

Abstract
The cellular cytoskeleton is crucial for many cellular functions such as cell motility and wound healing, as well as other processes that require shape change or force generation. Actin is one cytoskeleton component that regulates cell mechanics. Important properties driving this regulation include the amount of actin, its level of cross-linking, and its coordination with the activity of specific molecular motors like myosin. While studies investigating the contribution of myosin activity to cell mechanics have been performed on cells attached to a substrate, we investigated mechanical properties of cells in suspension. To do this, we used multiple probes for cell mechanics including a microfluidic optical stretcher, a microfluidic microcirculation mimetic, and real-time deformability cytometry. We found that nonadherent blood cells, cells arrested in mitosis, and naturally adherent cells brought into suspension, stiffen and become more solidlike upon myosin inhibition across multiple timescales (milliseconds to minutes). Our results hold across several pharmacological and genetic perturbations targeting myosin. Our findings suggest that myosin II activity contributes to increased whole-cell compliance and fluidity. This finding is contrary to what has been reported for cells attached to a substrate, which stiffen via active myosin driven prestress. Our results establish the importance of myosin II as an active component in modulating suspended cell mechanics, with a functional role distinctly different from that for substrate-adhered cells.
Introduction
The actin polymer network in cells is responsible for many cellular functions: it is involved in determining cell shape, in cell migration and in generating forces (1–4). Actin forms different types of networks in forms of branched or bundled fibers and provides mechanical stability to cells. The latter can be controlled by the amount of filamentous actin and the cross-linking between these filaments (5), which are both tightly regulated by the cell according to its functions (1). The actin cytoskeleton is fundamentally different in adherent cells compared to cells in suspension, mainly reflected in the absence of stress fibers in suspended cells. Therefore, it is not surprising that differences in mechanical properties can be found which reflect the different functional requirements of these cells (6). It has even been shown that adherent cells change mechanical properties directly after they are detached from substrates and brought into suspension (7). Apart from the filaments themselves and their cross-linkers, there are also molecular motors, such as myosin II, which can interact with and modify the actin cytoskeleton (1). Molecular motors are micromachines that can transport cargo within cells along microtubules (kinesin, dynein) or slide two actin filaments along each other (myosin). Filament sliding is important when the cell needs to change its shape or to exert forces, as it does during cell migration, mitosis, (muscle) cell contraction, wound closure, or mechanosensing of its environment. The activity of myosin II can also influence the mechanical properties of cells by prestressing the actin cortex underlying the plasma membrane (8) and by generating tension along actomyosin stress fibers (9). Adherent cells have anchoring points to the substrate, called focal adhesions, and actin stress fibers are spanning through the cell from one focal adhesion point to another. Attachment to a substrate is thus a prerequisite for the formation of stress fibers and their abundance decreases with the stiffness of the substrate (10).
Interestingly, the progressive disappearance of stress fibers on compliant substrates correlates with a softening of the cells, presumably due to decreasing prestress in the actin network (10,11). This modulation of cell mechanical properties by the state of attachment needs to be taken into account when choosing a cell mechanics measurement technique. There are several ways of measuring mechanical properties of adherent cells. A popular method is atomic force microscopy, which can probe viscoelastic properties of cells with high spatial resolution by indenting the surface of the cell with a nano- to micrometer-sized probe (7,12). A different approach is magnetic twisting cytometry, where magnetic beads are bound to the surface of cells and forces are applied by external magnetic fields (13). Techniques for measuring mechanical properties of cells in suspension are less common. This can be done by micropipette aspiration (14) or optical tweezers, which trap and move beads bound to the surface of cells (15). Most of these methods have low throughput and still require physical contact with the cell surface, which can still induce focal adhesions and stress fibers (16). One way of measuring pure mechanical properties of cells in absence of stress fibers is by means of a microfluidic optical stretcher, where no physical contact is required because forces are generated via momentum transfer of light (17,18). Another measurement technique is based on the concept of measuring advection times of cells on passage through microchannels, which has been shown to correlate with whole-cell compliance (6,18). Finally, hydrodynamic stretching of cells through a microfluidic channel allows high throughput quantification of cell deformation by shear stresses and pressure gradients, and provides an alternative technique to characterize cell deformability in suspension on shorter timescales (19).
Measuring cells adhered to rigid substrates and cells that are completely in suspension represents the two extremes of cellular attachment. In this study, we investigated the role of myosin motors on the rheological properties of cells in suspension. We focused on non-muscle myosin II, which specifically cross-links two actin filaments and exerts tension (20). There are several drugs to interfere with myosin II activity, such as blebbistatin, Y-27632, ML7, or phorbol 12,13-dibutyrate. Blebbistatin is a small molecule inhibitor that selectively inhibits non-muscle myosin II in a state where the myosin head is not attached to the actin filament (21). Y-27632 also inhibits myosin activity but via the inhibition of the Rho-associated kinase (22). ML7 directly targets the myosin light chain kinase pathway by inhibiting its activity (23). Phorbol 12,13-dibutyrate, on the other hand, activates the protein kinase C (24,25) and therefore increases myosin II activity. So far, there exists a range of data on mechanical property measurements of adherent cells treated with these drugs (26–29), which have shown that inhibition of myosin activity leads to a loss of cell prestress and a subsequent lower measured elastic modulus of the cells.
Because no systematic study of the effect of these pharmacological interventions for cells in a fully suspended state (i.e., without physical attachment to the probe) has been conducted (to our knowledge), we focused on studying the effect of myosin inhibition on the mechanical compliance of cells in suspension, using a microfluidic optical stretcher (OS), a microfluidic microcirculation mimetic (MMM), and a real-time deformability cytometer (RT-DC). To compare with existing reports on adherent cells, we specifically included studies of 3T3 fibroblasts, HeLa cells, and TNGA mouse embryonic stem cells detached from the substrate before the measurement, as well as cells in naturally nonadherent states such as mitotic cells. To assure that the observed effect is not specific to adherent cells, we carried out further studies on two distinct hematopoietic cell lines, NB4 and HL60 leukemia cells (both nonadherent cells growing naturally in suspension), as well as HL60-differentiated lineages: neutrophils, monocytes, and macrophages. Our results showed that apart from the macrophages and neutrophils (to a lesser extent), all the tested cell lines in suspension showed a significant decrease in cell compliance and fluidity, and a concomitant increase in steady-state viscosity with myosin inhibition on a deformation timescale of seconds, opposite to what has been reported for cells attached to substrates. Our work provides unambiguous evidence that myosin II plays a fundamentally different role in modulating cell mechanical properties of suspended cells as compared to adherent cells, possibly through myosin-induced actin disassembly.
Materials and Methods
Microfluidic optical stretcher
The principle and setup of the microfluidic OS have been described extensively elsewhere (17,18). Essentially, the device is a dual beam laser trap capable of trapping and deforming cells through optically induced stress acting on the cell surface. The microfluidic flow chamber consists of two coaxial optical fibers aligned perpendicular to a square glass capillary, which delivers the cells in suspension. The chamber was mounted on an inverted microscope (Eclipse TE2000U) equipped with a Plan Fluor ELWD 40× /0.60 NA objective (both from Nikon, Tokyo, Japan). A charge-coupled device camera (AVT MARLIN F-146B, 50 frames/s; Firstsight Vision, CA) was attached to the microscope for image acquisition. The flow of cell suspension was adjusted through the relative difference in heights of an inlet and outlet reservoir connected to the capillary. The laser used was a single-mode, continuous-wave fiber laser at a wavelength of λ = 1064 nm (YLM-5-1070-LP; IPG Photonics, Oxford, MA).
For imaging and data analysis, a custom-built LABVIEW software (National Instruments, Austin, TX) was used to track the cell shape during the stretching. Data of the cell deformation along the major axis r(t) was stored for every time-frame, while r0 was the measured length of the cell during the initial trapping period. The time-varying axial strain,
was then evaluated accordingly. The optically induced stress was computed using the generalized Lorenz-Mie model (Boyde et al. (30)). A general stress profile modeled for a sphere is shown in Fig. S1 in the Supporting Material. Once the peak stress σo along the laser axis was evaluated, the time-dependent creep compliance J(t) could be readily obtained as
| (1) |
where FG is a geometric factor that takes into account the cell size and stress distribution (31). To further extract cell viscoelastic parameters, one can employ either a power law, or a simple mechanical model incorporating combinations of dashpots and springs (6,7,32). The equation for the power-law model is
| (2) |
Here, β = 0 corresponds to Hooke’s law, where the cells behave like a perfectly elastic material, while β = 1 corresponds to Newton’s law of viscous deformations, with the cells behaving like a fluid. The β is therefore a measure of the cell fluidity. The prefactor Jo characterizes the compliance of the material, while the time is normalized by a timescale τo, which is set to 1 s. Because the power-law model assumes timescale independence, which may not generally hold for biological cells, we also fit the compliance data with the standard linear liquid (SLL) model, which has been recently shown to characterize cell deformation for blood precursor cells (6). The creep compliance for such a model takes the form
| (3) |
where Et represents the transient elastic response within the cytoskeleton while ηt represents the transient viscosity, possibly arising from the poroelastic effect (33). The value ηs represents the steady-state viscosity at long timescales, possibly originating from molecular slips between the transient cross-linkers in the cell cortex. A transition time, τ = ηt/Et, can also be defined as the characteristic timescale beyond which the steady-state viscous behavior starts to dominate.
For each cell type under different pharmacological treatment, the number of selected cells per OS experiment was n ≥ 30. The creep compliance data are depicted as mean ± SE. Representative compliance data were selected from two or more independent measurements. All measurements were performed at room temperature. All plotting, statistical analysis, and curve fitting were performed using ORIGIN 8.5 software (OriginLab, Northampton, MA) and MATLAB (The MathWorks, Natick, MA). All box plots include whiskers at 5–95% range, horizontal box lines at 25–75% range, median line, and the mean. For parametric fitting, the extracted rheological parameters were computed within 95% confidence bounds using the nonlinear least-squares regression algorithm, and presented as the best fit values ± SE of the estimates.
Microfluidic microcirculation mimetic
To validate changes in suspended cell mechanical properties free of possible heating artifacts as in the case of OS experiments (34), we developed a microfluidic microcirculation mimetic (MMM). A MMM is a microfluidic device consisting of a long microchannel with many successive constrictions smaller than the cell diameter. The passage of cells driven through a MMM should be inversely correlated with the compliance of the cells because the timescales of passing through the full device is comparable with the timescales of optical stretching. The production of MMMs was done using standard photolithographic techniques as reported in our previous work (6). In short, PDMS (Sylgard 184; Dow Corning, MI) was degassed by centrifugation (1200 rpm for 20 min), poured onto molds, and baked for ∼20 min at 100°C. Inlet and outlet holes were punched (1.5 mm hole; Harris Uni-Core, Sigma-Aldrich, Munich, Germany) in the PDMS chip. The chips were bonded to 22-mm glass cover slides (No. 1; Marienfeld-Superior, Lauda-Königshofen, Germany) using air plasma (Plasma Cleaner PDC-32G; Harrick Plasma, Ithaca, NY). MMMs with 5 and 7 μm as the smallest constriction widths (maximum width 15 μm) were made with a constant height of 15 μm. The inlet and outlet of the devices were connected to a computerized air pressure control system (MFCS-FLEX; Fluigent, Villejuif France) using custom-cut pipette tips (Eppendorf, Hamburg, Germany) in which ∼20–40 μL of cell suspension was introduced for each experiment. The device was mounted on a model No. IX-70 inverted microscope with 40× objective for measuring cell size and 5 objective for measuring the advection time through the entire device (both objectives from Olympus, Tokyo, Japan). A camera (DMK 21BF04; The Imaging Source, Bremen, Germany) was used to record advection videos at a frame-rate of 60 frames/s. Advection times were extracted from the videos using custom-made written codes in MATLAB (The MathWorks, Natick, MA). For each cell type under different pharmacological treatment, the number of selected cells per MMM experiment was n ≥ 30, and the advection times are representative of two independent experiments.
Real-time deformability cytometry
The basic idea of RT-DC is to quantify the deformation of cells on shorter timescales (approximately milliseconds) by shear stresses and pressure gradients while flowing through a narrow microfluidic channel (19). The microfluidic chip is made of PDMS (Sylgard 184; VWR, Darmstadt, Germany) using standard soft-lithography methods. After cross-linking the polymer for 45 min at 70°C and opening an inlet and outlet with a 1.5-mm biopsy puncher (Harris Uni-Core; Sigma-Aldrich), the bottom of the chip is sealed with a glass cover slide (thickness 2; Hecht, Pfaffenhofen an der Ilm, Germany) after plasma surface activation (PDC 32-G; Harrick Plasma, Ithaca, NY) of the PDMS. The microfluidic chip consists of two reservoirs connected by a 300-μm-long narrow channel with a 20 × 20 μm (as for HL60 cells) or 30 × 30 μm square cross section (for 3T3 fibroblasts). After fabrication, the chip is assembled on an Axiovert 200M inverted microscope (Carl Zeiss, Oberkochen, Germany) and connected to a syringe pump (NemeSyS; Cetoni, Korbussen, Germany). A high-power LED (CBT-120, 462 nm; Luminus Devices, Woburn, MA) is operated with a pulsed current for sample illumination. Image acquisition of cells is done using a high-speed CMOS camera (MC1362; Mikrotron, Unterschleissheim, Germany).
Cells are resuspended in a solution of PBS (phosphate-buffered saline) and 0.5% (w/v) methylcellulose (Sigma-Aldrich) to a final concentration of 106 cells/mL. Addition of methylcellulose helps prevent sedimentation of the cells. The cell suspension is drawn into a 1-mL syringe connected to the inlet of the chip. As the cell suspension is pumped through the chip, it gets deformed from a spherical into a bulletlike shape within the channel. An image analysis algorithm continuously analyzes the images from the camera in real-time, detects the presence of a cell inside the region of interest, and finds the cell contour. The quantified cell deformation (1, circularity) and cell size (cross-sectional area) are then displayed as a scatter plot and the average deformation is defined as the extracted mode (peak) of the fitted distribution. All data acquisition is carried out at the rear part of the 300-μm-long channel where the cell shape has reached steady state. As a reference, the deformation of the nondeformed cells in the reservoir is also measured. In a typical experiment, ∼2000 cells are analyzed at a particular flow rate, and more than three repeat experiments are performed.
Cell culture and differentiation
3T3 fibroblasts and HeLa cells were cultured in DMEM medium containing 10% FBS (fetal bovine serum) and 100 U/mL penicillin-streptomycin (all from Life Technologies, Carlsbad, CA, Germany). The TNGA mouse embryonic stem cell line was a kind gift from the lab of Austin Smith, and were cultured in GMEM-medium (Sigma-Aldrich) supplemented with 10% FBS, 2 mM GlutaMax, 100 U/mL penicillin-streptomycin, 1× MEM nonessential amino acids, 100 μM 2-mercaptoethanol (all from Life Technologies), 1 mM sodium pyruvate (Sigma-Aldrich), and 1000 U/mL LIF (Millipore, Billerica, MA). NB4 and HL60 cells were cultured in RPMI-1640 medium containing 10% FBS, 2 mM L-glutamine, and 100 U/mL penicillin-streptomycin (all from Life Technologies). HL60 cells were induced to differentiate along the myeloid lineage following well-established protocols (6,35). In essence, mature neutrophil- and monocyte-like cells were obtained from HL60 cells 96 h after induction of differentiation. Macrophage-like cells were obtained 24 h after induction of differentiation. For experiments on pharmacological interventions, cells were treated with 50 μM of blebbistatin or 10 μM of ML7 for 20 min, with 10 μM of Y-27632 for 30 min, and 2 μM of cytochalasin D for 10 min, all before the experiments. Jasplakinolide was added to a final concentration of 1 μM and incubated for 30 min before the experiments, while phorbol 12,13-dibutyrate was added to a final concentration of 100 nM and incubated for 1 h before experiments. All pharmacological drugs were obtained from Sigma-Aldrich unless otherwise stated.
For myosin II knock-down experiments, MYH9 siRNA plasmids (SMARTpool ON-TARGETplus; GE Dharmacon, Lafayette, CO) or negative controls (ON-TARGETplus nontargeting Pool; GE Dharmacon) were transiently transfected into 3T3 fibroblasts using the Lipofectamine2000 reagent (Life Technologies) following the manufacturer’s instructions. In brief, transfections were performed in six-well plates. For each well, 2.5 μg of plasmid DNA was added to 250 μL of Opti-MEM (Life Technologies) reduced serum medium, while a separate mixture of 5 μL Lipofectamine2000 and 250 μL Opti-MEM was being prepared. Both mixtures were incubated for 5 min at room temperature before being combined and left for another 20 min at room temperature. The entire volume was then added to each well with cells already added with 2 mL cell medium. The cells were incubated for another 72 h before being assayed for OS experiments. For OS experiments on mitotic HeLa cells, cell synchronization was achieved by a standard double thymidine (Sigma-Aldrich) block and release protocol, and 10 μM dimethylenastron (VWR) was applied for 10 h to keep the cells in the mitotic phase.
Fluorescence imaging of suspended cells
For actin staining of cells in suspension, trypsinized cells were first washed with prewarmed PBS, before being treated with 50 μM of blebbistatin or 10 μM of ML7 and incubated for 20 min in a cell culture incubator. The cells were then fixed with 4% formaldehyde in PBS solution for 20 min at room temperature. This way, the cells were “frozen” in the suspended state before they had any time to start adhering to the substrate. A quantity of 100 μL of the suspended fixed cells was then seeded onto a PLL (poly-L-lysine)-coated glass slide for 20 min at room temperature. For permeabilization, we incubated the cells with 0.1% Triton X-100 diluted in PBS for 5 min. After that, the cells were stained with phalloidin-Alexa-Fluor 488 (Life Technologies) diluted in a 1% BSA solution in PBS for a final concentration of 165 nM, and incubated for 20 min at room temperature in the dark. The sample was then washed three times with PBS. Cells stained for actin were imaged in confocal mode using a model No. LSM 700 confocal microscope (Carl Zeiss) with a C-Apochromat 40× /1.2 NA water objective (Carl Zeiss). Gain settings were kept the same for all samples of a given experiment. For statistical comparison of morphological change in actin cortical structures between the control and blebbistatin-treated cells, multiple images from different regions of the dish were collected in each experiment.
Ultrastructural imaging using scanning electron microscope
Cells were washed in PBS once before being trypsinized and resuspended in 4% PFA (paraformaldehyde) in PBS. The cells were left in fixatives for 20 min at room temperature. The cells were then resuspended in PBS and seeded onto PLL-coated cover glass. After 10 min, PBS was removed and 1% Triton X-100 in PBS buffer was added and the permeabilization was performed at room temperature for 5 min. The cover glass was then washed three times with PBS. After that, the cells were dehydrated in a graded series of ethanol before being critical-point-dried with liquid carbon dioxide, and sputter-coated with gold (5–10 nm in thickness). The specimens were later examined under a scanning electron microscope (JSM-7500F; JEOL, Eching, Germany) operated at an accelerating voltage of 5 kV.
Results
Suspended adherent cells stiffen with myosin II inhibition
To investigate changes to suspended cell mechanical properties when subjected to pharmacological interventions, an OS was employed (Fig. 1 A) to measure the whole-cell compliance in the suspended state, with or without drugs in the suspension medium. As discussed in Materials and Methods, the time-dependent strain of the cells is extracted and normalized by the peak stress and a geometric factor to obtain the creep compliance for the cell, as shown in Fig. 1 B. We define the peak compliance as the compliance at the end of the stretch period (t = 4 or 3 s). Suspended cells typically respond to the optical stress in a viscoelastic manner, with cell fluidity β ranging from 0.3 to 0.6 (32,34). A fit of the data with the SLL model allows us to further extract meaningful viscoelastic parameters at transient and long timescales. Because the deformation response at very small timescales can vary significantly among different drugs and cell types, we focused on first extracting ηs, the relevant fitting parameter at long timescales. To do this, we fit a linear function to the compliance curves for t > 2 s. The inverse slope yields ηs and the inverse intercept Et. We subsequently fit the model to the entire data and minimized with respect to ηt to determine its value, while keeping ηs and Et fixed. We note that although the SLL model may not render the best fits to all compliance data for different cell lines under various treatments, which attests to their fundamental rheological differences, we opt to settle on the SLL model in order to draw meaningful comparison of the viscoelastic parameters.
Figure 1.
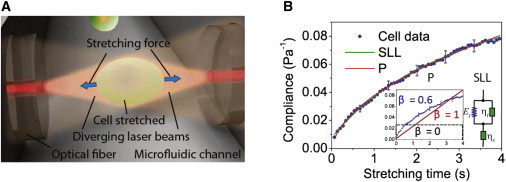
Schematic of an optical stretcher experiment and data analysis. (A) Cells flowing through the microfluidic channel are serially trapped and stretched by two diverging, counterpropagating laser beams emanating from single-mode optical fibers. The momentum transfer of light to the surface of the cell results in (mainly axial) optically induced surface stresses, leading to cellular deformation along the optical axis. (B) Creep compliance curve for a representative HL60 cell stretched at 0.9 Pa. The creep compliance curve can be fitted to a power-law (P) model or a SLL mechanical model. In the power-law model, the creep response follows a power law, with β = 0 for purely elastic materials, unity for a purely viscous fluid, and 0 < β < 1 for viscoelastic materials, such as cells (see inset). The SLL model comprises the transient elastic (Et) and viscous components (ηt), and the steady-state viscous component (ηs). To see this figure in color, go online.
Given that adherent cells have been shown to soften with myosin II inhibition (26–29), we seek to address the following question: will the same effect be observed for such cells when they are in a fully suspended state in the absence of any stress fibers? As such, we detached 3T3 fibroblasts from the substrates using trypsin/EDTA, before measuring their deformability in the OS. As depicted in Fig. 2 A, the untreated 3T3 fibroblasts, when stretched by a constant optically induced stress of 0.85 Pa, showed a time-dependent creep compliance that appeared to increase linearly with time, indicative of a fluidlike behavior. When the cells were stretched in the presence of a myosin II inhibitor such as blebbistatin, the entire creep compliance curve changed significantly (Fig. 2 A, red). Specifically, the curve consisted of an initial rapid increase in creep compliance, followed by a gradual decrease in the rate of compliance. In view of recent evidence of phototoxicity and photoinactivation of blebbistatin by UV and blue light (36,37), we tested the effect of another myosin-inhibitor, Y-27632, and affirmed a similar change in the creep compliance profile (Fig. 2 A, green). The same effect was also observed for two additional adherent cell lines, HeLa cells and TNGA mouse embryonic stem cells (Fig. 2, B and C), thereby ruling out a cell-specificity issue.
Figure 2.
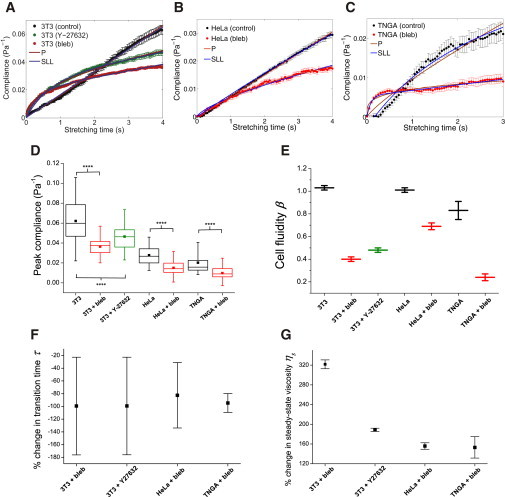
Influence of myosin inhibitors on the compliance of adherent cell lines brought into suspension. (A) Compliance of 3T3 fibroblasts when treated with blebbistatin (n = 89) or Y-27632 (n = 72), compared to controls (n = 65). (B and C) Compliance of blebbistatin-treated HeLa cells (n = 94) and TNGA mouse embryonic stem cells (n = 45) compared to their respective controls (n = 89, 30). (Brown and blue lines) Fits of the power-law model and SLL model to the compliance data, respectively. (D) Box plots of the peak compliance for various cell lines under blebbistatin or Y-27632 treatments. Statistical significance is indicated by asterisks: ∗∗∗∗p < 0.0001. (E–G) Cell fluidity, % change in transition times, and % change in steady-state viscosity, respectively, for various cell lines treated with blebbistatin or Y-27632. Error bars are standard errors of the fit parameters. To see this figure in color, go online.
Furthermore, OS measurements for HeLa cells up to 60 s (Fig. S2) confirm that the effect of myosin II inhibition does not reverse or become nonlinear at longer timescales. As shown in Fig. 2 D, all three cell lines exhibited a significantly lower peak compliance for the blebbistatin or Y-27632-treated cases than those of the controls. When we fit the power-law model to the data, we observed a dramatic decrease in cell fluidity β for cells treated with blebbistatin or Y-27632, indicating that the cells became more solidlike after inhibition of myosin II activity (Fig. 2 E and Table S1 in the Supporting Material). Further fitting with the SLL model showed that for cells treated with blebbistatin or Y-27632, there was a significant decrease in the transition time τ (Fig. 2 F; Table S2), suggesting that the cells reached their steady-state viscous state quicker. Furthermore, this effect is accompanied by a significant increase in the steady-state viscosity ηs for all cells treated with blebbistatin or Y-27632 (Fig. 2 G), indicating that these cells flow at a much lower rate at long timescales.
To ascertain that the effect of cell stiffening is not an experimental artifact due to photodestabilization of the drugs during optical stretching, we also performed OS experiments on myosin II knocked-down cells and control cells. The results are depicted in Fig. 3 A. Indeed, 3T3 fibroblasts treated with RNAi against MYH9, the heavy chain subunit of myosin IIA, showed a strong decrease in deformability compared to the control cells, as reflected by the significant drop in peak compliance (inset). We hypothesize that this stiffening phenomenon may be attributed to specific actions of myosin on actin filaments. Hence, we carried out independent OS experiments on cells treated with Jasplakinolide, an actin-stabilizing drug that promotes actin polymerization (Fig. 3 B). Interestingly, we found that the compliance curve of Jasplakinolide-treated 3T3 fibroblasts closely resembled that of the blebbistatin-treated cells, providing hints that myosin II may play a critical role in actin turnover dynamics on suspended cells free of any focal adhesions or stress fibers.
Figure 3.
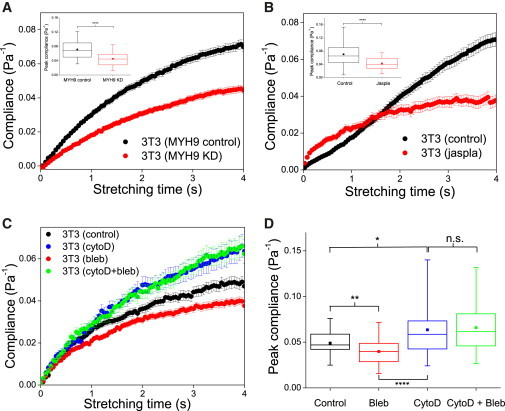
Role of actin cortex in compliance of 3T3 fibroblasts brought into suspension. (A) Compliance curves for 3T3 fibroblasts with RNAi against MYH9 (n = 114), compared to controls (n = 95). (B) Compliance curves for 3T3 fibroblasts after treatment with Jasplakinolide (n = 38), compared to controls (n = 78). (Insets in A and B) Box plots of peak compliance for control versus knockdown or drug treatments. (C) Compliance curves for 3T3 fibroblasts after treatment with cytochalasin D only (n = 45), cytochalasin D plus blebbistatin (n = 38), or with blebbistatin only (n = 80), compared to controls (n = 60). All experiments were performed on cells of the same passage on the same day. (D) Box plots of the peak compliance for cells under various treatments as in (C). ∗∗∗∗p < 0.0001, ∗∗p < 0.01 and ∗p < 0.05. To see this figure in color, go online.
Finally, as a control to confirm the structural integrity of actin filaments in regulating such behavior, we conducted further “no-cortex” controls for 3T3 cells in which cells were treated with 2 μM cytochalasin D, which depolymerizes the actin cortex, before further treatment with blebbistatin. We observed a significant stiffening of 3T3s treated with blebbistatin (Fig. 3 C, red) compared to the controls cells, confirming previous results. When cells were treated with cytochalasin D (Fig. 3 C, blue), they were significantly more compliant than the control cells, confirming the role of actin in cell deformability. However, we observed no significant difference in the creep compliance profile (Fig. 3 C, green) and the peak compliance (Fig. 3 D) between cells treated with only cytochalasin D and those treated with both cytochalasin D and blebbistatin. This supports our hypothesis that indeed myosin inactivation-induced stiffening is attributed to disruption of specific action of myosin on the actin cortex. As a positive control, we also carried out OS experiments on 3T3 fibroblasts treated with phorbol 12,13-dibutyrate, which is known to enhance myosin II activity (Fig. S3). Here we observed just the opposite effect: the phorbol 12,13-dibutyrate-treated cells were more compliant compared to the control cells, as indicated by a slightly higher compliance curve throughout the stretch period (Fig. S3 A), although the increase in peak compliance was less significant compared to the degree of cell stiffening for blebbistatin-treated cells (Fig. S3 B).
To gain insights into the role of actin cytoskeletal structures in the observed cell stiffening effect, we imaged F-actin in both the untreated cells and blebbistatin-treated cells using confocal microscopy. We observed a marked surface morphological difference in the cortical actin structures between the untreated and blebbistatin-treated 3T3 fibroblasts (Fig. S4, A and B). Specifically, the untreated cells showed a dense actin cortical layer underneath the plasma membrane, usually accompanied by many blebs and spherical protuberances at the cell periphery, while the blebbistatin-treated cells exhibited numerous microvilli projections on the cell surface. A similar effect was observed for cells imaged using a scanning electron microscope (Fig. S4, C and D) and, for cells treated with ML7, a further myosin inhibitor (Fig. S4, E and F). This phenotypic difference was highly reproducible and observed across the entire population (Fig. S5).
Nonadherent cell lines exhibit similar stiffening effect with myosin II inhibition
To ensure that the observed cell stiffening effect with myosin inhibition is not specific to normally adherent cell lines in suspension, we also investigated the effect of blebbistatin on two distinct hematopoietic cell lines, NB4 leukemia and HL60/S4 myeloid precursor cells, both of which can be differentiated into other lineages. Unlike 3T3 fibroblasts, these two cell lines grow naturally in suspension, which makes the study of myosin II activity in modulating suspended cell stiffness even more pertinent. As shown in Fig. 4 A, when NB4 cells were treated with blebbistatin and stretched by a constant stress of 0.90 Pa, we observed a significantly less compliant response than that of the controls throughout the period of stretch. A similar stiffening effect was observed for blebbistatin-treated, undifferentiated HL60/S4 cells (Fig. 4 B).
Figure 4.
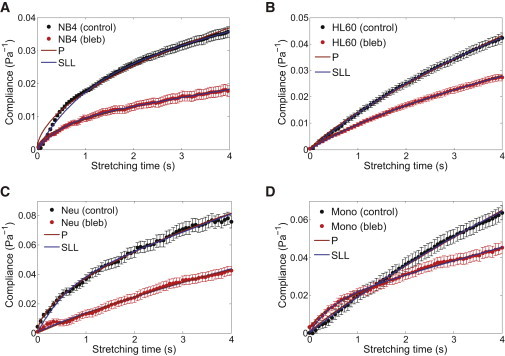
Influence of blebbistatin on the compliance of naturally suspended cells. Compliance of (A) NB4 cells (n = 44), (B) HL60 cells (n = 94), (C) HL60-derived neutrophils (n = 73), and (D) HL60-derived monocytes (n = 72), compared to their respective controls (n = 165, 89, 75, 85). (Brown and blue lines) Fits of the power-law model and SLL model to the compliance data, respectively. To see this figure in color, go online.
An interesting question arises as to whether inhibition of myosin II activity causes a similar cell stiffening effect for the differentiated lineages and, more specifically, whether there is a lineage- or function-specificity of myosin II activity in modulating cell mechanical properties. To address this question, we differentiated the HL60 cells in vitro into neutrophils and monocytes (see Materials and Methods for protocol), and investigated their creep behavior in response to blebbistatin treatment under optical stretching. The results for neutrophils and monocytes are shown in Fig. 4, C and D. Clearly, these cells also exhibited a significantly less compliant response when treated with blebbistatin. Of note is the compliance curve for blebbistatin-treated neutrophils, which appeared to grow linearly in time, suggestive of a more fluidlike behavior. This is in contrast to all other cell lines that showed a gradual decrease in the rate of compliance with time. For the sake of completeness, we also included studies on HL60-differentiated macrophages. Macrophages are tissue-resident cells that are adherent in nature, fundamentally different compared to neutrophils and monocytes, which are nonadherent. As such, the results may not be directly comparable to that of neutrophils and monocytes. As shown in Fig. S6, we observed that blebbistatin-treated macrophages showed little, if any, changes in the compliance curve, as compared to the control case.
A quantitative analysis of the creep compliance curves is shown in Fig. 5. For NB4 cells, undifferentiated HL60 cells, and neutrophils, the peak compliance for the blebbistatin-treated cases was significantly lower than those of the controls (p < 0.0001), while for monocytes we observed a less, albeit still significant difference between both cases (p < 0.001) (Fig. 5 A). A comparison of the extracted power-law exponents before and after blebbistatin treatment is depicted in Fig. 5 B. Apart from the HL60-derived neutrophils, which seem to have become more fluidlike, all cell lines exhibit a drop in the cell fluidity β after treatment with blebbistatin, with the effect on monocytes being the most significant (see also Table S3). This is consistent with the previous observation of suspended 3T3 fibroblasts becoming more solidlike or elastic after blebbistatin treatment. A similar trend of solidification was observed using the SLL model. As shown in Fig. 5 C and Table S4, there is a general decrease in the transition time for all cases, again with the exception of neutrophils. More importantly, we observed a marked increase in the steady-state viscosity ηs after blebbistatin treatment for all cell lines (Fig. 5 D; Table S4), again similar to our observation for adherent cell lines brought into suspension.
Figure 5.
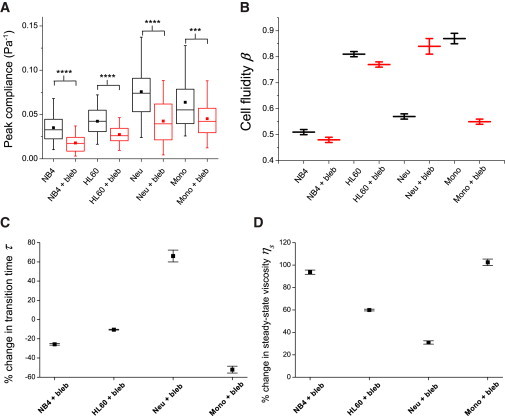
Changes in viscoelastic parameters of naturally suspended cells after myosin inhibition by blebbistatin. (A) Box plots of the peak compliance for various cell lines. For all cell lines tested, the peak compliance for blebbistatin-treated cases was significantly lower than for controls. ∗∗∗∗p < 0.0001 and ∗∗∗p < 0.001. (B–D) Cell fluidity, % change in transition times, and % change in steady-state viscosity, respectively, for various cell lines treated with blebbistatin compared to their controls. Error bars are standard errors of the fit parameters. To see this figure in color, go online.
Of note, confocal imaging of suspended HL60 cells stained for actin revealed vast numbers of branched microvilli at the cell surface, compared to the controls that displayed more surface blebs at the cell periphery (data not shown), similar to what was observed for the 3T3 fibroblasts. This suggests that the formation of a more filamentous, microvillus actin cytoskeleton with blebbistatin treatment likely contributes to a stiffer actin cortex and a reduced whole-cell compliance.
Cell stiffening correlates with longer advection time through microconstrictions
Owing to the fact that the OS, operated at a wavelength of 1064 nm, might introduce additional heating effects that can impact cell rheology (34,38,39), we employed a MMM to confirm the impact of changes to suspended cell mechanical properties free from heating artifacts (see Materials and Methods). In this setup, cells were advected sequentially through a long microchannel with many constrictions smaller than the cell diameter using driving pressures within physiological range (50 mbar) (Fig. 6 A and Movie S1 in the Supporting Material). We hypothesize that because cell compliance may be the main limiting factor controlling the rapid passage of cells through narrow constrictions (6), cells treated with blebbistatin, being stiffer, will take longer to pass through the device. Indeed, we found that blebbistatin-treated HL60 cells required a longer average advection time than the control cells (Fig. 6 B), when advected through the MMM with 5-μm wide constrictions. To test the opposite effect to the measured cell stiffening, we treated HL60 cells with 1 μM cytochalasin D, a drug that inhibits actin polymerization and which has previously been shown to soften the cells significantly (6). This led to a highly significant decrease in the advection time, as expected because cell softening should facilitate the passage of cells through the narrow constrictions. In the case of 3T3 fibroblasts, we found that most untreated cells flowed through the MMM (5 μm constriction width) with no hindrance, but a majority of cells treated with blebbistatin and Y-27632 were trapped even at the first constriction (data not shown). We then replaced the MMM with 7-μm constriction widths. In this case, cells previously trapped at the first constriction were able to passage through the first constriction, before eventually being trapped between the fifth and the 10th constriction. We therefore conclude that the blebbistatin-treated 3T3 fibroblasts became considerably stiffer, similar to the observed cell stiffening seen in HL60s. Our results from MMM measurements support the findings from OS measurements, and unambiguously confirm that cells, once in suspension, stiffen with inhibition of myosin II activity.
Figure 6.
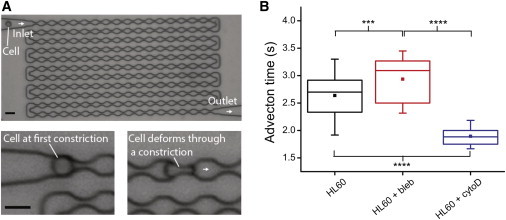
Stiffening of blebbistatin-treated HL60 cells in MMM. (A) Phase contrast image of an MMM consisting of a large number of microconstrictions. A constant pressure difference, maintained between the inlet and the outlet, is used to advect cells through the device. The minimum gaps at the constrictions (5 μm) are smaller than the diameter of cells, ensuring that each cell is sequentially deformed during the advection. The driving pressure used was 50 mbar. Scale bar is 15 μm. (B) Box plots of the advection time (from entry to exit) for HL60 cells treated with blebbistatin or cytochalasin D, as compared to controls. ∗∗∗∗p < 0.0001 and ∗∗∗p < 0.001. To see this figure in color, go online.
Hydrodynamic stretching of cells at millisecond timescales
To test the effect of myosin inhibition on cellular deformability at shorter timescales than probed in the OS and MMM, we used a technique based on the concept of hydrodynamic stretching. Unlike the OS and MMM, which measure cell mechanical properties with throughput rates of 100–1000 cells/h, RT-DC offers a much higher throughput of ∼100 cells/s 19. As shown in Fig. 7 A (also see Materials and Methods), a cell suspension is pumped through a microfluidic chip illuminated with a pulsed LED. Due to shear stress, a flowing cell is typically deformed into a bulletlike shape within the channel. An image analysis algorithm then captures and analyzes the cell contour continuously in real-time (shown as red outline over the cell). The quantified cell deformation and cell size are then displayed as a scatter plot, with histograms plotted, which allows us to extract the average deformation at a particular flow rate. We measured the changes in the average cell deformation (from the reservoir to the microchannel) for the drug-treated cells, and normalized this value to that of the control cells. The obtained ratio is then defined as the relative deformation that essentially gives a good measure of the cell deformability of the drug-treated cells compared to that of the control cells.
Figure 7.
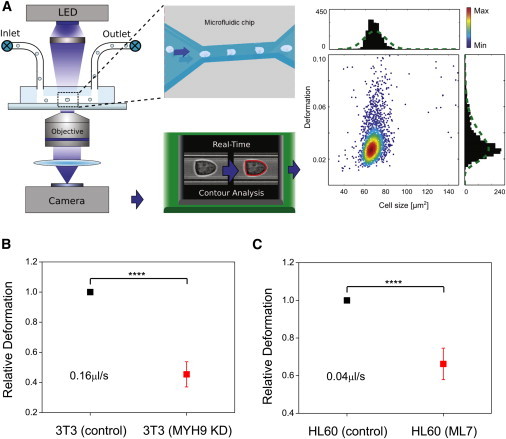
Hydrodynamic stretching technique measures cell stiffness on short timescales of milliseconds. (A) Schematic illustration of measurement principle. A cell suspension is pumped through a microfluidic chip with a narrow constriction (see inset for top-view) using a computer-controlled syringe pump. The cell deforms inside the channel and imaged, and the contour (red) is determined by image analysis algorithm in real-time. A scatter plot of cell size versus deformation is then obtained (each dot representing a single-cell event). (B and C) Comparisons of the relative deformation of control cells versus 3T3 cells with RNAi against MYH9, and ML7-treated HL60 cells, respectively. The flow rates are indicated in the plots. ∗∗∗∗p < 0.0001. To see this figure in color, go online.
At these short deformation timescales in the range of milliseconds, we observed that 3T3 fibroblasts with RNAi against MYH9 were deformed significantly less than the control cells (Fig. 7 B), with a relative deformation of 0.454 indicating cell stiffening associated with myosin inactivation. As a further check, we used another pharmacological inhibitor of myosin, ML7, which specifically blocks the activity of myosin light chain kinase, and measured its effect on HL60 cell stiffness using both the OS and the RT-DC. Similar to the results of blebbistatin-treated cells in OS experiments, the ML7-treated cells showed a significant decrease in cell deformability as shown by the change in the creep compliance profile and peak compliance (Fig. S7, A and B). RT-DC experiments also revealed >30% decrease in cell deformability for ML7-treated cells compared to the controls (Fig. 7 C), consistent with the OS results. In both the genetic knockdown and ML7 treatment experiments, the observed cell-stiffening effect was reproducible over three repeat experiments.
Discussion
In this study, we measured and characterized the effects of pharmacological inhibitions or genetic knockdown, targeting myosin II activity on the deformability of suspended cells, using an optical stretcher, a microfluidic microcirculation mimetic, and real-time deformability cytometry. We showed that cells, be it adherent cells brought into suspension or nonadherent cell lines growing naturally in suspension, exhibit a drop in cell compliance measured on a seconds-timescale after the inhibition of myosin II activity. By fitting a power law and a SLL model to the cell compliance curves, we found that in general, suspended cells became more solidlike with a concomitant increase in steady-state viscosity after inhibition of myosin activity. We have verified that the results were not too sensitive to the choice of the fitting range. To do this, the compliance curves were fitted with a linear function for t > tc, where tc is the critical time to be tested. To test the sensitivity of our findings on tc, we performed the full analysis for different times between 0.8 s and 2.5 s. Fig. S8 shows ηs scaled by its average, as a function of tc. For most cell lines and drugs tested, ηs changes by <10% for 1.1 s < tc < 2.2 s. Most of our compliance data ranges from 0 to 4 s. We thus choose half of the maximum time, 2 s, as the critical time, knowing that our results depend weakly on the choice of tc. Complementary studies using MMM, which also measures mechanical properties in their suspended state, showed similar evidence of cell stiffening for blebbistatin-treated cells compared to the corresponding controls (Fig. 6). The timescale of mechanical testing in OS is between 0.1 and 10 s, while in MMM it lies between 0.01 s, the estimated passage time of a single constriction, and 4 s, the estimated time of passing through the entire device. In this sense, the timescales are overlapping and the results can be compared.
A possible explanation for the change in overall cell deformability after myosin II inhibition is that the actin cortex of suspended cells behaves more like a polymer melt or gel. In this case, active myosin motors can help disentangle the actin filaments by actively pushing the actin filaments along each other, effectively dissipating stress that results in a more viscous deformation response. Such a mechanism has been demonstrated in purified actin gels mixed with myosin (40). In this study, the gel only flowed out of an angled cuvette after myosin being activated via ATP, thus fluidizing the actin gel. In particular, it was reported that such fluidlike response only occurs at timescales >8 s, consistent with our observed changes in viscous behavior at timescales of seconds up to one minute (Fig. S2). Further, the gel exhibited a much shorter relaxation time and lower viscosity after myosin activation, consistent with our observation of a lower whole-cell viscosity for control cells. This hypothesis of fluidization was further confirmed by actin gliding assays where the speed of single actin filaments in vitro was monitored. This speed decreased significantly after the addition of blebbistatin (41). In the same work, Medeiros et al. (41) further showed that actin filaments associated with filopodia in neuronal growth cones became longer and thicker after the addition of blebbistatin. Such an effect could be related to a decreased rate in actin bundle severing after myosin II inhibition. However, it is not known if similar processes take place in suspended cells.
However, RT-DC results indicate that a decrease in cell deformability after myosin inhibition (Fig. 7) can be detected even at very short deformation timescales of milliseconds, suggesting that the myosin-inactivated cells are structurally stiffer even before being deformed. A simple estimate of myosin dynamics might help elucidate this phenomenon. The speed of actin in in vitro assays has been estimated to be ∼5 μm/s (42), which is higher than what it will likely be in a cell. The deformation timescale in RT-DC is in the range of milliseconds, so during this time a myosin motor would only walk ∼50 nm (corresponding to ∼18 subunits), which is not enough to generate sufficient force on actin filaments and cause significant disentanglement in an actin gel at densities and mesh sizes found in the actin cortex. This finding argues against myosin-induced actin fluidization as the main mechanism for cell stiffening upon myosin inhibition. We therefore propose an alternative mechanism based on myosin-induced actin depolymerization, inspired by recent experiments that demonstrated the role of myosin II in active disassembly of actin filaments (43–45). In these publications, actin depolymerization was attributed to an increased bending of actin fibers by active myosin motors. Hence, a decrease in myosin activity due to blebbistatin would result in less actin depolymerization compared to the control cells. Because F-actin is a main contributor to cell elasticity (17,31), this would result in stiffer cells. Of note is the recent work by Vogel et al. (46), which reported the direct observation of buckling and breaking of membrane-bound actin filaments by myosin motors using total internal reflection fluorescence microscopy. Such a mechanism of filament fragmentation by myosin may contribute to actin turnover and possibly a lower cortex elasticity, and hence a higher whole-cell compliance.
Our hypothesis of myosin-induced actin depolymerization is further supported by the observation of a general decrease in compliance for 3T3 fibroblasts treated with Jasplakinolide, which stabilizes actin filaments (Fig. 3 B). The striking resemblance in compliance curves between the Jasplakinolide-treated cells and those treated with blebbistatin (Fig. 2 A) points toward the picture that disabling myosin activity leads to an effective F-actin stabilization. Our observations are consistent with previous reports showing F-actin disassembly upon stimulation of myosin II phosphorylation in fish keratocytes (47), and the stabilization of an equatorial actin network during late cytokinesis arising from decreased F-actin turnover dynamics due to blebbistatin treatment (48). Finally, according to the reptation model (40,49), the time for an actin filament in an entangled actin solution to disengage from its tube confinement scales as L3 where L is the actin filament length. Hence, if myosin participates in active actin depolymerization, it will presumably lead to a decrease in L and allow the actin filaments to escape out of the confined tube more easily. The overall result is a lower viscosity of the actin network at long timescales.
Further insights came from fluorescence imaging of the actin cytoskeleton in suspended cells under different pharmacological treatments. As shown in Fig. S3, cells treated with blebbistatin and ML7 tend to exhibit a microvillus cell surface and even thick bundles of actin filaments, while the untreated cells displayed a blebbing surface morphology and a more homogeneous, isotropic actin cortical layer underneath the plasma membrane. Here we note that the measured higher compliance of untreated cells are not artifacts due to edge detection of a more bleblike cell membrane, because the same effect of cell stiffening has been confirmed with MMM, which does not require the detection of a cell edge and is thus insensitive to this aspect. Our observations echo findings by Maloney et al. (7) that showed a clear correlation between effective stiffening of human mesenchymal stem cells after prolonged suspension and a significant reduction of blebbing cells. Furthermore, reports by Blaser et al. (50) showed that a transient increase in myosin activity could lead to increased separation of the acto-myosin cortex from the plasma membrane and the subsequent formation of blebs, while the opposite case of a low myosin II level has been shown to inhibit blebbing and increases filopodia/lamellipodia formation in amoeboid movement (51). However, the exact molecular-scale interactions between myosin II and the plasma membrane-cortex structure remains poorly understood, as summarized by a recent review (52). Several regulatory proteins have been found to function as membrane-to-cortex attachment, such as the ezrin/radixin/moesin proteins and Arp2/3 (53). It has been found that reducing these membrane-to-cortex attachment proteins could lead to an increase in the formation of blebs (54,55). Future works might involve the specific knockout of ezrin/radixin/moesin or Arp2/3 proteins, and measure any corresponding changes in cell stiffness due to such weaker coupling of the membrane to cortex. Taken together, our results suggest that after blebbistatin treatment, suspended cells may undergo cytoskeletal remodeling involving the reattachment and strengthening of cortex-membrane links, which might also contribute to cell stiffening.
Our observation of cell stiffening for myosin-inhibited cells deformed at timescales of seconds stands in clear contrast to data published on cells adherent to a substrate (26–29). In these publications, the cells were reported to soften upon myosin inhibition. Martens and Radmacher (27) attributed this result to the cellular prestress that occurs when myosin pulls on the actin stress fibers. Reducing the activity of myosin therefore decreases this prestress and the cells become more compliant and more fluidlike. The opposite effect has also been observed for studies using phorbol 12,13-dibutyrate to activate myosin II activity. Moy et al. (26) applied magnetic twisting rheology to adherent cells and found an increase in cell stiffness, also possibly attributed to the buildup of cellular prestress. In our study, all cell types tested (with the exception of macrophages) became less compliant and less fluidlike after inhibition of myosin II, and vice versa. Because all our measurements are carried out while the cells are in suspension, no focal adhesions and therefore no stress fibers are present during the measurements. Due to the nonexistence of these cellular structures, myosin could not exert the same effect of prestress on cells in suspension as in adherent cells. Hence, it stands to reason that cells in our measurements do not display a greater compliance after myosin inhibition. Such hypothesis is supported by the work of Rico et al. (56), who varied the adherence of cells by changing the ambient temperature and measured cell compliance using atomic force microscopy. They reported an increase in cell compliance with the loss of adhesion, which is highly connected to the loss of stress fibers in less adherent cells. Notably, even when two beads were attached to otherwise suspended 3T3 fibroblasts so that their mechanical properties could be measured using optical tweezers, the effect of blebbistatin was cell softening, just as with cells attached to a substrate (57). This suggests that even if the cells are in suspension, as long as they remain connected to the environment via focal adhesions, the dominant role of myosin II seems to be cell stiffening.
The lack of a change in cell compliance and cell fluidity for macrophages upon myosin inhibition (Fig. S6) has been reported in previous studies using magnetic twisting cytometry (58). This effect is not likely due to a low expression level of myosin IIA in macrophages (59,60). Instead, the different expression levels of nesprin 1 and 2, SUN1, vimentin, plectin, and lamin A/C in macrophages compared to neutrophils and monocytes (61,62), as well as a more adherent nature of macrophages that reside normally in the tissue, might play a role. Alternatively, the concentration of blebbistatin used in this study was too low to impact myosin activity in macrophages.
There are several reports on the study of myosin activity on the mechanical properties of cells in a suspended state confined by specific boundary conditions. Using micropipette aspiration, Tinevez et al. (63) showed that myosin inhibition using blebbistatin and Y-27632 can lead to a decrease in cortical tension. A decrease in cortical tension does not necessarily equate with a decrease in cortex stiffness, because the latter also depends inversely on the cell radius (64,65), which was not explicitly addressed in the work of Tinevez et al. (63). Using an electromechanical deformation technique, MacQueen et al. (66) found a linear dependence of the cell stiffness (Young’s modulus) and the cortical actin thickness. Although their setup allows characterization of cell stiffness in a fully suspended state free of any attachment to the experimental probe (similar to OS), they did not explicitly investigate the effects of myosin II activity on cell stiffness. Interestingly, the correlation between cortical tension and the steady-state viscosity of neutrophils has been studied by Tsai et al. (67) using a micropipette aspiration technique. Their work showed that with the addition of cytochalasin B, which induces actin depolymerization, both the cortical tension and the steady-state viscosity drop, consistent with our results on cell softening with cytochalasin D treatments (Fig. 3 C).
In the context of development, there are several cell systems where actomyosin can exist primarily as cortices without stress fibers or focal adhesions. Examples of these include embryonic progenitor cells, egg cells, and meiotic cells. Another closely related system is a cell undergoing mitosis. During this process, a global increase of myosin activity results in an increase in cortical stiffness and the dissolution of focal adhesions, leading to cell rounding (4,64,65,68). Extrapolating these results would mean that a decrease of myosin activity results in an effective decrease of cell stiffness, an effect contrary to our data. However, the exact effects of myosin inhibition on mitotic cells are less studied. A recent study shows that mitotic HeLa cells become more compliant when myosin II activity is inhibited with the knockdown of Ect2, a central regulator of mitotic rounding (69). This result is contrary to our presented findings. To clarify the apparent discrepancy, we carried out OS experiments on arrested mitotic HeLa cells with and without blebbistatin treatment, as shown in Fig. S9. We observed the mitotic cells to be stiffer than the interphase cells, consistent with previous work (4,69). Surprisingly, the addition of blebbistatin to mitotic cells led to a further increase in cell stiffness, confirming our main finding that myosin inhibition leads to cell stiffening as long as the cells are in a suspended, nonattached state. This also suggests that the effect of Ect2 inhibition and blebbistatin treatment are not equivalent or comparable, possibly due to the fact that Ect2 inhibition does lower the amount of F-actin and decreases the activity of myosin II, both of which exert opposite effects on cell stiffness. As such, it is plausible that the effect of a lower amount of F-actin dominates the effect of reduced myosin activity, leading to an overall increase in cell deformability in this particular case.
Apart from its specific actions on the cortical actin structures, myosin II activity can also exert secondary effects on other cellular structures. For example, recent work has shown that molecular motor activity can lead to constant agitation of cytoplasm (70), which may influence the assembly of cytoskeletal architecture, although the direct influence of myosin motors in enhancing the fluctuations of actin filaments has not been shown empirically. Furthermore, there is recent evidence for cytoskeletal motors and cross-linking proteins synergistically sharing the force applied to the cell cortex (71). Notably, in the absence of myosin II, α-actinin cross-linkers can accumulate at the sites of highest dilation of the cortex to counteract the applied force. Therefore, it is conceivable that inhibiting myosin activity may lead to an increased binding rate of these cross-linkers, which results in a form of compensation effect and increased cell stiffness.
In summary, cells, once suspended and free from any physical attachment to the surrounding, stiffen and become more solidlike after myosin II inhibition—contrary to reports of cell softening with adherent cells (26–29). Measurements on adherent cells are generally done on cells adhering to plastic tissue culture dishes. In living tissue, there is no stiff material such as plastic or glass to which cells adhere. Nonetheless, the other extreme—cells being entirely in suspension as in our study—is not the normal case either. Only specific cell types, such as nonactivated blood cells, entirely lack attachments. For most cell types, however, the in vivo environment is situated in between these two extremes. Cells growing in a compliant, three-dimensional environment display fewer focal adhesions compared to cells grown on stiff substrates (10). The increased number of focal adhesions in cells on rigid substrates caused more and stronger stress fibers (10), which then lead to an increase in cellular prestress. Therefore, the stiffness of the cell environment and the adhesion to that substrate need to be considered when extrapolating to in vivo situations from cell mechanical measurements carried out in vitro. The most realistic results would be obtained by measuring the effect of myosin II inhibition on cells in their in vivo environment. However, such a measurement is not feasible with any technique, as of this writing, known to be available.
Conclusion
To conclude, we find that myosin II inhibition unequivocally leads to a significant decrease in whole-cell compliance and a more solidlike behavior for cells in suspension at a timescale of milliseconds to 1 min, irrespective of whether they are normally adhered to a substrate or naturally in suspension. We have confirmed our findings with a number of cell lines and myosin inhibitors, using a number of cell mechanical measurement techniques such as the OS, MMM, and RT-DC. Our work highlights the fundamentally different role that myosin II plays in modulating mechanical properties of suspended cells devoid of any stress fibers or focal adhesions, as compared to cells adhered to a substrate. In particular we have identified fingerprints of myosin-regulated actin depolymerization and active remodeling of the membrane-cortex attachment. Our work also suggests the use of myosin II activity as a potential pharmacological target for impacting cellular advective function of immune cells. This may have wide medical implications such as in treatments of circulatory disorders.
Acknowledgments
The authors are grateful for insightful discussions with Guillaume Charras, Florian Rehfeldt, Christoph Schmidt, Ulrich Schwarz, Jacques Prost, Valerie Weaver, and Sanjay Kumar. The HL60/S4 cell line was a generous gift from Don and Ada Olins, University of New England. The authors acknowledge Maik Herbig for technical assistance in RT-DC experiments, and Gianaurelio Cuniberti and Larysa Baraban for technical assistance in photolithography.
The authors acknowledge financial support from the European Community’s Seventh Framework Program (grant No. FP7/2007-2013) under grant agreement No. TRANSPOL 264399 (to C.J.C.), Cambridge Gates Scholarship (to F.L.), Cambridge Commonwealth Bursary and Duke of Edinburgh Scholarship (to A.E.E.), and the LightTouch Starting Investigator Grant of the European Research Council and the Humboldt-Professorship from the Alexander-von-Humboldt Foundation (to J.G.).
Footnotes
Chii J. Chan and Andrew E. Ekpenyong contributed equally to this work.
Supporting Material
Blebbistatin-treated HL60 cells require significantly longer times to deform before they can slip through the constrictions, as compared to the untreated cells when tested at the same driving pressure of 50 mbar. Video presented is in real time.
References
- 1.Alberts B., Johnson A., Walter P. 4th Ed. Garland Science; New York: 2002. Molecular Biology of the Cell. [Google Scholar]
- 2.Théry M., Bornens M. Cell shape and cell division. Curr. Opin. Cell Biol. 2006;18:648–657. doi: 10.1016/j.ceb.2006.10.001. [DOI] [PubMed] [Google Scholar]
- 3.Mogilner A., Keren K. The shape of motile cells. Curr. Biol. 2009;19:R762–R771. doi: 10.1016/j.cub.2009.06.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stewart M.P., Helenius J., Hyman A.A. Hydrostatic pressure and the actomyosin cortex drive mitotic cell rounding. Nature. 2011;469:226–230. doi: 10.1038/nature09642. [DOI] [PubMed] [Google Scholar]
- 5.Gardel M.L., Shin J.H., Weitz D.A. Elastic behavior of cross-linked and bundled actin networks. Science. 2004;304:1301–1305. doi: 10.1126/science.1095087. [DOI] [PubMed] [Google Scholar]
- 6.Ekpenyong A.E., Whyte G., Guck J. Viscoelastic properties of differentiating blood cells are fate- and function-dependent. PLoS ONE. 2012;7:e45237. doi: 10.1371/journal.pone.0045237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Maloney J.M., Nikova D., van Vliet K.J. Mesenchymal stem cell mechanics from the attached to the suspended state. Biophys. J. 2010;99:2479–2487. doi: 10.1016/j.bpj.2010.08.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ingber D.E. Tensegrity: the architectural basis of cellular mechanotransduction. Annu. Rev. Physiol. 1997;59:575–599. doi: 10.1146/annurev.physiol.59.1.575. [DOI] [PubMed] [Google Scholar]
- 9.Vale R.D. The molecular motor toolbox for intracellular transport. Cell. 2003;112:467–480. doi: 10.1016/s0092-8674(03)00111-9. [DOI] [PubMed] [Google Scholar]
- 10.Yeung T., Georges P.C., Janmey P.A. Effects of substrate stiffness on cell morphology, cytoskeletal structure, and adhesion. Cell Motil. Cytoskeleton. 2005;60:24–34. doi: 10.1002/cm.20041. [DOI] [PubMed] [Google Scholar]
- 11.Tee S.Y., Fu J., Janmey P.A. Cell shape and substrate rigidity both regulate cell stiffness. Biophys. J. 2011;100:L25–L27. doi: 10.1016/j.bpj.2010.12.3744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Radmacher M., Tillamnn R.W., Gaub H.E. From molecules to cells: imaging soft samples with the atomic force microscope. Science. 1992;257:1900–1905. doi: 10.1126/science.1411505. [DOI] [PubMed] [Google Scholar]
- 13.Bausch A.R., Möller W., Sackmann E. Measurement of local viscoelasticity and forces in living cells by magnetic tweezers. Biophys. J. 1999;76:573–579. doi: 10.1016/S0006-3495(99)77225-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hochmuth R.M. Micropipette aspiration of living cells. J. Biomech. 2000;33:15–22. doi: 10.1016/s0021-9290(99)00175-x. [DOI] [PubMed] [Google Scholar]
- 15.Taylor P., Molloy J.E., Padgett M.J. Lights, action: optical tweezers. Contemp. Phys. 2002;43:241–258. [Google Scholar]
- 16.Gittes F., Schnurr B., Schmidt C. Microscopic viscoelasticity: shear moduli of soft materials determined from thermal fluctuations. Phys. Rev. Lett. 1997;79:3286–3289. [Google Scholar]
- 17.Guck J., Schinkinger S., Bilby C. Optical deformability as an inherent cell marker for testing malignant transformation and metastatic competence. Biophys. J. 2005;88:3689–3698. doi: 10.1529/biophysj.104.045476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lautenschläger F., Paschke S., Guck J. The regulatory role of cell mechanics for migration of differentiating myeloid cells. Proc. Natl. Acad. Sci. USA. 2009;106:15696–15701. doi: 10.1073/pnas.0811261106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Otto O., Rosendahl P., Guck J. Real-time deformability cytometry: on-the-fly cell mechanical phenotyping. Nat. Methods. 2015;12:199–202. doi: 10.1038/nmeth.3281. [DOI] [PubMed] [Google Scholar]
- 20.Lauffenburger D.A., Horwitz A.F. Cell migration: a physically integrated molecular process. Cell. 1996;84:359–369. doi: 10.1016/s0092-8674(00)81280-5. [DOI] [PubMed] [Google Scholar]
- 21.Kovács M., Tóth J., Sellers J.R. Mechanism of blebbistatin inhibition of myosin II. J. Biol. Chem. 2004;279:35557–35563. doi: 10.1074/jbc.M405319200. [DOI] [PubMed] [Google Scholar]
- 22.Maekawa M., Ishizaki T., Narumiya S. Signaling from Rho to the actin cytoskeleton through protein kinases ROCK and LIM-kinase. Science. 1999;285:895–898. doi: 10.1126/science.285.5429.895. [DOI] [PubMed] [Google Scholar]
- 23.Bain J., McLauchlan H., Cohen P. The specificities of protein kinase inhibitors: an update. Biochem. J. 2003;371:199–204. doi: 10.1042/BJ20021535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Blumberg P.M., Delclos K.B., Yeh E. Phorbol ester receptors and the in vitro effects of tumor promoters. Ann. N. Y. Acad. Sci. 1983;407:303–315. doi: 10.1111/j.1749-6632.1983.tb47836.x. [DOI] [PubMed] [Google Scholar]
- 25.Yanagita T., Kobayashi H., Wada A. Protein kinase C and the opposite regulation of sodium channel α- and β1-subunit mRNA levels in adrenal chromaffin cells. J. Neurochem. 1999;73:1749–1757. doi: 10.1046/j.1471-4159.1999.731749.x. [DOI] [PubMed] [Google Scholar]
- 26.Moy A.B., Blackwell K., English A. Phorbol ester-mediated pulmonary artery endothelial barrier dysfunction through regulation of actin cytoskeletal mechanics. Am. J. Physiol. Lung Cell. Mol. Physiol. 2004;287:L153–L167. doi: 10.1152/ajplung.00292.2003. [DOI] [PubMed] [Google Scholar]
- 27.Martens J.C., Radmacher M. Softening of the actin cytoskeleton by inhibition of myosin II. Pflugers Arch. 2008;456:95–100. doi: 10.1007/s00424-007-0419-8. [DOI] [PubMed] [Google Scholar]
- 28.Nijenhuis N., Zhao X., Derby B. Combining AFM and acoustic probes to reveal changes in the elastic stiffness tensor of living cells. Biophys. J. 2014;107:1502–1512. doi: 10.1016/j.bpj.2014.07.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.MacKay J.L., Keung A.J., Kumar S. A genetic strategy for the dynamic and graded control of cell mechanics, motility, and matrix remodeling. Biophys. J. 2012;102:434–442. doi: 10.1016/j.bpj.2011.12.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Boyde L., Chalut K.J., Guck J. Interaction of Gaussian beam with near-spherical particle: an analytic-numerical approach for assessing scattering and stresses. J. Opt. Soc. Am. A Opt. Image Sci. Vis. 2009;26:1814–1826. doi: 10.1364/josaa.26.001814. [DOI] [PubMed] [Google Scholar]
- 31.Ananthakrishnan R., Guck J., Käs J. Quantifying the contribution of actin networks to the elastic strength of fibroblasts. J. Theor. Biol. 2006;242:502–516. doi: 10.1016/j.jtbi.2006.03.021. [DOI] [PubMed] [Google Scholar]
- 32.Maloney J.M., Lehnhardt E., van Vliet K.J. Mechanical fluidity of fully suspended biological cells. Biophys. J. 2013;105:1767–1777. doi: 10.1016/j.bpj.2013.08.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Moeendarbary E., Valon L., Charras G.T. The cytoplasm of living cells behaves as a poroelastic material. Nat. Mater. 2013;12:253–261. doi: 10.1038/nmat3517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chan C.J., Whyte G., Guck J. Impact of heating on passive and active biomechanics of suspended cells. Interface Focus. 2014;4:20130069. doi: 10.1098/rsfs.2013.0069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Olins A.L., Buendia B., Olins D.E. Retinoic acid induction of nuclear envelope-limited chromatin sheets in HL-60. Exp. Cell Res. 1998;245:91–104. doi: 10.1006/excr.1998.4210. [DOI] [PubMed] [Google Scholar]
- 36.Kolega J. Phototoxicity and photoinactivation of blebbistatin in UV and visible light. Biochem. Biophys. Res. Commun. 2004;320:1020–1025. doi: 10.1016/j.bbrc.2004.06.045. [DOI] [PubMed] [Google Scholar]
- 37.Sakamoto T., Limouze J., Sellers J.R. Blebbistatin, a myosin II inhibitor, is photoinactivated by blue light. Biochemistry. 2005;44:584–588. doi: 10.1021/bi0483357. [DOI] [PubMed] [Google Scholar]
- 38.Ebert S., Travis K., Guck J. Fluorescence ratio thermometry in a microfluidic dual-beam laser trap. Opt. Express. 2007;15:15493–15499. doi: 10.1364/oe.15.015493. [DOI] [PubMed] [Google Scholar]
- 39.Kiessling T.R., Stange R., Fritsch A.W. Thermorheology of living cells—impact of temperature variations on cell mechanics. New J. Phys. 2013;15:045026. [Google Scholar]
- 40.Humphrey D., Duggan C., Käs J. Active fluidization of polymer networks through molecular motors. Nature. 2002;416:413–416. doi: 10.1038/416413a. [DOI] [PubMed] [Google Scholar]
- 41.Medeiros N.A., Burnette D.T., Forscher P. Myosin II functions in actin-bundle turnover in neuronal growth cones. Nat. Cell Biol. 2006;8:215–226. doi: 10.1038/ncb1367. [DOI] [PubMed] [Google Scholar]
- 42.Borejdo J., Burlacu S. Velocity of movement of actin filaments in in vitro motility assay. Measured by fluorescence correlation spectroscopy. Biophys. J. 1992;61:1267–1280. doi: 10.1016/S0006-3495(92)81935-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Haviv L., Gillo D., Bernheim-Groswasser A. A cytoskeletal demolition worker: myosin II acts as an actin depolymerization agent. J. Mol. Biol. 2008;375:325–330. doi: 10.1016/j.jmb.2007.09.066. [DOI] [PubMed] [Google Scholar]
- 44.Matsui T.S., Kaunas R., Deguchi S. Non-muscle myosin II induces disassembly of actin stress fibers independently of myosin light chain dephosphorylation. Interface Focus. 2011;1:754–766. doi: 10.1098/rsfs.2011.0031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schmoller K.M., Semmrich C., Bausch A.R. Slow down of actin depolymerization by cross-linking molecules. J. Struct. Biol. 2011;173:350–357. doi: 10.1016/j.jsb.2010.09.003. [DOI] [PubMed] [Google Scholar]
- 46.Vogel S.K., Petrasek Z., Schwille P. Myosin motors fragment and compact membrane-bound actin filaments. eLife. 2013;2:e00116. doi: 10.7554/eLife.00116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wilson C.A., Tsuchida M.A., Theriot J.A. Myosin II contributes to cell-scale actin network treadmilling through network disassembly. Nature. 2010;465:373–377. doi: 10.1038/nature08994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Guha M., Zhou M., Wang Y.-L. Cortical actin turnover during cytokinesis requires myosin II. Curr. Biol. 2005;15:732–736. doi: 10.1016/j.cub.2005.03.042. [DOI] [PubMed] [Google Scholar]
- 49.Doi M., Edwards S.F. Clarendon; Oxford, UK: 1986. The Theory of Polymer Dynamics. [Google Scholar]
- 50.Blaser H., Reichman-Fried M., Raz E. Migration of zebrafish primordial germ cells: a role for myosin contraction and cytoplasmic flow. Dev. Cell. 2006;11:613–627. doi: 10.1016/j.devcel.2006.09.023. [DOI] [PubMed] [Google Scholar]
- 51.Yoshida K., Soldati T. Dissection of amoeboid movement into two mechanically distinct modes. J. Cell Sci. 2006;119:3833–3844. doi: 10.1242/jcs.03152. [DOI] [PubMed] [Google Scholar]
- 52.Clark A.G., Wartlick O., Paluch E.K. Stresses at the cell surface during animal cell morphogenesis. Curr. Biol. 2014;24:R484–R494. doi: 10.1016/j.cub.2014.03.059. [DOI] [PubMed] [Google Scholar]
- 53.Yang Q., Zhang X.-F., Forscher P. Arp2/3 complex-dependent actin networks constrain myosin II function in driving retrograde actin flow. J. Cell Biol. 2012;197:939–956. doi: 10.1083/jcb.201111052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Diz-Muñoz A., Krieg M., Heisenberg C.P. Control of directed cell migration in vivo by membrane-to-cortex attachment. PLoS Biol. 2010;8:e1000544. doi: 10.1371/journal.pbio.1000544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Beckham Y., Vasquez R.J., Gardel M.L. Arp2/3 inhibition induces amoeboid-like protrusions in MCF10A epithelial cells by reduced cytoskeletal-membrane coupling and focal adhesion assembly. PLoS ONE. 2014;9:e100943. doi: 10.1371/journal.pone.0100943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rico F., Chu C., Moy V.T. Temperature modulation of integrin-mediated cell adhesion. Biophys. J. 2010;99:1387–1396. doi: 10.1016/j.bpj.2010.06.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schlosser F., Rehfeldt F., Schmidt C.F. Force fluctuations in three-dimensional suspended fibroblasts. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2015;370:20140028. doi: 10.1098/rstb.2014.0028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Patel N.R., Bole M., Koziel H. Cell elasticity determines macrophage function. PLoS ONE. 2012;7:e41024. doi: 10.1371/journal.pone.0041024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bhuwania R., Cornfine S., Linder S. Supervillin couples myosin-dependent contractility to podosomes and enables their turnover. J. Cell Sci. 2012;125:2300–2314. doi: 10.1242/jcs.100032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Valerius N.H., Stendahl O.I., Stossel T.P. Distribution of actin-binding protein and myosin in neutrophils during chemotaxis and phagocytosis. Adv. Exp. Med. Biol. 1982;141:19–28. doi: 10.1007/978-1-4684-8088-7_3. [DOI] [PubMed] [Google Scholar]
- 61.Olins A.L., Hoang T.V., Olins D.E. The LINC-less granulocyte nucleus. Eur. J. Cell Biol. 2009;88:203–214. doi: 10.1016/j.ejcb.2008.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Beneš P., Macecková V., Šmarda J. Role of vimentin in regulation of monocyte/macrophage differentiation. Differentiation. 2006;74:265–276. doi: 10.1111/j.1432-0436.2006.00077.x. [DOI] [PubMed] [Google Scholar]
- 63.Tinevez J.-Y., Schulze U., Paluch E. Role of cortical tension in bleb growth. Proc. Natl. Acad. Sci. USA. 2009;106:18581–18586. doi: 10.1073/pnas.0903353106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Salbreux G., Charras G., Paluch E. Actin cortex mechanics and cellular morphogenesis. Trends Cell Biol. 2012;22:536–545. doi: 10.1016/j.tcb.2012.07.001. [DOI] [PubMed] [Google Scholar]
- 65.Clark A.G., Paluch E. Mechanics and regulation of cell shape during the cell cycle. In: Kubiak J.Z., editor. Cell Cycle in Development (Results and Problems in Cell Differentiation) Springer; Berlin, Germany: 2011. pp. 31–73. [DOI] [PubMed] [Google Scholar]
- 66.MacQueen L.A., Thibault M., Wertheimer M.R. Electromechanical deformation of mammalian cells in suspension depends on their cortical actin thicknesses. J. Biomech. 2012;45:2797–2803. doi: 10.1016/j.jbiomech.2012.08.040. [DOI] [PubMed] [Google Scholar]
- 67.Tsai M.A., Frank R.S., Waugh R.E. Passive mechanical behavior of human neutrophils: effect of cytochalasin B. Biophys. J. 1994;66:2166–2172. doi: 10.1016/S0006-3495(94)81012-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Levayer R., Lecuit T. Biomechanical regulation of contractility: spatial control and dynamics. Trends Cell Biol. 2012;22:61–81. doi: 10.1016/j.tcb.2011.10.001. [DOI] [PubMed] [Google Scholar]
- 69.Matthews H.K., Delabre U., Baum B. Changes in Ect2 localization couple actomyosin-dependent cell shape changes to mitotic progression. Dev. Cell. 2012;23:371–383. doi: 10.1016/j.devcel.2012.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Brangwynne C.P., Koenderink G.H., Weitz D.A. Cytoplasmic diffusion: molecular motors mix it up. J. Cell Biol. 2008;183:583–587. doi: 10.1083/jcb.200806149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Luo T., Mohan K., Robinson D.N. Molecular mechanisms of cellular mechanosensing. Nat. Mater. 2013;12:1064–1071. doi: 10.1038/nmat3772. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Blebbistatin-treated HL60 cells require significantly longer times to deform before they can slip through the constrictions, as compared to the untreated cells when tested at the same driving pressure of 50 mbar. Video presented is in real time.