

Abstract
Statins competitively inhibit hepatic 3-hydroxy-3-methylglutaryl-coenzyme A reductase, resulting in reduced plasma total and low-density lipoprotein cholesterol levels. Recently, it has been shown that statins exert additional ‘pleiotropic’ effects by increasing expression levels of the membrane water channels aquaporin 2 (AQP2). AQP2 is localized mainly in the kidney and plays a critical role in determining cellular water content. This additional effect is independent of cholesterol homoeostasis, and depends on depletion of mevalonate-derived intermediates of sterol synthetic pathways, i.e. farnesylpyrophosphate and geranylgeranylpyrophosphate. By up-regulating the expression levels of AQP2, statins increase water reabsorption by the kidney, thus opening up a new avenue in treating patients with nephrogenic diabetes insipidus (NDI), a hereditary disease that yet lacks high-powered and limited side effects therapy. Aspects related to water balance determined by AQP2 in the kidney, as well as standard and novel therapeutic strategies of NDI are discussed.
Keywords: apical membrane, aquaporin, cholesterol-lowering drugs, hypercholesterolaemia, HMG-CoA, kidney, nephrogenic diabetes insipidus, vasopressin, water channels
Introduction
Water balance and AQP2 regulation by vasopressin
- Nephrogenic diabetes insipidus (NDI)
- –Hereditary NDI
- –Acquired NDI
Vasopressin-independent signals regulating AQP2 trafficking and potential use for NDI treatment
- Current treatment of NDI and use of statins
- –Statins and AQP2
Advantages and disadvantages of statins in the treatment of NDI
Conclusions
Introduction
Statins are the first-line recommended pharmacological therapy in patients with dyslipidemias and for both primary 1 and secondary 2 prevention of coronary heart disease 3–6 (Table1). Statins are widely used to reduce risks for atherosclerotic cardiovascular disease 7,8 and associated morbidity and mortality, by decreasing plasma total and low-density lipoprotein cholesterol (LDL-C) concentrations 9,10. Statins occupy part of the active binding site of 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) 11 and inhibit its enzymatic activity, a key step leading to the reduction in cellular sterol pool. Expression levels of LDL receptors are increased by a compensatory mechanism, leading to increased hepatic LDL uptake and decreased plasma cholesterol 12,13. Statins decrease biliary cholesterol output by reducing availability of biliary cholesterol 14–16 in both healthy individuals and hypercholesterolaemic patients 14–17. Statins have also beneficial effects on the vascular wall by stabilizing the atherosclerotic plaques, ameliorating impaired endothelial function, and reducing vascular inflammation 18.
Table 1.
Multiple effects of statins
| Effect(s) | Underlying mechanism(s) |
|---|---|
| Orthodox effects | |
|
|
| Pleiotropic effects | |
| Established (atherosclerotic diseases) | |
| Improved endothelial dysfunction 209 |
|
| Significant reduction of inflammatory markers (CRP) 210,211 |
|
| Decreased plaque growth 211 |
|
| Stimulation of angiogenesis 213 |
|
| Decreased plaque rupture or fissuration 214 |
|
| Prevention of thrombosis 215 |
|
| Potential (non-atherosclerotic diseases) | |
| Prevention of dementia 216,217 |
|
| Preserved renal function 174,218 |
|
| Improved bone metabolism 219–221 |
|
| Improved outcome in chronic obstructive pulmonary disease (COPD) 222,223 |
|
| Improved erectile dysfunction 224,225 |
|
| Prevention of gallstone diseases 226,227 |
|
| Increased expression of AQP2 in the apical membrane of the kidney collecting duct principal cells [146 ] (see text and Fig.3 for details) |
|
A recently identified ‘pleiotropic’ effect of statins is the increased expression levels of the renal membrane water channels Aquaporin 2 (AQP2). This effect is independent of classical cholesterol homoeostasis 19,20, but rather depends on depletion of mevalonate-derived intermediates of sterol synthetic pathways, i.e. isoprenoid intermediates, including farnesylpyrophosphate (FPP) and geranylgeranylpyrophosphate (GGPP).
This review will summarize aspects related to water balance, renal AQP2, vasopressin and nephrogenic diabetes insipidus (NDI), as well as current treatment of NDI and possible use of statins with respect to AQP2 trafficking.
Water balance and AQP2 regulation by vasopressin
Water balance results from the equilibrium between daily water intake and urine excretion, in accord with daily changes of body and environmental factors 21. The kidney has a central role in preserving water balance: hypovolaemia and increased plasma osmolality stimulate aortic/carotid baroreceptors and hypothalamic osmoreceptors, respectively, to promote antidiuresis. The hypothalamus subsequently stimulates secretion of antidiuretic peptide hormone arginine vasopressin (AVP) from the pituitary gland. The terminal renal tubules at the level of connecting tubules and collecting ducts are characterized by variable permeability to water which is regulated by AVP and its interaction with the type 2 vasopressin receptor (AVPR2). The ultimate step in water reabsorption in the kidney is regulated by the interactions among AVP, AVPR2 and specific water channels, namely aquaporins (AQPs), playing critical roles in determining the cellular water content, and water balance in the body (See also http://www.nobelprize.org/nobel_prizes/chemistry/laureates/2003/) (access 09, 2014) 22–26.
Aquaporins are widely distributed in all kingdoms of life from bacteria to plants and to mammals 27. There are 13 known mammalian AQPs, nine expressed in the kidney (AQP1, 2, 3, 4, 5, 6, 7, 8 and 11) 28–32. AQP1, 2, 3, 4 are involved in water transport across the epithelia of the renal tubule 33,34 (Fig.1). The role of AQP5 in type-B intercalated cells is still being investigated 35. AQP1 is found in proximal tubules and descending thin limbs of kidney. AQP2 is localized predominately in the intracellular vesicles and the apical plasma membrane of connecting tubule cells and collecting duct cells, while AQP3 and AQP4 are expressed in the basolateral plasma membrane of these cells 36–40. Water permeability and osmotic transport in the renal collecting duct depends upon the amount of active AQP2 (the principal AVP-sensitive water channel) in the apical plasma membrane of collecting duct principal cells. AQP2, normally stored in the cytosol 41,42 during diuresis, is re-directed and fused to the apical membrane of collecting duct principal cells following AVP stimulation 43,44. As homotetramer, AQP2 initiates water reabsorption within a favourable osmotic gradient between the lumen of the tubule and the interstitium. Electron microscopy studies confirmed the presence of intramembranous particle aggregates associated with enhanced water permeability 44,45. Intracellular movement of water is followed by rapid flux of water towards the basolateral membrane of collecting duct principal cells. After AVP stimulation has subsided, AQP2 water channels are removed from the apical membrane and returned to the cytoplasm by endocytosis 44,45.
Fig 1.

Anatomic structure of the nephron and collecting duct system, and localization of different aquaporins (AQPs) in the kidneys with vasopressin (AVP) effect. Sites of reabsorption of water and sodium chloride (NaCl) are shown. AQP6 is localized in the intracellular vesicle membranes of type-A intercalated cells of the collecting duct.
Binding of AVP (the polypeptide originating from the hypothalamus and migrating to the posterior pituitary through the supraopticohypophyseal tract 46) to AVPR2 results in COOH-terminal phosphorylation of the AVPR2. β-arrestin recruitment is followed by AVPR2 internalization, which implies the negative regulation of AVPR2 47. Upon AVPR2 activation, however, the signalling sequence involves Gsα dissociation, adenylyl cyclase activation, increased intracellular cAMP, activation of protein kinase type A (PKA), and phosphorylation of AQP2 at serine 256 plus other residues in the COOH terminus 48–50 (Fig.2). Thus, AQP2-bearing vesicles translocation to the plasma membrane is a combined effect of exocytosis and endocytosis 41,51–55 (Fig.3A and B). The process of intracellular vesicular trafficking is complex and requires several proteins. G proteins and subunits G1 and G0 assist exocytosis and endocytosis and heterotrimeric G proteins from the Gi family are involved in cAMP-dependent trafficking of AQP2 26. Monomeric GTP-binding proteins belonging to the Rab family also play a key role in the context of intracellular vesicle trafficking of AQP2 56. The Ras superfamily of small GTP-binding proteins is also involved in vesicle trafficking and regulates actin cytoskeleton organization and actin polymerization 57. Activation of proteins of the Rho-family occurs: Rac1 (formation of lamellipodia), Rho (formation of actin-based structures of filopodia, regulation of stress fibres and formation of focal adhesion complexes 58) and Cdc42 (activator of Rac1 and Rho). GTP-binding proteins from the Rho-family fluctuate from active GTP-bound status (when Rho is bound to its putative effectors, the Rho kinases 59) to inactive GDP-bound form; this interconversion is regulated by factors including GEP (GDP/GTP exchange protein), GAP (GTPase activating protein, which binds to the GTP-form and stimulates the intrinsic GTPase activity of monomeric G proteins) and GDI (GDP dissociation inhibitor which inhibits GDP dissociation, prevents GTP hydrolysis and maintains the Rho-family members in a soluble form) 60,61. In particular, translocating the membrane-associated active Rho form to a soluble compartment implies inactivation via Rho-GDI interaction. Decreasing Rho activity implies depolymerization of F-actin, which is considered a physical barrier preventing AQP2-containing vesicles exocytosis, and greater insertion of AQP2 into the apical plasma membrane 62. This step is clearly shown for RhoA, following phosphorylation by PKA at Serine 188 63, a regulatory mechanism also operating in the case of AQP2 trafficking (see below and Table2) 62. A short-term regulation (5–15 min.), mainly dependent on AVP 51, is the one which affects the trafficking of AQP2-containing membrane vesicles to and from the apical membrane. The long-term regulation (>24 hrs) of renal water permeability implies the overall effect on AQP2 gene and AQP2 protein abundance in the cell, also under the AVP control 43,54,64. In the latter case, dysregulation of such mechanisms is responsible for clinical conditions characterized by disturbed water balance (Table3). Furthermore, AQP2 recycles constitutively between cell surface and intracellular vesicles, independently of AVP stimulation 65–67.
Fig 2.
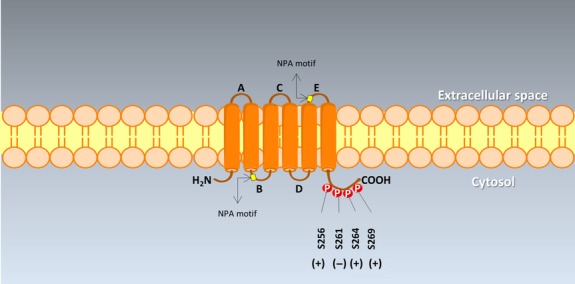
The topology of AQP2 with the COOH-terminal phosphorylation sites. AQP2 is a tetramer consisting of four identical protein subunits placed in the plasma membrane. Six transmembrane α-helices are arranged in a right-handed bundle and are represented by cylinders, with the amino (NH2-) and the carboxyl (COOH-) termini located on the cytoplasmic surface of the membrane. Five interhelical loop regions (A–E) form the extracellular and cytoplasmic vestibules. Loops B and E are hydrophobic loops that contain the highly, although not completely conserved, asparagine–proline–alanine (NPA) motifs. Such motifs appear to dip and overlap into the membrane, to construct the water pore 33,90. Serine residues at potential phosphorylation sites are labelled with their amino acid numbers at the carboxyl-terminal tail. AVP mediated increased (+) phosphorylation at S256, S264 and S269, and decreased (−) phosphorylation at S261. Both S269 and S256 phosphorylation are involved in AQP2 accumulation in the plasma membrane 50,246,247.
Fig 3.

Molecular pathways involved in AQP2-mediated water transport in the kidney. (A) Signalling cascades and molecular pathways involved in AQP2-mediated water transport in relation to vasopressin (AVP) and vasopressin receptor (AVPR2) in the principal cells of the collecting ducts 22,33,37,115. The increased influx of water by AQP2 tetramer at the apical site requires a complex cascade of intracellular processes in concert with efflux of water by AQP3 and AQP4 tetramers at the basolateral membrane. The AVPR2 is composed of 7 membrane-spanning helices. Upon binding of AVP within the transmembrane helices II–IV, allosteric structural changes occur 78,79, the G-alpha-s heterodimeric protein is stimulated, and activates the adenylyl cyclase. This step results in increased intracellular levels of cyclic adenosine monophosphate (cAMP), activation of protein kinase A (PKA), phosphorylation of AQP2 in intracellular vesicles at serine 256 and other residues in the AQP2 OOH terminal 49,50 (see also Fig.2), trafficking of endocytic vesicles to the apical plasma membrane, and fusion of AQP2-containing vesicles with the apical membrane. As stated in the text, PKA is also responsible for phosphorylation of the membrane-associated RhoA, association with GDI to form the inactive complex RhoA-GDI, a step facilitating AQP2 insertion into the plasma membrane during VP/PKA/cAMP-induced AQP2 translocation 62. The docking system for vesicles might include specific receptors in the collecting duct cells which are associated with certain membrane domains housing AQP2 (e.g. syntaxin-4). Abbreviation: PDEs, phosphodiesterases. See also 33,37,247,248. (B) Proposed model of transcytotic trafficking of AQP2 from basolateral to apical membrane in principal cell of the collecting ducts. At least eight steps are involved: (1) Synthesis in the endoplasmic reticulum and transport to the trans-Golgi network; (2) rapid insertion of AQP2 into the basolateral membrane; (3) rapid internalization by clathrin-dependent endocytosis which is responsible for limited expression of basolateral AQP2. This step is blockable by low temperature (4°C); (5) AQP2 transcytosis to the perinuclear recycling compartment and the apical recycling endosomes via the microtubule-dependent mechanism. This step is inhibitable by colchicine; (7) exocytosis of AQP2 at the apical membrane; (8) recycling of AQP2 towards the apical recycling endosomes via the clathrin-dependent endocytosis. Thin dotted arrows show alternative pathways (?) of AQP2. Asterisks indicate where vasopressin (AVP) stimulus is inducing increased exocytosis and recycling of AQP2 with effect on transepithelial water flux (apical side) and cell migration, tubulogenesis, and likely transepithelial water flux (basolateral side). See also 69,70.
Table 2.
Pathways involved in AQP2 trafficking in the kidney
| Pathway | Mechanism(s) |
|---|---|
| Activation of the G-coupled V2 receptor 21,26,43,49,51–54,64,68,71,74,83 |
|
| Nitric oxide/cGMP pathway 48,128,130,131,228 |
|
| COX/prostaglandin E2 pathway 134,229,230,155 |
|
| Modulation of actin cytoskeleton network | |
|
|
|
|
Table 3.
Disorders of water balance associated with dysregulation of AQP2
| Disorder | Description |
|---|---|
| Polyuric syndromes | |
|
Associated with low levels circulating vasopressin and decreased amount of AQP2 in collecting duct cells 115 |
|
Polyuria associated with depletion of renal AQP2 protein from the collecting ducts and connecting tubules |
|
Impaired trafficking of AQP2 Lack of fusion with the apical membrane and/or Decreased channel function |
Extracellular fluid volume (ECF)-expanded states
|
Oedematous disorders 231 |
AQP, aquaporin; NDI, nephrogenic diabetes insipidus.
Aquaporin 2 is constitutively targeted to the basolateral membrane in canine polarized (MDCK)- kidney cells, and is retrieved by clathrin-mediated endocytosis into Rab5-positive vesicles. The microtubule-dependent (colchicine-sensitive) transcytosis of AQP2 might involve intracellular organelles, i.e. the endoplasmic reticulum, trans-Golgi network, perinuclear recycling compartment, and apical recycling endosomes within Rab11-positive vesicles (for continuous recycling between the apical membrane and the perinuclear region). Thus, a novel role for AQP2 has been suggested, i.e. cell migration, tubulogenesis, epithelial morphogenesis and, possibly, transepithelial water flux 68–70 (Fig.3A and B). AVP, aldosterone and hypertonicity also enhance AQP2 expression at the basolateral membrane, as shown by both in vitro and in vivo studies 71–73. Moreover, AVP leads to increased urine osmolality to about 1200 mOsm/kg with decreased urine output to 0.5 ml/min. The opposite is seen without AVP (i.e. urine osmolality decreased to about 50 mOsmol/kg and urine flow rate to 20 ml/min.) 74.
Nephrogenic diabetes insipidus
Diabetes insipidus is characterized by polyuria and compensatory polydipsia and encompasses four types: (i) the central form (the congenital familial neurohypophyseal diabetes insipidus or the acquired form); (ii) the NDI; (iii) the gestational diabetes insipidus; and (iv) the primary polydipsia (see review 21 for details). NDI is a syndrome in which the kidneys fail to conserve water because of variable degrees of resistance to AVP and can be hereditary or acquired.
Hereditary NDI
The hereditary NDI is a rare disorder appearing in infancy characterized by resistance to ADH, polyuria and polydipsia 75,76. This disease is caused by mutations in either the AVPR2 or AQP2 genes 37,75,77–79. About 90% of the patients with congenital NDI are diagnosed because of the presence of AVPR2 gene mutations (X-chromosome at Xq28) 80 leading to a dysfunctional AVPR2. Over 220 mutations have been identified so far, including missense/nonsense, splicing, small deletions, small insertions, small indels, gross deletions, gross insertions/duplications and complex rearrangements. Mutations in L1CAM, a gene close to the AVPR2 gene, may also account for some rare cases of NDI 21,81 (refer to http://www.ndif.org) (access 09, 2014). Five classes of AVPR2 gene mutations have been described 82 and comprise: a truncated receptor protein, a misfolded receptor (retained in the endoplasmic reticulum), a receptor unable to elicit cAMP production or to interact with AVP at the cell surface, and a receptor protein misrouted to intracellular organelles. Mouse models have been produced for X-NDI to better understand compensatory changes in the kidney and innovative treatments 83–85. Mild phenotypes of NDI have been also identified and are consistent with a number of additional mutations (e.g. p.Arg104Cys or p.Ser329Arg) 21. The X-linked inheritance implies that more pronounced polyuria is observed in males. Patients do not improve even after administration of exogenous AVP 86. The defect is present at birth with significant variability because of partial or incomplete NDI; patients have large volumes (more than 30 ml/kg/day, i.e. >3 l/day in adults or >2 l/m2 in children) of dilute urine (less than 250 mOsmol/kg) produced and associated with exaggerated thirst. Thus, typical symptoms of NDI include polydipsia, polyuria, hypernatremia and dehydration 37,51. Hypernatremia is usually associated with reduced feeding and weight loss, irritability, dry skin, and recessed eyeballs 87. Potential long-term complications of NDI are mental retardation, megacystis, hydroureter, hydronephrosis and renal failure 87–89.
Another hereditable form of NDI involves the autosomal recessive or the autosomal dominant forms conferring the mutations in the AQP2 gene on chromosome 12q13 encoding a 271-amino acid protein 33,55,90. More than 50 mutations in the AQP2 gene have been described, so far, including missense/nonsense, splicing, small deletions and small insertions. The mutations imply decreased channel function and/or defective fusion of the AQP2 (retained in the intracellular space) 91 with the apical membrane 42,92. The autosomal recessive NDI is seen in patients who are homozygous or compound heterozygous for mutations in the AQP2 gene. As a result, abnormalities consist of AQP2 misfolding, retention in the endoplasmic reticulum, or rapid degradation of the water channel protein 82. This NDI variant is encountered equally in both genders and starts at birth with a severe clinical picture, although partial NDI is rarely seen 74. The dominant form of NDI accounts for 10% of autosomal cases, and is because of the mutations involving the carboxyl tail of AQP2 and therefore the water channel intracellular routing 21. Abnormalities include AQP2 misrouting 93, intra-Golgi retention, or routing of AQP2 to lysosomes, late endosomes, or basolateral plasma membrane, where AQP3 an AQP4 should be, instead 94.
Acquired NDI
The acquired NDI syndromes are the commonest clinical conditions (Table3). All forms are characterized by decreased expression of AQP2 or abnormal trafficking of AQP2 to the apical plasma membrane. Reduced expression of AQP2 is encountered in both acute and chronic renal failure 21. Either bilateral or monolateral sustained ureteral obstruction is associated with persistently decreased AQP2 mRNA and protein levels in the inner medullary collecting ducts 95,96. Abnormal transcriptional pathways or regulation of mRNA degradation might be involved 96, as AQP2 trafficking to the apical plasma membrane of collecting duct principal cells is still functional after ureteral obstruction 95–97. The vasopressin receptor or its coupling to adenylyl cyclase also appears to be affected by the obstruction 98. During ureteral obstruction, a role for intrarenal angiotensin II generation in inhibiting vasopressin signalling and cyclooxygenase-2 (COX-2) in impairing renal handling of sodium and water has been advocated. Pharmacological manipulation with angiotensin receptor blockers (e.g. candesartan) 99 or COX-2 inhibitors 100,101 might prevent the reduction in AQP2 down-regulation and post-obstructive polyuria, as seen in animal models of ureteral obstruction.
Following treatment with lithium salts in bipolar affective disorders, up to 40% of patients may develop lithium-induced NDI 102–104. In rats, long-term treatment of lithium is associated with >90% decrease of AQP2 protein levels in the kidneys and severe polyuria, partly reversible 105. Decreased AQP2 mRNA abundance has been advocated to explain reduced AQP2 protein levels 106. The effects of lithium on the kidneys impact the calcium-sensing receptor and the calmodulin-dependent pathways 107,108, and COX-2 function 109. Some proteins involved in a myriad of functions, i.e. regulation of gene expression, signal transduction, cytoskeletal organization, cellular reorganization, cell proliferation and apoptosis, might also be affected 21,110. Antibiotics (e.g. demeclocycline 111 and foscarnet 112), antifungals (e.g. amphotericin B 113), and antineoplastic drugs (e.g. ifosfamide 114) might also cause reversible forms of acquired NDI. Hypokalaemia-induced NDI with polyuria and defective urinary concentrating ability may follow inappropriate diuretic therapy or primary aldosteronism 115. Central mechanisms in the brain might be also involved (e.g. inhibition of vasopressin secretion 116, or primary polydipsia 117). Reduced AQP2 expression levels in the inner medulla and cortex and decreased urinary concentrating capacity are found in rats following a potassium-deficient diet 118, and could follow an early (12–24 hrs) hypokalaemic effect on AQP2 protein and mRNA concentrations 119. Hypercalcaemia is also associated with decreased AQP2 expression and the mechanism is likely mediated by hypercalciuria, the calcium-sensing receptor and calcium-dependent activation of the proteolytic enzyme calpain 108,120,121. Inflammatory conditions are associated with polyuria and impaired renal concentrating ability, as shown in dogs and cats with pyometra 122,123. This is likely because of activation of inflammatory cytokine signalling pathways resulting in decreased expression levels of AQP2 and V2 receptors in the renal medulla 124. Also, NF-kB, interleukin-1β, and bacterial species (Escherichia coli, Klebsiella)-dependent endotoxins might influence AQP2 gene expression, vasopressin binding, vasopressin V2 receptors, and AQP2 protein concentrations 124–127.
Vasopressin-independent signals regulating AQP2 trafficking and potential use for NDI treatment
In 90% of the cases, NDI is transmitted as an X-linked recessive trait caused by mutations in the V2R gene. To rescue the inactivation of the V2R-elicited cAMP pathway, alternative intracellular pathways might be activated, which promotes AQP2 trafficking towards the plasma membrane. Different intracellular pathways appear to be involved in regulating AQP2 translocation (Table2), besides the classical regulation which is mediated by the specific G protein-coupled AVPR2 21.
Arginine vasopressin-independent pathways could lead to AQP2 expression at the plasma membrane in renal cells. The nitric oxide/cGMP pathway is one of the most interesting pathways 128 and implies the formation of nitric oxide from L-arginine, activation of soluble guanylate cyclase (GC), and increased intracellular cGMP concentration. Activated PKG can phosphorylate AQP2 directly or indirectly through PKA activation 63. Indeed, mice lacking all the nitric oxide synthase isoforms developed NDI 129. Moreover, the cGMP phosphodiesterase inhibitor sildenafil (Viagra), increased insertion of AQP2 in the apical membrane of renal cells both in vivo and in vitro 130 and reduced polyuria in rats with lithium-induced NDI 131. Prostaglandins, in particular E2 (PGE2), are abundantly expressed in the kidney and are considered modulators of AQP2 plasma membrane expression. The EP1-4 receptors have the 4 receptor subtypes through which PGE2 exerts its pharmacological actions 132,133. EP1 receptors preferentially couple to an increase in cell calcium. EP2 and EP4 receptors stimulate cyclic AMP through a Gs subunit, whereas the EP3 receptor preferentially couples to Gi, inhibiting cyclic AMP generation. COX inhibitors decrease PGE2 production and counteract the inhibitory role of EP3 receptor on cAMP production, thus increasing AQP2 exocytosis. Pharmacological stimulation of EP2 and EP4 alleviates NDI in the mouse and rat experimental models of the disease 84,134. With a similar mechanism calcitonin, the hormone produced by parafollicular cells, increases AQP2 apical targeting in vitro and in vivo by activating its Gs-coupled cognate receptor expressed in collecting duct renal cells and markedly ameliorates polyuria in vasopressin-deficient Brattleboro rats 135.
A therapeutic approach based on one of the molecules listed above might achieve a positive clinical outcome in patients affected by NDI.
Current treatment of NDI and use of statins
Exogenous administration of the AVP analogue desmopressin is used to treat central diabetes insipidus 136 and nocturnal enuresis 137. This approach, however, is ineffective in patients with congenital NDI because mutations in the V2R or AQP2 genes inactivate these proteins.
Gene therapy to cure NDI remains experimental and highly speculative 138. Acquired NDI may benefit from treatment of the underlying condition, and revision of dosage/discontinuation of an inciting drug. Treatment of hereditary NDI, however, remains a significant challenge, mainly because of the lack of function of AVPR2 and the lack of effect by desmopressin (Table4). To prevent severe complications, treatment of congenital NDI must start in infancy; high doses of desmopressin may be effective in patients with partial NDI or in heterozygous females with polyuria, when some AVPR2 function is retained. In the other cases, water intake must be appropriate to counteract water loss causing polydipsia and polyuria. The quality of life, however, is negatively affected by excessive drinking and urination and by potential complications. Low sodium diet and drugs such as diuretics and NSAIDs might have additional benefits (i.e. increased urine osmolality and 30–70% decrease of urine volume) 139–141.
Table 4.
Standard and experimental therapeutic approaches to hereditary nephrogenic diabetes insipidus
| Regimen | Notes |
|---|---|
Standard
|
|
|
|
|
|
Experimental
|
|
|
|
|
DMSO, dimethylsulfoxide; EP, prostaglandin E; NSAIDS, non-steroidal anti-inflammatory drugs.
Urine output could be reduced by ∽70% when hydrochlorothiazide diuretic (25 mg daily) is used with very low sodium-restricted diet of 9 mEq/day 142. Potassium sparing agents such as amiloride, might have an additive effect with thiazide diuretics, via mechanisms likely including the inhibition of potassium loss induced by thiazides 143. Diuretics in NDI are likely to reduce urine output by promoting proximal reabsorption of sodium and water. In this condition, less water is delivered to the AVP-sensitive tract of the nephron, the collecting duct.
Renal prostaglandin synthesis (mediated by the prostaglandin synthetase) is inhibited by NSAIDs. The effect of NSAIDs in NDI is based on the inhibition of the antagonizing effect of prostaglandins on AVP. A better urinary concentration is achieved with NSAIDs, and output in NDI can be reduced by 25–50% 144,145.
Because of poor therapeutic outcome and potential persistently severe side effects (e.g. renal and gastrointestinal complications), the attention has moved from the above-mentioned therapeutic regimens to novel strategies (Table4). Statins, the cholesterol-lowering agents acting on HMG-CoA reductase, have promising effects by working on mechanisms totally different from AVP and cAMP.
Statins and AQP2
Recent investigations have shown that statins increase AQP2 expression in the apical membrane of the collecting duct principal cells in the kidneys 49,146–149. Early in vitro experiments on renal MCD4 cells have shown that long-term treatment (3 days) of lovastatin might do so by reducing plasma membrane cholesterol 147 (also see below). The same group reported that fluvastatin acts on mouse kidney collecting duct cells by a vasopressin-independent mechanism, and this effect leads to water retention, reduces urine volume, and increases urine osmolality in mice 148.
Li et al. 49 used cell cultures and in vitro kidney slice from Brattleboro rats to assess AQP2 trafficking in response to incubation with simvastatin. Short-term exposure to simvastatin produces no change in cholesterol plasma membrane levels, but increases AQP2 accumulation in the apical membrane of principal cells of kidney slices from Brattleboro rats. At variance with VP effect, the action of statins is not associated with increased intracellular cAMP or inhibited by the PKA inhibitor H-89. Instead, the mechanism of action of simvastatin appears to be independent from cAMP/PKA activation and the phosphorylation of AQP2 at Ser256 which represents the classical pathway of VP-regulated AQP2 trafficking (Fig.2). Mechanisms of decreased constitutive endocytosis and/or increased constitutive exocytosis of AQP2 might be also affected by statin treatment 147,148. Clathrin plays a major role in the formation of coated vesicles and is involved in endocytosis 67,66. Li et al. 49 showed that simvastatin induces membrane accumulation of AQP2 in LLCPK-1 cells because of reduced clathrin-mediated endocytosis, rather than increased exocytosis. Same effects on AQP2 endocytosis in MCD4 cells were shown in a parallel study by Procino et al. 121.
The statin-mediated inhibition of the early step in the cholesterol biosynthetic pathway in any targeted tissue (i.e. catalyzation of HMG-CoA to mevalonate), leads in turn to the inhibition of the synthesis of isoprenoid intermediates such as FPP and GGPP 13. FPP and GGPP act as lipid anchors required for membrane tethering and activation of several proteins, such as heterotrimeric G proteins and small GTP-binding proteins (in particular the family of Ras from FPP, and the families of Rho, Rap and Rab GTPases from GGPP). Finally, the effect of early inhibition of mevalonic acid synthesis will be the downstream inhibition of several intracellular signalling molecules, accounting for the so-called ‘pleiotropic effects’ of statins. This scenario also applies to AQP2 trafficking in the kidneys. The above-mentioned effect of statins on isoprenoid intermediates might partly explain the lack of posttranslational changes of several signalling proteins (e.g. small GTP-binding proteins), as such molecules assist a number of cellular functions including cytoskeletal assembly as well as trafficking of proteins and lipids 150.
A previous in vitro study 151 has found that the statin-mediated inhibition of isoprenylation of Rho GTPase decreases the endocytosis of fluorescein isothiocyanate (FITC)-labelled albumin in kidney cells. The activation of this pathway results in the actin cytoskeletal reorganization and plays a role in protein trafficking and intracellular transport processes. Moreover, statins influence Rho GTPases which regulate the cytoskeleton 152,153. Both elements likely regulate vesicle trafficking and endocytosis 154,155.
Procino et al. 148, demonstrated that both fluvastatin and isoprenylation inhibitors significantly reduced the amount of active membrane-tethered RhoA and Rab5 GTPases with a parallel increase of AQP2 plasma membrane expression in vivo and in vitro. The study of Li et al. 49 confirmed that the clathrin-dependent effect of statins on AQP2 endocytosis involves the down-regulation of Rho GTPase (specifically RhoA) activity in a dose-dependent manner, and is already evident at concentrations as low as 10 μM. In particular, simvastatin-dependent accumulation of AQP2 at the plasma membrane could be prevented in transfected cells by overexpressing the constitutively active RhoA G14V, but not by the dominant negative RhoA T19N. They concluded that, among the family of Rho GTPases, RhoA is involved in simvastatin-mediated membrane trafficking of AQP2 49.
In wild-type C57BL/6 mice intraperitoneal injection of different classes of statins showed that fluvastatin was as effective as AVP in promoting AQP2 apical accumulation in the kidney collecting ducts 148. In the same work, prolonged treatment of fluvastatin induced a significant reduction of the diuresis and increase of urine osmolality with no effect on glomerular filtration rate 148.
Brattleboro rats lacking AVP because of spontaneous mutation of the AVP gene 66,156, were treated with intraperitoneal administration of simvastatin to a final plasma concentration of 200 μM, without any visible side effect 49. Simvastatin caused a decrease in urinary volume associated with consistently increased urine osmolality. Immunofluorescence staining of AQP2 revealed a significant increase in the apical membrane of the principal cells of the collecting duct in the cortex and outer medulla of the kidney of simvastatin-treated animals. More recently, it has been shown that a administration of a combination of secretin and fluvastatin dramatically reduced the polyuria and increased urine osmolality in the mouse model of X-linked NDI 149.
It is unclear whether additional membrane transporters might be influenced by statins, inducing AQP2 trafficking 146. Subcellular distribution of A and B subunits of V-ATPase, a protein showing membrane recycling, is not affected by simvastatin 49. In the study by Procino et al. 148, additional basolateral and apical membrane Na+ transporters (Na+/K+-ATPase and NKCC2) were up-regulated in the kidney membrane fraction by fluvastatin, suggesting that these transporters might contribute to Na+ and water reabsorption.
The statin-dependent inhibition of isoprenylation might affect additional Rho GTPases (e.g. Rac1 and Cdc42) and lead to an acute effect on AQP2 trafficking 154,157. Li and colleagues 49 demonstrated an acute effect of statins (within 60 min.) after a single injection with disappearance in 5–6 hrs. Likely, the simvastatin-mediated effect would be rapid modulation of RhoA GTPase activity, rather than cholesterol depletion 158, since a longer time (more than 35 hrs) is required for statins to induce 50% depletion of cholesterol membrane and influence trafficking of proteins and vesicles 159,160.
Studies on the effect of statins on AQP2 trafficking in animal models 49,147–149 used statins doses that are commonly used in rat/murine studies 161–163. However, because of the rapid up-regulation of HMG-CoA reductase observed in rodents during statin treatment 164, these doses are higher than those used in humans. The doses used in these studies are not therefore predictive of those needed in humans to achieve the same result. In addition, personal unpublished observations from these authors indicate that administration of different statins doses increases AQP2 plasma membrane expression in patients requiring hypocholesterolaemic therapy. Therefore, statins doses in the range of the currently used to reduce blood cholesterol, might be beneficial for NDI patients.
Statin-independent mechanisms might also promote AQP2 accumulation at the plasma membrane of kidney cells. For example, decreasing plasma membrane cholesterol by the cholesterol-depleting drug methyl-β-cyclodextrin (mβCD) 66,165, a blocker of clathrin-mediated endocytosis 160,166,167 including AQP2 67,66, is associated with rapid accumulation of AQP2 in cultured kidney epithelial cells and in principal cells of the intact perfused kidney. Furthermore, functioning of Rho-family GTPases (including RhoA) might follow an isoprenylation-independent pathway, i.e. phosphorylation of RhoA by PKA at Ser188. This step is a key event for cytoskeletal dynamics controlling cAMP-induced AQP2 translocation, and would lead to increased association with GDI (RhoA-GDI) and reduced RhoA membrane association and activity 168. The attenuation of Rho activity results in depolymerization of F-actin, facilitating AQP2 insertion into the plasma membrane during VP/PKA/cAMP-induced AQP2 translocation 62.
A different pathway leading to increased AQP2 abundance in the apical membrane might involve the nuclear receptor peroxisomal proliferator-activated receptor subtype γ (PPAR-γ). Indeed, the synthetic PPAR-γ agonist rosiglitazone, besides improving insulin resistance, is associated with fluid retention and oedema. This side effect appears to be mediated by an increase in sodium and water retention (via increased abundance of AQP2, and AQP3) in the kidney 169,170.
Advantages and disadvantages of statins in the treatment of NDI
The effects of statins with respect to AQP2 trafficking and water reabsorption in the kidneys have been raising much interest about their potential therapeutic pleiotropic effects in patients with NDI. Pilot studies from our group suggest that simvastatin increases AQP2 plasma membrane expression in humans treated for hypercholesterolaemia. The dose effect of different statins, however, needs to be tested in clinical trials with respect to duration of treatment, pharmacokinetics and lipophilic properties of different molecules 171.
The possibility of adverse reactions during long-term use and high-dosage statin therapy needs to be addressed. This aspect is of interest in patients with NDI who will likely require chronic treatment of statins.
In healthy individuals, atorvastatin treatment leads to modest and transitory decrease in sodium excretion and no change in renal function. In the same study, no change was documented in glomerular filtration rate, vasoactive hormones, tubular function and renal plasma flow 172. Some statins (simvastatin or rosuvastatin 173), might induce tubular inhibition of small-molecular-weight proteins and transient low-molecular-weight proteinuria 151,174,175. Hyperlipidemic patients administered with rosuvastatin 10 or 20 mg/day for 3 months, for example, show a dose-dependent increase in urinary low-molecular protein α-1 microglobulin 176. A plausible explanation might be the inhibition of HMG-CoA reductase in the proximal tubule cells. This step leads to a depletion of the cellular geranylgeranyl pyrophosphate pool (an intermediate of the sterol pathway) and reduced function of one or more GTP-binding proteins, which are known to be involved in the process of proximal tubular endocytosis 151,177–180. There is evidence suggesting that increased transient low-molecular-weight proteinuria following statin treatment is rather a benign outcome 181. Renal failure has been rarely reported with high doses (80 mg/day) of rosuvastatin. Renal adverse events have also been reported with other statins 182–185. By contrast, patients taking statins often suffer from underlying chronic kidney disease and still, statins reduce proteinuria and glomerular filtration rate 186, without aggravating renal failure 187,188 or aggravating proteinuria 189. The use of statins is also advised to persons with chronic renal insufficiency 190.
A recent study investigating the short-term (13 days) effect of statins on the urinary protein concentration and proteome in healthy volunteers found that either rosuvastatin (40 mg/day) or pravastatin (80 mg/day) did not induce major changes in the urinary protein concentration/proteome (on a background of high variability in the baseline urinary proteome/proteins among volunteers 191). In the animal model, statins prevented the development of renal injury and enhanced renal perfusion 192,193. A simvastatin-dependent increase in nitric oxide mediated the amelioration of glomerular filtration rate, renal plasma flow and endothelial function in patients with autosomal dominant polycystic kidney disease 194. Improved renal function was observed in statin-treated patients with ischaemic heart disease 195. In patients with already impaired glomerular filtration rate, statins did not change or slightly increased urinary albumin excretion, independently on dose or type of statins 196.
Muscle injury ranging to myalgias (up to 10%) 197 even with normal creatine kinase concentration, to myositis (0.5%) to rhabdomyolysis (<0.1%) eventually evolving to acute renal failure from myoglobinuria have been reported in some patients using statins with a median time of 1 month. Pravastatin and fluvastatin have the lowest rate of muscle side effects. Statin-associated myopathy is enhanced in patients with decreased thyroid function, acute and chronic renal failure, and obstructive liver disease.
Statin-induced liver injury disclosed by mild persistent elevations in aminotransferases has been reported in up to 3% of patients receiving statins (1.2 episode/100,000 users), usually during the first 3 months in a dose-dependent fashion 198. The true importance of such possibility has been questioned by several studies comparing statin use with placebo or with the general population 199–202.
Forms of reversible cognitive dysfunction and memory loss have been associated particularly with lipophilic simvastatin, pravastatin and atorvastatin 203.
Reports have associated the use of some statins with the increased risk of developing diabetes mellitus in non-diabetics. In diabetic patients, furthermore, the glycaemic control might become more problematic with the intensive use of some statins 204.
Statin use must be also discontinued during pregnancy (increased risk of congenital central nervous system and limb abnormalities) and breastfeeding.
Whether longer treatment periods might change such outcome is currently unknown. Also, the ultimate interaction between statin-dependent proteinuria and AQP2 (as well as other kidney AQPs) will be the focus of further clinical research.
Conclusions
The regulation of AQP2 expression in the kidney tubule is a key step in maintaining water balance. NDI represents a severe disturbance of water homoeostasis, exposing to polydipsia, polyuria, hypernatremia and dehydration. A better knowledge about NDI has recently emerged with genetic, clinical, molecular and pathophysiological perspectives. Statins improve cardiovascular outcome, and evidence shows that statins modulate the expression of AQP2 mRNA and protein in the kidneys, thereby increasing water reabsorption. This non-lipid dependent pleiotropic property of statins, if proven to be effective and well-tolerated, will open new venues to the treatment of hereditable NDI. It is possible that the beneficial effects of statins on NDI will outweigh the overall limited risk of adverse effects.
Acknowledgments
This work was supported in part by a research grant MRAR08P011-2012 (to P.P. and M.S.) from Italian Agency of Drug (AIFA), by Telethon GGP12040 (to M.S.) and by a research grant DK73917 (to D.Q.-H.W.) from the National Institutes of Health (US Public Health Service).
Conflicts of interest
None declared.
Author contribution
LB and GP discussed the general outlines of the article, performed the literature search, wrote the first draft and contributed to improve the following versions; DQHW gave important conceptual contribution and reviewed the manuscript; MS designed the outlines, gave important conceptual contribution, improved the final version of the manuscript and provided further conceptual suggestions; PP designed the outlines, gave important conceptual contribution, designed tables and figures, reviewed the final version of the paper. All authors reviewed and approved the final version of the paper.
References
- Shepherd J, Cobbe SM, Ford I, et al. Prevention of coronary heart disease with pravastatin in men with hypercholesterolemia. West of Scotland Coronary Prevention Study Group. N Engl J Med. 1995;333:1301–7. doi: 10.1056/NEJM199511163332001. [DOI] [PubMed] [Google Scholar]
- Jukema JW, Bruschke AV, van Boven AJ, et al. Effects of lipid lowering by pravastatin on progression and regression of coronary artery disease in symptomatic men with normal to moderately elevated serum cholesterol levels. The Regression Growth Evaluation Statin Study (REGRESS) Circulation. 1995;91:2528–40. doi: 10.1161/01.cir.91.10.2528. [DOI] [PubMed] [Google Scholar]
- Davignon J. The cardioprotective effects of statins. Curr Atheroscler Rep. 2004;6:27–35. doi: 10.1007/s11883-004-0113-7. [DOI] [PubMed] [Google Scholar]
- The Long-Term Intervention with Pravastatin in Ischaemic Disease (LIPID) Study Group. Prevention of cardiovascular events and death with pravastatin in patients with coronary heart disease and a broad range of initial cholesterol levels. N Engl J Med. 1998;339:1349–57. doi: 10.1056/NEJM199811053391902. [DOI] [PubMed] [Google Scholar]
- Jones PH, Davidson MH, Stein EA, et al. Comparison of the efficacy and safety of rosuvastatin versus atorvastatin, simvastatin, and pravastatin across doses (STELLAR* Trial) Am J Cardiol. 2003;92:152–60. doi: 10.1016/s0002-9149(03)00530-7. [DOI] [PubMed] [Google Scholar]
- Jones P, Kafonek S, Laurora I, et al. Comparative dose efficacy study of atorvastatin versus simvastatin, pravastatin, lovastatin, and fluvastatin in patients with hypercholesterolemia (the CURVES study) Am J Cardiol. 1998;81:582–7. doi: 10.1016/s0002-9149(97)00965-x. [DOI] [PubMed] [Google Scholar]
- Goff DC, Lloyd-Jones DM, Bennett G, et al. 2013 ACC/AHA guideline on the assessment of cardiovascular risk: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol. 2014;63:2935–59. doi: 10.1016/j.jacc.2013.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone NJ, Robinson J, Lichtenstein AH, et al. 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults. J Am Coll Cardiol. 2014;63:2889–934. doi: 10.1016/j.jacc.2013.11.002. [DOI] [PubMed] [Google Scholar]
- Baigent C, Keech A, Kearney PM, et al. Efficacy and safety of cholesterol-lowering treatment: prospective meta-analysis of data from 90,056 participants in 14 randomised trials of statins. Lancet. 2005;366:1267–78. doi: 10.1016/S0140-6736(05)67394-1. [DOI] [PubMed] [Google Scholar]
- Josan K, Majumdar SR, McAlister FA. The efficacy and safety of intensive statin therapy: a meta-analysis of randomized trials. CMAJ. 2008;178:576–84. doi: 10.1503/cmaj.070675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Istvan ES, Deisenhofer J. Structural mechanism for statin inhibition of HMG-CoA reductase. Science. 2001;292:1160–4. doi: 10.1126/science.1059344. [DOI] [PubMed] [Google Scholar]
- Kovanen PT, Bilheimer DW, Goldstein JL, et al. Regulatory role for hepatic low density lipoprotein receptors in vivo in the dog. Proc Natl Acad Sci USA. 1981;78:1194–8. doi: 10.1073/pnas.78.2.1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein JL, Brown MS. Regulation of the mevalonate pathway. Nature. 1990;343:425–30. doi: 10.1038/343425a0. [DOI] [PubMed] [Google Scholar]
- Kallien G, Lange K, Stange EF, et al. The pravastatin-induced decrease of biliary cholesterol secretion is not directly related to an inhibition of cholesterol synthesis in humans. Hepatology. 1999;30:14–20. doi: 10.1002/hep.510300119. [DOI] [PubMed] [Google Scholar]
- Duane WC, Hunninghake DB, Freeman ML, et al. Simvastatin, a competitive inhibitor of HMG-CoA reductase, lowers cholesterol saturation index of gallbladder bile. Hepatology. 1988;8:1147–50. doi: 10.1002/hep.1840080531. [DOI] [PubMed] [Google Scholar]
- Loria P, Bertolotti M, Cassinadri MT, et al. Short-term effects of simvastatin on bile acid synthesis and bile lipid secretion in human subjects. Hepatology. 1994;19:882–8. [PubMed] [Google Scholar]
- Logan GM, Duane WC. Lovastatin added to ursodeoxycholic acid further reduces biliary cholesterol saturation. Gastroenterology. 1990;98:1572–6. doi: 10.1016/0016-5085(90)91092-k. [DOI] [PubMed] [Google Scholar]
- Takemoto M, Liao JK. Pleiotropic effects of 3-hydroxy-3-methylglutaryl coenzyme a reductase inhibitors. Arterioscler Thromb Vasc Biol. 2001;21:1712–9. doi: 10.1161/hq1101.098486. [DOI] [PubMed] [Google Scholar]
- McFarlane SI, Muniyappa R, Francisco R, et al. Clinical review 145: pleiotropic effects of statins: lipid reduction and beyond. J Clin Endocrinol Metab. 2002;87:1451–8. doi: 10.1210/jcem.87.4.8412. [DOI] [PubMed] [Google Scholar]
- McKenney JM. Potential nontraditional applications of statins. Ann Pharmacother. 2003;37:1063–71. doi: 10.1345/aph.1C499. [DOI] [PubMed] [Google Scholar]
- Moeller HB, Rittig S, Fenton RA. Nephrogenic diabetes insipidus: essential insights into the molecular background and potential therapies for treatment. Endocr Rev. 2013;34:278–301. doi: 10.1210/er.2012-1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agre P, King LS, Yasui M, et al. Aquaporin water channels–from atomic structure to clinical medicine. J Physiol. 2002;542:3–16. doi: 10.1113/jphysiol.2002.020818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Portincasa P, Palasciano G, Svelto M, et al. Aquaporins in the hepatobiliary tract. Which, where and what they do in health and disease. Eur J Clin Invest. 2008;38:1–10. doi: 10.1111/j.1365-2362.2007.01897.x. [DOI] [PubMed] [Google Scholar]
- Calamita G, Ferri D, Gena P, et al. Water transport into bile and role in bile formation. Curr Drug Targets Immune Endocr Metabol Disord. 2005;5:137–42. doi: 10.2174/1568008054064850. [DOI] [PubMed] [Google Scholar]
- Portincasa P, Calamita G. Water channel proteins in bile formation and flow in health and disease: when immiscible becomes miscible. Mol Aspects Med. 2012;33:651–64. doi: 10.1016/j.mam.2012.03.010. [DOI] [PubMed] [Google Scholar]
- Valenti G, Procino G, Liebenhoff U, et al. A heterotrimeric G protein of the Gi family is required for cAMP-triggered trafficking of aquaporin 2 in kidney epithelial cells. J Biol Chem. 1998;273:22627–34. doi: 10.1074/jbc.273.35.22627. [DOI] [PubMed] [Google Scholar]
- Marples D. Water channels: who needs them anyway? The Lancet. 2000;355:1571–2. doi: 10.1016/S0140-6736(00)02209-1. [DOI] [PubMed] [Google Scholar]
- Nichols R. Polyuria and polydipsia. Diagnostic approach and problems associated with patient evaluation. Vet Clin North Am Small Anim Pract. 2001;31:833–44. doi: 10.1016/s0195-5616(01)50001-7. [DOI] [PubMed] [Google Scholar]
- Wilson JL, Miranda CA, Knepper MA. Vasopressin and the regulation of aquaporin-2. Clin Exp Nephrol. 2013;17:751–64. doi: 10.1007/s10157-013-0789-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takata K, Matsuzaki T, Tajika Y. Aquaporins: water channel proteins of the cell membrane. Prog Histochem Cytochem. 2004;39:1–83. doi: 10.1016/j.proghi.2004.03.001. [DOI] [PubMed] [Google Scholar]
- Nagase H, Agren J, Saito A, et al. Molecular cloning and characterization of mouse aquaporin 6. Biochem Biophys Res Commun. 2007;352:12–6. doi: 10.1016/j.bbrc.2006.10.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Procino G, Mastrofrancesco L, Sallustio F, et al. AQP5 is expressed in type-B intercalated cells in the collecting duct system of the rat, mouse and human kidney. Cell Physiol Biochem. 2011;28:683–92. doi: 10.1159/000335762. [DOI] [PubMed] [Google Scholar]
- Nielsen S, Frokiaer J, Marples D, et al. Aquaporins in the kidney: from molecules to medicine. Physiol Rev. 2002;82:205–44. doi: 10.1152/physrev.00024.2001. [DOI] [PubMed] [Google Scholar]
- Nielsen S, Kwon TH, Christensen BM, et al. Physiology and pathophysiology of renal aquaporins. J Am Soc Nephrol. 1999;10:647–63. doi: 10.1681/ASN.V103647. [DOI] [PubMed] [Google Scholar]
- Hadchouel J, Busst C, Procino G, et al. Regulation of extracellular fluid volume and blood pressure by pendrin. Cell Physiol Biochem. 2011;28:505–12. doi: 10.1159/000335116. [DOI] [PubMed] [Google Scholar]
- Fushimi K, Uchida S, Hara Y, et al. Cloning and expression of apical membrane water channel of rat kidney collecting tubule. Nature. 1993;361:549–52. doi: 10.1038/361549a0. [DOI] [PubMed] [Google Scholar]
- Nielsen S, Kwon TH, Frokiaer J, et al. Regulation and dysregulation of aquaporins in water balance disorders. J Intern Med. 2007;261:53–64. doi: 10.1111/j.1365-2796.2006.01760.x. [DOI] [PubMed] [Google Scholar]
- Ecelbarger CA, Terris J, Frindt G, et al. Aquaporin-3 water channel localization and regulation in rat kidney. Am J Physiol. 1995;269:F663–72. doi: 10.1152/ajprenal.1995.269.5.F663. [DOI] [PubMed] [Google Scholar]
- Terris J, Ecelbarger CA, Marples D, et al. Distribution of aquaporin-4 water channel expression within rat kidney. Am J Physiol. 1995;269:F775–85. doi: 10.1152/ajprenal.1995.269.6.F775. [DOI] [PubMed] [Google Scholar]
- Fenton RA, Pedersen CN, Moeller HB. New insights into regulated aquaporin-2 function. Curr Opin Nephrol Hypertens. 2013;22:551–8. doi: 10.1097/MNH.0b013e328364000d. [DOI] [PubMed] [Google Scholar]
- Deen PM, Verdijk MA, Knoers NV, et al. Requirement of human renal water channel aquaporin-2 for vasopressin-dependent concentration of urine. Science. 1994;264:92–5. doi: 10.1126/science.8140421. [DOI] [PubMed] [Google Scholar]
- Deen PM, Croes H, van Aubel RA, et al. Water channels encoded by mutant aquaporin-2 genes in nephrogenic diabetes insipidus are impaired in their cellular routing. J Clin Invest. 1995;95:2291–6. doi: 10.1172/JCI117920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen S, DiGiovanni SR, Christensen EI, et al. Cellular and subcellular immunolocalization of vasopressin-regulated water channel in rat kidney. Proc Natl Acad Sci USA. 1993;90:11663–7. doi: 10.1073/pnas.90.24.11663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris HW, Jr, Strange K, Zeidel ML. Current understanding of the cellular biology and molecular structure of the antidiuretic hormone-stimulated water transport pathway. J Clin Invest. 1991;88:1–8. doi: 10.1172/JCI115263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown D. Membrane recycling and epithelial cell function. Am J Physiol. 1989;256:F1–12. doi: 10.1152/ajprenal.1989.256.1.F1. [DOI] [PubMed] [Google Scholar]
- Zimmerman EA, Nilaver G, Hou-Yu A, et al. Vasopressinergic and oxytocinergic pathways in the central nervous system. Fed Proc. 1984;43:91–6. [PubMed] [Google Scholar]
- Oakley RH, Laporte SA, Holt JA, et al. Association of beta-arrestin with G protein-coupled receptors during clathrin-mediated endocytosis dictates the profile of receptor resensitization. J Biol Chem. 1999;274:32248–57. doi: 10.1074/jbc.274.45.32248. [DOI] [PubMed] [Google Scholar]
- Brown D. The ins and outs of aquaporin-2 trafficking. Am J Physiol Renal Physiol. 2003;284:F893–901. doi: 10.1152/ajprenal.00387.2002. [DOI] [PubMed] [Google Scholar]
- Li W, Zhang Y, Bouley R, et al. Simvastatin enhances aquaporin-2 surface expression and urinary concentration in vasopressin-deficient Brattleboro rats through modulation of Rho GTPase. AJP Renal Physiol. 2011;301:F309–18. doi: 10.1152/ajprenal.00001.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffert JD, Pisitkun T, Wang G, et al. Quantitative phosphoproteomics of vasopressin-sensitive renal cells: regulation of aquaporin-2 phosphorylation at two sites. Proc Natl Acad Sci USA. 2006;103:7159–64. doi: 10.1073/pnas.0600895103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen S, Chou CL, Marples D, et al. Vasopressin increases water permeability of kidney collecting duct by inducing translocation of aquaporin-CD water channels to plasma membrane. Proc Natl Acad Sci USA. 1995;92:1013–7. doi: 10.1073/pnas.92.4.1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klussmann E, Maric K, Rosenthal W. The mechanisms of aquaporin control in the renal collecting duct. Rev Physiol Biochem Pharmacol. 2000;141:33–95. doi: 10.1007/BFb0119577. [DOI] [PubMed] [Google Scholar]
- Knepper MA, Inoue T. Regulation of aquaporin-2 water channel trafficking by vasopressin. Curr Opin Cell Biol. 1997;9:560–4. doi: 10.1016/s0955-0674(97)80034-8. [DOI] [PubMed] [Google Scholar]
- Hayashi M, Sasaki S, Tsuganezawa H, et al. Expression and distribution of aquaporin of collecting duct are regulated by vasopressin V2 receptor in rat kidney. J Clin Invest. 1994;94:1778–83. doi: 10.1172/JCI117525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki S, Fushimi K, Saito H, et al. Cloning, characterization, and chromosomal mapping of human aquaporin of collecting duct. J Clin Invest. 1994;93:1250–6. doi: 10.1172/JCI117079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liebenhoff U, Rosenthal W. Identification of Rab3-, Rab5a- and synaptobrevin II-like proteins in a preparation of rat kidney vesicles containing the vasopressin-regulated water channel. FEBS Lett. 1995;365:209–13. doi: 10.1016/0014-5793(95)00476-p. [DOI] [PubMed] [Google Scholar]
- Ren XD, Kiosses WB, Schwartz MA. Regulation of the small GTP-binding protein Rho by cell adhesion and the cytoskeleton. EMBO J. 1999;18:578–85. doi: 10.1093/emboj/18.3.578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nobes CD, Hall A. Rho, rac, and cdc42 GTPases regulate the assembly of multimolecular focal complexes associated with actin stress fibers, lamellipodia, and filopodia. Cell. 1995;81:53–62. doi: 10.1016/0092-8674(95)90370-4. [DOI] [PubMed] [Google Scholar]
- Dong JM, Leung T, Manser E, et al. cAMP-induced morphological changes are counteracted by the activated RhoA small GTPase and the Rho kinase ROKalpha. J Biol Chem. 1998;273:22554–62. doi: 10.1074/jbc.273.35.22554. [DOI] [PubMed] [Google Scholar]
- Sasaki T, Takai Y. The Rho small G protein family-Rho GDI system as a temporal and spatial determinant for cytoskeletal control. Biochem Biophys Res Commun. 1998;245:641–5. doi: 10.1006/bbrc.1998.8253. [DOI] [PubMed] [Google Scholar]
- Van Aelst L, D'Souza-Schorey C. Rho GTPases and signaling networks. Genes Dev. 1997;11:2295–322. doi: 10.1101/gad.11.18.2295. [DOI] [PubMed] [Google Scholar]
- Tamma G, Klussmann E, Procino G, et al. cAMP-induced AQP2 translocation is associated with RhoA inhibition through RhoA phosphorylation and interaction with RhoGDI. J Cell Sci. 2003;116:1519–25. doi: 10.1242/jcs.00355. [DOI] [PubMed] [Google Scholar]
- Forget MA, Desrosiers RR, Gingras D, et al. Phosphorylation states of Cdc42 and RhoA regulate their interactions with Rho GDP dissociation inhibitor and their extraction from biological membranes. Biochem J. 2002;361:243–54. doi: 10.1042/0264-6021:3610243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiGiovanni SR, Nielsen S, Christensen EI, et al. Regulation of collecting duct water channel expression by vasopressin in Brattleboro rat. Proc Natl Acad Sci USA. 1994;91:8984–8. doi: 10.1073/pnas.91.19.8984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustafson CE, Katsura T, McKee M, et al. Recycling of AQP2 occurs through a temperature- and bafilomycin-sensitive trans-Golgi-associated compartment. Am J Physiol Renal Physiol. 2000;278:F317–26. doi: 10.1152/ajprenal.2000.278.2.F317. [DOI] [PubMed] [Google Scholar]
- Lu H, Sun TX, Bouley R, et al. Inhibition of endocytosis causes phosphorylation (S256)-independent plasma membrane accumulation of AQP2. Am J Physiol Renal Physiol. 2004;286:F233–43. doi: 10.1152/ajprenal.00179.2003. [DOI] [PubMed] [Google Scholar]
- Sun TX, Van Hoek A, Huang Y, et al. Aquaporin-2 localization in clathrin-coated pits: inhibition of endocytosis by dominant-negative dynamin. Am J Physiol Renal Physiol. 2002;282:F998–1011. doi: 10.1152/ajprenal.00257.2001. [DOI] [PubMed] [Google Scholar]
- Chen Y, Rice W, Gu Z, et al. Aquaporin 2 promotes cell migration and epithelial morphogenesis. J Am Soc Nephrol. 2012;23:1506–17. doi: 10.1681/ASN.2012010079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yui N, Lu HAJ, Chen Y, et al. Basolateral targeting and microtubule-dependent transcytosis of the aquaporin-2 water channel. AJP Cell Physiol. 2012;304:C38–48. doi: 10.1152/ajpcell.00109.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto CT. Caring about the other 47% of the water channels. Focus on “Basolateral targeting and microtubule-dependent transcytosis of the aquaporin-2 water channel”. AJP Cell Physiol. 2012;304:C33–5. doi: 10.1152/ajpcell.00348.2012. [DOI] [PubMed] [Google Scholar]
- van Balkom BW, van Raak M, Breton S, et al. Hypertonicity is involved in redirecting the aquaporin-2 water channel into the basolateral, instead of the apical, plasma membrane of renal epithelial cells. J Biol Chem. 2003;278:1101–7. doi: 10.1074/jbc.M207339200. [DOI] [PubMed] [Google Scholar]
- Coleman RA, Wu DC, Liu J, et al. Expression of aquaporins in the renal connecting tubule. Am J Physiol Renal Physiol. 2000;279:F874–83. doi: 10.1152/ajprenal.2000.279.5.F874. [DOI] [PubMed] [Google Scholar]
- de Seigneux S, Nielsen J, Olesen ET, et al. Long-term aldosterone treatment induces decreased apical but increased basolateral expression of AQP2 in CCD of rat kidney. Am J Physiol Renal Physiol. 2007;293:F87–99. doi: 10.1152/ajprenal.00431.2006. [DOI] [PubMed] [Google Scholar]
- Babey M, Kopp P, Robertson GL. Familial forms of diabetes insipidus: clinical and molecular characteristics. Nat Rev Endocrinol. 2011;7:701–14. doi: 10.1038/nrendo.2011.100. [DOI] [PubMed] [Google Scholar]
- Bichet DG. Hereditary polyuric disorders: new concepts and differential diagnosis. Semin Nephrol. 2006;26:224–33. doi: 10.1016/j.semnephrol.2006.02.004. [DOI] [PubMed] [Google Scholar]
- Devonald MA, Karet FE. Renal epithelial traffic jams and one-way streets. J Am Soc Nephrol. 2004;15:1370–81. doi: 10.1097/01.asn.0000123804.18566.69. [DOI] [PubMed] [Google Scholar]
- Deen PM, Marr N, Kamsteeg EJ, et al. Nephrogenic diabetes insipidus. Curr Opin Nephrol Hypertens. 2000;9:591–5. doi: 10.1097/00041552-200011000-00001. [DOI] [PubMed] [Google Scholar]
- Slusarz MJ, Gieldon A, Slusarz R, et al. Analysis of interactions responsible for vasopressin binding to human neurohypophyseal hormone receptors-molecular dynamics study of the activated receptor-vasopressin-G(alpha) systems. J Pept Sci. 2006;12:180–9. doi: 10.1002/psc.714. [DOI] [PubMed] [Google Scholar]
- Slusarz MJ, Slusarz R, Ciarkowski J. Molecular dynamics simulation of human neurohypophyseal hormone receptors complexed with oxytocin-modeling of an activated state. J Pept Sci. 2006;12:171–9. doi: 10.1002/psc.713. [DOI] [PubMed] [Google Scholar]
- Bichet DG, Razi M, Lonergan M, et al. Hemodynamic and coagulation responses to 1-desamino[8-D-arginine] vasopressin in patients with congenital nephrogenic diabetes insipidus. N Engl J Med. 1988;318:881–7. doi: 10.1056/NEJM198804073181403. [DOI] [PubMed] [Google Scholar]
- Knops NB, Bos KK, Kerstjens M, et al. Nephrogenic diabetes insipidus in a patient with L1 syndrome: a new report of a contiguous gene deletion syndrome including L1CAM and AVPR2. Am J Med Genet A. 2008;146A:1853–8. doi: 10.1002/ajmg.a.32386. [DOI] [PubMed] [Google Scholar]
- Robben JH, Knoers NV, Deen PM. Cell biological aspects of the vasopressin type-2 receptor and aquaporin 2 water channel in nephrogenic diabetes insipidus. Am J Physiol Renal Physiol. 2006;291:F257–70. doi: 10.1152/ajprenal.00491.2005. [DOI] [PubMed] [Google Scholar]
- Yun J, Schoneberg T, Liu J, et al. Generation and phenotype of mice harboring a nonsense mutation in the V2 vasopressin receptor gene. J Clin Invest. 2000;106:1361–71. doi: 10.1172/JCI9154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li JH, Chou CL, Li B, et al. A selective EP4 PGE2 receptor agonist alleviates disease in a new mouse model of X-linked nephrogenic diabetes insipidus. J Clin Invest. 2009;119:3115–26. doi: 10.1172/JCI39680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schliebe N, Strotmann R, Busse K, et al. V2 vasopressin receptor deficiency causes changes in expression and function of renal and hypothalamic components involved in electrolyte and water homeostasis. Am J Physiol Renal Physiol. 2008;295:F1177–90. doi: 10.1152/ajprenal.00465.2007. [DOI] [PubMed] [Google Scholar]
- Knoers NV, Deen PM. Molecular and cellular defects in nephrogenic diabetes insipidus. Pediatr Nephrol. 2001;16:1146–52. doi: 10.1007/s004670100051. [DOI] [PubMed] [Google Scholar]
- Wesche D, Deen PM, Knoers NV. Congenital nephrogenic diabetes insipidus: the current state of affairs. Pediatr Nephrol. 2012;27:2183–204. doi: 10.1007/s00467-012-2118-8. [DOI] [PubMed] [Google Scholar]
- Bichet DG. Nephrogenic diabetes insipidus. Am J Med. 1998;105:431–42. doi: 10.1016/s0002-9343(98)00301-5. [DOI] [PubMed] [Google Scholar]
- Knoers N, Monnens LA. Nephrogenic diabetes insipidus: clinical symptoms, pathogenesis, genetics and treatment. Pediatr Nephrol. 1992;6:476–82. doi: 10.1007/BF00874020. [DOI] [PubMed] [Google Scholar]
- Heinke F, Labudde D. Membrane protein stability analyses by means of protein energy profiles in case of nephrogenic diabetes insipidus. Comput Math Methods Med. 2012;2012:790281. doi: 10.1155/2012/790281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki S. Nephrogenic diabetes insipidus: update of genetic and clinical aspects. Nephrol Dial Transplant. 2004;19:1351–3. doi: 10.1093/ndt/gfh172. [DOI] [PubMed] [Google Scholar]
- Hochberg Z, Van Lieburg A, Even L, et al. Autosomal recessive nephrogenic diabetes insipidus caused by an aquaporin-2 mutation. J Clin Endocrinol Metab. 1997;82:686–9. doi: 10.1210/jcem.82.2.3781. [DOI] [PubMed] [Google Scholar]
- Kamsteeg EJ, Bichet DG, Konings IB, et al. Reversed polarized delivery of an aquaporin-2 mutant causes dominant nephrogenic diabetes insipidus. J Cell Biol. 2003;163:1099–109. doi: 10.1083/jcb.200309017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boone M, Deen PM. Congenital nephrogenic diabetes insipidus: what can we learn from mouse models? Exp Physiol. 2009;94:186–90. doi: 10.1113/expphysiol.2008.043000. [DOI] [PubMed] [Google Scholar]
- Frokiaer J, Marples D, Knepper MA, et al. Bilateral ureteral obstruction downregulates expression of vasopressin-sensitive AQP-2 water channel in rat kidney. Am J Physiol. 1996;270:F657–68. doi: 10.1152/ajprenal.1996.270.4.F657. [DOI] [PubMed] [Google Scholar]
- Frokiaer J, Christensen BM, Marples D, et al. Downregulation of aquaporin-2 parallels changes in renal water excretion in unilateral ureteral obstruction. Am J Physiol. 1997;273:F213–23. doi: 10.1152/ajprenal.1997.273.2.F213. [DOI] [PubMed] [Google Scholar]
- Li C, Wang W, Knepper MA, et al. Downregulation of renal aquaporins in response to unilateral ureteral obstruction. Am J Physiol Renal Physiol. 2003;284:F1066–79. doi: 10.1152/ajprenal.00090.2002. [DOI] [PubMed] [Google Scholar]
- Kim SW, Cho SH, Oh BS, et al. Diminished renal expression of aquaporin water channels in rats with experimental bilateral ureteral obstruction. J Am Soc Nephrol. 2001;12:2019–28. doi: 10.1681/ASN.V12102019. [DOI] [PubMed] [Google Scholar]
- Jensen AM, Li C, Praetorius HA, et al. Angiotensin II mediates downregulation of aquaporin water channels and key renal sodium transporters in response to urinary tract obstruction. Am J Physiol Renal Physiol. 2006;291:F1021–32. doi: 10.1152/ajprenal.00387.2005. [DOI] [PubMed] [Google Scholar]
- Norregaard R, Jensen BL, Li C, et al. COX-2 inhibition prevents downregulation of key renal water and sodium transport proteins in response to bilateral ureteral obstruction. Am J Physiol Renal Physiol. 2005;289:F322–33. doi: 10.1152/ajprenal.00061.2005. [DOI] [PubMed] [Google Scholar]
- Norregaard R, Jensen BL, Topcu SO, et al. COX-2 activity transiently contributes to increased water and NaCl excretion in the polyuric phase after release of ureteral obstruction. Am J Physiol Renal Physiol. 2007;292:F1322–33. doi: 10.1152/ajprenal.00394.2006. [DOI] [PubMed] [Google Scholar]
- Stone KA. Lithium-induced nephrogenic diabetes insipidus. J Am Board Fam Pract. 1999;12:43–7. doi: 10.3122/15572625-12-1-43. [DOI] [PubMed] [Google Scholar]
- Garofeanu CG, Weir M, Rosas-Arellano MP, et al. Causes of reversible nephrogenic diabetes insipidus: a systematic review. Am J Kidney Dis. 2005;45:626–37. doi: 10.1053/j.ajkd.2005.01.008. [DOI] [PubMed] [Google Scholar]
- Grunfeld JP, Rossier BC. Lithium nephrotoxicity revisited. Nat Rev Nephrol. 2009;5:270–6. doi: 10.1038/nrneph.2009.43. [DOI] [PubMed] [Google Scholar]
- Marples D, Christensen S, Christensen EI, et al. Lithium-induced downregulation of aquaporin-2 water channel expression in rat kidney medulla. J Clin Invest. 1995;95:1838–45. doi: 10.1172/JCI117863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laursen UH, Pihakaski-Maunsbach K, Kwon TH, et al. Changes of rat kidney AQP2 and Na, K-ATPase mRNA expression in lithium-induced nephrogenic diabetes insipidus. Nephron Exp Nephrol. 2004;97:e1–16. doi: 10.1159/000077593. [DOI] [PubMed] [Google Scholar]
- Sands JM, Naruse M, Baum M, et al. Apical extracellular calcium/polyvalent cation-sensing receptor regulates vasopressin-elicited water permeability in rat kidney inner medullary collecting duct. J Clin Invest. 1997;99:1399–405. doi: 10.1172/JCI119299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bustamante M, Hasler U, Leroy V, et al. Calcium-sensing receptor attenuates AVP-induced aquaporin-2 expression via a calmodulin-dependent mechanism. J Am Soc Nephrol. 2008;19:109–16. doi: 10.1681/ASN.2007010092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao R, Patel S, Hao C, et al. GSK3beta mediates renal response to vasopressin by modulating adenylate cyclase activity. J Am Soc Nephrol. 2010;21:428–37. doi: 10.1681/ASN.2009060672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen J, Hoffert JD, Knepper MA, et al. Proteomic analysis of lithium-induced nephrogenic diabetes insipidus: mechanisms for aquaporin 2 down-regulation and cellular proliferation. Proc Natl Acad Sci USA. 2008;105:3634–9. doi: 10.1073/pnas.0800001105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth H, Becker KL, Shalhoub RJ, et al. Nephrotoxicity of demethylchlortetracycline hydrochloride. A prospective study. Arch Intern Med. 1967;120:433–5. [PubMed] [Google Scholar]
- Navarro JF, Quereda C, Quereda C, et al. Nephrogenic diabetes insipidus and renal tubular acidosis secondary to foscarnet therapy. Am J Kidney Dis. 1996;27:431–4. doi: 10.1016/s0272-6386(96)90369-8. [DOI] [PubMed] [Google Scholar]
- Metzger NL, Varney Gill KL. Nephrogenic diabetes insipidus induced by two amphotericin B liposomal formulations. Pharmacotherapy. 2009;29:613–20. doi: 10.1592/phco.29.5.613. [DOI] [PubMed] [Google Scholar]
- Skinner R. Chronic ifosfamide nephrotoxicity in children. Med Pediatr Oncol. 2003;41:190–7. doi: 10.1002/mpo.10336. [DOI] [PubMed] [Google Scholar]
- Radin MJ, Yu MJ, Stoedkilde L, et al. Aquaporin-2 regulation in health and disease. Vet Clin Pathol. 2012;41:455–70. doi: 10.1111/j.1939-165x.2012.00488.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutecki GW, Cox JW, Robertson GW, et al. Urinary concentrating ability and antidiuretic hormone responsiveness in the potassium-depleted dog. J Lab Clin Med. 1982;100:53–60. [PubMed] [Google Scholar]
- Berl T, Linas SL, Aisenbrey GA, et al. On the mechanism of polyuria in potassium depletion. The role of polydipsia. J Clin Invest. 1977;60:620–5. doi: 10.1172/JCI108813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marples D, Frokiaer J, Dorup J, et al. Hypokalemia-induced downregulation of aquaporin-2 water channel expression in rat kidney medulla and cortex. J Clin Invest. 1996;97:1960–8. doi: 10.1172/JCI118628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amlal H, Krane CM, Chen Q, et al. Early polyuria and urinary concentrating defect in potassium deprivation. Am J Physiol Renal Physiol. 2000;279:F655–63. doi: 10.1152/ajprenal.2000.279.4.F655. [DOI] [PubMed] [Google Scholar]
- Procino G, Carmosino M, Tamma G, et al. Extracellular calcium antagonizes forskolin-induced aquaporin 2 trafficking in collecting duct cells. Kidney Int. 2004;66:2245–55. doi: 10.1111/j.1523-1755.2004.66036.x. [DOI] [PubMed] [Google Scholar]
- Procino G, Mastrofrancesco L, Tamma G, et al. Calcium-sensing receptor and aquaporin 2 interplay in hypercalciuria-associated renal concentrating defect in humans. An in vivo and in vitro study. PLoS ONE. 2012;7:e33145. doi: 10.1371/journal.pone.0033145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pretzer SD. Clinical presentation of canine pyometra and mucometra: a review. Theriogenology. 2008;70:359–63. doi: 10.1016/j.theriogenology.2008.04.028. [DOI] [PubMed] [Google Scholar]
- Kenney KJ, Matthiesen DT, Brown NO, et al. Pyometra in cats: 183 cases (1979-1984) J Am Vet Med Assoc. 1987;191:1130–2. [PubMed] [Google Scholar]
- Grinevich V, Knepper MA, Verbalis J, et al. Acute endotoxemia in rats induces down-regulation of V2 vasopressin receptors and aquaporin-2 content in the kidney medulla. Kidney Int. 2004;65:54–62. doi: 10.1111/j.1523-1755.2004.00378.x. [DOI] [PubMed] [Google Scholar]
- Hasler U, Leroy V, Jeon US, et al. NF-kappaB modulates aquaporin-2 transcription in renal collecting duct principal cells. J Biol Chem. 2008;283:28095–105. doi: 10.1074/jbc.M708350200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hocherl K, Schmidt C, Kurt B, et al. Inhibition of NF-kappaB ameliorates sepsis-induced downregulation of aquaporin-2/V2 receptor expression and acute renal failure in vivo. Am J Physiol Renal Physiol. 2010;298:F196–204. doi: 10.1152/ajprenal.90607.2008. [DOI] [PubMed] [Google Scholar]
- Stone EA, Littman MP, Robertson JL, et al. Renal dysfunction in dogs with pyometra. J Am Vet Med Assoc. 1988;193:457–64. [PubMed] [Google Scholar]
- Bouley R, Breton S, Sun T, et al. Nitric oxide and atrial natriuretic factor stimulate cGMP-dependent membrane insertion of aquaporin 2 in renal epithelial cells. J Clin Invest. 2000;106:1115–26. doi: 10.1172/JCI9594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morishita T, Tsutsui M, Shimokawa H, et al. Nephrogenic diabetes insipidus in mice lacking all nitric oxide synthase isoforms. Proc Natl Acad Sci. 2005;102:10616–21. doi: 10.1073/pnas.0502236102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouley R, Pastor-Soler N, Cohen O, et al. Stimulation of AQP2 membrane insertion in renal epithelial cells in vitro and in vivo by the cGMP phosphodiesterase inhibitor sildenafil citrate (Viagra) Am J Physiol Renal Physiol. 2005;288:F1103–12. doi: 10.1152/ajprenal.00337.2004. [DOI] [PubMed] [Google Scholar]
- Sanches TR, Volpini RA, Massola Shimizu MH, et al. Sildenafil reduces polyuria in rats with lithium-induced NDI. AJP Renal Physiol. 2011;302:F216–25. doi: 10.1152/ajprenal.00439.2010. [DOI] [PubMed] [Google Scholar]
- Sugimoto Y, Narumiya S. Prostaglandin E receptors. J Biol Chem. 2006;282:11613–7. doi: 10.1074/jbc.R600038200. [DOI] [PubMed] [Google Scholar]
- Breyer MD, Breyer RM. G Protein–coupled prostanoid receptors and the kidney. Annu Rev Physiol. 2001;63:579–605. doi: 10.1146/annurev.physiol.63.1.579. [DOI] [PubMed] [Google Scholar]
- Olesen ET, Rutzler MR, Moeller HB, et al. Vasopressin-independent targeting of aquaporin-2 by selective E-prostanoid receptor agonists alleviates nephrogenic diabetes insipidus. Proc Natl Acad Sci USA. 2011;108:12949–54. doi: 10.1073/pnas.1104691108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouley R, Lu HA, Nunes P, et al. Calcitonin has a vasopressin-like effect on aquaporin-2 trafficking and urinary concentration. J Am Soc Nephrol. 2011;22:59–72. doi: 10.1681/ASN.2009121267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson AG. DDAVP in the treatment of central diabetes insipidus. N Engl J Med. 1976;294:507–11. doi: 10.1056/NEJM197603042941001. [DOI] [PubMed] [Google Scholar]
- Muller D, Marr N, Ankermann T, et al. Desmopressin for nocturnal enuresis in nephrogenic diabetes insipidus. Lancet. 2002;359:495–7. doi: 10.1016/S0140-6736(02)07667-5. [DOI] [PubMed] [Google Scholar]
- Yoshida M, Iwasaki Y, Asai M, et al. Gene therapy for central diabetes insipidus: effective antidiuresis by muscle-targeted gene transfer. Endocrinology. 2004;145:261–8. doi: 10.1210/en.2003-0366. [DOI] [PubMed] [Google Scholar]
- Soylu A, Kasap B, Ogun N, et al. Efficacy of COX-2 inhibitors in a case of congenital nephrogenic diabetes insipidus. Pediatr Nephrol. 2005;20:1814–7. doi: 10.1007/s00467-005-2057-8. [DOI] [PubMed] [Google Scholar]
- Pattaragarn A, Alon US. Treatment of congenital nephrogenic diabetes insipidus by hydrochlorothiazide and cyclooxygenase-2 inhibitor. Pediatr Nephrol. 2003;18:1073–6. doi: 10.1007/s00467-003-1195-0. [DOI] [PubMed] [Google Scholar]
- Hochberg Z, Even L, Danon A. Amelioration of polyuria in nephrogenic diabetes insipidus due to aquaporin-2 deficiency. Clin Endocrinol. 1998;49:39–44. doi: 10.1046/j.1365-2265.1998.00426.x. [DOI] [PubMed] [Google Scholar]
- Earley LE, Orloff J. The mechanism of antidiuresis associated with the administration of hydrochlorothiazide to patients with vasopressin-resistant diabetes insipidus. J Clin Invest. 1962;41:1988–97. doi: 10.1172/JCI104657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knoers N, Monnens LA. Amiloride-hydrochlorothiazide versus indomethacin-hydrochlorothiazide in the treatment of nephrogenic diabetes insipidus. J Pediatr. 1990;117:499–502. doi: 10.1016/s0022-3476(05)81106-0. [DOI] [PubMed] [Google Scholar]
- Monnens L, Jonkman A, Thomas C. Response to indomethacin and hydrochlorothiazide in nephrogenic diabetes insipidus. Clin Sci. 1984;66:709–15. doi: 10.1042/cs0660709. [DOI] [PubMed] [Google Scholar]
- Libber S, Harrison H, Spector D. Treatment of nephrogenic diabetes insipidus with prostaglandin synthesis inhibitors. J Pediatr. 1986;108:305–11. doi: 10.1016/s0022-3476(86)81010-1. [DOI] [PubMed] [Google Scholar]
- Wade JB. Statins affect AQP2 traffic. AJP Renal Physiol. 2011;301:F308. doi: 10.1152/ajprenal.00248.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Procino G, Barbieri C, Carmosino M, et al. Lovastatin-induced cholesterol depletion affects both apical sorting and endocytosis of aquaporin-2 in renal cells. Am J Physiol Renal Physiol. 2010;298:F266–78. doi: 10.1152/ajprenal.00359.2009. [DOI] [PubMed] [Google Scholar]
- Procino G, Barbieri C, Carmosino M, et al. Fluvastatin modulates renal water reabsorption in vivo through increased AQP2 availability at the apical plasma membrane of collecting duct cells. Pflugers Arch. 2011;462:753–66. doi: 10.1007/s00424-011-1007-5. [DOI] [PubMed] [Google Scholar]
- Procino G, Milano S, Carmosino M, et al. Combination of secretin and fluvastatin ameliorates the polyuria associated with X-linked nephrogenic diabetes insipidus in mice. Kidney Int. 2014;86:127–38. doi: 10.1038/ki.2014.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takai Y, Sasaki T, Matozaki T. Small GTP-binding proteins. Physiol Rev. 2001;81:153–208. doi: 10.1152/physrev.2001.81.1.153. [DOI] [PubMed] [Google Scholar]
- Sidaway JE, Davidson RG, McTaggart F, et al. Inhibitors of 3-hydroxy-3-methylglutaryl-CoA reductase reduce receptor-mediated endocytosis in opossum kidney cells. J Am Soc Nephrol. 2004;15:2258–65. doi: 10.1097/01.ASN.0000138236.82706.EE. [DOI] [PubMed] [Google Scholar]
- Maher BM, Dhonnchu TN, Burke JP, et al. Statins alter neutrophil migration by modulating cellular Rho activity-a potential mechanism for statins-mediated pleotropic effects? J Leukoc Biol. 2009;85:186–93. doi: 10.1189/jlb.0608382. [DOI] [PubMed] [Google Scholar]
- Rikitake Y, Liao JK. Rho GTPases, statins, and nitric oxide. Circ Res. 2005;97:1232–5. doi: 10.1161/01.RES.0000196564.18314.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridley AJ. Rho GTPases and actin dynamics in membrane protrusions and vesicle trafficking. Trends Cell Biol. 2006;16:522–9. doi: 10.1016/j.tcb.2006.08.006. [DOI] [PubMed] [Google Scholar]
- Zelenina M, Christensen BM, Palmer J, et al. Prostaglandin E(2) interaction with AVP: effects on AQP2 phosphorylation and distribution. Am J Physiol Renal Physiol. 2000;278:F388–94. doi: 10.1152/ajprenal.2000.278.3.F388. [DOI] [PubMed] [Google Scholar]
- Laycock JF, Lee J, Lewis AF. The effect of chlorpropamide on water balance in pitressin-treated Brattleboro rats. Br J Pharmacol. 1974;52:253–63. doi: 10.1111/j.1476-5381.1974.tb09708.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall A. Rho GTPases and the control of cell behaviour. Biochem Soc Trans. 2005;33:891–5. doi: 10.1042/BST20050891. [DOI] [PubMed] [Google Scholar]
- Brown LM, Rogers SJ, Cello JP, et al. Cost-effective treatment of patients with symptomatic cholelithiasis and possible common bile duct stones. J Am Coll Surg. 2011;212:1049–60. doi: 10.1016/j.jamcollsurg.2011.02.017. e1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pediconi MF, Gallegos CE, De Los Santos EB, et al. Metabolic cholesterol depletion hinders cell-surface trafficking of the nicotinic acetylcholine receptor. Neuroscience. 2004;128:239–49. doi: 10.1016/j.neuroscience.2004.06.007. [DOI] [PubMed] [Google Scholar]
- Hansen GH, Niels-Christiansen LL, Thorsen E, et al. Cholesterol depletion of enterocytes. Effect on the Golgi complex and apical membrane trafficking. J Biol Chem. 2000;275:5136–42. doi: 10.1074/jbc.275.7.5136. [DOI] [PubMed] [Google Scholar]
- Sparrow CP, Burton CA, Hernandez M, et al. Simvastatin has anti-inflammatory and antiatherosclerotic activities independent of plasma cholesterol lowering. Arterioscler Thromb Vasc Biol. 2001;21:115–21. doi: 10.1161/01.atv.21.1.115. [DOI] [PubMed] [Google Scholar]
- Ni W, Egashira K, Kataoka C, et al. Antiinflammatory and antiarteriosclerotic actions of HMG-CoA reductase inhibitors in a rat model of chronic inhibition of nitric oxide synthesis. Circ Res. 2001;89:415–21. doi: 10.1161/hh1701.096614. [DOI] [PubMed] [Google Scholar]
- Dimmeler S, Aicher A, Vasa M, et al. HMG-CoA reductase inhibitors (statins) increase endothelial progenitor cells via the PI 3-kinase/Akt pathway. J Clin Invest. 2001;108:391–7. doi: 10.1172/JCI13152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kita T, Brown MS, Goldstein JL. Feedback regulation of 3-hydroxy-3-methylglutaryl coenzyme A reductase in livers of mice treated with mevinolin, a competitive inhibitor of the reductase. J Clin Invest. 1980;66:1094–100. doi: 10.1172/JCI109938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo LM, McKee M, Brown D. Methyl-beta-cyclodextrin induces vasopressin-independent apical accumulation of aquaporin-2 in the isolated, perfused rat kidney. Am J Physiol Renal Physiol. 2006;291:F246–53. doi: 10.1152/ajprenal.00437.2005. [DOI] [PubMed] [Google Scholar]
- Wang Y, Thiele C, Huttner WB. Cholesterol is required for the formation of regulated and constitutive secretory vesicles from the trans-Golgi network. Traffic. 2000;1:952–62. doi: 10.1034/j.1600-0854.2000.011205.x. [DOI] [PubMed] [Google Scholar]
- Heiniger HJ, Kandutsch AA, Chen HW. Depletion of L-cell sterol depresses endocytosis. Nature. 1976;263:515–7. doi: 10.1038/263515a0. [DOI] [PubMed] [Google Scholar]
- Krukoff TL, Calaresu FR. Exogenous vasopressin reverses hyperactivity in the hypothalamus of Brattleboro rats. Am J Physiol. 1984;247:R932–5. doi: 10.1152/ajpregu.1984.247.5.R932. [DOI] [PubMed] [Google Scholar]
- Tiwari S, Blasi ER, Heyen JR, et al. Time course of AQP-2 and ENaC regulation in the kidney in response to PPAR agonists associated with marked edema in rats. Pharmacol Res. 2008;57:383–92. doi: 10.1016/j.phrs.2008.03.013. [DOI] [PubMed] [Google Scholar]
- Song J, Knepper MA, Hu X, et al. Rosiglitazone activates renal sodium- and water-reabsorptive pathways and lowers blood pressure in normal rats. J Pharmacol Exp Ther. 2004;308:426–33. doi: 10.1124/jpet.103.058008. [DOI] [PubMed] [Google Scholar]
- McKenney JM. Pharmacologic characteristics of statins. Clin Cardiol. 2003;26:III32–8. doi: 10.1002/clc.4960261507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulsen L, Holm C, Bech JN, et al. Effects of statins on renal sodium and water handling. Nephrol Dial Transplant. 2008;23:1556–61. doi: 10.1093/ndt/gfm807. [DOI] [PubMed] [Google Scholar]
- Alsheikh-Ali AA, Ambrose MS, Kuvin JT, et al. The safety of rosuvastatin as used in common clinical practice: a postmarketing analysis. Circulation. 2005;111:3051–7. doi: 10.1161/CIRCULATIONAHA.105.555482. [DOI] [PubMed] [Google Scholar]
- Vidt DG, Cressman MD, Harris S, et al. Rosuvastatin-induced arrest in progression of renal disease. Cardiology. 2004;102:52–60. doi: 10.1159/000077704. [DOI] [PubMed] [Google Scholar]
- Brewer HB., Jr Benefit-risk assessment of Rosuvastatin 10 to 40 milligrams. Am J Cardiol. 2003;92:23K–9K. doi: 10.1016/s0002-9149(03)00779-3. [DOI] [PubMed] [Google Scholar]
- Kostapanos MS, Milionis HJ, Saougos VG, et al. Dose-dependent effect of rosuvastatin treatment on urinary protein excretion. J Cardiovasc Pharmacol Ther. 2007;12:292–7. doi: 10.1177/1074248407306676. [DOI] [PubMed] [Google Scholar]
- Wang L, Ellis MJ, Gomez JA, et al. Mechanisms of the proteinuria induced by Rho GTPases. Kidney Int. 2012;81:1075–85. doi: 10.1038/ki.2011.472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis S, Mellor H. Regulation of endocytic traffic by rho family GTPases. Trends Cell Biol. 2000;10:85–8. doi: 10.1016/s0962-8924(99)01710-9. [DOI] [PubMed] [Google Scholar]
- Pizon V, Desjardins M, Bucci C, et al. Association of Rap1a and Rap1b proteins with late endocytic/phagocytic compartments and Rap2a with the Golgi complex. J Cell Sci. 1994;107(Pt 6):1661–70. doi: 10.1242/jcs.107.6.1661. [DOI] [PubMed] [Google Scholar]
- Verhulst A, D'Haese PC, De Broe ME. Inhibitors of HMG-CoA reductase reduce receptor-mediated endocytosis in human kidney proximal tubular cells. J Am Soc Nephrol. 2004;15:2249–57. doi: 10.1097/01.ASN.0000136778.32499.05. [DOI] [PubMed] [Google Scholar]
- Grundy SM. The issue of statin safety: where do we stand? Circulation. 2005;111:3016–9. doi: 10.1161/CIRCULATIONAHA.105.557652. [DOI] [PubMed] [Google Scholar]
- Garcia-Rodriguez LA, Masso-Gonzalez EL, Wallander MA, et al. The safety of rosuvastatin in comparison with other statins in over 100,000 statin users in UK primary care. Pharmacoepidemiol Drug Saf. 2008;17:943–52. doi: 10.1002/pds.1603. [DOI] [PubMed] [Google Scholar]
- Garcia-Rodriguez LA, Gonzalez-Perez A, Stang MR, et al. The safety of rosuvastatin in comparison with other statins in over 25,000 statin users in the Saskatchewan Health Databases. Pharmacoepidemiol Drug Saf. 2008;17:953–61. doi: 10.1002/pds.1602. [DOI] [PubMed] [Google Scholar]
- Guthrie RM, Martin DR. The safety of rosuvastatin: effects on renal and hepatic function. Expert Opin Drug Saf. 2007;6:573–81. doi: 10.1517/14740338.6.5.573. [DOI] [PubMed] [Google Scholar]
- McAfee AT, Ming EE, Seeger JD, et al. The comparative safety of rosuvastatin: a retrospective matched cohort study in over 48,000 initiators of statin therapy. Pharmacoepidemiol Drug Saf. 2006;15:444–53. doi: 10.1002/pds.1281. [DOI] [PubMed] [Google Scholar]
- Kassimatis TI, Konstantinopoulos PA. The role of statins in chronic kidney disease (CKD): friend or foe? Pharmacol Ther. 2009;122:312–23. doi: 10.1016/j.pharmthera.2009.03.008. [DOI] [PubMed] [Google Scholar]
- McKenney JM, Davidson MH, Jacobson TA, et al. Final conclusions and recommendations of the National Lipid Association Statin Safety Assessment Task Force. Am J Cardiol. 2006;97:89C–94C. doi: 10.1016/j.amjcard.2006.02.030. [DOI] [PubMed] [Google Scholar]
- Shepherd J, Vidt DG, Miller E, et al. Safety of rosuvastatin: update on 16,876 rosuvastatin-treated patients in a multinational clinical trial program. Cardiology. 2007;107:433–43. doi: 10.1159/000100908. [DOI] [PubMed] [Google Scholar]
- Sandhu S, Wiebe N, Fried LF, et al. Statins for improving renal outcomes: a meta-analysis. J Am Soc Nephrol. 2006;17:2006–16. doi: 10.1681/ASN.2006010012. [DOI] [PubMed] [Google Scholar]
- Tonelli M, Moye L, Sacks FM, et al. Pravastatin for secondary prevention of cardiovascular events in persons with mild chronic renal insufficiency. Ann Intern Med. 2003;138:98–104. doi: 10.7326/0003-4819-138-2-200301210-00010. [DOI] [PubMed] [Google Scholar]
- Verhulst A, Geryl H, D'Haese P. No evidence for statin-induced proteinuria in healthy volunteers as assessed by proteomic analysis. J Biomed Biotechnol. 2011;2011:456076. doi: 10.1155/2011/456076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson SH, Chade AR, Feldstein A, et al. Lipid-lowering-independent effects of simvastatin on the kidney in experimental hypercholesterolaemia. Nephrol Dial Transplant. 2003;18:703–9. doi: 10.1093/ndt/gfg143. [DOI] [PubMed] [Google Scholar]
- Kido M, Ando K, Oba S, et al. Renoprotective effect of pravastatin in salt-loaded Dahl salt-sensitive rats. Hypertens Res. 2005;28:1009–15. doi: 10.1291/hypres.28.1009. [DOI] [PubMed] [Google Scholar]
- van Dijk MA, Kamper AM, van Veen S, et al. Effect of simvastatin on renal function in autosomal dominant polycystic kidney disease. Nephrol Dial Transplant. 2001;16:2152–7. doi: 10.1093/ndt/16.11.2152. [DOI] [PubMed] [Google Scholar]
- Athyros VG, Mikhailidis DP, Liberopoulos EN, et al. Effect of statin treatment on renal function and serum uric acid levels and their relation to vascular events in patients with coronary heart disease and metabolic syndrome: a subgroup analysis of the GREek Atorvastatin and Coronary heart disease Evaluation (GREACE) Study. Nephrol Dial Transplant. 2007;22:118–27. doi: 10.1093/ndt/gfl538. [DOI] [PubMed] [Google Scholar]
- Atthobari J, Brantsma AH, Gansevoort RT, et al. The effect of statins on urinary albumin excretion and glomerular filtration rate: results from both a randomized clinical trial and an observational cohort study. Nephrol Dial Transplant. 2006;21:3106–14. doi: 10.1093/ndt/gfl244. [DOI] [PubMed] [Google Scholar]
- Bruckert E, Hayem G, Dejager S, et al. Mild to moderate muscular symptoms with high-dosage statin therapy in hyperlipidemic patients–the PRIMO study. Cardiovasc Drugs Ther. 2005;19:403–14. doi: 10.1007/s10557-005-5686-z. [DOI] [PubMed] [Google Scholar]
- Bjornsson E, Jacobsen EI, Kalaitzakis E. Hepatotoxicity associated with statins: reports of idiosyncratic liver injury post-marketing. J Hepatol. 2012;56:374–80. doi: 10.1016/j.jhep.2011.07.023. [DOI] [PubMed] [Google Scholar]
- Cohen DE, Anania FA, Chalasani N National Lipid Association Statin Safety Task Force Liver Expert P. An assessment of statin safety by hepatologists. Am J Cardiol. 2006;97:77C–81C. doi: 10.1016/j.amjcard.2005.12.014. [DOI] [PubMed] [Google Scholar]
- Scandinavian Simvastatin Survival Study Group. Randomised trial of cholesterol lowering in 4444 patients with coronary heart disease: the Scandinavian Simvastatin Survival Study (4S) Lancet. 1994;344:1383–9. [PubMed] [Google Scholar]
- Pfeffer MA, Keech A, Sacks FM, et al. Safety and tolerability of pravastatin in long-term clinical trials: prospective Pravastatin Pooling (PPP) Project. Circulation. 2002;105:2341–6. doi: 10.1161/01.cir.0000017634.00171.24. [DOI] [PubMed] [Google Scholar]
- Smith CC, Bernstein LI, Davis RB, et al. Screening for statin-related toxicity: the yield of transaminase and creatine kinase measurements in a primary care setting. Arch Intern Med. 2003;163:688–92. doi: 10.1001/archinte.163.6.688. [DOI] [PubMed] [Google Scholar]
- Wagstaff LR, Mitton MW, Arvik BM, et al. Statin-associated memory loss: analysis of 60 case reports and review of the literature. Pharmacotherapy. 2003;23:871–80. doi: 10.1592/phco.23.7.871.32720. [DOI] [PubMed] [Google Scholar]
- Preiss D, Seshasai SR, Welsh P, et al. Risk of incident diabetes with intensive-dose compared with moderate-dose statin therapy: a meta-analysis. JAMA. 2011;305:2556–64. doi: 10.1001/jama.2011.860. [DOI] [PubMed] [Google Scholar]
- Ness GC, Zhao Z, Lopez D. Inhibitors of cholesterol biosynthesis increase hepatic low-density lipoprotein receptor protein degradation. Arch Biochem Biophys. 1996;325:242–8. doi: 10.1006/abbi.1996.0030. [DOI] [PubMed] [Google Scholar]
- Conde K, Vergara-Jimenez M, Krause BR, et al. Hypocholesterolemic actions of atorvastatin are associated with alterations on hepatic cholesterol metabolism and lipoprotein composition in the guinea pig. J Lipid Res. 1996;37:2372–82. [PubMed] [Google Scholar]
- Arad Y, Ramakrishnan R, Ginsberg HN. Lovastatin therapy reduces low density lipoprotein apoB levels in subjects with combined hyperlipidemia by reducing the production of apoB-containing lipoproteins: implications for the pathophysiology of apoB production. J Lipid Res. 1990;31:567–82. [PubMed] [Google Scholar]
- Collins R, Armitage J, Parish S, et al. MRC/BHF Heart Protection Study of cholesterol-lowering with simvastatin in 5963 people with diabetes: a randomised placebo-controlled trial. Lancet. 2003;361:2005–16. doi: 10.1016/s0140-6736(03)13636-7. [DOI] [PubMed] [Google Scholar]
- Marchesi S, Lupattelli G, Siepi D, et al. Short-term atorvastatin treatment improves endothelial function in hypercholesterolemic women. J Cardiovasc Pharmacol. 2000;36:617–21. doi: 10.1097/00005344-200011000-00011. [DOI] [PubMed] [Google Scholar]
- Cannon CP, Braunwald E, McCabe CH, et al. Intensive versus moderate lipid lowering with statins after acute coronary syndromes. N Engl J Med. 2004;350:1495–504. doi: 10.1056/NEJMoa040583. [DOI] [PubMed] [Google Scholar]
- Zwaka TP, Hombach V, Torzewski J. C-reactive protein-mediated low density lipoprotein uptake by macrophages: implications for atherosclerosis. Circulation. 2001;103:1194–7. doi: 10.1161/01.cir.103.9.1194. [DOI] [PubMed] [Google Scholar]
- Kleemann R, Verschuren L, de Rooij BJ, et al. Evidence for anti-inflammatory activity of statins and PPARalpha activators in human C-reactive protein transgenic mice in vivo and in cultured human hepatocytes in vitro. Blood. 2004;103:4188–94. doi: 10.1182/blood-2003-11-3791. [DOI] [PubMed] [Google Scholar]
- Kaski JC. Infection, endothelial dysfunction, and atherogenesis. Circulation. 2003;108:E171–2. doi: 10.1161/01.CIR.0000108169.26553.D7. [DOI] [PubMed] [Google Scholar]
- Callahan AS., 3rd Vascular pleiotropy of statins: clinical evidence and biochemical mechanisms. Curr Atheroscler Rep. 2003;5:33–7. doi: 10.1007/s11883-003-0066-2. [DOI] [PubMed] [Google Scholar]
- Levy Y. Beyond cholesterol lowering: effect of statins on markers of cardiovascular disease. Isr Med Assoc J. 2004;6:490–1. [PubMed] [Google Scholar]
- Fassbender K, Simons M, Bergmann C, et al. Simvastatin strongly reduces levels of Alzheimer's disease beta -amyloid peptides Abeta 42 and Abeta 40 in vitro and in vivo. Proc Natl Acad Sci USA. 2001;98:5856–61. doi: 10.1073/pnas.081620098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snowdon DA, Greiner LH, Mortimer JA, et al. Brain infarction and the clinical expression of Alzheimer disease. The Nun Study. JAMA. 1997;277:813–7. [PubMed] [Google Scholar]
- Tonelli M, Moye L, Sacks FM, et al. Effect of pravastatin on loss of renal function in people with moderate chronic renal insufficiency and cardiovascular disease. J Am Soc Nephrol. 2003;14:1605–13. doi: 10.1097/01.asn.0000068461.45784.2f. [DOI] [PubMed] [Google Scholar]
- Ruan F, Zheng Q, Wang J. Mechanisms of bone anabolism regulated by statins. Biosci Rep. 2012;32:511–9. doi: 10.1042/BSR20110118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan KA, Andrade SE, Boles M, et al. Inhibitors of hydroxymethylglutaryl-coenzyme A reductase and risk of fracture among older women. Lancet. 2000;355:2185–8. doi: 10.1016/S0140-6736(00)02400-4. [DOI] [PubMed] [Google Scholar]
- Wang PS, Solomon DH, Mogun H, et al. HMG-CoA reductase inhibitors and the risk of hip fractures in elderly patients. JAMA. 2000;283:3211–6. doi: 10.1001/jama.283.24.3211. [DOI] [PubMed] [Google Scholar]
- Lawes CM, Thornley S, Young R, et al. Statin use in COPD patients is associated with a reduction in mortality: a national cohort study. Prim Care Respir J. 2012;21:35–40. doi: 10.4104/pcrj.2011.00095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young RP, Hopkins R, Eaton TE. Pharmacological actions of statins: potential utility in COPD. Eur Respir Rev. 2009;18:222–32. doi: 10.1183/09059180.00005309. [DOI] [PubMed] [Google Scholar]
- Gokce MI, Gulpinar O, Ozturk E, et al. Effect of atorvastatin on erectile functions in comparison with regular tadalafil use. A prospective single-blind study. Int Urol Nephrol. 2012;44:683–7. doi: 10.1007/s11255-012-0126-z. [DOI] [PubMed] [Google Scholar]
- La Vignera S, Condorelli RA, Vicari E, et al. Statins and erectile dysfunction: a critical summary of current evidence. J Androl. 2012;33:552–8. doi: 10.2164/jandrol.111.015230. [DOI] [PubMed] [Google Scholar]
- Di Ciaula A, Wang DQ, Wang HH, et al. Targets for current pharmacologic therapy in cholesterol gallstone disease. Gastroenterol Clin North Am. 2010;39:245–64. doi: 10.1016/j.gtc.2010.02.005. viii-ix. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodmer M, Brauchli YB, Krahenbuhl S, et al. Statin use and risk of gallstone disease followed by cholecystectomy. JAMA. 2009;302:2001–7. doi: 10.1001/jama.2009.1601. [DOI] [PubMed] [Google Scholar]
- Bouley R, Hasler U, Lu HA, et al. Bypassing vasopressin receptor signaling pathways in nephrogenic diabetes insipidus. Semin Nephrol. 2008;28:266–78. doi: 10.1016/j.semnephrol.2008.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SW, Kim JW, Choi KC, et al. Indomethacin enhances shuttling of aquaporin-2 despite decreased abundance in rat kidney. J Am Soc Nephrol. 2004;15:2998–3005. doi: 10.1097/01.ASN.0000145877.28811.82. [DOI] [PubMed] [Google Scholar]
- Shaw S, Marples D. N-ethylmaleimide causes aquaporin-2 trafficking in the renal inner medullary collecting duct by direct activation of protein kinase A. Am J Physiol Renal Physiol. 2005;288:F832–9. doi: 10.1152/ajprenal.00041.2004. [DOI] [PubMed] [Google Scholar]
- Schrier RW, Ohara M, Rogachev B, et al. Aquaporin-2 water channels and vasopressin antagonists in edematous disorders. Mol Genet Metab. 1998;65:255–63. doi: 10.1006/mgme.1998.2765. [DOI] [PubMed] [Google Scholar]
- Batlle DC, von Riotte AB, Gaviria M, et al. Amelioration of polyuria by amiloride in patients receiving long-term lithium therapy. N Engl J Med. 1985;312:408–14. doi: 10.1056/NEJM198502143120705. [DOI] [PubMed] [Google Scholar]
- Kirchlechner V, Koller DY, Seidl R, et al. Treatment of nephrogenic diabetes insipidus with hydrochlorothiazide and amiloride. Arch Dis Child. 1999;80:548–52. doi: 10.1136/adc.80.6.548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim GH, Lee JW, Oh YK, et al. Antidiuretic effect of hydrochlorothiazide in lithium-induced nephrogenic diabetes insipidus is associated with upregulation of aquaporin-2, Na-Cl co-transporter, and epithelial sodium channel. J Am Soc Nephrol. 2004;15:2836–43. doi: 10.1097/01.ASN.0000143476.93376.04. [DOI] [PubMed] [Google Scholar]
- Stokes JB. Integrated actions of renal medullary prostaglandins in the control of water excretion. Am J Physiol. 1981;240:F471–80. doi: 10.1152/ajprenal.1981.240.6.F471. [DOI] [PubMed] [Google Scholar]
- Berl T, Raz A, Wald H, et al. Prostaglandin synthesis inhibition and the action of vasopressin: studies in man and rat. Am J Physiol. 1977;232:F529–37. doi: 10.1152/ajprenal.1977.232.6.F529. [DOI] [PubMed] [Google Scholar]
- Bernier V, Morello JP, Zarruk A, et al. Pharmacologic chaperones as a potential treatment for X-linked nephrogenic diabetes insipidus. J Am Soc Nephrol. 2006;17:232–43. doi: 10.1681/ASN.2005080854. [DOI] [PubMed] [Google Scholar]
- Morello JP, Salahpour A, Laperriere A, et al. Pharmacological chaperones rescue cell-surface expression and function of misfolded V2 vasopressin receptor mutants. J Clin Invest. 2000;105:887–95. doi: 10.1172/JCI8688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jean-Alphonse F, Perkovska S, Frantz MC, et al. Biased agonist pharmacochaperones of the AVP V2 receptor may treat congenital nephrogenic diabetes insipidus. J Am Soc Nephrol. 2009;20:2190–203. doi: 10.1681/ASN.2008121289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robben JH, Kortenoeven ML, Sze M, et al. Intracellular activation of vasopressin V2 receptor mutants in nephrogenic diabetes insipidus by nonpeptide agonists. Proc Natl Acad Sci USA. 2009;106:12195–200. doi: 10.1073/pnas.0900130106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robben JH, Sze M, Knoers NV, et al. Functional rescue of vasopressin V2 receptor mutants in MDCK cells by pharmacochaperones: relevance to therapy of nephrogenic diabetes insipidus. Am J Physiol Renal Physiol. 2007;292:F253–60. doi: 10.1152/ajprenal.00247.2006. [DOI] [PubMed] [Google Scholar]
- Los EL, Deen PM, Robben JH. Potential of nonpeptide (ant)agonists to rescue vasopressin V2 receptor mutants for the treatment of X-linked nephrogenic diabetes insipidus. J Neuroendocrinol. 2010;22:393–9. doi: 10.1111/j.1365-2826.2010.01983.x. [DOI] [PubMed] [Google Scholar]
- Yang B, Zhao D, Verkman AS. Hsp90 inhibitor partially corrects nephrogenic diabetes insipidus in a conditional knock-in mouse model of aquaporin-2 mutation. FASEB J. 2009;23:503–12. doi: 10.1096/fj.08-118422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohara E, Rai T, Yang SS, et al. Pathogenesis and treatment of autosomal-dominant nephrogenic diabetes insipidus caused by an aquaporin 2 mutation. Proc Natl Acad Sci USA. 2006;103:14217–22. doi: 10.1073/pnas.0602331103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Rouffignac C, Elalouf JM. Effects of calcitonin on the renal concentrating mechanism. Am J Physiol. 1983;245:F506–11. doi: 10.1152/ajprenal.1983.245.4.F506. [DOI] [PubMed] [Google Scholar]
- Hoffert JD, Fenton RA, Moeller HB, et al. Vasopressin-stimulated increase in phosphorylation at Ser269 potentiates plasma membrane retention of aquaporin-2. J Biol Chem. 2008;283:24617–27. doi: 10.1074/jbc.M803074200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bichet DG. Nephrogenic and central diabetes insipidus. In: Coffman TM, Falk RJ, Molitoris BA, Neilson EG, editors. Schrier's diseases of the kidney. Philadelphia: Lippincott Williams & Wilkins; 2012. pp. 2055–81. 9th ed. [Google Scholar]
- Eckardt KU, Coresh J, Devuyst O, et al. Evolving importance of kidney disease: from subspecialty to global health burden. Lancet. 2013;382:158–69. doi: 10.1016/S0140-6736(13)60439-0. [DOI] [PubMed] [Google Scholar]