

Abstract
Equinatoxin II (EqtII) is a soluble, 20 kDa pore-forming protein toxin isolated from the sea anemone Actinia equina. Although pore formation has long been known to occur in distinct stages, including monomeric attachment to phospholipid membranes followed by detachment of the N-terminal helical domain and oligomerization into the final pore assembly, atomistic-level detail of the protein-lipid interactions underlying these events remains elusive. Using high-resolution solution state NMR of uniformly-15N-labeled EqtII at the critical micelle concentration of dodecylphosphocholine, we have mapped the lipid-binding site through chemical shift perturbations. Subsequent docking of an EqtII monomer onto a dodecylphosphocholine micelle, followed by 400 ns of all-atom molecular dynamics simulation, saw several high-occupancy lipid-binding pockets stabilized by cation-π, hydrogen bonding, and hydrophobic interactions; and stabilization of the loop housing the conserved arginine-glycine-aspartate motif. Additional simulation of EqtII with an N-acetyl sphingomyelin micelle, for which high-resolution NMR data cannot be obtained due to aggregate formation, revealed that sphingomyelin specificity might occur via hydrogen bonding to the 3-OH and 2-NH groups unique to the ceramide backbone by side chains of D109 and Y113; and main chains of P81 and W112. Furthermore, a binding pocket formed by K30, K77, and P81, proximate to the hinge region of the N-terminal helix, was identified and may be implicated in triggering pore formation.
Introduction
Equinatoxin II (EqtII) is a soluble pore-forming toxin produced by the sea anemone Actinia equina (1). EqtII belongs to actinoporins—a family of potent hemolytic proteins isolated from the venoms of sea anemones, found to have a high isoelectric point (>9), receptor-like affinity to sphingomyelin (SM), and a well-conserved β-sandwich core flanked by two α-helical domains (2,3). Crystal and solution-state NMR structures have been solved for EqtII (4,5) and homologous sticholysins StI and StII from Stichodactyla helianthus (6,7) and Fragaceatoxin FraC from Actinia fragacea (8). A crystal structure of a binary complex of StII and the phosphocholine (POC) headgroup moiety, common to SM and phosphatidylcholine (PC) lipids, suggested the lipid-binding site consisted of a conserved patch rich in solvent-exposed aromatic amino acids (7), which has since been corroborated by site-directed mutagenesis and solution NMR with vesicle, micelle, and bicelle systems (9–13). The lipid-binding site of actinoporins is essential for hemolytic activity and membrane permeation as attachment of monomeric species is required to initiate translocation of the N-terminus to the hydrophobic interior of the membrane and oligomerization into a trimeric/tetrameric pore (14–19).
Although EqtII maintains nM affinity to PC membrane models, affinity for SM is an order of magnitude greater and essential for lytic activity (9,20). Furthermore, the potency of actinoporins against SM has been described as a coadaptive trait, whereby sea anemones have evolved resistant phosphono analogs of SM that are either nonmethylated or monomethylated at the headgroup amino position to avert self-selectivity (21). SM selectivity of EqtII and the interactions responsible have not been defined at atomic detail. Nor have they been for other pore-forming toxins such as lysenin, produced by the earthworm Eisenia fetida (22), and cholesterol-dependent hemolysins, listeriolysin O, and perfringolysin O from bacteria Listeria monocytogenes and Clostridium perfringens, respectively (23,24), that target eukaryotic membranes. This receptor-like specificity toward lipids has been exploited for probing SM and cholesterol-enriched domains in eukaryotic cell membranes (25–29).
Understanding lipid-specific interactions at atomic-level detail, however, is difficult to achieve by current experimental methods. Although x-ray structures collected for StII-POC and, more recently, POC-lysenin-SM complexes have shed some light (7,22), the lack of a surrounding membrane environment remains a significant limitation. SM selectivity has been explored by chemical modification (methylation) of the ceramide backbone, unique to SM over PC lipids, and has implicated the 2-NH and 3-OH positions as essential for binding and membrane permeabilization of actinoporins (30). Furthermore, biophysical studies of several mutants of EqtII and StII, particularly at (or equivalent to) W112 and Y113 positions, show considerable impact on lytic activity against SM-containing membranes (9,10,31), whereas 19F NMR studies identify W112 as important for SM selectivity (11).
Several NMR methods exist to derive essential information on protein interactions with model membrane systems (i.e., vesicles, micelles, bicelles, etc.), such as 31P solid-state experiments (32–34), diffusion measurements (35), nuclear Overhauser effects, paramagnetic relaxation enhancement (36), and chemical shift perturbations (CSPs) (11,13,37). However, a high degree of ambiguity in determining the specific nature of the interactions responsible for the observed effects often remains.
In this work, we have used CSP from solution NMR of uniformly 15N-labeled EqtII in dodecylphosphocholine (DPC) micelles to map out the residues making up the interfacial lipid-binding site. With these data, we were able to perform all-atom molecular dynamics (MD) simulation of EqtII docked to a characteristic DPC micelle model (38) to identify the specific interactions underlying general PC recognition. To understand the SM-dependence of actinoporin pore formation, the same docking mode was also tested with an N-acetyl sphingomyelin (Ac-SM) micelle, which could not be investigated by NMR because of formation of large aggregates, to identify additional interactions unique to the ceramide backbone.
Materials and Methods
NMR spectroscopy
Uniformly 15N- and 13C15N-labeled EqtII was prepared from expressing the EqtII gene in M9 minimal media supplemented with ISOGRO (Sigma-Aldrich, St. Louis, MO), purified and concentrated according to protocols reported previously (39), and then lyophilized for storage at −20°C. Yields of 3.6 and 4.2 mg/L of growth medium were determined for 15N and 13C15N protein, respectively. NMR samples were prepared to 10 mM sodium phosphate, 20 mM NaCl, 0.02% NaN3 in 90% H2O/10% D2O at pH 5.00; and protein concentration confirmed by 280 nm absorbance with an extinction coefficient of 36,100 M−1 cm−1 (40). 1H-15N heteronuclear single quantum correlation (HSQC) (41) spectra were recorded at 30°C on a 500 MHz Bruker Avance spectrometer (Rheinstetten, Germany) equipped with a cryoprobe. Data were processed with NMRPipe (42) and analyzed using CCPNmr Analysis (43). Chemical shifts were referenced to the Trp-116 Hε1-Nε1 crosspeak, found throughout our study to be unaffected by either pH or salt conditions, and compared to assignments deposited in the BioMagResBank database (44). Heavily shifted crosspeaks were confirmed by a 3D-HNCA spectrum (45). DPC stock (1 M in water) was titrated directly into the NMR tube and resulting selective shifts to 1H15N crosspeaks were averaged according to the equation ΔδHN = ((ΔδH2 + αΔδN2)/2)1/2, where ΔδH and ΔδN are the chemical shift changes induced to the 1H and 15N dimensions, respectively, and α is a weighting factor of 0.14 (37).
MD parameters
Restrained electrostatic potentials for DPC atoms were taken directly from validated micelle models (38). All other bonded and nonbonded parameters were obtained from the general AMBER force field (GAFF) (46) except for acyl chain carbon and hydrogen, which were assigned revised Lennard-Jones and torsion parameters from the GAFFlipid force field (47). Charges for Ac-SM (18:1, 2:0) were derived by an analogous scheme as for DPC (Fig. S1 in the Supporting Material), using fragments ethylmethylphosphate and choline from geometry-optimized Protein Data Bank (PDB) structures from the F-92 project deposited in the R.E.D. database (38). 1-pentadecene and the ceramide-backbone fragment were geometry optimized at the Hartree-Fock level of theory with a 6-31G∗ basis set using Gaussian09 (rev. C) as part of the R.E.D. web server (48) and starting structures in extended conformation. Charges and atom types assigned to DPC and Ac-SM are detailed in Tables S1 and S2.
MD simulations
All simulations were run using NAMD 2.9 (49) scaled over 256 IBM Blue Gene/Q CPU cores (Rochester, MN). An initial 5000 steps of conjugate gradient minimization on all systems was performed and productions run using a 12 Å (first 220 ns) cutoff, or 9 Å (final 200 ns), with smoothing functions disabled for electrostatics and van der Waals terms. Long-range electrostatics were treated using the particle mesh Ewald method. The SETTLE algorithm was applied to water hydrogen atoms and the SHAKE algorithm applied to all other hydrogen atoms with a 10−8 tolerance. For compatibility with the AMBER force field, 1–3 bonded interactions were ignored, 1–4 electrostatics were scaled by 0.833333333, and 1–4 van der Waals terms divided by 2. Temperature was maintained at 303.15 K using the Langevin thermostat with a 5 ps−1 dampening coefficient and decoupled from hydrogen atoms. The Langevin piston Nosé-Hoover method was used to control pressure at 1.01325 bar using a 100 fs oscillation period and 50 fs dampening time. Electrostatics were evaluated in 2 fs time steps and remaining nonbonded interactions every 1 fs.
EqtII(5-179) starting coordinates were taken from chain A of the crystal structure (PDB 1IAZ, 1.9 Å) (5). Solvent and sulfate ions were removed, and all histidines protonated at Nε positions. Docking onto an equilibrated DPC micelle structure of 54 molecules (DPC54) (38) was performed with rough guidance from NMR CSPs. AMBER99SB parameters (50) were assigned to all protein atoms and the system neutralized with seven chloride ions, and then solvated in a truncated octahedron of TIP3P water molecules (8 Å, first 220 ns; or 11 Å shell final 200 ns) using the LEAP program part of AmberTools12 software. Distance restraints (10 kcal.mol−1) on atoms Y113 Oη to DPC P8 (4.40 Å); Y113 Cγ to DPC N4 (5.10 Å); Y113 Cζ to DPC N4 (4.76 Å); and Y138 Oη to DPC P8 (3.56 Å), measured from equivalent positions of the crystal structure of an StII-POC complex (PDB 1O72) (7), were applied to the nearest DPC for the initial 20 ns of simulation. Unrestrained production runs followed for 200 ns, and then periodic boundaries (water shells) and the cutoff for force evaluation were expanded and reduced, respectively, for a further 200 ns to minimize risks of periodic boundary artifacts. An identical approach was applied for docking to an equilibrated Ac-SM54 micelle structure obtained from a 60 ns simulation. Further system details are outlined in Table S3.
MD trajectories were analyzed every 10 ps using VMD 1.9.1 (51) with scripts (available on request) capable of detecting contacts (including hydrophobic), H-bonds, and cation-π complexes; and sorting by frame, residue (or other attribute), and pairwise association. Cation-π interactions were analyzed between six-membered ring systems of Tyr, Trp, and Phe residues and the choline nitrogen cation of DPC and Ac-SM. All distances between ring carbons and the central nitrogen had to be within 7 Å, and all distances measured within a 1.5 Å range to satisfy cation-π criteria (52). H-bonds were defined by a D-H—A angle (donor atom, D; acceptor atom, A; and hydrogen, H) <20°, and a D-A distance <3 Å. Hydrophobic contacts were considered only for nonpolar protein and micelle atoms (i.e., atoms involved in C-H and S-H bonds) within 3 Å.
Results
Refinement of the lipid-binding site by NMR with DPC
Micelle models, despite limitations such as high curvature, denaturing effects, and thinner hydrophobic region compared to membrane bilayers (53), are amenable for high-resolution NMR and are a convenient size for MD simulation. Solution NMR of uniformly 15N-labeled EqtII with DPC was used to provide additional information on the lipid-binding site to details obtained from 19F-NMR (11) and site-directed mutagenesis experiments (9,31). DPC was titrated incrementally in 1.5 mM additions and perturbations of crosspeaks observed from 1H-15N HSQC spectra were used to identify lipid interactions along the entire backbone and some side-chain positions. The first titration point, at approximately the critical micelle concentration (CMC) of DPC (∼1.1 mM) (54), provided the best signal, whereas considerable broadening was observed above CMC (1H-15N HSQC spectra are depicted in Fig. S2). Line-broadened spectra have been problematic for previous 2D-NMR studies of EqtII and StI with DPC (11,12), and may be the result of oligomerization.
Residues S54, G85, A87, D109, Y110, W112, Y113, W116, N118, Y138, and N139—consistent with the lipid-binding site—and distant regions surrounding V60 and H169, were perturbed at backbone amide positions upon titrating 1.5 mM DPC. Several unassigned perturbations were also observed in 1H-15N HSQC spectra, which may correspond to neighboring residues E135, L136, Y137, R161, N165, and E173. Downfield shifts observed for W116 Hε (+0.04 ppm) and Nε (+0.02 ppm) resonances are consistent with previous 19F NMR studies of EqtII with 5-19F-labeled tryptophans, which suggested that the side chain of W116 was located at the polar interface of the DPC micelle (11). 19F CSPs to W112 also suggested the side chain to be embedded within the hydrophobic interior (11), and is supported by shifts to the Hε-Nε crosspeak reported for StI with dihexanoylphosphatidylcholine (DHPC) micelles (13), but not observed in our work. This discrepancy may reflect our use of DPC close to the CMC, where a relatively high concentration of free monomer will compete with fully formed micelles for EqtII binding. In our experience and in reports of StI with DHPC, binding occurs with both monomeric and micellar forms, albeit greater NMR perturbations occur to more residues with DHPC present (13). Thus in our system, the perturbations represent a hybrid of the two scenarios for StI and maybe why nonspecific binding is more apparent at distal V60 and H169 positions.
NMR-guided MD with a DPC micelle
Although perturbations obtained by NMR provide detailed mapping of residues affected at their backbone amide positions, ambiguity remains in knowing the exact interactions responsible for the perturbations. All-atom MD simulations were, therefore, used to derive explicit details of the dynamics, hydrogen bonds, cation-π interactions, hydrophobic contacts, and penetration of the protein upon binding to a micelle model lipid environment. The initial configuration of the EqtII-DPC54 complex was chosen so that affected residues observed by NMR were located at the protein-micelle interface (Fig. 1 A), whereas restraints were applied over the initial 20 ns of equilibration to hold a POC headgroup in a position consistent with the StII-POC crystal structure (7). The DPC micelle model and charge parameters chosen have been shown to accurately replicate experimentally known characteristics of the DPC micelle, including size and ellipsoidal shape, although the AMBER99SB force field was described to overestimate trans character of the acyl chains relative to the other force fields tested (38). In this work the GAFFlipid force field (47) reduced trans populations of acyl chains to 73.5% compared to GAFF (80.7%) and AMBER99SB (83.8%; 84.3% by Abel et al. (38)) (see Table S4 for further details).
Figure 1.
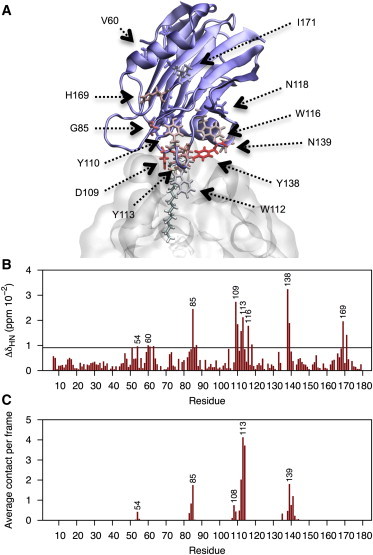
(A) Initial configuration of the EqtII-DPC54 complex before MD simulation. The single DPC molecule represented with bonds was restrained at the POC headgroup over the initial 20 ns of equilibration. Residues heavily affected in NMR experiments are represented in bonds and color-coded (blue-red) according to their CSPs. (B) Normalized perturbations for backbone 1H15N crosspeaks in the presence of 1.5 mM DPC. Dashed line specifies significance where values are greater than the mean plus the standard deviation. Side-chain perturbations are not plotted. (C) Number of intermolecular contacts (<5 Å) experienced between backbone nitrogen positions and heavy atoms of DPC over the last 200 ns of simulation. To see this figure in color, go online.
Over 400 ns of unrestrained simulation, only a very minor change from the original docked position was observed for the EqtII-DPC54 complex. Regions showing NMR effects correlated qualitatively well to intermolecular contacts experienced to the backbone nitrogen position during simulation (Fig. 1). The magnitudes, however, between these measurements varied noticeably, most likely from differential weighting of CSPs depending on the interactions responsible (i.e., side chain/backbone hydrogen bonding, hydrophobic interactions, structural transitions, etc.) (37) and/or micelle instability at the experimental concentration. Simulations identify that loops β7-β8 (N111–Y113) and some residues of the α2 helix (E134, Y137–N139) are inserted into the micelle headgroup region (Fig. 2 A). The deep insertion of W112 into the nonpolar upper acyl region and polar interfacial location of W116 is also consistent with 19F NMR shifts seen with DPC micelles (11).
Figure 2.
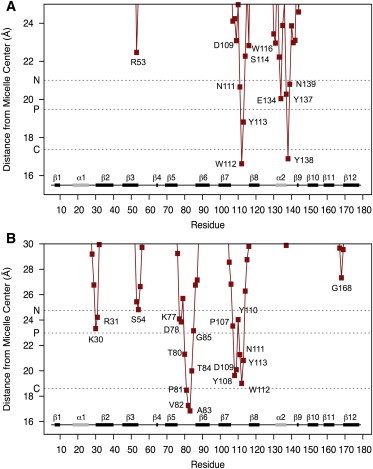
Average radii from the micelle center of mass measured for each protein residue over the final 200 ns of simulation with (A) DPC, and (B) Ac-SM micelle. Dashed lines indicate the average radii measured for all micelle nitrogen, phosphorous, and first acyl carbon atoms of the micelles. Secondary structure elements displayed at the bottom of each plot are labeled according to (5). To see this figure in color, go online.
Multiple binding pockets for DPC (based on occupancy) were identified in the conserved aromatic region. The longest-lived interaction was to the DPC that had restraints applied during the initial equilibration phase. The interaction persisted for the entire simulation and involved stable coordination of the choline by a cation-π box formed between W116, Y137, and Y138 (or Y113) (Fig. 3). S114 and R144 additionally provided H-bonding to this site, but mostly participated in a more exchangeable interaction adjacent to W112. Hydrogen bonding from R53 and Y138, and loose cation-π interactions from Y113, also created a high-occupancy DPC binding site (Fig. 3).
Figure 3.
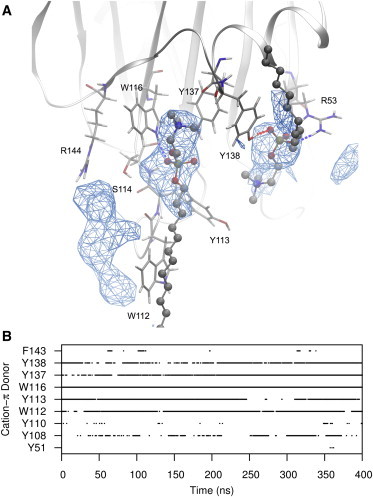
(A) Representative snapshot of a DPC residue engaged in a long-timescale interaction by a cation-π box formed by aromatic rings of W116, Y137, and Y138. A second DPC is also shown in a high-occupancy site formed from hydrogen bonding from R53 and Y138, with weak cation-π association from Y113. Regions of high occupancy are represented in wireframe and calculated over the final 200 ns of simulation. (B) Time-evolution profiles of all EqtII residues engaging cation-π interactions with DPC residues. Fig. S4 includes time-evolution profiles for hydrogen bonds. To see this figure in color, go online.
Y113 and W116 are completely conserved throughout the actinoporin family (Table 1), whereas natural variants of R53 (K), W112 (L, F), S114 (T), Y133 (S), Y138 (N), and R144 (K, E) are conservative for the dominant type of interaction provided. Conserved residues S54, G85, Y108 (F), and N111 also provided significant interactions, but were much less defined due to greater DPC exchange at peripheral zones.
Table 1.
Conservation and summary of intermolecular interactions of EqtII residues with DPC and Ac-SM micelles over the final 200 ns of MD simulation
| H-bonds (102 frame−1)a |
Hydrophobic (frame−1)b |
Cation-π (%)c |
Natural Variantsd | ||||
|---|---|---|---|---|---|---|---|
| DPC | Ac-SM | DPC | Ac-SM | DPC | Ac-SM | ||
| V29 | 1.9 | I (4,5,15–18) | |||||
| K30 | 42, 49(b) | 3.9 | S (4,5,7–14,18); N (6); D (15–17) | ||||
| R31 | 50 | ||||||
| R53 | 60 | 102 | 1.4 | 1.2 | K (15–17) | ||
| S54 | 19 | 28 | 0.9 | 1.6 | |||
| G55 | 20(b) | ||||||
| K77 | 49 | ||||||
| R79 | 47 | T (7–10,12,13); S (14); K (15–18) | |||||
| P81 | 24(b) | 11.5 | |||||
| V82 | 1.4 | 11.6 | D (11) | ||||
| A83 | 0.8 | 8.3 | T (11) | ||||
| T84 | 5 | 2.6 | 4.2 | R (15–17) | |||
| G85 | 14(b) | 2 | |||||
| V87 | 0.8 | 1.7 | |||||
| P107 | 0.3 | 1.1 | |||||
| Y108 | 18, 6(b) | 19 | 3.7 | 4.4 | 28 | F (5,7,9–18) | |
| D109 | 37 | 0.4 | 2.9 | ||||
| Y110 | 15, 2(b) | 1.5 | 3.1 | 1 | 42 | ||
| N111 | 27 | 15 | 0.4 | 1.5 | |||
| W112 | 6 | 8, 10(b) | 24.9 | 17.5 | 42 | 7 | L (5–8,10,11,15–17); F (12,13,18) |
| Y113 | 24 | 25 | 8.3 | 8.5 | 27 | 4 | |
| S114 | 15, 5(b) | 13, 10(b) | T (16,17) | ||||
| W116 | 32 | 19 | 7.3 | 2.4 | 86 | 14 | |
| Q130 | 11 | 4 | E (15–17); Y (18) | ||||
| R131 | 22 | 6 | S (6); A (7,10,11); G (8,9,12–14); K (15–17) | ||||
| Y133 | 63 | 15 | S (8) | ||||
| E134 | 2.4 | 0.1 | N (15–17) | ||||
| Y137 | 27 | 23 | 5.9 | 2.6 | 81 | 60 | L (7); S (8) |
| Y138 | 58 | 30 | 12.9 | 2.2 | 17 | 22 | N (15–17) |
| N139 | 15 | 2.6 | H (3,6); Y (4); G (7–10,12–14); D (18) | ||||
| S141 | 17, 1(b) | I (7); N (8–18) | |||||
| P142 | 5.6 | 1.5 | |||||
| R144 | 50 | 18 | K (6,15–17); G (7,8); E (18) | ||||
Displayed if total H-bonds per frame >5 in either simulation and not zero. Hydrogen bonds involving backbone atoms are specified by (b).
Displayed if >1 in either simulation and not zero.
Displayed if >5 in either simulation and not zero.
Actinoporins are indexed in parentheses according to sequence identity to EqtII (1, 100.0%, P61914). Indices (bold), sequence identity and UniProt accession ID (italics) for each protein are as follows: TenC (2, 100.0%, P61915), FraC (3, 89.9%, B9W5G6), EqtIV (4, 87.2%, Q9Y1U9), EqtV (5, 82.1%, Q93109), BpI (6, 74.3%, C5NSL2), OrG (7, 72.3%, Q5I2B1), OrA (8, 70.9%, Q5I4B8), StII (9, 67.4%, P07845), RTX-SII (10, 67.2%, P0C1F8), RTX-A (11, 66.9%, P58691), HmgIII (12, 66.7%, Q9U6X1), HmT (13, 65.5% (55)), StI (14, 65.3%, P81662), AvtI (15, 61.2%, Q5R231), PsTX-20A (16, 60.7%, Q8IAE2), AvtII (17, 60.7%, D2YZQ3), and SrcI (18, 60.7%, Q86FQ0). Propeptide sequences were manually removed from sequences and alignments performed using the Clustal Omega web server. Sequence alignments included in Fig. S3.
Simulations with an Ac-SM micelle
Ac-SM charge parameters were derived from an identical fragment-based approach used for DPC (38), which kept charges at the choline and phosphate atoms comparable between lipids. Because Ac-SM micelles have not received the same extensive experimental characterization as DPC, an aggregation number of 54 was also applied. Dynamic light scattering of Ac-SM in water detected particles consistent with micelles above 4 μM. The hydrodynamic radius was estimated at 35.7 ± 3.3 Å compared to 25.6 ± 0.3 Å measured for DPC micelles. The hydrodynamic radius of the DPC micelle compared well to the simulated distance to the choline nitrogen atoms (Fig. 2 B) and the first hydration shell (25.4 ± 0.7 Å; see Fig. S5), although simulated Ac-SM radii (29.5 ± 0.5 Å) were noticeably lower than the dynamic light-scattering measurement. Consequently, the seemingly greater curvature in the Ac-SM simulation may expose the headgroups to greater hydration and stress the outermost regions of the lipid-binding site compared to that in an experimental system.
NMR chemical shifts of EqtII bound to an Ac-SM micelle could not be obtained due to suspected formation of large aggregates, which has been noted for StI with mixed micelles of 16C-lysoPC and lyso-SM (13). In addition, Ac-SM having a CMC of only several micromolar precluded titrations being performed at submicelle concentration (due to sensitivity limitations) where such aggregates would unlikely form—unlike DPC, which give workable spectra up to 1.5 mM. The initial starting configuration for simulation of EqtII with DPC was, therefore, used also for Ac-SM (Fig. 4 A). This binding mode, however, did not hold with the Ac-SM micelle. Interactions developed rapidly at the N-terminal hinge (K30, R31) with the insertion of loops β3-β4 (S54) and β5-β6 (K77 to G85) and slight detachment of the α2 helix (Fig. 2 B). As a consequence, cation-π interactions were less prominent in the conserved aromatic region (Table 1; Fig S4 includes per-residue time-evolution profiles for H-bonds and cation-π interactions). Two of several binding modes observed in the POC binding site are shown in Fig. 4, B and C, to highlight hydrogen bonding to the 3-OH ceramide position by W112 (main chain) and Y113. These were the only interactions in this region that could be considered as selective to unique functional groups of SM. Hydrophobic contribution from W112, and hydrogen bonding from the main chain, is consistent with natural leucine and phenylalanine mutations reported to have wild-type (WT) activity (9).
Figure 4.
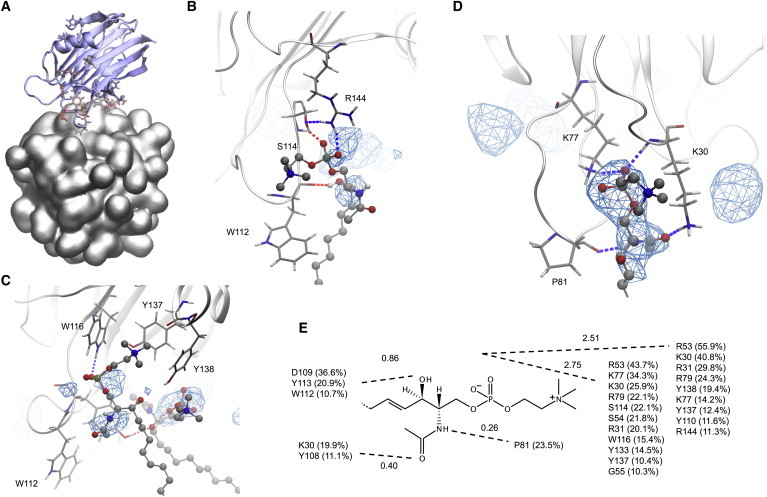
(A) Starting configuration for EqtII bound to an Ac-SM micelle. (B) Ac-SM bound specifically at 3-OH by the main chain of W112 and supported by additional hydrogen bonding from R144 and S114. (C) An Ac-SM bound weakly by cation-π interactions to W116, Y137, and Y138, and hydrogen bonding to W116. An adjacent Ac-SM is bound at the 3-OH by the hydroxyl of Y113. (D) Representative snapshot of Ac-SM bound by hydrogen bonds from K30 (main chain and side chain), K77, and P81 (main chain). Sites of high occupancy were also observed from hydrogen bonding to S54 and G85 (left zone), and to K30, Y108, and D109 (right zone). (E) Total occupancy of hydrogen bonding (side chain, main chain, donor, and acceptor) provided by EqtII residues for polar groups of Ac-SM (values above dashed lines are the average number of hydrogen bonds per frame to the polar positions). Values were calculated over the last 200 ns of simulation and shown only if >10%. To see this figure in color, go online.
Interestingly, the longest-lived interaction for a single Ac-SM was at the hinge of the N-terminal helix, which persisted from ∼20 to 400 ns of the simulation. Ac-SM was held in place by fluctuating hydrogen bonds from K30, K77, P81, and Y108. Ac-SM-selective H-bonding toward the 2-NH position occurred from the main-chain carbonyl of P81, whereas the other residues bound at nonunique phosphate and carbonyl positions. Cysteine mutations have identified by surface plasmon resonance, slightly reduced binding affinities (to 68–90% of WT) at R31, S54, and K77 positions, but disproportionate losses to lytic activity (to 6–8% of WT), which suggested that these residues might be involved in subsequent oligomerization and structural rearrangements of the N-terminus required for pore formation (31,56).
Effects of binding on EqtII structure and dynamics
Backbone root mean-square deviation values measured over the course of simulation fluctuated discretely between 0.9 and 1.6 Å over a long timescale (∼100 ns), but showed no sign of any major restructuring (not shown). Although any significant changes would likely require significantly longer simulations to observe, based on past experimental (NMR, circular dichroism) work (6,11,12,57), they are unlikely to occur with highly curved micelle models. Large fluctuations in root mean-square deviation values coincided with periodic swinging of the unstructured N-terminal residues having the highest root mean-square fluctuation (RMSF) values (Fig. 5 A), and a rare, reversible, unwinding event of the upper α1 helix (residues 11 to 17) occurred at ∼300 ns with DPC (Fig. 5 B). High RMSF values of N147, at the apex of loop β9-β10, for EqtII alone and with Ac-SM, are of interest due to the close proximity of the conserved R144-G145-D146 motif that has suggested roles in integrin binding (2) and/or oligomerization of sticholysin in solution (58). Large fluctuations occurred over long periods (once in 100 ns) in which loop β9-β10 peeled from the protein (compare Fig. 5, B and C). Simultaneous separation of three transient long-range hydrogen bonds between residues N147–N165, G145–S166, S114–R144, and N147–S166 is required for the separation, whereas further modulation may occur from hydrogen bonding observed between N165–S167, D146–H150, D146–R144, and D146–G148. Only S114 and R144 had any direct interactions to micelle lipids, where contributions from R144 were much lower for the Ac-SM simulation due to the α2 helix slightly detaching from the micelle. Disproportionate reduction in lytic activity (93% compared to only 20% loss in binding affinity) previously described for a S114C mutant is consistent with roles beyond membrane binding (i.e., oligomerization), whereas R144C mutation resulted in ∼40% loss in both activity and binding (31). Furthermore, equivalent G145A mutation of StII severely reduces activity and alters oligomerization behavior (58).
Figure 5.
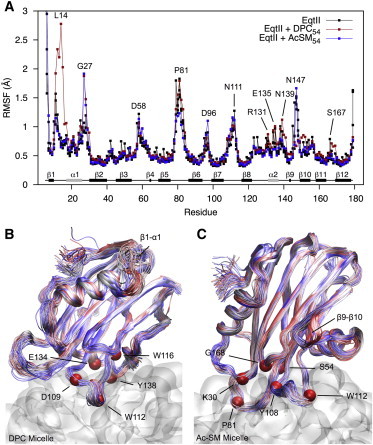
(A) RMSF profile calculated at Cα positions of EqtII alone (100 ns) and bound to micelles (last 200 ns). Overlay of backbone structures taken every 2 ns over the last 200 ns of simulation with DPC (B) and Ac-SM (C) micelles. Structures are color scaled according to simulation time (blue-red). Deeply inserted residues (local minima of Fig. 2) are identified by space-filled Cα positions. To see this figure in color, go online.
Discussion
A combination of NMR and MD simulation was used to define interactions of EqtII with DPC in atomistic detail. NMR CSPs of backbone residues provided simple but effective means of mapping the lipid-binding site, whereas DPC micelles were a conveniently small system for direct comparison with all-atom simulation. Several perturbations to a conserved patch of aromatic amino acids on two solvent-exposed loops and the α2 helix were consistent with the POC binding site seen in an x-ray structure of StII from S. helianthus (7). Mutagenesis (9–11,31), tryptophan fluorescence (59), and NMR (11–13) studies of EqtII and StI, have also provided evidence that this site is responsible for general affinity toward lipids with POC headgroups. In this work, 400 ns of MD simulation identified that coordination of the trimethylammonium group of POC within a cation-π box (60), formed by the aromatic rings of Y113, W116, Y137, and Y138, constituted the primary interaction with DPC. Cooperation from nearby hydrogen bond donors (S114 and R144) also contributed to a chelation effect that led to a central DPC occupying this region for the entire simulation. A high occupancy for DPC bridging S114 and R144 (depicted in Fig. 3 A) may offer an enhanced long-range interaction to stabilize loop β9-β10 or, perhaps, hydrogen bonding of R144 directly to S114 and D146 may destabilize the region to prevent significant oligomerization before membrane attachment.
Cation-π box motifs have recently been described as mediating PC affinity of phosphatidylinositol-specific phospholipase C from Bacillus thuringiensis (52,60). Furthermore, cation-π interactions may explain why a conserved P-[WYF]-D in zebrafish actinoporin-like protein is responsible for PC-binding properties (61). Residues involved in DPC binding are conserved among actinoporins, although Y137L/S variations in OrA and OrG actinoporins from Oulactis orientalis are notable exceptions. OrA and OrG are ∼100-fold less hemolytic (against mammalian erythrocytes) than other actinoporins (62), which may be attributed partly to the loss of Y137 involvement in the cation-π box (>80% occupancy). Furthermore, anchorage of W112 into the acyl region of the micelle correlates with 19F-NMR data of EqtII with DPC micelles (11). W112 is commonly substituted for L and F among actinoporins. According to the Wimley-White hydrophobicity scale, these residues are the next best candidates for being located in acyl environments based on water-octanol transfer free energies (63). The lower hydrophobicity of these variants may be compensated by commonly coinciding Y108F substitutions, which may reorient the protein slightly and possibly explain why StI has greater relative perturbation for loop β5-β6 (13).
SM selectivity has long been assumed to occur at the POC binding site based on nonselectivity of W112A and Y113A mutants (9), and SM-induced perturbation to W112 observed by 19F-NMR (11). SM differs from PC by the 2-NH and 3-OH groups on its ceramide backbone. Molecular modeling studies of a single SM docked at the POC binding site of StII proposed that the phenolic hydroxyl of Y137 (EqtII numbering) might selectively H-bond these positions (30). In this work, MD provided the capability of more accurately representing the water-lipid interface and exploring multiple binding sites. Furthermore, MD could more effectively equilibrate to an optimal binding configuration, which was particularly important because NMR was unable to obtain experimental guidance for initial setup. Instead, by applying the same starting configuration used for the DPC micelle, simulations predicted that D109, W112 (main chain), and Y113 were the only residues in proximity to the POC site to H-bond the 3-OH of SM. Although consistent with lack of SM selectivity for W112A and Y113A mutants (9), and 19F NMR perturbations to W112 (11), long-timescale binding to Ac-SM was not observed in this region. Furthermore, H-bonding from Y113 does not explain why phenylalanine mutations maintain WT activity (9), considering that MD simulation indicated cation-π interactions as less important for binding to the Ac-SM micelle. Alternatively, W112 and Y113 may play a role in interfacial positioning, which would explain the conservative nature of the Y113F mutation (9) given that tyrosine and phenylalanine provide almost equally favorable transfer free energies to the PC-membrane interface (63).
Interactions toward Ac-SM occurred at the hinge of the α1 helix via H-bonds from K30, K77, and P81. K77 is entirely conserved in actinoporins and is essential for proper oligomerization (56). P81 is also completely conserved, perhaps for maintaining structure of the loop β5-β6, but K30 can vary between serine, asparagine and aspartic acid. Serine and asparagine are conservative variants for H-bond donation to the carbonyl of Ac-SM. K30D variants (AvtI, PsTX-20A, and AvtII), however, may instead act as a H-bond acceptor to the 3-OH. The location of this binding site at the hinge of the N-terminal helix is intriguing based on well-documented structural rearrangements required for pore lytic activity (14–19,40,57,59), and is consistent with a tightly bound lipid, assigned to be SM, recently identified in a crystallized pore of FraC (64). The lipid was noted to stabilize the pore by bridging adjacent protein chains (64) but our data also imply that binding may occur as early as the monomeric-binding phase of pore formation. Direct interaction to the N-terminal hinge, or reorientation to bring loops β3-β4 and β5-β6 to the membrane interface, might trigger the conformational changes necessary for pore formation. However, these would not be observed during simulation due to the long timescales required (∼seconds (16)) and is not expected for highly curved model systems (6,11,12,57).
Conclusions
NMR studies of backbone dynamics of StI with DHPC micelles revealed increased dynamics over the ps-ns timescale in loops α1-β2 and β5-β6 (EqtII nomenclature), whereas loops β7-β8 and α2-β9 displayed conformational exchange in the ms-μs regime (13). MD of EqtII with DPC did not suggest similar behavior based on backbone RMSF calculations, although such dynamics would reflect roughly the lipid-protein exchange rate occurring in these regions. Changes in backbone dynamics were, however, observed at the conserved R144-G145-D146 motif, which is suggested to play a part in oligomerization in solution (58) and integrin binding (2). The direct involvement of R144 in DPC binding suppressed periodic detachment of loop β9-β10 (at N147) from the β-sandwich core, whereas the motion was reintroduced with Ac-SM due to R144 having reduced roles in binding. The implications of the dynamics observed for loop β9-β10, and its variability between DPC and Ac-SM, is difficult to assess with limited experimental data available. For mixed lipid systems PC could possibly bind in this region while Ac-SM is attached to the N-terminal hinge. However, the slow exchange of some lipids precluded testing by MD simulations, but may explain why phase-boundaries between liquid-disordered PC and liquid-ordered SM or SM/cholesterol phases promote EqtII activity in model systems (16,65–67).
Author Contributions
D.K.W.: experimental design, acquisition, analysis and interpretation of data, drafting of manuscript; SY.: acquisition, analysis and interpretation of NMR data, critical revision; N.R.: protein expression and labeling, analysis and functional study; G.A.: contributed analytic tool, interpretation of data, critical revision; T.P.L., M.T.D., and J.W.: design and analysis of MD study, critical revision; F.S.: study conception and design, interpretation of data, writing sections and critical revision.
Acknowledgments
This work was supported by a University of Melbourne Dyason – Vanderbilt Partnership grant, VLSCI Resource Allocation Scheme and grants from the Slovenian Research Agency. D.W. is a recipient of an Australian Postgraduate Award and a Dowd Foundation Postgraduate Research Scholarship.
Supporting Material
References
- 1.Ferlan I., Lebez D. Equinatoxin, a lethal protein from Actinia equina—I. Purification and characterization. Toxicon. 1974;12:57–61. doi: 10.1016/0041-0101(74)90099-3. [DOI] [PubMed] [Google Scholar]
- 2.Anderluh G., Maček P. Cytolytic peptide and protein toxins from sea anemones (Anthozoa: Actiniaria) Toxicon. 2002;40:111–124. doi: 10.1016/s0041-0101(01)00191-x. [DOI] [PubMed] [Google Scholar]
- 3.Kristan K.C., Viero G., Anderluh G. Molecular mechanism of pore formation by actinoporins. Toxicon. 2009;54:1125–1134. doi: 10.1016/j.toxicon.2009.02.026. [DOI] [PubMed] [Google Scholar]
- 4.Hinds M.G., Zhang W., Norton R.S. Solution structure of the eukaryotic pore-forming cytolysin equinatoxin II: implications for pore formation. J. Mol. Biol. 2002;315:1219–1229. doi: 10.1006/jmbi.2001.5321. [DOI] [PubMed] [Google Scholar]
- 5.Athanasiadis A., Anderluh G., Turk D. Crystal structure of the soluble form of equinatoxin II, a pore-forming toxin from the sea anemone Actinia equina. Structure. 2001;9:341–346. doi: 10.1016/s0969-2126(01)00592-5. [DOI] [PubMed] [Google Scholar]
- 6.García-Linares S., Castrillo I., Gavilanes J.G. Three-dimensional structure of the actinoporin sticholysin I. Influence of long-distance effects on protein function. Arch. Biochem. Biophys. 2013;532:39–45. doi: 10.1016/j.abb.2013.01.005. [DOI] [PubMed] [Google Scholar]
- 7.Mancheño J.M., Martín-Benito J., Hermoso J.A. Crystal and electron microscopy structures of sticholysin II actinoporin reveal insights into the mechanism of membrane pore formation. Structure. 2003;11:1319–1328. doi: 10.1016/j.str.2003.09.019. [DOI] [PubMed] [Google Scholar]
- 8.Mechaly A.E., Bellomio A., Guérin D.M.A. Structural insights into the oligomerization and architecture of eukaryotic membrane pore-forming toxins. Structure. 2011;19:181–191. doi: 10.1016/j.str.2010.11.013. [DOI] [PubMed] [Google Scholar]
- 9.Bakrač B., Gutiérrez-Aguirre I., Anderluh G. Molecular determinants of sphingomyelin specificity of a eukaryotic pore-forming toxin. J. Biol. Chem. 2008;283:18665–18677. doi: 10.1074/jbc.M708747200. [DOI] [PubMed] [Google Scholar]
- 10.Alegre-Cebollada J., Cunietti M., Martínez-del-Pozo A. Calorimetric scrutiny of lipid binding by sticholysin II toxin mutants. J. Mol. Biol. 2008;382:920–930. doi: 10.1016/j.jmb.2008.07.053. [DOI] [PubMed] [Google Scholar]
- 11.Anderluh G., Razpotnik A., Norton R.S. Interaction of the eukaryotic pore-forming cytolysin equinatoxin II with model membranes: 19F NMR studies. J. Mol. Biol. 2005;347:27–39. doi: 10.1016/j.jmb.2004.12.058. [DOI] [PubMed] [Google Scholar]
- 12.Castrillo I., Araujo N.A., Bruix M. Specific interactions of sticholysin I with model membranes: an NMR study. Proteins. 2010;78:1959–1970. doi: 10.1002/prot.22712. [DOI] [PubMed] [Google Scholar]
- 13.López-Castilla A., Pazos F., Pires J.R. Solution NMR analysis of the interaction between the actinoporin sticholysin I and DHPC micelles—correlation with backbone dynamics. Proteins. 2014;82:1022–1034. doi: 10.1002/prot.24475. [DOI] [PubMed] [Google Scholar]
- 14.Hong Q., Gutiérrez-Aguirre I., Anderluh G. Two-step membrane binding by Equinatoxin II, a pore-forming toxin from the sea anemone, involves an exposed aromatic cluster and a flexible helix. J. Biol. Chem. 2002;277:41916–41924. doi: 10.1074/jbc.M204625200. [DOI] [PubMed] [Google Scholar]
- 15.Kristan K., Podlesek Z., Anderluh G. Pore formation by equinatoxin, a eukaryotic pore-forming toxin, requires a flexible N-terminal region and a stable β-sandwich. J. Biol. Chem. 2004;279:46509–46517. doi: 10.1074/jbc.M406193200. [DOI] [PubMed] [Google Scholar]
- 16.Rojko N., Kristan K.C., Anderluh G. Membrane damage by an α-helical pore-forming protein, Equinatoxin II, proceeds through a succession of ordered steps. J. Biol. Chem. 2013;288:23704–23715. doi: 10.1074/jbc.M113.481572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gutiérrez-Aguirre I., Barlič A., González-Mañas J.M. Membrane insertion of the N-terminal α-helix of equinatoxin II, a sea anemone cytolytic toxin. Biochem. J. 2004;384:421–428. doi: 10.1042/BJ20040601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Malovrh P., Viero G., Anderluh G. A novel mechanism of pore formation: membrane penetration by the N-terminal amphipathic region of equinatoxin. J. Biol. Chem. 2003;278:22678–22685. doi: 10.1074/jbc.M300622200. [DOI] [PubMed] [Google Scholar]
- 19.Kristan K., Viero G., Anderluh G. The equinatoxin N-terminus is transferred across planar lipid membranes and helps to stabilize the transmembrane pore. FEBS J. 2007;274:539–550. doi: 10.1111/j.1742-4658.2006.05608.x. [DOI] [PubMed] [Google Scholar]
- 20.Belmonte G., Pederzolli C., Menestrina G. Pore formation by the sea anemone cytolysin equinatoxin II in red blood cells and model lipid membranes. J. Membr. Biol. 1993;131:11–22. doi: 10.1007/BF02258530. [DOI] [PubMed] [Google Scholar]
- 21.Meinardi E., Florin-Christensen M., Florin-Christensen J. The molecular basis of the self/nonself selectivity of a coelenterate toxin. Biochem. Biophys. Res. Commun. 1995;216:348–354. doi: 10.1006/bbrc.1995.2630. [DOI] [PubMed] [Google Scholar]
- 22.De Colibus L., Sonnen A.F.P., Gilbert R.J.C. Structures of lysenin reveal a shared evolutionary origin for pore-forming proteins and its mode of sphingomyelin recognition. Structure. 2012;20:1498–1507. doi: 10.1016/j.str.2012.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Anderluh G., Lakey J.H. Disparate proteins use similar architectures to damage membranes. Trends Biochem. Sci. 2008;33:482–490. doi: 10.1016/j.tibs.2008.07.004. [DOI] [PubMed] [Google Scholar]
- 24.Bavdek A., Kostanjšek R., Anderluh G. pH dependence of listeriolysin O aggregation and pore-forming ability. FEBS J. 2012;279:126–141. doi: 10.1111/j.1742-4658.2011.08405.x. [DOI] [PubMed] [Google Scholar]
- 25.Yachi R., Uchida Y., Arai H. Subcellular localization of sphingomyelin revealed by two toxin-based probes in mammalian cells. Genes Cells. 2012;17:720–727. doi: 10.1111/j.1365-2443.2012.01621.x. [DOI] [PubMed] [Google Scholar]
- 26.Garcia P.S., Chieppa G., Rufini S. Sticholysin II: a pore-forming toxin as a probe to recognize sphingomyelin in artificial and cellular membranes. Toxicon. 2012;60:724–733. doi: 10.1016/j.toxicon.2012.05.018. [DOI] [PubMed] [Google Scholar]
- 27.Bakrač B., Kladnik A., Anderluh G. A toxin-based probe reveals cytoplasmic exposure of Golgi sphingomyelin. J. Biol. Chem. 2010;285:22186–22195. doi: 10.1074/jbc.M110.105122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Abe M., Kobayashi T. Imaging local sphingomyelin-rich domains in the plasma membrane using specific probes and advanced microscopy. Biochim. Biophys. Acta. 2014;1841:720–726. doi: 10.1016/j.bbalip.2013.07.003. [DOI] [PubMed] [Google Scholar]
- 29.Ohno-Iwashita Y., Shimada Y., Iwashita S. Perfringolysin O, a cholesterol-binding cytolysin, as a probe for lipid rafts. Anaerobe. 2004;10:125–134. doi: 10.1016/j.anaerobe.2003.09.003. [DOI] [PubMed] [Google Scholar]
- 30.Maula T., Isaksson Y.J.E., Slotte J.P. 2NH and 3OH are crucial structural requirements in sphingomyelin for sticholysin II binding and pore formation in bilayer membranes. Biochim. Biophys. Acta. 2013;1828:1390–1395. doi: 10.1016/j.bbamem.2013.01.018. [DOI] [PubMed] [Google Scholar]
- 31.Anderluh G., Barlič A., Menestrina G. Cysteine-scanning mutagenesis of an eukaryotic pore-forming toxin from sea anemone: topology in lipid membranes. Eur. J. Biochem. 1999;263:128–136. doi: 10.1046/j.1432-1327.1999.00477.x. [DOI] [PubMed] [Google Scholar]
- 32.Roberts M.F., Redfield A.G. Phospholipid bilayer surface configuration probed quantitatively by (31)P field-cycling NMR. Proc. Natl. Acad. Sci. USA. 2004;101:17066–17071. doi: 10.1073/pnas.0407565101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sani M.A., Weber D.K., Gehman J.D. A practical implementation of de-Pake-ing via weighted Fourier transformation. PeerJ. 2013;1:e30. doi: 10.7717/peerj.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sani M.A., Separovic F., Gehman J.D. Disentanglement of heterogeneous dynamics in mixed lipid systems. Biophys. J. 2011;100:L40–L42. doi: 10.1016/j.bpj.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yao S., Weber D.K., Keizer D.W. Measuring translational diffusion coefficients of peptides and proteins by PFG-NMR using band-selective RF pulses. Eur. Biophys. J. 2014;43:331–339. doi: 10.1007/s00249-014-0965-x. [DOI] [PubMed] [Google Scholar]
- 36.Papavoine C.H.M., Konings R.N.H., van de Ven F.J.M. Location of M13 coat protein in sodium dodecyl sulfate micelles as determined by NMR. Biochemistry. 1994;33:12990–12997. doi: 10.1021/bi00248a007. [DOI] [PubMed] [Google Scholar]
- 37.Williamson M.P. Using chemical shift perturbation to characterize ligand binding. Prog. Nucl. Magn. Reson. Spectrosc. 2013;73:1–16. doi: 10.1016/j.pnmrs.2013.02.001. [DOI] [PubMed] [Google Scholar]
- 38.Abel S., Dupradeau F.Y., Marchi M. Molecular dynamics simulations of a characteristic DPC micelle in water. J. Chem. Theory Comput. 2012;8:4610–4623. doi: 10.1021/ct3003207. [DOI] [PubMed] [Google Scholar]
- 39.Anderluh G., Pungerčar J., Gubenšek F. Cloning, sequencing, and expression of equinatoxin II. Biochem. Biophys. Res. Commun. 1996;220:437–442. doi: 10.1006/bbrc.1996.0391. [DOI] [PubMed] [Google Scholar]
- 40.Anderluh G., Pungercar J., Macek P. N-terminal truncation mutagenesis of equinatoxin II, a pore-forming protein from the sea anemone Actinia equina. Protein Eng. 1997;10:751–755. doi: 10.1093/protein/10.7.751. [DOI] [PubMed] [Google Scholar]
- 41.Palmer A.G., III, Cavanagh J., Rance M. Sensitivity improvement in proton-detected two-dimensional heteronuclear correlation NMR spectroscopy. J. Magn. Reson. 1991;93:151–170. [Google Scholar]
- 42.Delaglio F., Grzesiek S., Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 43.Vranken W.F., Boucher W., Laue E.D. The CCPN data model for NMR spectroscopy: development of a software pipeline. Proteins. 2005;59:687–696. doi: 10.1002/prot.20449. [DOI] [PubMed] [Google Scholar]
- 44.Zhang W., Hinds M.G., Norton R.S. Letter to the editor: Sequence-specific resonance assignments of the potent cytolysin equinatoxin II. J. Biomol. NMR. 2000;18:281–282. doi: 10.1023/a:1026793926271. [DOI] [PubMed] [Google Scholar]
- 45.Grzesiek S., Bax A. Improved 3D triple-resonance NMR techniques applied to a 31 kDa protein. J. Magn. Reson. 1992;96:432–440. [Google Scholar]
- 46.Wang J., Wolf R.M., Case D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004;25:1157–1174. doi: 10.1002/jcc.20035. [DOI] [PubMed] [Google Scholar]
- 47.Dickson C.J., Rosso L., Gould I.R. GAFFlipid: a general amber force field for the accurate molecular dynamics simulation of phospholipid. Soft Matter. 2012;8:9617–9627. [Google Scholar]
- 48.Vanquelef E., Simon S., Dupradeau F.Y. R.E.D. Server: a web service for deriving RESP and ESP charges and building force field libraries for new molecules and molecular fragments. Nucleic Acids Res. 2011;39:W511–W517. doi: 10.1093/nar/gkr288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Phillips J.C., Braun R., Schulten K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hornak V., Abel R., Simmerling C. Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins. 2006;65:712–725. doi: 10.1002/prot.21123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Humphrey W., Dalke A., Schulten K. VMD: visual molecular dynamics. J. Mol. Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. 27–28. [DOI] [PubMed] [Google Scholar]
- 52.Grauffel C., Yang B., Reuter N. Cation-π interactions as lipid-specific anchors for phosphatidylinositol-specific phospholipase C. J. Am. Chem. Soc. 2013;135:5740–5750. doi: 10.1021/ja312656v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Warschawski D.E., Arnold A.A., Marcotte I. Choosing membrane mimetics for NMR structural studies of transmembrane proteins. Biomembranes. 2011;1808:1957–1974. doi: 10.1016/j.bbamem.2011.03.016. [DOI] [PubMed] [Google Scholar]
- 54.Stafford R.E., Fanni T., Dennis E.A. Interfacial properties and critical micelle concentration of lysophospholipids. Biochemistry. 1989;28:5113–5120. doi: 10.1021/bi00438a031. [DOI] [PubMed] [Google Scholar]
- 55.Samejima Y., Yanagisawa M., Mebs D. Amino acid sequence studies on cytolytic toxins from sea anemone Heteractis magnifica, Entacmaea quadricolor and Stichodactyla mertensii (Anthozoa) Toxicon. 2000;38:259–264. doi: 10.1016/s0041-0101(99)00154-3. [DOI] [PubMed] [Google Scholar]
- 56.Anderluh G., Barlič A., Menestrina G. Lysine 77 is a key residue in aggregation of equinatoxin II, a pore-forming toxin from sea anemone Actinia equina. J. Membr. Biol. 2000;173:47–55. doi: 10.1007/s002320001006. [DOI] [PubMed] [Google Scholar]
- 57.Miles A.J., Drechsler A., Separovic F. The effects of lipids on the structure of the eukaryotic cytolysin equinatoxin II: a synchrotron radiation circular dichroism spectroscopic study. Biochim. Biophys. Acta. 2008;1778:2091–2096. doi: 10.1016/j.bbamem.2008.04.001. [DOI] [PubMed] [Google Scholar]
- 58.García-Linares S., Richmond R., Martínez-Del-Pozo A. The sea anemone actinoporin (Arg-Gly-Asp) conserved motif is involved in maintaining the competent oligomerization state of these pore-forming toxins. FEBS J. 2014;281:1465–1478. doi: 10.1111/febs.12717. [DOI] [PubMed] [Google Scholar]
- 59.Malovrh P., Barlič A., Anderluh G. Structure-function studies of tryptophan mutants of equinatoxin II, a sea anemone pore-forming protein. Biochem. J. 2000;346:223–232. [PMC free article] [PubMed] [Google Scholar]
- 60.Cheng J., Goldstein R., Roberts M.F. The cation-π box is a specific phosphatidylcholine membrane targeting motif. J. Biol. Chem. 2013;288:14863–14873. doi: 10.1074/jbc.M113.466532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gutiérrez-Aguirre I., Trontelj P., Anderluh G. Membrane binding of zebrafish actinoporin-like protein: AF domains, a novel superfamily of cell membrane binding domains. Biochem. J. 2006;398:381–392. doi: 10.1042/BJ20060206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Monastyrnaya M., Leychenko E., Kozlovskaya E. Actinoporins from the sea anemones, tropical Radianthus macrodactylus and northern Oulactis orientalis: comparative analysis of structure-function relationships. Toxicon. 2010;56:1299–1314. doi: 10.1016/j.toxicon.2010.07.011. [DOI] [PubMed] [Google Scholar]
- 63.Wimley W.C., White S.H. Experimentally determined hydrophobicity scale for proteins at membrane interfaces. Nat. Struct. Biol. 1996;3:842–848. doi: 10.1038/nsb1096-842. [DOI] [PubMed] [Google Scholar]
- 64.Tanaka K., Caaveiro J.M.M., Tsumoto K. Structural basis for self-assembly of a cytolytic pore lined by protein and lipid. Nat. Commun. 2015;6:6337. doi: 10.1038/ncomms7337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Schön P., García-Sáez A.J., Schwille P. Equinatoxin II permeabilizing activity depends on the presence of sphingomyelin and lipid phase coexistence. Biophys. J. 2008;95:691–698. doi: 10.1529/biophysj.108.129981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Barlič A., Gutiérrez-Aguirre I., González-Mañas J.M. Lipid phase coexistence favors membrane insertion of equinatoxin-II, a pore-forming toxin from Actinia equina. J. Biol. Chem. 2004;279:34209–34216. doi: 10.1074/jbc.M313817200. [DOI] [PubMed] [Google Scholar]
- 67.Drechsler A., Anderluh G., Separovic F. Solid-state NMR study of membrane interactions of the pore-forming cytolysin, equinatoxin II. Biochim. Biophys. Acta. 2010;1798:244–251. doi: 10.1016/j.bbamem.2009.10.012. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.