

Abstract
Rationale: Pseudomonas aeruginosa, the predominant cause of chronic airway infections of patients with cystic fibrosis, exhibits extensive phenotypic diversity among isolates within and between sputum samples, but little is known about the underlying genetic diversity.
Objectives: To characterize the population genetic structure of transmissible P. aeruginosa Liverpool Epidemic Strain in chronic infections of nine patients with cystic fibrosis, and infer evolutionary processes associated with adaptation to the cystic fibrosis lung.
Methods: We performed whole-genome sequencing of P. aeruginosa isolates and pooled populations and used comparative analyses of genome sequences including phylogenetic reconstructions and resolution of population structure from genome-wide allele frequencies.
Measurements and Main Results: Genome sequences were obtained for 360 isolates from nine patients. Phylogenetic reconstruction of the ancestry of 40 individually sequenced isolates from one patient sputum sample revealed the coexistence of two genetically diverged, recombining lineages exchanging potentially adaptive mutations. Analysis of population samples for eight additional patients indicated coexisting lineages in six cases. Reconstruction of the ancestry of individually sequenced isolates from all patients indicated smaller genetic distances between than within patients in most cases.
Conclusions: Our population-level analysis demonstrates that coexistence of distinct lineages of P. aeruginosa Liverpool Epidemic Strain within individuals is common. In several cases, coexisting lineages may have been present in the infecting inoculum or assembled through multiple transmissions. Divergent lineages can share mutations via homologous recombination, potentially aiding adaptation to the airway during chronic infection. The genetic diversity of this transmissible strain within infections, revealed by high-resolution genomics, has implications for patient segregation and therapeutic strategies.
Keywords: bacteria, population genetics, genomics, homologous recombination
At a Glance Commentary
Scientific Knowledge on the Subject
Chronic lung infections caused by Pseudomonas aeruginosa remain the major cause of morbidity and mortality among patients with cystic fibrosis. Transmissible strains, such as the widespread (United Kingdom and North America) Liverpool Epidemic Strain (LES), are especially problematic and require segregation of affected patients within cystic fibrosis units to prevent cross-infection. Surprisingly high levels of phenotypic diversity within individual patient sputum samples have been demonstrated, and genome sequencing of sequential isolates suggests that the pathogen accumulates mutations over time. However, little is known about the underlying genetic diversity within infecting populations or the distribution of diversity among patients.
What This Study Adds to the Field
Using genomic analysis of sequential isolates, others have found evidence for rare replacements of one P. aeruginosa lineage by another. Here we show, using large-scale population genomic analyses, that the coexistence of distinct lineages of the P. aeruginosa LES is typical, occurring in seven of nine chronically infected patients with cystic fibrosis sampled. Genetic divergence between lineages within patients was greater than between, implying acquisition of diverse P. aeruginosa populations and potentially acquisition of distinct lineages among the LES-infected cohort. Furthermore, evidence for homologous recombination between divergent lineages within a patient provides evidence for a novel mode of potentially adaptive evolution by P. aeruginosa during chronic infection.
Pseudomonas aeruginosa is the most common cause of airway infection in cystic fibrosis (CF) (1) and once established in the chronic stage is notoriously resistant to clearance by chemotherapy (2). Chronic-stage infections exhibit both adaptation and diversification. The genetic mechanisms underlying some chronic-stage adaptations, such as the switch to mucoid phenotype, have been well established for many years (3). Other common adaptations include mutations in the gene encoding the key quorum sensing regulator LasR (4), loss of motility (5), auxotrophy (6), hypermutability (7), and increasing resistance to antibiotics (8). It has been shown that evolutionary adaptation can occur rapidly in the airways of patients with CF (9, 10). Although most patients acquire their infecting P. aeruginosa from environmental sources with subsequent adaptation to the CF airway (11), there have been a number of transmissible strains identified (12). Notable among these is the Liverpool Epidemic Strain (LES), which is the most abundant clone of P. aeruginosa isolated from patients with CF in the United Kingdom (13, 14), and has been reported in North America (15, 16).
The genetic basis of diversification in chronic infections remains poorly understood. Whereas a number of studies have reported on the genetic adaptation of P. aeruginosa during CF lung infections by targeting specific genes (7) or whole genomes for sequencing (17, 18), these studies have generally been optimized to capture genetic changes over time. At the expense of sampling depth within individuals, these studies sampled sequential isolates from individual patients with CF or single isolates from many different patients with CF. Consequently, it is essential that investigations of population-scale genetic diversity of P. aeruginosa be extended to consider diversity both within and between multiple chronically infected patients with CF. Evidence from phenotypic studies suggests widespread heterogeneity (19–22), and genome sequencing of paired P. aeruginosa isolates from three individuals revealed genetic diversity (23). A recent study implicated spatial separation within the CF lung as causing diversification into distinct lineages in a single patient with CF (24). A report of a less common CF pathogen, Burkholderia dolosa, described unexpectedly high genetic diversity within patients using genome sequencing of pooled population samples (25). To characterize the population structure of the P. aeruginosa LES populations in chronically infected patients with CF, we assayed the genome sequence diversity among 40 isolates from a sputum sample for each of nine adults attending the same CF unit.
Methods
Acquisition of Samples and Isolation of P. aeruginosa
Samples were collected from nine adult patients with CF, each chronically infected with the P. aeruginosa LES, as described previously (19). Briefly, a sputum sample was collected from each patient at a routine visit to the Regional Adult Cystic Fibrosis Unit in Liverpool, United Kingdom during January 2009. Sputum was treated with an equal volume of Sputasol (Oxoid, Basingstoke, UK), incubated at room temperature with shaking at 200 rpm for 15 minutes, and then cultured on Pseudomonas-selective agar under aerobic conditions with CN supplement (Oxoid)as described previously (19). Forty LES colonies were selected to maximize colony morphology diversity and identified as described previously (19). Details concerning age, sex, and clinical status of patients CF01 and CF03–CF10 were given in our previous study (19). Based on information available since this previous study, CF01, CF03, CF05, and CF07 are now known to have been LES positive since at least 1995; CF04, CF06, CF09, and CF10 since 1995 but before 2004; and CF08 since at least 2008. This study was approved by the local research ethics committee (REC reference 08/H1006/47).
Genomic DNA Preparation and Sequencing
Details of DNA extraction are outlined in the online supplement. Library preparation and whole-genome shotgun sequencing was performed by the Centre for Genomic Research at the University of Liverpool, United Kingdom using Illumina short read sequencing technology. Details of quality control of sequenced read data is outlined in the online supplement. The European Nucleotide Archive accession number for the study is PRJEB6642.
Variant Calling and De Novo Genome Assemblies
Reads were aligned to the P. aeruginosa LESB58 reference genome sequence (National Center for Biotechnology Information accession number, NC_011770) with the BWA-MEM aligner (26). For individually sequenced isolates, paired-end reads were assembled de novo using SPAdes Genome Assembler version 3.0 (27). Details of single-nucleotide polymorphism (SNP), insertion and deletion discovery, prediction of genetic variant effects on protein sequences, and de novo genome assembly are outlined in the online supplement. Genome assemblies were used to double-check homoplasies indicated by analysis of read alignments. The European Nucleotide Archive accession numbers for the assemblies of sequenced isolates are ERZ021677-716.
Phylogenetic Reconstruction and Hypothesis Testing
Variable sites identified among the aligned sequencing reads for each genome sequence were combined into a multiple alignment for phylogenetic analysis. Genome phylogenies were reconstructed using the BIONJ algorithm (28) implemented in the APE version 3.1–2 (29) for the R statistical computing environment version 3.1.0. Statistical support for edges in phylogenies were split frequencies among a nonparametric bootstrap replicate sample of maximum-likelihood phylogenies inferred using Garli version 2.01 (30) and a Bayesian sample of phylogenies inferred using MrBayes version 3.2.2 (31) and counted using methods of the DendroPy library for phylogenetic computing version 3.12.0 (32) in Python version 2.7.8. The HKY85 nucleotide substitution model was selected for use in Garli with JModelTest2 version 2.1.5 (33). P. aeruginosa LESB58 genome sequence was used as an outgroup for rooting, the suitability of which was tested by phylogenetic reconstruction including sequences of distantly related LESlike 4, 5, 7, DK2, and PAO1 P. aeruginosa isolates. The scale-boot R package version 0.3–3 was used to perform the Approximately Unbiased (AU) test on a multiscale bootstrap for the alternative and maximum likelihood topologies against the sequence alignment (34). Details of phylogenetic reconstructions, homoplasy identification, and inference of homologous recombination are outlined in the online supplement.
Inference of Divergent Lineages from SNP Frequencies
For each patient sputum sample, we tested for the coexistence of a pair of divergent lineages. A lineage is defined as a group of isolates with high genetic similarity. Divergent coexisting lineages in a sample are defined as each exhibiting more genetic differences from their most recent common ancestor (MRCA) than their MRCA has to the MRCA of all cohort patient isolates (i.e., of this epidemic) inferred from all samples. An alternative hypothesis is all isolates in a sample forming a single lineage where genetic divergence within the sample is less than to other samples. These hypotheses concern the deepest part of the phylogeny in each sample: the length of the root edge must be shorter than the lengths of the edges descending from the deepest bifurcation (the primary edges) for the coexisting, divergent lineages hypothesis to be supported. The root and primary edges can be inferred from their corresponding root and primary peaks in the SNP frequency distributions. Details of inference of divergent lineages from SNP frequencies are outlined in more detail in the online supplement. Monte Carlo simulation of nucleotide sequence evolution over the hypothesized phylogenies for corroboration was achieved using the PhyloSim R package version 2.1.1 (35).
Results
A Single Chronic CF Infection Sputum Sample Contains Divergent, Recombining P. aeruginosa Lineages
Among 40 P. aeruginosa LES isolates from a sputum sample from a patient with CF collected in 2009 (CF03) that were individually genome sequenced, we identified between 71 and 130 SNPs relative to the complete genome sequence of P. aeruginosa LESB58, isolated in 1988. Despite these 40 isolates being obtained from a single patient sample, reconstructions of their shared evolutionary history, by different methods, revealed two divergent lineages (clades) (Figure 1). One lineage had 13 members (CF03 lineage A) and the other had 27 (CF03 lineage B).
Figure 1.

Pseudomonas aeruginosa Liverpool Epidemic Strain population structure in cystic fibrosis sputum sample CF03 consists of two divergent, recombining lineages. Rooted neighbor-joining (BIONJ) phylogenetic reconstruction of 40 isolate genome sequences obtained from a single cystic fibrosis sputum sample (CF03, collected 2009) calculated from a distance matrix of single-nucleotide polymorphism (SNP) counts. SNPs among whole-genome sequence short reads mapped to the P. aeruginosa LESB58 reference genome sequence (collected 1988), which also serves as an outgroup for rooting. The three support values for each edge are the percent split frequency among a nonparametric bootstrap replicate sample of BIONJ and maximum-likelihood phylogenies and among a Bayesian sample of phylogenies. Only edges with at least 80% support by all three measures are labeled with the respective split frequencies. The isolate sequences sharing homoplasies are indicated with circles in the right-most columns, which correspond to circled variants in Figure 2; the column numbers relate to row numbers in Table 1.
CF03 lineage A was characterized by 55 shared SNPs, and lineage B had 24 shared SNPs. There were 79 SNPs separating the coexisting CF03 lineages A and B but, by contrast, only 42 SNPs separate their MRCA from the LESB58 reference sequence and phylogenetic outgroup. The suitability of LESB58 as an outgroup is demonstrated by a phylogenetic reconstruction in which it is partitioned with distantly related isolates from the study sequences (Figure 1; see online supplement). CF03 lineage B genome sequences were more similar to LESB58 than to lineage A genome sequences, yet both lineages were from the same CF chronic infection sputum sample (Figure 2). Mutations exclusive to each lineage included those predicted to alter proteins associated with virulence factors (see Tables E1 and E2 in the online supplement). For example, the 13 members of lineage A are predicted to have a truncated MexB multidrug efflux transporter (PLES_04241), whereas the 27 members of lineage B are predicted to have a truncated antisigma factor MucA (PLES_45801) involved in the regulation of alginate biosynthesis.
Figure 2.

Chromosome positions and predicted effects of mutations within and between CF03 lineages A and B. Genome map of mutations in 40 isolate genome sequences (outer lanes) obtained from a single sputum sample (CF03, collected 2009). Lanes are ordered by phylogenetic relationships in the neighbor-joining phylogeny from Figure 1, adapted and plotted at the lower left, and rooted using the LESB58 reference genome (collected 1988) as outgroup. The outer 13 lanes correspond to CF03 lineage A and the 27 lanes inward to CF03 lineage B. The next, wider, lane corresponds to the LESB58 reference genome sequence with prophage (light brown) and genomic island (dark green) regions indicated at their positions relative to the origin of replication indicated by the outer scale. The innermost lane is a plot of percent guanine-cytosine (GC) in 5-kbp regions calculated every 2.5 kbp and filled red above and below 50% GC. For each isolate genome sequence, the mutations identified from sequenced reads aligned to the reference sequence were classified as single-nucleotide polymorphisms (SNP), small insertions, or small deletions. Those that occurred in protein-coding regions are further classified by the predicted effects on transcription to mRNA and translation to protein sequences. Mutation classes are plotted on the genome map in different colors indicated in the key. Regions in which no reads mapped to the reference chromosome for an isolate sample are indicated by gaps in the corresponding lane. Homoplasies are circled and correspond to those described in Figure 1 and Table 1. ORF = open reading frame.
Although mutually exclusive mutations define the two coexisting CF03 lineages, numerous mutations shared between a minority of each lineage (homoplasies) provide evidence of DNA transfer between cells via homologous recombination (Figures 1 and 2). An alternative hypothesis excluding recombination between cells requires coincidental, parallel mutations to explain the phylogenetic distribution of these mutations among isolates, the probabilities of which are low (<1 × 10−7) and listed in Table 1. Two SNPs were common between a minority of isolates in each lineage indicating horizontal genetic transfer between the CF03 lineages. One of these is a nonsynonymous (protein altering) replacement in lysC, which codes for an aspartate kinase in LESB58 (locus ID, PLES_44121; P. aeruginosa PAO1 locus ID, PA0904; homoplasy 3 in Figure 1 and Table 1). Two homoplasies were detected within the larger clade indicating horizontal genetic transfer among members of CF03 lineage B. One of these intralineage transfers was a deletion predicted to cause the truncation of mpl, an open reading frame encoding a Mur family ligase in LESB58 (PLES_09561; PA4020; homoplasy 1 in Figure 1 and Table 1). The other intralineage transfer is a deletion of two codons within glpT, which encodes a glycerol-3-phosphate transporter (PLES_56291; PA5235; homoplasy 4 in Figure 1 and Table 1).
Table 1.
Incidence of Homoplasies Likely to Be Involved in Homologous Recombination among Sputum Sample CF03 Isolates with Predicted Effects on Transcription of Open Reading Frames and Translation to Polypeptides Where Applicable
| Homoplasy ID* | Incidence | Description | Annotation | Probability of Independent Mutations† | Position (bp)‡ |
|---|---|---|---|---|---|
| 1 | Lineage B, isolates 13 and 24; isolates 14, 20, and 31 | Deletion of one nucleotide in PLES_09561 (mpl) causing nonsense mutations and loss of stop codon extending the ORF | UDP-N-acetylmuramate:l-alanyl-γ-d-glutamyl-meso-diaminopimelate ligase (Mur ligase family) | 3.18 × 10−10 | 1037925 |
| 2 | Lineage A, isolate 25; lineage B, isolate 35; isolate 31 | SNP in nonprotein coding region upstream of ORF PLES_37671 (acnA) on the forward strand and PLES_37661 (ygdE) on reverse strand | None. Between operons including ORFs coding for a putative RNA 2′-O-ribose methyltransferase and aconitate hydratase | 1.84 × 10−8§ | 4165056 |
| 3 | Lineage A, isolate 4; lineage B, isolate 8 | Nonsynonymous SNP in PLES_44121 (lysC) | Aspartate kinase | 1.84 × 10−8 | 4847728 |
| 4 | Lineage B, isolates 13 and 24; isolates 14 and 20 | Deletion of two codons in PLES_56291 (glpT) | sn-glycerol-3-phosphate transporter | 2.87 × 10−10 | 6229046 |
Definition of abbreviations: ID = identification; ORF = open reading frame; SNP = single-nucleotide polymorphism.
Homoplasy IDs correspond to phylogenetic distribution in Figure 1 and are referred to in the text.
Probability that a pair of homoplasies are caused by independent mutations occurring in different lineages at the same position in the genome (parallel evolution), as opposed to a single ancestral mutation transferred between lineages with chromosomal integration by homologous recombination. Calculation considers only fourfold degenerate sites in protein coding regions of LESB58 reference chromosome (13) as a conservative estimate of total sites not under strong selection that would tolerate mutations. Insertions, deletions, and SNPs are included even though the nearly neutral sites considered only concern SNPs. Consequently, the calculation is more conservative with respect to favoring a single mutation (see Methods).
Positions are according to the LESB58 reference chromosome.
The most recent common ancestor of isolates 31 and 35 was considered to have the homoplasy to simplify calculation. Consequently, the estimate is more conservative.
Coexisting, Divergent Lineages in Chronic CF Infections Are Typical for the Liverpool Epidemic Strain
To assess the prevalence of divergent P. aeruginosa LES lineages within chronic infections, we investigated a further eight patients with CF. We used SNP frequencies, derived from sequencing an equimolar pool of genomic DNA from 40 isolates per patient, to estimate the genetic distance between the inferred MRCA of the epidemic and the MRCA of the patient sample (SNPs fixed in each sample minus SNPs fixed in all samples, root edge in a phylogeny) and then the genetic distances between the lineages descending from the patient sample MRCA (primary edges to each lineage). Thus, if both primary lineage edges are longer than the root edge, divergent lineages are present. To validate the root and lineage edges in the phylogenies deduced from SNP frequencies within each pooled data sample, we first sequenced a pair of isolates from each patient to confirm that SNPs inferred as lineage-specific were always in linkage (orange and turquoise in Figures 4 and 5) and then simulated sequence evolution along each phylogeny to ensure simulated SNP frequencies (red, blue, and purple peaks in Figures 4 and 5) agreed with observed SNP frequencies. Finally, we used the sequences of all 40 isolates from CF03 to validate these approaches. Both observed and simulated SNP frequency distributions for CF03 correspond to the phylogeny shape: two deeply divergent lineages descending from the sample MRCA (Figure 3).
Figure 4.

Pseudomonas aeruginosa Liverpool Epidemic Strain populations in six of eight other sputum samples from a patient with cystic fibrosis (CF) consist of two divergent lineages. A further eight patients provided a sputum sample from each of which genomic DNA of 40 isolates was sequenced in equimolar pools. Single-nucleotide polymorphism (SNP) analysis revealed that six samples consisted of two divergent lineages shown in A–F (CF01, CF04, CF05, CF07, CF08, and CF09). (Left) Histograms of SNP frequency distributions observed among pooled genome sequences of 40 isolates. (Center) Hypotheses of root edge and lineage edges plotted as phylogenies deduced from the observed SNP frequency distributions. Only the root and lineage edges are relevant to the hypothesis of two divergent lineages. (Right) Histogram of SNP frequencies among sequences simulated along each phylogeny, recapitulating observed SNP peaks corresponding to the root and deepest edges (indicated by purple, red, and blue). For each sample two isolates were sequenced separately, the observed SNP frequencies for which are indicated among the 40 in each pool with green, turquoise, and orange (left). Edges within clades of lineages are not relevant to the hypotheses. The SNP mutations in the root edges are shared and derived in all descendants (i.e., fixed in the population), and thus appear at the maximum frequency of 40: both the root edge and corresponding peak in the simulated SNP frequency histograms (right) are colored purple. The red and blue peaks in the simulation histograms correspond to the lineage edges arising from the patient’s most recent common ancestor (MRCA) at the deepest bifurcation and should be at frequencies that sum to the total isolates (40). In all of these samples the deepest pair of lineages were considered divergent because they had more mutations since their MRCA (red and blue peaks) than their MRCA had to that of all samples in the study (the outgroup; peak at frequency 40, purple on the right). Except for CF04 (B), the pairs of isolates were representatives of each divergent lineage so that the highest-frequency peak in the distribution of observed SNPs exclusive to each isolate (orange or turquoise) corresponds to one of the lineage peaks in the distribution of simulated SNPs (red or blue).
Figure 5.
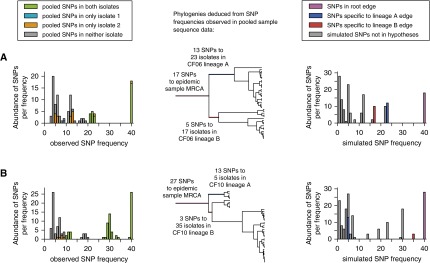
Pseudomonas aeruginosa Liverpool Epidemic Strain populations in two of eight other sputum samples from patients with cystic fibrosis (CF) consist of a single lineage. The sampling approach, analysis, and plotting is as described for Figure 4. For these two samples (CF06 and CF10) the deepest pair of lineages were not considered divergent because both had fewer mutations since their most recent common ancestor (MRCA; red and blue edges and peaks) than their MRCA had to that of all samples in the study (purple peak edges and peaks): the purple peak was larger than the red and blue peaks. SNP = single-nucleotide polymorphism.
Figure 3.

Simulation of sequence evolution is useful for corroboration of phylogenetic hypotheses deduced from single-nucleotide polymorphism (SNP) frequency distributions. (Left) Histogram of observed SNP frequencies among individual genome sequences of 40 isolates in two divergent lineages. (Center) Neighbor-joining (BIONJ) phylogenetic reconstruction of the 40 isolates adapted from Figure 1. (Right) Histogram of SNP frequencies among sequences simulated along the phylogeny in the center. The phylogeny is rooted using the inferred most recent common ancestor (MRCA) of all isolates in this study as an outgroup. Edge highlight colors correspond to histogram peak colors (solid) according to the SNPs represented: frequency of peak (position on x-axis) corresponds to edge length, whereas area of peak (abundance) corresponds to the number of tips descendant from the edge. The simulation accurately reproduces the SNP frequency distribution (right) observed in the sequence data (left) and is thus useful to corroborate phylogenetic hypotheses deduced from SNP frequencies among isolates (see Figure 4). The red and blue hatched peaks in the histograms represent diversity within, not between, each of the two deepest lineages (A and B) and are not relevant to a hypothesis of two divergent, coexisting lineages.
In addition to CF03, the presence of two divergent lineages was detected in six of the eight other patients (Figure 4). Thus, chronic infections within these patients harbor a pair of P. aeruginosa lineages that differ from their MRCA by more SNPs than their MRCA differs from the inferred epidemic origin (epidemic MRCA inferred from the whole dataset). All members of one of the two lineages in sample CF09 shared an abundance of mutations including a 3-bp out-of-frame deletion in mutS, and nonsynonymous SNPs in mutM and uvrB. Mutations at these loci have been shown to cause the “hypermutator” phenotype (36, 37) that in a previous report was identified in 36% of CF airway, but in no non-CF, P. aeruginosa infections (38). Less structure was observed for the remaining two patients (Figure 5), such that the most divergent lineages share more SNPs with each other than differentiates them from the epidemic MRCA. Thus, the edges arising from the first bifurcation at the patient sample MRCA were shorter than the root edge to the epidemic MRCA.
Comparisons of P. aeruginosa Lineages among Chronically Infected Patients with CF Suggests Transmissions of Diverse Populations or Multiple Lineages
We next investigated whether our principal finding of divergent coexisting lineages can be explained exclusively by diversification within a patient following an initial infection. Alternative explanations include a single transmission of a diverse LES population from which lineages diverge, or multiple transmissions of different lineages to a patient causing superinfection. To elucidate patterns of diversification, we reconstructed the ancestry and evolutionary relationships among the 16 distinct P. aeruginosa LES lineages identified among the nine patients with CF. All of the individually sequenced isolates were included in the reconstruction using SNPs relative to the LESB58 genome: 40 isolates from patient CF03 and two each from the other eight patients (Figure 6).
Figure 6.

Complex patterns of Pseudomonas aeruginosa Liverpool Epidemic Strain transmission among chronically infected patients with cystic fibrosis (CF). Neighbor-joining (BIONJ) phylogenetic reconstruction of 40 P. aeruginosa isolate genome sequences from a patient CF03 sputum sample and two P. aeruginosa isolate genome sequences from eight other patient sputum samples, all collected in 2009. The distance matrix consisted of raw counts of shared single-nucleotide polymorphisms (SNP). Mutations are relative to the P. aeruginosa LESB58 genome, collected in 1988, which also serves as an outgroup for rooting. Support for each edge is as described in Figure 1. The 13 sequences representing CF03 lineage A and the 27 sequences representing CF03 lineage B each form a monophyletic clade and are represented as blue and red triangles, respectively. The edges to CF05 isolate 2 and CF09 isolate 2 are not to scale because they represent many more mutations than other edges. Patient sample isolates CF03, CF05, and CF07–9 are paraphyletic, consistent with some patient infections being from diverse inocula and/or acquisitions of multiple lineages.
Within-host diversification was clearly supported for patients CF06 and CF10. Thus, their isolates each grouped together to the exclusion of others (i.e., formed monophyletic clades with high bootstrap support). Furthermore, the relatively small diversity in their pooled samples was well represented in the sequenced isolate pairs (Figure 5), indicating all isolates would group in their respective CF06 or CF10 clades (Figure 6). The CF04 patient isolates are the only others that group together, but according to the population data represent only one of two lineages in CF04 (Figure 4A). The phylogenetic placement of CF04 isolates implies a transmission of one lineage from CF01. The other CF04 lineage, containing 47 SNPs absent from CF01, supports either superinfection from another patient or transfer of a genetically diverse innoculum from CF01 and subsequent loss of a lineage from CF01. Elsewhere in the phylogeny, isolates did not seem to group within patients. To confirm this, given that the phylogeny was poorly resolved in the region where lineages diverged, we performed an explicit test of whether specific groupings were supported by the data using the Shimodaira AU test (see online supplement for details and discussion). Exclusive groupings of isolates within a patient were rejected by the AU test for patients CF03 (P = 0.0019), CF05 (P = 0.0006), and CF07 (P = 0.0087) but not CF08 (P = 0.1423) or CF09 (P = 0.3679). CF03, CF05, and CF07 are therefore consistent with either superinfection or with transmission of genetically diverse innocula.
Discussion
We observed high population genetic structure within P. aeruginosa infections such that in seven out of nine patients, divergent LES lineages were identified. These coexisting lineages were typically more closely related to lineages in other patients than to each other and include the broadest nonhypermutator P. aeruginosa genetic diversity within a single patient yet reported. In one such case that we examined in more detail, genetic transfer between the divergent lineages by homologous recombination was evident. Genetic exchange between coexisting but divergent lineages can increase genetic variation and provides a mechanism by which adaptation to the lung environment may be accelerated.
Our study is the first to observe that multiple P. aeruginosa lineages coexisting within individual patients usually arise from genetic diversity acquired from other patients and that multiple coexisting lineages may be a general feature of CF chronic infection by the LES. This might reflect a unique trait of this strain or transmissible P. aeruginosa strains more generally, rather than a widespread characteristic of CF infections. Alternatively, such divergent lineages within P. aeruginosa infections may be relatively common, but would only be detectable using a systematic approach as used here to characterize genetic diversity within infections.
To date, no other studies have been designed to quantify the contemporary genetic diversity within each patient of a cohort. A recent report described coexisting lineages in an individual with CF with evidence of spatial separation between the nasopharynx and the lower lung correlating with genetic distance (24). Another recent report described coexisting lineages in two patients dominated by hypermutators (39). Other studies have generated data consistent with, but not conclusive of, coexisting, divergent lineages. Evidence for transmission of the abundant P. aeruginosa clone C lineage among siblings, with the possibility of subsequent coexistence of two clone C lineages, has been reported (40), whereas Chung and coworkers (23) found 54 SNPs and 38 indels differentiating a pair of nonhypermutator isolates from a single patient. This latter evolutionary distance is comparable with the divergences between coexisting lineages in our study. Our results are also consistent with a recent study of Burkholderia dolosa, a relatively rare CF airway pathogen, which included numerous isolates from single sputum samples and identified coexisting divergent lineages (25). However, unlike the current report, comparative analyses between patient samples have not been performed and diversification has been suggested to have proceeded exclusively within each patient since initial infection, as opposed to acquisition of diverse inocula or divergent lineages.
Despite the extensive phenotypic diversity present among isolates (19), our genomic analysis indicates that the presence of discrete lineages, rather than a continuum of diversity within a sputum sample, is typical for LES infecting populations. Given the complexity and spatial heterogeneity of the CF airway, maintenance of the interlineage diversity may simply reflect the availability of sufficient niches to accommodate newly acquired invading P. aeruginosa populations. The apparent restriction to two distinct coexisting lineages in any one patient may be a reflection of physiologic compartmentalization within the lungs, for example between the two lungs, or the lungs and paranasal sinuses (24). An alternative explanation is a skewed community composition among lineages: one could be numerically dominant, whereas many others are rare.
In the present study, we demonstrate that homologous recombination can occur among pathogenic P. aeruginosa during chronic infection. Evidence for recombination has been previously reported in sequence data from large P. aeruginosa isolate collections from environmental, animal, and human sources (41, 42). When chronic infections contain significant genetic diversity as reported here, the potential for homologous recombination to generate novel genotypes is increased because of greater differences in genetic backgrounds within the patient. Epistatic effects, such as a mutation being neutral to the CF airway in one P. aeruginosa genetic background, but adaptive in another (43, 44), may be intensified by the greater genetic differences between coexisting lineages.
The cohort segregation policy adopted in Liverpool (United Kingdom) was designed to prevent transmission of the LES to patients free from P. aeruginosa, or infected with other strains (45), and as such the patients sampled in this study were not segregated from each other. Although cohort segregation has been proved successful in halting the spread of the strain to new patients, our data indicate that it may not have prevented further transmission events among the LES-infected cohort. Although patients with CF infected with LES are known to have a higher rate of mortality than those with other P. aeruginosa strains (46), at present it is not known whether having multiple distinct lineages of LES is worse than having a single lineage. Nor can we be sure that these sublineages remain stable within patients. Our previous study, based on isolate phenotyping, suggested that populations were dynamic over a period of several months (19). Further studies are needed to address the issue of lineage stability over time and to determine the clinical consequences of the coexistence of different lineages.
The accurate and rapid sequencing of bacterial genome sequences, made possible by the most recently available bench-top DNA sequencing platforms, provides numerous advantages over conventional methods for diagnosing hospital outbreaks and tracing transmission routes (47). However, the potential for infections to be composed of multiple, divergent lineages has generally not been considered in diagnostics, where it is still typical for a single clone to be taken as representative of an infection. Our results demonstrate that this will, at least for chronic infections, vastly underestimate the diversity within infections. In particular, it is known that conventional antimicrobial susceptibility tests are not good predictors for response to therapy (48), which may be explicable in part by the high genetic diversity harbored within an infection, including at loci encoding antimicrobial resistance, and which would be missed by sampling only one or a few clones. Our use of whole-genome sequencing and analysis of the infection-specific population-level data from individual patients to diagnose infection with multiple distinct lineages therefore holds promise for diagnostic clinical microbiology and lineage-targeted therapies (48).
Acknowledgments
Acknowledgment
The authors acknowledge the technical assistance provided by staff at the Centre for Genomic Research, University of Liverpool, United Kingdom, and are grateful to staff and patients at the Regional Adult Cystic Fibrosis Unit, Liverpool, United Kingdom.
Footnotes
Supported by The Wellcome Trust (award 093306/Z/10).
Author Contributions: M.A.B., C.W., and S.P. conceived the study, obtained funding, and contributed resources. D.W., M.A.B., C.W., and S.P. designed the study, interpreted the data, and wrote the manuscript. D.W. performed evolutionary analyses. M.J.W. acquired clinical samples. B.E. prepared DNA samples for sequencing. S.H. prepared sequence data and performed variant discovery. All authors read and approved the manuscript.
This article has an online supplement, which is accessible from this issue's table of contents at www.atsjournals.org
Originally Published in Press as DOI: 10.1164/rccm.201409-1646OC on January 15, 2015
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1.Driscoll JA, Brody SL, Kollef MH. The epidemiology, pathogenesis and treatment of Pseudomonas aeruginosa infections. Drugs. 2007;67:351–368. doi: 10.2165/00003495-200767030-00003. [DOI] [PubMed] [Google Scholar]
- 2.Murray TS, Egan M, Kazmierczak BI. Pseudomonas aeruginosa chronic colonization in cystic fibrosis patients. Curr Opin Pediatr. 2007;19:83–88. doi: 10.1097/MOP.0b013e3280123a5d. [DOI] [PubMed] [Google Scholar]
- 3.Govan JR, Deretic V. Microbial pathogenesis in cystic fibrosis: mucoid Pseudomonas aeruginosa and Burkholderia cepacia. Microbiol Rev. 1996;60:539–574. doi: 10.1128/mr.60.3.539-574.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hoffman LR, Richardson AR, Houston LS, Kulasekara HD, Martens-Habbena W, Klausen M, Burns JL, Stahl DA, Hassett DJ, Fang FC, et al. Nutrient availability as a mechanism for selection of antibiotic tolerant Pseudomonas aeruginosa within the CF airway. PLoS Pathog. 2010;6:e1000712. doi: 10.1371/journal.ppat.1000712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Goodman AL, Kulasekara B, Rietsch A, Boyd D, Smith RS, Lory S. A signaling network reciprocally regulates genes associated with acute infection and chronic persistence in Pseudomonas aeruginosa. Dev Cell. 2004;7:745–754. doi: 10.1016/j.devcel.2004.08.020. [DOI] [PubMed] [Google Scholar]
- 6.Thomas SR, Ray A, Hodson ME, Pitt TL. Increased sputum amino acid concentrations and auxotrophy of Pseudomonas aeruginosa in severe cystic fibrosis lung disease. Thorax. 2000;55:795–797. doi: 10.1136/thorax.55.9.795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ciofu O, Mandsberg LF, Bjarnsholt T, Wassermann T, Høiby N. Genetic adaptation of Pseudomonas aeruginosa during chronic lung infection of patients with cystic fibrosis: strong and weak mutators with heterogeneous genetic backgrounds emerge in mucA and/or lasR mutants. Microbiology. 2010;156:1108–1119. doi: 10.1099/mic.0.033993-0. [DOI] [PubMed] [Google Scholar]
- 8.Ashish A, Shaw M, Winstanley C, Ledson MJ, Walshaw MJ. Increasing resistance of the Liverpool Epidemic Strain (LES) of Pseudomonas aeruginosa (Psa) to antibiotics in cystic fibrosis (CF)—a cause for concern? J Cyst Fibros. 2012;11:173–179. doi: 10.1016/j.jcf.2011.11.004. [DOI] [PubMed] [Google Scholar]
- 9.Wilder CN, Allada G, Schuster M. Instantaneous within-patient diversity of Pseudomonas aeruginosa quorum-sensing populations from cystic fibrosis lung infections. Infect Immun. 2009;77:5631–5639. doi: 10.1128/IAI.00755-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rau MH, Hansen SK, Johansen HK, Thomsen LE, Workman CT, Nielsen KF, Jelsbak L, Høiby N, Yang L, Molin S. Early adaptive developments of Pseudomonas aeruginosa after the transition from life in the environment to persistent colonization in the airways of human cystic fibrosis hosts. Environ Microbiol. 2010;12:1643–1658. doi: 10.1111/j.1462-2920.2010.02211.x. [DOI] [PubMed] [Google Scholar]
- 11.Römling U, Fiedler B, Bosshammer J, Grothues D, Greipel J, von der Hardt H, Tümmler B. Epidemiology of chronic Pseudomonas aeruginosa infections in cystic fibrosis. J Infect Dis. 1994;170:1616–1621. doi: 10.1093/infdis/170.6.1616. [DOI] [PubMed] [Google Scholar]
- 12.Fothergill JL, Walshaw MJ, Winstanley C. Transmissible strains of Pseudomonas aeruginosa in cystic fibrosis lung infections. Eur Respir J. 2012;40:227–238. doi: 10.1183/09031936.00204411. [DOI] [PubMed] [Google Scholar]
- 13.Winstanley C, Langille MG, Fothergill JL, Kukavica-Ibrulj I, Paradis-Bleau C, Sanschagrin F, Thomson NR, Winsor GL, Quail MA, Lennard N, et al. Newly introduced genomic prophage islands are critical determinants of in vivo competitiveness in the Liverpool Epidemic Strain of Pseudomonas aeruginosa. Genome Res. 2009;19:12–23. doi: 10.1101/gr.086082.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Martin K, Baddal B, Mustafa N, Perry C, Underwood A, Constantidou C, Loman N, Kenna DT, Turton JF. Clusters of genetically similar isolates of Pseudomonas aeruginosa from multiple hospitals in the UK. J Med Microbiol. 2013;62:988–1000. doi: 10.1099/jmm.0.054841-0. [DOI] [PubMed] [Google Scholar]
- 15.Aaron SD, Vandemheen KL, Ramotar K, Giesbrecht-Lewis T, Tullis E, Freitag A, Paterson N, Jackson M, Lougheed MD, Dowson C, et al. Infection with transmissible strains of Pseudomonas aeruginosa and clinical outcomes in adults with cystic fibrosis. JAMA. 2010;304:2145–2153. doi: 10.1001/jama.2010.1665. [DOI] [PubMed] [Google Scholar]
- 16.Jeukens J, Boyle B, Kukavica-Ibrulj I, Ouellet MM, Aaron SD, Charette SJ, Fothergill JL, Tucker NP, Winstanley C, Levesque RC. Comparative genomics of isolates of a Pseudomonas aeruginosa epidemic strain associated with chronic lung infections of cystic fibrosis patients. PLoS One. 2014;9:e87611. doi: 10.1371/journal.pone.0087611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Smith EE, Buckley DG, Wu Z, Saenphimmachak C, Hoffman LR, D’Argenio DA, Miller SI, Ramsey BW, Speert DP, Moskowitz SM, et al. Genetic adaptation by Pseudomonas aeruginosa to the airways of cystic fibrosis patients. Proc Natl Acad Sci USA. 2006;103:8487–8492. doi: 10.1073/pnas.0602138103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Marvig RL, Johansen HK, Molin S, Jelsbak L. Genome analysis of a transmissible lineage of Pseudomonas aeruginosa reveals pathoadaptive mutations and distinct evolutionary paths of hypermutators. PLoS Genet. 2013;9:e1003741. doi: 10.1371/journal.pgen.1003741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mowat E, Paterson S, Fothergill JL, Wright EA, Ledson MJ, Walshaw MJ, Brockhurst MA, Winstanley C. Pseudomonas aeruginosa population diversity and turnover in cystic fibrosis chronic infections. Am J Respir Crit Care Med. 2011;183:1674–1679. doi: 10.1164/rccm.201009-1430OC. [DOI] [PubMed] [Google Scholar]
- 20.Ashish A, Paterson S, Mowat E, Fothergill JL, Walshaw MJ, Winstanley C. Extensive diversification is a common feature of Pseudomonas aeruginosa populations during respiratory infections in cystic fibrosis. J Cyst Fibros. 2013;12:790–793. doi: 10.1016/j.jcf.2013.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Workentine ML, Sibley CD, Glezerson B, Purighalla S, Norgaard-Gron JC, Parkins MD, Rabin HR, Surette MG. Phenotypic heterogeneity of Pseudomonas aeruginosa populations in a cystic fibrosis patient. PLoS One. 2013;8:e60225. doi: 10.1371/journal.pone.0060225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mayer-Hamblett N, Ramsey BW, Kulasekara HD, Wolter DJ, Houston LS, Pope CE, Kulasekara BR, Armbruster CR, Burns JL, Retsch-Bogart G, et al. Pseudomonas aeruginosa phenotypes associated with eradication failure in children with cystic fibrosis. Clin Infect Dis. 2014;59:624–631. doi: 10.1093/cid/ciu385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chung JC, Becq J, Fraser L, Schulz-Trieglaff O, Bond NJ, Foweraker J, Bruce KD, Smith GP, Welch M. Genomic variation among contemporary Pseudomonas aeruginosa isolates from chronically infected cystic fibrosis patients. J Bacteriol. 2012;194:4857–4866. doi: 10.1128/JB.01050-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Markussen T, Marvig RL, Gómez-Lozano M, Aanæs K, Burleigh AE, Høiby N, Johansen HK, Molin S, Jelsbak L. Environmental heterogeneity drives within-host diversification and evolution of Pseudomonas aeruginosa. MBio. 2014;5:e01592–e14. doi: 10.1128/mBio.01592-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lieberman TD, Flett KB, Yelin I, Martin TR, McAdam AJ, Priebe GP, Kishony R. Genetic variation of a bacterial pathogen within individuals with cystic fibrosis provides a record of selective pressures. Nat Genet. 2014;46:82–87. doi: 10.1038/ng.2848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 2012;19:455–477. doi: 10.1089/cmb.2012.0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gascuel O. BIONJ: an improved version of the NJ algorithm based on a simple model of sequence data. Mol Biol Evol. 1997;14:685–695. doi: 10.1093/oxfordjournals.molbev.a025808. [DOI] [PubMed] [Google Scholar]
- 29.Paradis E, Claude J, Strimmer K. APE: analyses of phylogenetics and evolution in R language. Bioinformatics. 2004;20:289–290. doi: 10.1093/bioinformatics/btg412. [DOI] [PubMed] [Google Scholar]
- 30.Zwickl DJ. Austin, TX: The University of Texas at Austin, School of Biological Sciences; 2006. Genetic algorithm approaches for the phylogenetic analysis of large biological sequence datasets under the maximum likelihood criterion [Ph.D. thesis] [Google Scholar]
- 31.Ronquist F, Huelsenbeck JP. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics. 2003;19:1572–1574. doi: 10.1093/bioinformatics/btg180. [DOI] [PubMed] [Google Scholar]
- 32.Sukumaran J, Holder MT. DendroPy: a Python library for phylogenetic computing. Bioinformatics. 2010;26:1569–1571. doi: 10.1093/bioinformatics/btq228. [DOI] [PubMed] [Google Scholar]
- 33.Darriba D, Taboada GL, Doallo R, Posada D. jModelTest 2: more models, new heuristics and parallel computing. Nat Methods. 2012;9:772. doi: 10.1038/nmeth.2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shimodaira H. Testing regions with nonsmooth boundaries via multiscale bootstrap. J Stat Plan Inference. 2008;138:1227–1241. [Google Scholar]
- 35.Sipos B, Massingham T, Jordan GE, Goldman N. PhyloSim - Monte Carlo simulation of sequence evolution in the R statistical computing environment. BMC Bioinformatics. 2011;12:104. doi: 10.1186/1471-2105-12-104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Oliver A, Sánchez JM, Blázquez J. Characterization of the GO system of Pseudomonas aeruginosa. FEMS Microbiol Lett. 2002;217:31–35. doi: 10.1111/j.1574-6968.2002.tb11452.x. [DOI] [PubMed] [Google Scholar]
- 37.Oliver A, Baquero F, Blázquez J. The mismatch repair system (mutS, mutL and uvrD genes) in Pseudomonas aeruginosa: molecular characterization of naturally occurring mutants. Mol Microbiol. 2002;43:1641–1650. doi: 10.1046/j.1365-2958.2002.02855.x. [DOI] [PubMed] [Google Scholar]
- 38.Oliver A, Cantón R, Campo P, Baquero F, Blázquez J. High frequency of hypermutable Pseudomonas aeruginosa in cystic fibrosis lung infection. Science. 2000;288:1251–1254. doi: 10.1126/science.288.5469.1251. [DOI] [PubMed] [Google Scholar]
- 39.Feliziani S, Marvig RL, Luján AM, Moyano AJ, Di Rienzo JA, Krogh Johansen H, Molin S, Smania AM. Coexistence and within-host evolution of diversified lineages of hypermutable Pseudomonas aeruginosa in long-term cystic fibrosis infections. PLoS Genet. 2014;10:e1004651. doi: 10.1371/journal.pgen.1004651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cramer N, Wiehlmann L, Tümmler B. Clonal epidemiology of Pseudomonas aeruginosa in cystic fibrosis. Int J Med Microbiol. 2010;300:526–533. doi: 10.1016/j.ijmm.2010.08.004. [DOI] [PubMed] [Google Scholar]
- 41.Wiehlmann L, Wagner G, Cramer N, Siebert B, Gudowius P, Morales G, Köhler T, van Delden C, Weinel C, Slickers P, et al. Population structure of Pseudomonas aeruginosa. Proc Natl Acad Sci USA. 2007;104:8101–8106. doi: 10.1073/pnas.0609213104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kidd TJ, Ritchie SR, Ramsay KA, Grimwood K, Bell SC, Rainey PB. Pseudomonas aeruginosa exhibits frequent recombination, but only a limited association between genotype and ecological setting. PLoS One. 2012;7:e44199. doi: 10.1371/journal.pone.0044199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rakhimova E, Munder A, Wiehlmann L, Bredenbruch F, Tümmler B. Fitness of isogenic colony morphology variants of Pseudomonas aeruginosa in murine airway infection. PLoS One. 2008;3:e1685. doi: 10.1371/journal.pone.0001685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Damkiær S, Yang L, Molin S, Jelsbak L. Evolutionary remodeling of global regulatory networks during long-term bacterial adaptation to human hosts. Proc Natl Acad Sci USA. 2013;110:7766–7771. doi: 10.1073/pnas.1221466110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ashish A, Shaw M, Winstanley C, Humphreys L, Walshaw MJ. Halting the spread of epidemic Pseudomonas aeruginosa in an adult cystic fibrosis centre: a prospective cohort study. JRSM Short Rep. 2013;4:1. doi: 10.1258/shorts.2012.012018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Al-Aloul M, Crawley J, Winstanley C, Hart CA, Ledson MJ, Walshaw MJ. Increased morbidity associated with chronic infection by an epidemic Pseudomonas aeruginosa strain in CF patients. Thorax. 2004;59:334–336. doi: 10.1136/thx.2003.014258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Reuter S, Ellington MJ, Cartwright EJ, Köser CU, Török ME, Gouliouris T, Harris SR, Brown NM, Holden MT, Quail M, et al. Rapid bacterial whole-genome sequencing to enhance diagnostic and public health microbiology. JAMA Intern Med. 2013;173:1397–1404. doi: 10.1001/jamainternmed.2013.7734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Smith AL, Fiel SB, Mayer-Hamblett N, Ramsey B, Burns JL. Susceptibility testing of Pseudomonas aeruginosa isolates and clinical response to parenteral antibiotic administration: lack of association in cystic fibrosis. Chest. 2003;123:1495–1502. doi: 10.1378/chest.123.5.1495. [DOI] [PubMed] [Google Scholar]