (to be deleted) and nptI (conferring kanamycin resistance) by using primer sets S1/S2, S3/S4, and U1/U2, respectively. The upstream and downstream regions of gene X and nptI are sequentially assembled by overlap extension PCR with primers S1 and U2. The blunt-end DNA fragments are directly cloned into FspI-digested pHKD01. The plasmid is integrated into the chromosome of A. baumannii by conjugation and homologous recombination. The excision of the plasmid region with nptI in the chromosome via a second single-crossover homologous-recombination event, by sucrose counterselection, on the side of the region opposite that of the first integration event leads to markerless gene deletion. The markerless deletion mutant is confirmed by PCR. sacB, levansucrase-encoding gene; cat, chloramphenicol resistance gene; Kanr, kanamycin resistant; Ampr, ampicillin resistant; Kans, kanamycin sensitive.
To validate the proposed method, basD and bauA, required for iron acquisition by A. baumannii (15, 16), were selected as the target genes to be deleted in A. baumannii ATCC 19606. In PCR-1, the upstream and downstream regions of each target gene and nptI were amplified by PCR with each locus-specific primer as indicated in Table 2 (Fig. 3A). In PCR-2, the approximately 3.2-kb DNA fragments, in which the upstream and downstream regions and nptI were combined by overlap extension PCR (Fig. 3A), were gel purified and cloned into FspI-digested pHKD01 to yield pHKD02 (ΔbasD::nptI) and pHKD03 (ΔbauA::nptI) (Table 1). E. coli S17-1 λpir containing each of the resulting plasmids was used as a conjugal donor to generate the isogenic mutants of A. baumannii ATCC 19606 by homologous recombination. As shown in Fig. 3B, a second single-crossover homologous-recombination event, by which each target gene was deleted, was confirmed by PCR. PCR analysis of the genomic DNA of the wild type using primers BASD01F and BASD02R produced a 4.9-kb fragment, whereas the genomic DNA from the basD mutant HKD01 resulted in an amplified DNA fragment of approximately 2.0 kb (Fig. 3B). The size of this 2.0-kb fragment was in good agreement with the projected size of the DNA fragment without basD (2.9 kb). In addition, identification of the mutant in which bauA was deleted was confirmed by PCR using primers BAUA01F and BAUA02R. PCR analysis of the genomic DNA of the wild type and of bauA mutant HKD02 indicated fragment sizes of 4.3 kb and 2.0 kb, respectively (Fig. 3B). Therefore, the combined results demonstrated that the basD and bauA genes were successfully deleted in the HKD01 and HKD02 strains, respectively. Furthermore, to examine the effects of basD and bauA mutation on the survival of A. baumannii under iron-depleted conditions, the growth of each mutant strain under iron-depleted conditions was compared to that of the parental wild-type strain (Fig. 3C). Under iron-replete conditions (LB medium), the growth of the mutants did not differ from that of the wild type. However, the growth of the mutant strains was significantly impaired when they were cultured in iron-depleted LB medium. This is consistent with the previous observation that BasD and BauA are essential for the survival of A. baumannii under iron-depleted conditions (15).
FIG 3.
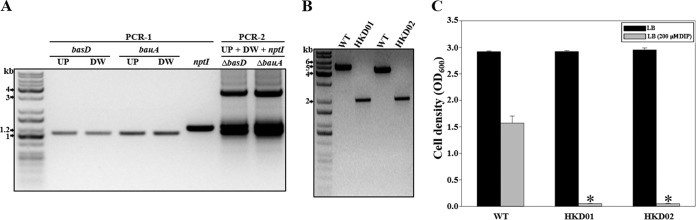
Construction of basD and bauA mutants. (A) In PCR-1, the downstream and upstream regions of basD and bauA were amplified by PCR using the genomic DNA of A. baumannii ATCC 19606. nptI, conferring kanamycin resistance, was also amplified by PCR using pUC4K as a template. In PCR-2, the DNA fragments with the target genes deleted were combined with nptI by overlap extension PCR. UP, upstream region of the target gene to be deleted; DW, downstream region of the target gene to be deleted. (B) PCR analysis of the wild-type and mutant strains generated by allelic exchange. Molecular size markers (1-kb DNA ladder; Kapa) are indicated. (C) The growth of the strains on LB medium without and with 0.2 mM DIP was measured after 10 h. Error bars represent SEM. Means and SEM were calculated from three independent experiments. Asterisks indicate significant differences (P < 0.0001) from the wild type cultured in LB medium with 0.2 mM DIP. WT, wild type; HKD01, basD mutant; HKD02, bauA mutant.
Functional analysis of siderophore gene cluster 1 in A. baumannii ATCC 19606 through markerless gene deletion.
A. baumannii strains have acquired different siderophore gene clusters (20). A. baumannii ATCC 19606 has siderophore gene cluster 1 on the bacterial genome. However, the functional aspects of these genes within the gene cluster have not been characterized. Therefore, the proposed method was used to analyze their roles in bacterial growth under iron-depleted conditions. Ten genes (rimL, piuB, cirA, menG, fhuF, rhbC1, rhbC2, araJ, iucD, and rhbC3) within the gene cluster (Fig. 4A) were selected as the target genes for deletion. Before proceeding with mutant construction, we investigated whether the expression of these genes is regulated by the level of iron. The expression levels of the genes in A. baumannii ATCC 19606 grown in either iron-depleted LB medium (with 0.2 mM DIP) or LB medium alone were analyzed by qRT-PCR (Fig. 4B). The expression levels of rimL, piuB, cirA, menG, fhuF, rhbC1, rhbC2, araJ, iucD, and rhbC3 in bacteria grown in the iron-depleted medium were significantly increased 196 (log10 2.29)-, 10 (log10 1.00)-, 27 (log10 1.43)-, 3 (log10 0.41)-, 11 (log10 1.02)-, 105 (log10 2.02)-, 16 (log10 1.20)-, 26 (log10 1.41)-, 28 (log10 1.45)-, and 95 (log10 1.98)-fold, respectively. These results indicated that each target gene is regulated by the level of iron in the growth medium. Hence, the 10 genes were inactivated separately by using the overlap extension PCR method proposed in this study. The disrupted genes were cloned into pHKD01 to result in pHKD04 (ΔrimL::nptI), pHKD05 (ΔpiuB::nptI), pHKD06 (ΔcirA::nptI), pHKD07 (ΔmenG::nptI), pHKD08 (ΔfhuF::nptI), pHKD09 (ΔrhbC1::nptI), pHKD10 (ΔrhbC2::nptI), pHKD11 (ΔaraJ::nptI), pHKD12 (ΔiucD::nptI), and pHKD13 (ΔrhbC3::nptI) (Table 1). Each of the genes was deleted using a conjugation-based gene transfer system and homologous recombination. Each mutant was confirmed by PCR using a pair of primers as indicated in Table 2 (Fig. 4C), and mutants were named HKD03, HKD04, HKD05, HKD06, HKD07, HKD08, HKD09, HKD10, HKD11, and HKD12 (Table 1). Moreover, to evaluate the efficiency of the proposed method in markerless gene deletion, PCR analysis was performed to examine the absence of the kanamycin resistance cassette in the bacterial strains obtained through sucrose treatment. Approximately 98% of the colonies were confirmed to be sucrose-resistant colonies in which the antibiotic resistance cassette with pHKD01 was eliminated by a second single-crossover homologous-recombination event (Table 4). The sucrose-resistant colonies were also used to evaluate the success rate of deletion of the target genes by using PCR analysis. The average success rate was approximately 48%, but for individual target genes, the success rate of deletion differed (20% to 77%, approximately) (Table 4).
FIG 4.

Functional analysis of siderophore gene cluster 1. (A) Siderophore gene cluster 1 (locus tags, HMPREF0010_00723 to -00734), selected as target genes for the construction of deletion mutants. The size of the open reading frame of each gene to be deleted is given. Locus tags are based on the database of the A. baumannii ATCC 19606 genome sequence that was retrieved from GenBank (accession number ACQB00000000). (B) The mRNA levels of rimL, piuB, cirA, menG, fhuF, rhbC1, rhbC2, araJ, iucD, and rhbC3 in A. baumannii ATCC 19606 were analyzed by qRT-PCR of the total RNA isolated from bacteria grown on LB medium containing 0.2 mM DIP or on LB medium alone. Each bar represents the mRNA level of a gene expressed in bacteria grown on LB medium containing 0.2 mM DIP relative to that in bacteria grown on LB medium alone. Error bars represent SEM. Means and SEM were calculated from three independent experiments. (C) PCR analysis of the wild type and the mutants generated by allelic exchanges. The PCR products from the mutants are approximately 2.0 kb smaller than those from the wild type. (D) The abilities of A. baumannii strains to grow on LB medium without or with 0.2 mM DIP were compared. The growth of the bacterial strains was monitored after 10 h. Error bars represent SEM. Means and SEM were calculated from three independent experiments. The asterisk indicates a significant change (P < 0.0001) from the result for the wild type cultured in LB medium with 0.2 mM DIP. WT, wild type; HKD03, rimL mutant; HKD04, piuB mutant; HKD05, cirA mutant; HKD06, menG mutant; HKD07, fhuF mutant; HKD08, rhbC1 mutant; HKD09, rhbC2 mutant; HKD10, araJ mutant; HKD11, iucD mutant; HKD12, rhbC3 mutant.
TABLE 4.
Efficiency of gene deletion by the proposed method
| Target gene | No. of merodiploids | No. (%) of colonies |
No. (%) of sucrose-resistant colonies |
||
|---|---|---|---|---|---|
| Sucrose sensitive | Sucrose resistant | WTa | Deletion mutant | ||
| basD | 50 | 0 (0) | 50 (100) | 30 (60) | 20 (40) |
| bauA | 50 | 2 (4) | 48 (96) | 35 (73) | 13 (27) |
| rimL | 50 | 1 (2) | 49 (98) | 11 (22.4) | 38 (77.6) |
| piuB | 50 | 2 (4) | 48 (96) | 18 (37.5) | 30 (62.5) |
| cirA | 50 | 0 (0) | 50 (100) | 12 (24) | 38 (76) |
| menG | 50 | 2 (4) | 48 (96) | 32 (66.7) | 16 (33.3) |
| fhuF | 50 | 0 (0) | 50 (100) | 25 (50) | 25 (50) |
| rhbC1 | 50 | 1 (2) | 49 (98) | 38 (77.6) | 11 (22.4) |
| rhbC2 | 50 | 0 (0) | 50 (100) | 30 (60) | 20 (40) |
| araJ | 50 | 1 (2) | 49 (98) | 17 (34.7) | 32 (65.3) |
| iucD | 50 | 0 (0) | 50 (100) | 15 (30) | 35 (70) |
| rhbC3 | 50 | 2 (4) | 48 (96) | 38 (79.2) | 10 (20.8) |
WT, wild type.
To examine the effects of the mutated genes on survival under iron-depleted conditions, the growth of the mutant strains cultured in either iron-replete or iron-depleted medium was compared with that of the wild-type strain. As shown in Fig. 4D, no difference in growth was observed among bacterial strains cultured in LB medium. However, the growth of the rimL mutant strain was significantly decreased in the iron-depleted medium, whereas the growth of other mutant strains was comparable to that of the wild type. These results indicated that RimL is likely to be essential for the survival of A. baumannii ATCC 19606 under iron-depleted conditions.
Double gene deletion in A. baumannii ATCC 19606.
To investigate the effectiveness of the proposed method in double gene deletion, we chose cirA as a target gene to be deleted in a single gene deletion mutant (the bauA mutant) of A. baumannii. To generate double deletion mutants, pHKD06 (ΔcirA::nptI) was integrated into the chromosome of HKD02 (bauA mutant) by conjugative transfer from E. coli S17-1 λpir. The merodiploid strain was selected on LB agar plates containing ampicillin and kanamycin. For excision of the plasmid with the kanamycin resistance cassette, a second single-crossover homologous-recombination event was introduced by sucrose counterselection. The deletion of the cirA gene in the double mutant (HKD13) (Table 1) was confirmed by PCR (Fig. 5). PCR analysis of the genomic DNA from the wild-type strain with primer pairs BAUA01F/BAUA02R and CIRA01F/CIRA02R revealed 4.3-kb fragments, whereas the genomic DNA from HKD13 resulted in amplified DNA fragments of approximately 2.0 kb. These results demonstrated that the markerless gene deletion method suggested in the present study is a useful tool for introducing an additional mutation into A. baumannii.
FIG 5.
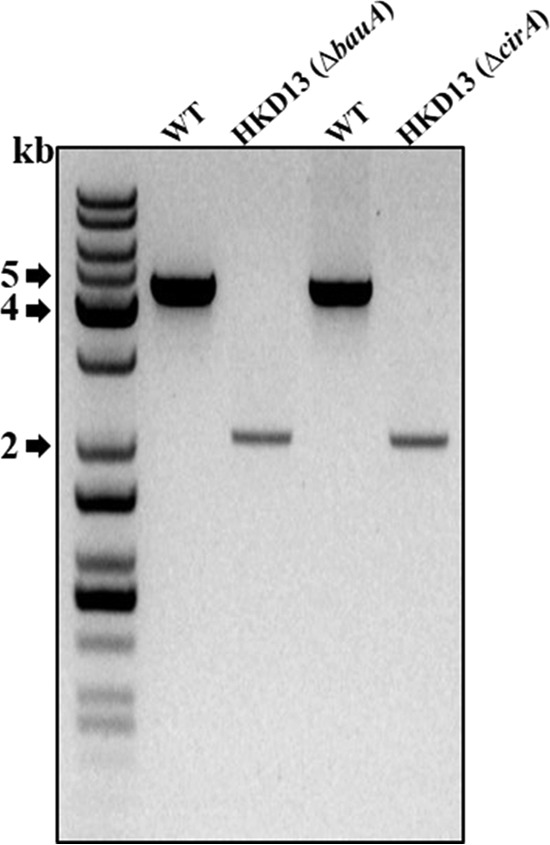
Construction of the double gene deletion mutant. PCR analysis was performed on the genomic DNA from the wild type and double deletion mutant HKD013 with primer pairs BAUA01F/BAUA02R and CIRA01F/CIRA02R. WT, wild type; HKD13, bauA cirA double deletion mutant.
Application of markerless gene deletion to A. baumannii strains other than ATCC 19606.
To demonstrate the applicability of the proposed method in two different clinical strains, A. baumannii ATCC 17978 and 1656-2, which exhibit different phenotypes with regard to antibiotic resistance and biofilm formation (18, 21), were selected. In order to choose a suitable antibiotic resistance cassette for the construction of mutants of these strains, the susceptibilities of these strains to chloramphenicol, kanamycin, and erythromycin were investigated by monitoring their growth on LB medium supplemented with different concentrations of these antibiotics (Fig. 6A). The growth of A. baumannii ATCC 17978 was not effectively suppressed by the addition of 30 μg/ml or 50 μg/ml of chloramphenicol to the medium. However, the growth of A. baumannii ATCC 17978 was significantly impaired in the presence of kanamycin (30 μg/ml or 50 μg/ml) or erythromycin (30 μg/ml or 50 μg/ml). On the other hand, the growth of A. baumannii 1656-2 was substantially inhibited only by erythromycin (30 μg/ml or 50 μg/ml). Therefore, for A. baumannii ATCC 17978 and 1656-2, an erythromycin resistance cassette was used as a selective marker for the construction of chimeric plasmids and the selection of their merodiploids.
FIG 6.
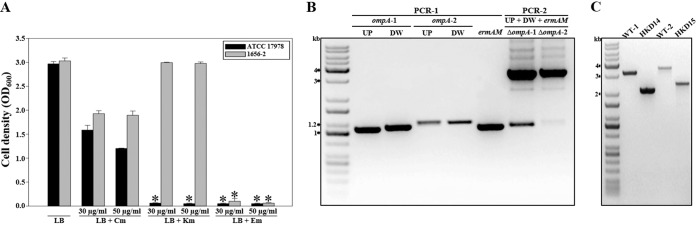
Gene deletion in A. baumannii strains other than ATCC 19606. (A) The susceptibilities of A. baumannii strains ATCC 17978 and 1656-2 to increasing amounts of antibiotics were explored by measuring their growth after 12 h on LB medium containing different concentrations of antibiotics. Asterisks indicate significant differences (P < 0.0001) from the wild-type strains cultured in LB medium. Cm, chloramphenicol; Km, kanamycin; Em, erythromycin. (B) Construction of ompA mutants. In PCR-1, the downstream and upstream regions of ompA were amplified by PCR using the genomic DNA of A. baumannii strain ATCC 17978 or 1656-2. ermAM, conferring erythromycin resistance, was amplified by PCR using pIL252 as the template. In PCR-2, each mutated DNA fragment was combined with ermAM by overlap extension PCR. ompA-1, ATCC 17978 ompA; ompA-2, 1656-2 ompA; UP, upstream region of the target gene to be deleted; DW, downstream region of the target gene to be deleted. (C) PCR analysis of each wild-type and mutant strain generated by allelic exchange. Molecular size markers (1-kb DNA ladder; Kapa) are indicated. WT-1, wild-type ATCC 17978; HKD14, ATCC 17978 ompA mutant; WT-2, wild-type 1656-2; HKD15, 1656-2 ompA mutant.
The ompA gene, essential for biofilm formation by A. baumannii ATCC 19606 (22), was chosen as the target gene to be deleted in A. baumannii ATCC 17978 and 1656-2. In PCR-1, the upstream and downstream regions of the ompA gene in the bacterial strains were amplified with the locus-specific primers shown in Table 2 (Fig. 6B). ermAM, conferring erythromycin resistance, was amplified using primer pair U3/U4 (Table 2) with pIL252 as the template. In PCR-2, the approximately 3.1-kb and 3.3-kb DNA fragments comprising the upstream and downstream regions of ompA and ermAM, respectively, were combined by overlap extension PCR (Fig. 6B); they were then gel purified and cloned into FspI-digested pHKD01 to result in pHKD14 (A. baumannii ATCC 17978 ΔompA::ermAM) and pHKD15 (A. baumannii 1656-2 ΔompA::ermAM) (Table 1). The ompA gene was deleted in each strain by using a conjugation-based gene transfer system and homologous recombination. In order to confirm the deletion of the target gene on the chromosomes of A. baumannii ATCC 17978 and 1656-2, PCR analysis was performed using primer pairs OMPA101F/OMPA102R and OMPA201F/OMPA202R (Table 2). PCR analysis of the genomic DNA of each wild-type strain indicated fragment sizes of 3.1 kb and 3.4 kb (Fig. 6C). However, the genomic DNAs from the ompA mutants, HKD14 and HKD15, yielded amplified DNA fragments of approximately 2.1 kb and 2.3 kb (Fig. 6C). These results indicated that the method proposed in the present study could be applied to A. baumannii clinical isolates that present different resistances to antibiotics without the construction of new vectors carrying suitable antibiotic resistance cassettes. Furthermore, the effects of ompA mutation on the biofilm-forming abilities of A. baumannii strains ATCC 17978 and 1656-2 were assessed using gentian violet staining (Fig. 7). In comparison to biofilm formation by the parental wild-type strains, the abilities of the ompA mutant strains to form biofilms were significantly reduced on polystyrene surfaces as visualized by using a digital imaging system (Fig. 7A). In quantitative biofilm analysis, the biofilm-forming abilities (calculated as OD570/OD600) of the wild-type strains, ATCC 17978 and 1656-2, were 0.8 and 3.6, whereas the abilities of the ompA mutant strains, HKD14 and HKD15, were 0.2 and 0.1, respectively (Fig. 7B). These results suggested that OmpA is likely to play an important role in biofilm formation in both strains.
FIG 7.
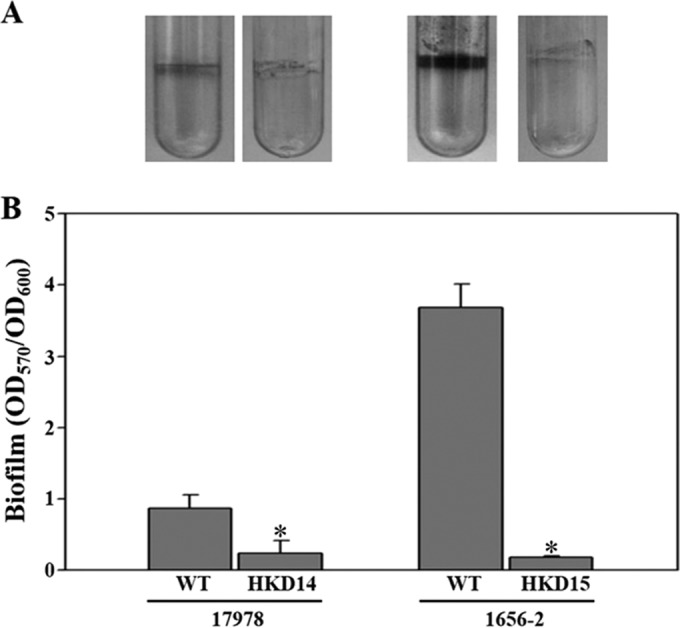
Effects of ompA mutation on biofilm formation by A. baumannii strains. (A) Biofilms formed on 5-ml polystyrene tubes were stained with gentian violet for 15 min. The remaining biofilms were photographed using a digital imaging system. (B) The amount of gentian violet eluted from the biofilms was quantitated as the OD570 normalized to total bacterial growth (OD600). Error bars represent SEM. Means and SEM were calculated from three independent experiments. Asterisks indicate significant differences (P < 0.005) from the wild-type strains. WT, wild type; HKD14, ATCC 17978 ompA mutant; HKD15, 1656-2 ompA mutant.
DISCUSSION
Unmarked gene deletion by suicide vectors has become an invaluable tool in the study of gene functions in the pathogenesis of A. baumannii (12–14), because gene disruption by the insertion of antibiotic resistance markers causes significant difficulties in the complementation of the mutant and the introduction of additional mutations into multidrug-resistant A. baumannii strains. However, in the process of DNA cloning, limited choices of restriction sites result in problems in the construction of chimeric plasmids. To eliminate this obstacle, commercially available kits (the Gibson Assembly cloning kit and the GeneArt Seamless assembly kit) have been developed. The kits are convenient because the methods enable cloning of the overlap DNA fragments directly into a vector linearized by restriction digestion, as well as successful assembly of multiple DNA fragments. However, this approach also has drawbacks. First, self-ligation of the digested plasmids results in a high background of false-positive colonies without the desired chimeric plasmid, making the selection of true colonies with the desired chimeric plasmid difficult. Second, the application of the method to markerless gene deletion in multidrug-resistant clinical isolates of A. baumannii is extremely restricted, because the construction of new vectors carrying a suitable antibiotic resistance marker is absolutely required for the selection of merodiploid strains.
To avoid these drawbacks, in this study, we have developed a rapid and easy cloning method for markerless gene deletion. This method is based on direct insertion of blunt-end PCR products, in which upstream and downstream regions of the target gene are consecutively fused with an antibiotic resistance cassette via overlap extension PCR, into a suicide vector where the same blunt ends have been created by restriction digestion. Importantly, the design is such that the antibiotic resistance cassette is inserted outside the downstream region of the target gene by overlap extension PCR to eliminate the background of false-positive colonies (without desired chimeric plasmids) and facilitate the selection of true colonies (with the desired chimeric plasmids) in DNA cloning. Moreover, this strategy includes removal of the antibiotic resistance cassette via homologous recombination and eliminates the necessity of constructing new vectors containing a suitable antibiotic resistance cassette for markerless gene deletion in multidrug-resistant clinical isolates of A. baumannii. To establish the direct cloning of the blunt-end DNA fragments, a new suicide vector was constructed by cloning the double-stranded oligomer containing the FspI blunt-end site into pDS132. For the validation of the proposed method, basD and bauA were selected as the target genes to be deleted, and markerless deletion of these genes was successfully achieved by using the proposed method.
Iron is one of the major limiting factors for pathogenic bacteria in host cells (23). For iron acquisition, pathogens regulate the expression of genes involved in the production of high-affinity iron-chelating compounds known as siderophores in response to the level of iron availability (24). Genes required for the biosynthesis, efflux, and uptake of siderophores are commonly clustered within bacterial genomes (23). Siderophore gene cluster 1, in which several genes likely to be involved in the biosynthesis (RimL, MenG, FhuF, RhbC1, RhbC2, IucD, and RhbC3), efflux (AraJ), and uptake (CirA and PiuB) of siderophores are clustered, has been found in A. baumannii strains (20). The expression of the genes within A. baumannii ATCC 17978 siderophore cluster 1 is significantly upregulated by iron-depleted environmental conditions (20). In agreement with this, we found that the expression of the genes within A. baumannii ATCC 19606 siderophore cluster 1 was substantially upregulated by iron-depleted conditions. These results indicated the potential importance of these genes in the biosynthesis, efflux, and uptake of A. baumannii siderophores under iron-depleted conditions. Therefore, in the present study, the roles of these genes in the survival of A. baumannii under iron-depleted conditions were examined through mutant construction by the proposed method and through phenotypic evaluation of the mutants for survival under iron-depleted conditions. We showed that rimL, encoding acetyltransferases, is likely to be essential for the survival of A. baumannii under iron-depleted conditions. However, further studies, including complementation testing and investigation of the regulatory mechanisms of rimL expression, are needed to clarify the role of rimL in the survival of A. baumannii under iron-depleted conditions. The effectiveness of the proposed method in double gene deletion was also demonstrated by the additional deletion of cirA in a bauA mutant.
Furthermore, the usefulness of the proposed method was proved by deletion of the ompA gene in A. baumannii strains ATCC 17978 and 1656-2. In particular, A. baumannii 1656-2 is a clinically important isolate, because its biofilm formation ability and resistance to various antibiotics are remarkable (18, 25). However, the genes involved in these phenotypes have not been characterized, due to the insufficiency of methods for the construction of A. baumannii mutants. Thus, the proposed method would help us gain insights into the roles of the genes associated with biofilm formation and antibiotic resistance in multidrug-resistant A. baumannii strains.
Taking into account the results presented here, the proposed method has several advantages. First, the availability of the restriction site is no longer limited to the cloning steps. Second, recombinant bacteria carrying a desired chimeric plasmid are readily selected on the medium containing the antibiotic corresponding to the antibiotic resistance cassette that is inserted outside the downstream region of the target gene by overlap extension PCR. Third, the antibiotic resistance cassette is easily excised by a second single-crossover homologous-recombination event without any scars. Finally, the method could be used for the introduction of gene deletion in multidrug-resistant clinical isolates of A. baumannii without the construction of new vectors containing a suitable antibiotic resistance marker, as well as for multiple gene deletion. Therefore, the proposed method is a useful tool for functional studies of the genes involved in the pathogenesis of multidrug-resistant A. baumannii.
ACKNOWLEDGMENTS
This research was supported by a grant from the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare, Republic of Korea (grant HI14C0257), and by the Basic Science Research Program through the National Research Foundation of Korea (NRF), funded by the Ministry of Education, Science and Technology (NRF-2013R1A1A1010353).
REFERENCES
- 1.Bergogne-Bérézin E, Towner KJ. 1996. Acinetobacter spp. as nosocomial pathogens: microbiological, clinical, and epidemiological features. Clin Microbiol Rev 9:148–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dijkshoorn L, Nemec A, Seifert H. 2007. An increasing threat in hospitals: multidrug-resistant Acinetobacter baumannii. Nat Rev Microbiol 5:939–951. doi: 10.1038/nrmicro1789. [DOI] [PubMed] [Google Scholar]
- 3.Seifert H, Strate A, Pulverer G. 1995. Nosocomial bacteremia due to Acinetobacter baumannii. Clinical features, epidemiology, and predictors of mortality. Medicine 74:340–349. [DOI] [PubMed] [Google Scholar]
- 4.Durante-Mangoni E, Zarrilli R. 2011. Global spread of drug-resistant Acinetobacter baumannii. Future Microbiol 6:407–422. doi: 10.2217/fmb.11.23. [DOI] [PubMed] [Google Scholar]
- 5.Smith MG, Gianoulis TA, Pukatzki S, Mekalanos JJ, Ornston LN, Gerstein M, Snyder M. 2007. New insights into Acinetobacter baumannii pathogenesis revealed by high-density pyrosequencing and transposon mutagenesis. Genes Dev 21:601–614. doi: 10.1101/gad.1510307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vallenet D, Nordmann P, Barbe VR, Poirel L, Mangenot S, Bataille E, Dossat C, Gas S, Kreimeyer A, Lenoble P, Oztas S, Poulain J, Segurens BA, Robert C, Abergel C, Claverie JM, Raoult D, Médigue C, Weissenbach J, Cruveiller SP. 2008. Comparative analysis of acinetobacters: three genomes for three lifestyles. PLoS One 3:e1805. doi: 10.1371/journal.pone.0001805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rumbo-Feal S, Gomez MJ, Gayoso C, Alvarez-Fraga L, Cabral MP, Aransay AM, Rodriguez-Ezpeleta N, Fullaondo A, Valle J, Tomas M, Bou G, Poza M. 2013. Whole transcriptome analysis of Acinetobacter baumannii assessed by RNA-sequencing reveals different mRNA expression profiles in biofilm compared to planktonic cells. PLoS One 8:e72968. doi: 10.1371/journal.pone.0072968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Roca I, Marti S, Espinal P, Martinez P, Gibert I, Vila J. 2009. CraA, a major facilitator superfamily efflux pump associated with chloramphenicol resistance in Acinetobacter baumannii. Antimicrob Agents Chemother 53:4013–4014. doi: 10.1128/AAC.00584-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Penwell WF, Arivett BA, Actis LA. 2012. The Acinetobacter baumannii entA gene located outside the acinetobactin cluster is critical for siderophore production, iron acquisition, and virulence. PLoS One 7:e36493. doi: 10.1371/journal.pone.0036493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Camarena L, Bruno V, Euskirchen G, Poggio S, Snyder M. 2010. Molecular mechanisms of ethanol-induced pathogenesis revealed by RNA-sequencing. PLoS Pathog 6:e1000834. doi: 10.1371/journal.ppat.1000834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Aranda J, Poza M, Pardo BG, Rumbo S, Rumbo C, Parreira JR, Rodriguez-Velo P, Bou G. 2010. A rapid and simple method for constructing stable mutants of Acinetobacter baumannii. BMC Microbiol 10:279. doi: 10.1186/1471-2180-10-279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bentancor LV, Camacho-Peiro A, Bozkurt-Guzel C, Pier GB, Maira-Litrán T. 2012. Identification of Ata, a multifunctional trimeric autotransporter of Acinetobacter baumannii. J Bacteriol 194:3950–3960. doi: 10.1128/JB.06769-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Amin IM, Richmond GE, Sen P, Koh TH, Piddock LJV, Chua KL. 2013. A method for generating marker-less gene deletions in multidrug-resistant Acinetobacter baumannii. BMC Microbiol 13:158. doi: 10.1186/1471-2180-13-158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tucker AT, Nowicki EM, Boll JM, Knauf GA, Burdis NC, Trent MS, Davies BW. 2014. Defining gene-phenotype relationships in Acinetobacter baumannii through one-step chromosomal gene inactivation. mBio 5(4):e01313-14. doi: 10.1128/mBio.01313-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zimbler DL, Penwell WF, Gaddy JA, Menke SM, Tomaras AP, Connerly PL, Actis LA. 2009. Iron acquisition functions expressed by the human pathogen Acinetobacter baumannii. Biometals 22:23–32. doi: 10.1007/s10534-008-9202-3. [DOI] [PubMed] [Google Scholar]
- 16.Gaddy JA, Arivett BA, McConnell MJ, Lopez-Rojas R, Pachon J, Actis LA. 2012. Role of acinetobactin-mediated iron acquisition functions in the interaction of Acinetobacter baumannii strain ATCC 19606T with human lung epithelial cells, Galleria mellonella caterpillars, and mice. Infect Immun 80:1015–1024. doi: 10.1128/IAI.06279-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sambrook J, Russell DW. 2001. Molecular cloning: a laboratory manual, 3rd ed Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 18.Lee HW, Koh YM, Kim J, Lee JC, Lee YC, Seol SY, Cho DT, Kim J. 2008. Capacity of multidrug-resistant clinical isolates of Acinetobacter baumannii to form biofilm and adhere to epithelial cell surfaces. Clin Microbiol Infect 14:49–54. doi: 10.1111/j.1469-0691.2007.01842.x. [DOI] [PubMed] [Google Scholar]
- 19.Philippe N, Alcaraz J-P, Coursange E, Geiselmann J, Schneider D. 2004. Improvement of pCVD442, a suicide plasmid for gene allele exchange in bacteria. Plasmid 51:246–255. doi: 10.1016/j.plasmid.2004.02.003. [DOI] [PubMed] [Google Scholar]
- 20.Eijkelkamp BA, Hassan KA, Paulsen IT, Brown MH. 2011. Investigation of the human pathogen Acinetobacter baumannii under iron limiting conditions. BMC Genomics 12:126. doi: 10.1186/1471-2164-12-126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Perez F, Hujer AM, Hujer KM, Decker BK, Rather PN, Bonomo RA. 2007. Global challenge of multidrug-resistant Acinetobacter baumannii. Antimicrob Agents Chemother 51:3471–3484. doi: 10.1128/AAC.01464-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gaddy JA, Tomaras AP, Actis LA. 2009. The Acinetobacter baumannii 19606 OmpA protein plays a role in biofilm formation on abiotic surfaces and in the interaction of this pathogen with eukaryotic cells. Infect Immun 77:3150–3160. doi: 10.1128/IAI.00096-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wandersman C, Delepelaire P. 2004. Bacterial iron sources: from siderophores to hemophores. Annu Rev Microbiol 58:611–647. doi: 10.1146/annurev.micro.58.030603.123811. [DOI] [PubMed] [Google Scholar]
- 24.Braun V. 2001. Iron uptake mechanisms and their regulation in pathogenic bacteria. Int J Med Microbiol 291:67–79. doi: 10.1078/1438-4221-00103. [DOI] [PubMed] [Google Scholar]
- 25.Lim YM, Shin KS, Kim J. 2007. Distinct antimicrobial resistance patterns and antimicrobial resistance-harboring genes according to genomic species of Acinetobacter isolates. J Clin Microbiol 45:902–905. doi: 10.1128/JCM.01573-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bouvet PJM, Grimont PAD. 1986. Taxonomy of the genus Acinetobacter with the recognition of Acinetobacter baumannii sp. nov., Acinetobacter haemolyticus sp. nov., Acinetobacter johnsonii sp. nov., and Acinetobacter junii sp. nov. and emended descriptions of Acinetobacter calcoaceticus and Acinetobacter lwoffii. Int J Syst Evol Microbiol 36:228–240. [Google Scholar]
- 27.Piechaud M, Second L. 1951. Studies of 26 strains of Moraxella lwoffii. Ann Inst Pasteur (Paris) 80:97–99. [PubMed] [Google Scholar]
- 28.Simon R, Priefer U, Pühler A. 1983. A broad host range mobilization system for in vivo genetic engineering: transposon mutagenesis in gram negative bacteria. Nat Biotechnol 1:784–791. doi: 10.1038/nbt1183-784. [DOI] [Google Scholar]
- 29.Simon D, Chopin A. 1988. Construction of a vector plasmid family and its use for molecular cloning in Streptococcus lactis. Biochimie 70:559–566. doi: 10.1016/0300-9084(88)90093-4. [DOI] [PubMed] [Google Scholar]