

Abstract
Proteins and peptides are widely indicated in many diseased states. Parenteral route is the most commonly employed method of administration for therapeutic proteins and peptides. However, requirement of frequent injections due to short in vivo half-life results in poor patient compliance. Non-invasive drug delivery routes such as nasal, transdermal, pulmonary, and oral offer several advantages over parenteral administration. Intrinsic physicochemical properties and low permeability across biological membrane limit protein delivery via non-invasive routes. One of the strategies to improve protein and peptide absorption is by delivering through nanostructured delivery carriers. Among nanocarriers, polymeric nanoparticles (NPs) have demonstrated significant advantages over other delivery systems. This article summarizes the application of polymeric NPs for protein and peptide drug delivery following oral, nasal, pulmonary, parenteral, transdermal, and ocular administrations.
Keywords: Brain, hydrophobic ion-pairing (HIP) complex, nasal, ocular, parenteral, polymeric nanoparticles, protein and peptide drug delivery, pulmonary, transdermal
Introduction
Recent advancements in biotechnology have resulted in the development of novel therapeutically active proteins and peptides. Some of the recently approved proteins and peptides are listed in Table 1. These potent therapeutics are indicated for several chronic conditions such as cancer, hepatitis, diabetes, rheumatoid arthritis, and leukemia [1]. However, several pharmaceutical and biopharmaceutical challenges limit their clinical application. Parenteral administration is the major route for administration of therapeutic proteins and peptides [2]. However, requirement of frequent injections due to short in vivo half-life of proteins and peptides is a major concern. Moreover, in some chronic conditions, frequent injections are required for a longer period of time leading to poor patient compliance [3]. Administration of proteins and peptides by oral, pulmonary, transdermal and nasal routes are few of the potential alternatives to parenteral injections. Among non-invasive routes, oral delivery is the primary route for administration of small molecules however; it is not preferable for proteins and peptides. Oral delivery of proteins and peptides is highly challenging due to presence of proteolytic enzymes in gastrointestinal tract (GIT) and their intrinsic physicochemical and biological properties like poor stability in lower pH of gastric fluid, large molecular size and poor permeation across gastrointestinal membrane [4]. Nasal and pulmonary administration received considerable attention because of low proteolytic activity relative to oral route, highly vascularized and large absorptive surfaces especially in the lungs resulting in improved absorption. Large size and proteolytic instability are major factors for poor absorption of therapeutic proteins across nasal and pulmonary mucosal surfaces. Moreover, physiological barriers such as mucociliary clearance may further limit protein and peptide absorption [4]. Similarly, hydrophilicity and large molecular size also limit transdermal protein delivery [4].
Table 1. Recently approved protein and peptide therapeutics [9].
| Protein or peptide | Brand name, manufacturer | Year of FDA approval | Molecular weight | Route of administration | Half-life | Target disease |
|---|---|---|---|---|---|---|
| Ziv-aflibercept | Zaltrap®, Regeneron and Sanofi | 2012 | 115 kDa | Intravenous infusion | 4-7 days | Metastatic colorectal cancer |
| Ocriplasmin | Jetrea®, ThromboGenics Inc. | 2012 | 27.2 kDa | Intravitreal injection | NA | Symptomatic vitreomacular adhesion |
| Raxibacumab | Abthrax®, GlaxoSmith Kline | 2012 | 146 kDa | Intravenous infusion | 16-19 days | Inhalational anthrax |
| Belimumab | Benlysta®, Human Genome Sciences, Inc. | 2011 | 147 kDa | Intravenous infusion | 19.4 days | Systemic lupus erythematosus |
| Ipilimumab | Yervoy ®, E.R. Squibb & Sons, L.L.C | 2011 | 148 kDa | Intravenous infusion | 15.4 days | Unresectable or metastatic melanoma |
| Belatacept | Nulojix®, E.R. Squibb & Sons, L.L.C | 2011 | 90 kDa | Intravenous infusion | 8-10 days | Prophylaxis of organ rejection (kidney transplant) |
| Brentuximab vedotin | Adcetris®, Seattle Genetics, Inc. | 2011 | 153 kDa | Intravenous infusion | 4-6 days | Hodgkin lymphoma and systemic anaplastic large cell lymphoma |
| Asparaginase Erwiniachryanthemi | Erwinaze, Jazz Pharmaceuticals, Inc. | 2011 | 35 kDa | Intramuscular injection | 16 h | Acute lymphoblastic leukemia |
| Aflibercept | Eylea®, Regeneron Pharmaceuticals, Inc. | 2011 | 115 kDa | Intravitreal injection | 5-6 days | Neovascular (Wet) age-related macular degeneration (AMD), Macular edema following central retinal vein occlusion (CRVO) |
| Velaglucerase alfa | Vpriv®, Shire US Manufacturing Inc. | 2010 | 63 kDa | Intravenous infusion | 11-12 min | Type 1 Gaucher disease |
| Tesamorelin | Egrifta®, EMD Serono, Inc. | 2010 | ∼5 kDa | Subcutaneous injection | 26-38 min | Lipodystrophy |
| Tocilizumab | Actemra®, Genen-tech, Inc. | 2010 | 148 kDa | Subcutaneous injection and Intravenous infusion | 6.3 days | Rheumatoid and systemic juvenile idiopathic arthritis |
| Collagenase clostridium histolyti-cum | Xiaflex®, Auxilium Pharmaceuticals, Inc. | 2010 | ∼113 kDa | Intralesional injection | NA | Dupuytren's contracture |
| Alglucosidase alfa | Lumizyme®, Genzyme Corporation | 2010 | 109 kDa | Intravenous infusion | 2.4 h | Pompe disease |
| Denosumab | Prolia®, Amgen Inc. | 2010 | 147 kDa | Subcutaneous injection | 25.4 days | Postmenopausal osteoporosis |
| IncobotulinumtoxinA | Xeomin, Merz Aesthetics. Inc. | 2010 | 150 kDa | Intramuscular injection | NA | Cervical dystonia |
| Pegloticase | Krystexxa®, Savient Pharmaceuticals, Inc. | 2010 | 540 kDa | Intravenous infusion | NA | Chronic gout |
NA: not available, Source: labels.fda.gov.
To overcome these challenges, proteins and peptides can be delivered efficiently by encapsulating in carrier systems such as microparticles, polymeric NPs, liposomes and solid lipid NPs. In this aspect, polymeric NPs offer unique advantages over other carrier systems. Smaller size relative to microspheres renders polymeric NPs a suitable drug carrier for parenteral administration. Moreover, it has been generally observed that NPs can translocate efficiently across epithelial surfaces relative to microparticles [5, 6]. Polymeric NPs also exhibit high stability in biological fluids compared to liposomes and solid lipid NPs. In addition, versatility of formulation, sustained release, subcellular size, protection of encapsulated proteins and peptides from enzymatic degradation and tissue biocompatibility render NPs as a promising delivery system for protein and peptide delivery [6]. Moreover, physicochemical properties (e.g. hydrophobicity, surface charge), drug release profile and biological behavior (e.g. bioadhesion, targeted drug delivery, improved cellular uptake) can be modulated by application of various polymeric materials and targeting ligands [7, 8].
In this review, we have made an attempt to summarize few of the recent (five to six years) development and application of polymeric NPs for the delivery of proteins and peptides via oral, transdermal, ocular, parenteral, pulmonary and nasal routes. Barriers to proteins and peptides delivery by various routes and advantages of polymeric NPs in overcoming delivery barriers have been summarized in Table 2.
Table 2. Barriers to protein and peptide delivery and advantages of polymeric NPs.
| Route of Administration | Advantages | Barriers | Advantages of polymeric NPs |
|---|---|---|---|
| Oral |
|
|
|
| Ocular |
|
|
|
| Transdermal |
|
|
|
| Parenteral |
|
|
|
| Pulmonary |
|
|
|
| Nasal |
|
|
|
GIT: Gastrointestinal tract.
Polymeric NPs as Carriers for Proteins and Peptides
Polymeric NPs are solid colloidal carriers composed of synthetic, semi-synthetic or natural polymers with size ranging from 10 to 1000 nm [10, 11]. These carriers are usually categorized as either nanospheres or nanocapsules. In nanospheres, drug is dispersed in polymeric matrix whereas nanocapsules are reservoir system in which drug is confined within a polymeric shell. Both polymeric nanospheres and nanocapsules have been explored for the delivery of protein and peptide therapeutics. Properties of polymeric NPs are significantly affected by nature of polymers either natural or synthetic and the method of preparation. A few examples of commonly employed natural polymers include chitosan (CS), gelatin and alginate [7]. These polymers are abundantly present in nature and have been extensively applied in oral proteins and peptides delivery. Among natural polymers, CS has shown most interesting potential, which is attributed to its better solubility at the intestinal pH, improving mucoadhesivness and permeation enhancement. In the small intestine, CS NPs can adhere to and infiltrate into mucus layer and open the tight junctions between contiguous epithelial cells [12]. Furthermore, pH-sensitive CS NPs can disintegrate and release the encapsulated drugs which then penetrate through the opened paracellular pathway. Examples of synthetic polymers investigated for proteins and peptides delivery are poly(DL-lactide co-glycolide) (PLGA), polycaprolactone (PCL), poly(lactic acid) (PLA), polyalkylcyanoacrylate, polyacrylic acid (PAA) and poly(methyl methacrylates) [7, 13, 14]. Among synthetic polymers, polyesters alone or in combination are the most relevant and most frequently studied for protein delivery. Contrary to natural polymers, synthetic polymers enable adjustable controlled drug release for a period of several days to weeks.
Proteins can be encapsulated, adsorbed or chemically linked to the surface of polymeric NPs. Protein incorporation in polymeric NPs can be achieved by various methods. Natural polymers are generally more sensitive to processing conditions. Therefore, NPs with natural polymers are generated using mild techniques including ionic gelation, polyelectrolyte complexation and coacervation [15-17]. NPs composed of synthetic polymers are normally prepared by more extensive techniques such as interfacial polymerization, emulsification–polymerization, emulsification-solvent evaporation, nanoprecipitation, salting out, supercritical fluids and emulsification solvent diffusion [18-24]. Preparation methods for NPs have been broadly reviewed in the literature and hence briefly discussed in the following section [18].
Emulsion Solvent Evaporation Method
Double emulsion solvent evaporation (w/o/w) is the most widely used method for loading biotherapeutics into polymeric NPs. In this method, an aqueous protein solution is first emulsified in polymer containing organic phase. Then, w/o/w emulsion is produced by adding w/o emulsion into an aqueous phase with emulsifier. The organic solvent is usually removed by evaporation. Emulsion solvent evaporation method provides unique advantages including control of particle size and release rate. However, major problems with this method are poor encapsulation and protein instability. Proteins are destabilized at aqueous-organic interface and shear stress which can lead to protein unfolding and aggregation [25]. Denatured and aggregated proteins are reported to produce immunogenic reactions and toxicity [26]. Addition of stabilizing excipients has been widely employed to stabilize protein at aqueous-organic interface. Excipients such as bovine serum albumin (BSA), sugars and polyethylene glycol (PEG) have demonstrated protein stabilization effect during NPs formulation [27-29]. As an alternative to w/o/w method, solid-in oil-in water (s/o/w) method is also used in order to avoid water-organic solvent interface and related protein denaturation. In the s/o/w method, the lyophilized protein powder is dispersed in an organic phase containing the dissolved polymer and this suspension is emulsified in an aqueous solution containing an emulsifying agent. Organic solvent is then evaporated and NPs are washed followed by freeze drying. Several investigators have reported that hydrophobic ion-pairing (HIP) complexation is an excellent strategy to stabilize protein during NPs fabrication and improve encapsulation efficiency. Recently, Gaudana et al. prepared BSA-loaded NPs by combining s/o/w method with HIP complexation approach [30]. In this study, BSA was ion-paired with dextran sulfate to form HIP complex. The complex was freeze dried and loaded into PLGA NPs by s/o/w method. Significantly high entrapment of BSA in NPs was achieved with this method. Also, secondary and tertiary structures of BSA maintained following HIP complexation and NPs preparation. Similarly, higher encapsulation of human IgG-Fab fragment in PLGA NPs was achieved following HIP complexation with dextran sulfate [31]. In another study, Rastogi et al. have prepared insulin-loaded PCL– PEG–PCL NPs utilizing modified s/o/w method in order to achieve higher protein loading. Insulin was hydrophobically modified with sodium deoxycholate. The lipophilicity of insulin following ion-pairing with sodium deoxycholate was enhanced by 5-fold resulting in an increase in entrapment efficiency by 10–50% compared to free insulin [32].
Nanoprecipitation Method
In this method, polymers and drugs are dissolved in a polar, water-miscible solvent (DMSO, acetone, or ethanol). The solution is added in a drop by drop manner into an aqueous solution with surfactant. NPs are formed instantaneously by rapid solvent diffusion. Main advantage of the method is that the drug molecules can be encapsulated without subjecting to shearing stress and high temperatures. The method was originally developed for encapsulation of hydrophobic molecules [7, 33]. Later on, Bilati et al. have modified this technique to encapsulate hydrophilic proteins and peptides into PLGA NPs [24]. NPs with small size and narrow size distribution can be easily obtained by this method. Similar to s/o/w method, protein in HIP complex form can also be incorporated into NPs by this technique. Combination of HIP complexation with nanoprecipitation have demonstrated enhancement in encapsulation of proteins such as human IgG-Fab fragment and lysozyme [31, 34]. In a recent study, Gaudana et al. have hydophobically ion-paired lysozyme with dextran sulfate and then the complex was loaded into PLGA NPs by nanoprecipitation [34]. Higher lysozyme encapsulation was achieved and released lysozyme maintained its biological activity.
Ionic Gelation and Cocervation Techniques
Ionic gelation and cocervation techniques are mainly employed for preparing NPs composed of natural polymers (CS, gelatin and sodium alginate). In ionic gelation, CS is dissolved in acetic acid (presence/absence of stabilizer) followed by the addition of polyanion or anionic polymer under mechanical stirring at room temperature [35]. The preparation conditions are mild and protein can be encapsulated without use of organic solvents or elevated temperature. However, it is difficult to achieve long term controlled release due to solubility of polymers. Some of the parameters affecting protein encapsulation by ionic gelation method are molecular weight of polymer, initial protein concentration and polymer concentration. It is reported that BSA encapsulation efficiency was enhanced around two times with increasing molecular weight of chitosan from 10 to 210 kDa. Protein and polymer concentration have demonstrated inverse effect on protein encapsulation efficiency [36].
Supercritical Fluids
Recently, NPs are also prepared utilizing supercritical fluids. In this technique, drug and polymer are first dissolved in supercritical fluid and the solution is expanded through a nozzle. The supercritical fluid is evaporated using spraying process which eventually leads to precipitation of solute particles. Advantages of this technique include processing of biolabile pharmaceuticals under mild operating conditions, flexibility in procedures, and elimination of organic solvent in the final product. Elvassore et al. produced insulin-loaded PLA NPs with high product yield and maintenance of >80% of the insulin hypoglycemic activity using this method [37]. The major problems observed with this method are requirement for special equipment, poor solubility of high molecular mass (10,000) polymers and strong polar substances in supercritical CO2 [38].
Application of Polymeric NPs in Delivery of Proteins and Peptides
Development and application of polymeric nanoparticulate carriers for the delivery of proteins and peptides via different delivery routes such as oral, pulmonary, nasal, parenteral, transdermal, and ocular have been discussed in this section.
Oral Delivery
Oral administration is the most preferred route due to its advantages such as patient convenience and compliance, avoidance of contamination and infections caused by the use of injections [39]. However, proteins and peptides exhibit poor oral bioavailability due to lower permeation across intestinal epithelium, aggregation and denaturation. Hindrance to oral administration of protein and peptide can be categorized as physical, chemical and enzymatic barriers. The physical barrier is attributed mainly to the continuous monolayer of intestinal epithelial cells which highly expresses intercellular tight junctions. Fig. 1 depicts the structure of intestinal epithelia. The intercellular tight junctions provide high physical integrity to intestinal epithelial cells [40]. It comprises of various transmembrane integral proteins, which form a continuous seal between adjacent intestinal cells. The porous structure of tight junctions contains fenestrate with the dimension of 3 to 10 Å [41]. Large molecular size and hydrophilic nature of proteins and peptides pose a challenge to passing through cellular membrane and also limit the passage through paracellular pathway [42, 43]. Physicochemical properties of polymeric NPs can be optimized to facilitate transport across intestinal epithelial cells [44]. Efflux proteins such as P-glycoprotein (P-gp) may act as a barrier and limit intestinal transport [45]. Polymeric NPs can be an excellent drug delivery systems to evade the efflux route as Pgp is incapable of recognizing NPs [46]. Moreover, rapid pH variations in GIT can induce degradation of orally administered proteins and peptides by oxidation, deamidation or hydrolysis. Proteins and peptides are vulnerable to acidic pH in the stomach [44]. Enzymatic degradation by pepsin in the stomach and/or pancreatic proteases in the intestine, and aminopeptidases existing in the brush-border membrane are other major barriers to oral protein and peptide delivery. In addition, pre-systemic degradation of peptides and proteins due to extensive first-pass metabolism may result in low dose fraction to enter the systemic circulation [47, 48]. Consequently, most proteins and peptides exhibit low absorption via oral administration. Polymeric NPs have been thoroughly explored to overcome these barriers and improve the oral absorption of proteins and peptides.
Figure 1.

Diagrammatic representation of intestinal epithelial cells with Peyer's patch, lymphoid follicle and overlying follicle-associated epithelium (FAE); Cells represented in Peyer's patch include enterocytes, goblet, lymphocytes and M cells.
NP Transport Mechanisms
Polymeric NPs can offer an alternative strategy to improve bioavailability of encapsulated proteins and peptides by providing protection towards degradation in gastrointestinal environment, enhancing cellular contact with intestinal membrane, and promoting absorption in the small intestine [49]. NPs could be trapped in the adherent mucus layer or translocated by intestinal epithelial cells. Possible interactions of NPs with the intestinal barrier have been depicted in (Fig. 2). There are two distinct mechanisms for NPs transport across the intestinal epithelium: paracellular pathway (between adjacent cells) and transcellular route.
Figure 2.

Possible interactions of NPs with the intestinal barrier and the different transport mechanisms of NPs uptake: (A) Non-receptor mediated transcellular transport (B) Paracellular transport (C) Receptor-mediated transcellular endocytosis (D) Receptor and M cell medicated phagocytosis (E) Non-receptor M cell mediated phagocytosis.
Paracellular Pathway
Paracellular space represents only about 0.01-0.1% of total absorptive intestinal surface with the dimension in the order of 10 Å. Therefore, transport of macromolecules as well as NPs is severely restricted from crossing mucosal epithelia through paracellular route (Fig. 2). To improve the paracelluar transport, various enhancers such as CS, starch, thiolated polymers and calcium chelators have been successfully employed to reversibly open tight junctions.
Transcellular Transport
Transcellular route for proteins and peptides includes passive transcellular diffusion, carrier-mediated transport, transcytosis by normal enterocytes (with/without receptor-mediated) and phagocytosis by M cells (with/without receptor-mediated). Transcellular transport of NPs across intestinal epithelium generally involves the latter two pathways (Fig. 2).
Transcytosis by Normal Enterocytes
Transcytosis of NPs across the intestinal epithelial cells involves two steps: first, an endocytic process occurring at the apical membrane which is followed by transport processes which allow particles to be delivered across the basolateral membrane [50]. Intestinal epithelium is primarily composed of enterocytes. Translocation of NPs through enterocytes is severely restricted owing to low endocytic capacity of the enterocytes [51, 52].
M Cell-Mediated Phagocytosis
M cells represent only 1% of the total intestinal cells and approximately 5% of the human follicle-associated epithelium (FAE) [53]. M cells mediated phagocytosis is a major route of translocation for orally administered proteins and peptides as well as NPs due to their high transcytotic activity. Several research studies have reported that the majority of particles are translocated in the FAE [54].
Application of Polymeric NPs as a Potential Oral Delivery System for Proteins and Peptides
The application of polymeric NPs for oral protein and peptide delivery has gained significant interest in the last few decades. Recent developments in polymeric NPs based oral delivery of proteins and peptides have been described in Table 3. Several factors need to be considered and optimized to develop efficient nanocarriers for oral delivery. Uptake of NPs through intestinal epithelium is dependent on particle size, surface charge, polymer hydrophobicity, mucoadhesivity, and the presence or absence of surface ligands. Studies on the effect of particle size have demonstrated that transcytosis of NPs is improved with the decrease in particles size [55, 56]. Particles with size ranging from 50 to 500 nm are generally favorable for transport of NPs in GIT [51]. Uptake of NPs by enterocytes or M cells is highly dependent on their size [57]. Smaller particles of less than 50-100 nm in size can be transcytosed by enterocytes, while the particles with size less than 1 μm are likely to be internalized by M cells [58]. NPs surface charge is also a key factor that may alter its entry in enterocytes or M cells. The surface charge can facilitate the proximity of particles to the intestinal epithelium, and may further enhance entry through Peyer's patches. Previously, it has been observed that negatively charged poly(styrene) NPs showed a limited uptake through M cells [59]. NPs with neutral charge and size of 130 and 950 nm were taken up through Peyer's patches. It has also been reported that the NPs with negative or neutral surface charge had higher transport efficiency across Peyer's patches relative to positive charged NPs [60]. Hence, dense coating the surface of NPs with the neutral charged polymer poly(ethylene glycol) (PEG) can minimize the binding of NPs with negatively charged mucin [61]. Particle hydrophobicity is one of the major factors that may affect particle absorption by intestinal epithelium, especially for Peyer's patches. Several reports have demonstrated NPs composed of hydrophobic polymer entered efficiently through Peyer's patches [62, 63]. In addition to change in surface properties, NPs transport across oral mucosa can be improved by application of bioadhesive polymers and surface modification with targeting ligands.
Table 3. tRecent developments in polymeric NPs based oral proteins and peptides delivery.
| Polymer | Protein or Peptide | Results | Ref. |
|---|---|---|---|
| PLGA | Insulin | In case of NPs, six fold higher oral bioavailability relative to plain insulin was noted in healthy rats. An equivalent blood glucose lowering effect from a 120 IU/kg oral dose of insulin NPs and 20 IU/kg subcutaneous dose of insulin solution in diabetic rats was observed. | [80] |
| PLGA-CS | Insulin | Positively charged (+43.1 ± 0.3 mV) CS-PLGA-NPs exhibited stronger bioadhesive potency than negatively charged PLGA-NPs and much higher relative pharmacological availability for orally delivered insulin with no increase in toxicity. | [81] |
| PLGA-eudragit | Salmon-calcitonin | Size ranged from 179.7 to 308.9 nm with a polydispersity index between 0.051 and 2.75, and surface charges ∼ -11 to +6 mV. Polymer type was an important factor influencing the release characteristics and the in vivo hypocalcemic effect. | [82] |
| PLA-F127-PLA | Insulin | Blood glucose concentration of oral insulin-loaded PLA-F127-29 NPs decreased from 18.5 to 5.3 mmol/L after 4.5 h; the minimum blood glucose concentration (about 4.5 mmol/L) was shown within about 5 h; the blood glucose concentration was retained at this level for additional 18.5 h. | [83] |
| PCL/eudragit RS | Insulin, aspart-insulin | Insulin-loaded NPs composed of PCL/Eudragit RS preserved the biological activity of aspart-insulin. The postprandial peak suppression was prolonged more than 24 h compared to regular insulin working only 6-8 h. | [84] |
| PAA | Leuprolide | NPs increased relative oral bioavailability of leuprolide by 4.2-fold. | [13] |
| Insulin | All polymers demonstrated great insulin complexation efficiency (78 and 93%). Quaternised PAA polymer showed lower cytotoxicity than PAA. | [85] | |
| Thiolated PAA | Insulin | Thiolated PAA improved AUC of insulin by 2.3-fold compared with unmodified PAA, which further contributed to a blood sugar reduction of 22%. | [86] |
| CS | Salmon calcitonin | CS nanocapsules significantly reduced the serum calcium levels, and delayed the reduction for more than 24 h, irrespective of the type and molecular weight of CS. | [87] |
| Insulin | The relative bioavailability of insulin was found to be approximately 20%. These results suggest that the formulation developed in the study might be employed as a potential approach for the oral delivery of insulin. | [88] | |
| Insulin | In the pharmacodynamic (PD) and pharmacokinetic (PK) evaluation in a diabetic rat model, the orally administered aspart-insulin-loaded NPs produced a slower hypoglycemic response for a prolonged period of time, whereas the SC injection of aspart-insulin produced a more pronounced hypoglycemic effect for a relatively shorter duration. | [89] | |
| CS/HPMCP | Insulin | In the pharmacodynamic (PD) and pharmacokinetic (PK) evaluation in a diabetic rat model, the orally administered aspart-insulin-loaded NPs produced a slower hypoglycemic response for a prolonged period of time, whereas the SC injection of aspart-insulin produced a more pronounced hypoglycemic effect for a relatively shorter duration. | [90] |
| CS/PGA-DTPA | Insulin | The oral intake of enteric-coated capsule containing CS/γPGA-DTPA NPs produced a prolonged reduction in blood glucose levels, with a maximum insulin concentration at 4 h and 20% relative oral bioavailability of insulin. | [91] |
| TMC-Cys | Insulin | TMC-Cys NPs improved insulin transport through rat intestine by 3.3-11.7 and 1.7-2.6 folds, enhanced Caco-2 cell internalization by 7.5-12.7 and 1.7-3.0 folds, and promoted uptake in Peyer's patches by 14.7-20.9 and 1.7-5.0 folds, as compared to insulin solution and TMC NPs, respectively. | [92] |
| Lauryl succinyl CS | Insulin | Modification of CS with both hydrophilic (succinyl) and hydrophobic (lauryl) moieties improved the release profiles, mucoadhesivity and the permeability of insulin. | [93] |
| Dextran-poloxamer-CS-albumin | Insulin | In the pharmacodynamic and pharmacokinetic studies with 50IU/kg nanoencapsulated insulin, 13% oral bioavailability indicated a 3- fold improvement in comparison to free insulin. | [94] |
| Dextran sulfate CS | rhHGF | rhHGF-loaded DS/CS NP showed potency of liver-targeting after oral administration. | [95] |
| Cholic acid (CA) modified dextran sulfate | Superoxide dismutase (SOD) | Higher degree of CA substitution in DS-CA can remarkably increase the cellular uptake of the loaded SOD. | [96] |
| Alginate/dextran sulfate/CS/albumin | Insulin | Permeation of insulin-loaded NPs was improved by 2.1-fold through Caco-2 cell monolayer, 3.7-fold through a mucus-secreting Caco-2/HT29 co-culture, and 3.9-fold through excised intestinal mucosa of Wistar rats, compared to insulin, respectively. | [97] |
| Alginate | Insulin | The association efficiency and loading capacities were optimized as high as 92% and 14.3%, respectively. Around 50% of the protein was partially retained by NPs in acidic gastric environment up to 24 h while around 75% of release was found under intestinal pH conditions. | [98] |
| Thiolated Eudragit | Insulin | The oral insulin-loaded Eul-cys NPs produced an increased and prolonged hypoglycemic action with 2.8-fold higher relative bioavailability of insulin (7.33 ± 0.33%) compared with Eul NPs (2.65 ± 0.63%). | [99] |
| PNIPPAm | Salmon calcitonin | All the NPs formulation (179.7 to 308.9 nm, -11 to +6 mV) showed efficient sCT encapsulation and release. | [100] |
PNIPPAm: Poly-N-isopropylacrylamide; CS: Chitosan; DTPA: diethylene triamine pentaacetic acid (DTPA); TMC-Cys: Trimethyl chitosan-cysteine conjugate; PASP: poly(L-aspartic acid); PMMA: poly(methyl methacrylate); SOD: Superoxide dismutase.
The use of bioadhesive synthetic (polyacrylates, cellulose derivatives) and natural polymers (alginate, CS derivatives) can provide prolonged interaction of NPs with the intestinal barrier, which extends the residence time for permeation across the intestinal epithelium. Among bioadhesive polymers, CS is most widely investigated for oral protein and peptide delivery. In a recent study, novel nanocarrier based on alginate-dextran sulfate core, complexed with a CS-PEG-albumin shell improved oral delivery of insulin [64]. NPs entered due to high adhesion to enterocytes and especially Peyer's patch region. In vivo oral studies displayed significant reduction of glycaemia for 24 h. In another study, carboxylated CS graft methyl methacrylate NPs have been developed for oral insulin delivery [65]. This nanocarrier exhibited sustained release at lower pH (2.0) relative to higher pH such as 6.8 and 7.4. At 25 IU/kg, the pharmacological bioavailability was 9.7% with desirable tissue and blood compatibility.
Modifying NPs by absorption or covalent attachment of targeting ligands on the surface is an alternative strategy for enhancing NPs transport across intestinal epithelial cells. These targeting ligands can interact with surface receptors on M cells, goblet cells, or enterocytes [66-68]. The targeting moieties studied include lectins, peptides, vitamins and wheat germ agglutinin [66, 67, 69, 70]. Targeted delivery was employed to improve bioavailability and hypoglycemic activity of insulin via oral administration. Insulin-loaded CS NPs were surface modified by conjugation with a synthetic CRTLTVRKC peptide [71]. The peptide significantly enhanced targeting to epithelium and translocation of CS NPs by interaction with a transmembrane protein stabilin-2. These interactions facilitate efficient delivery and higher transport of NPs. Yoo et al. reported the enhancement of oral drug absorption via M cell-targeting CS NPs. NPs were linked with a targeting peptide such as CKSTHPLSC (CKS9). The modified CS NPs linked with CKS9 peptide improved uptake at FAE area of Peyer's patch. In the in vitro transcytosis study across rate small intestine, the CKS9 peptide-modified CS NPs showed more effective transport ability and more specific accumulation into Peyer's patch regions compared with non-modified CS NPs. In another study, goblet cell-targeting NPs of insulin were developed by coupling cell targeting peptide CSKSSDYQC (CSK) with the trimethyl CS chloride surface to augment insulin oral absorption [68]. CSK peptide modification was observed to enhance transport of NPs in villi and facilitate the transition of drug molecules across the cellular membrane. CSK peptide also induced greater internalization of protein/peptides through clathrin and caveolae dependent endocytosis in HT29-MTX cells. CSK peptide modified NPs by oral administration in diabetic rats showed 1.5-fold higher bioavail-ability and greater hypoglycemic effect relative to unmodified NPs.
Recently, drug delivery through colon has emerged as an attractive strategy for delivering large molecular drugs into systemic circulation. The colon is the preferred absorption site for proteins and peptides due to following reasons: (i) less proteolytic activity of colon mucosa compared with small intestine; (ii) less endogenous metabolic enzymes; (iii) longer retention time of colon with 5 days and (iv) significant enhancement by absorption enhancers due to slow transit and stirring [72-75]. However, the colonic drug delivery system of protein and peptide is also complicated and limited due to (i) need of advanced technology, multiple formulation steps and skills of manufacturing; (ii) difficulty in accessing the colon at the distal portion of the alimentary canal; (iii) harsh conditions such as different pH and enzymes in GIT; (iv) drug loss due to binding to dietary residues, intestinal secretions, mucus in colon; (v) possible involvement of microflora in colon which could affect colonic performance via metabolic degradation of protein and peptide drugs; (vi) smaller surface area and tight junctions in the colon restrict drug transport across the mucosa into the systemic circulation [76, 77]. To overcome these challenges, polymeric NPs have been utilized for colon targeted protein and peptide delivery. Recently, Coco et al. formulated three different polymeric NPs such as pH-sensitive Eudragit® NPs, mucoadhesive trimethylchitosan (TMC) NPs, and PLGA-based NPs with targeting ligands to deliver ovalbumin (OVA) to colon for treating inflammatory bowel disease (IBD) [78]. TMC NPs exhibited highest apparent permeability for OVA in the untreated Caco-2 monolayer model, however, there was no difference among three NPs formulation in the inflamed model. Moreover, targeted PLGA NPs presented the highest accumulation of OVA in inflamed mouse colon. These results demonstrate the potential of active targeting of the colon for inflammatory bowel treatment. Laroui et al. has demonstrated the efficacy of tripeptide Lys-Pro-Val (KPV)-loaded NPs of size 400 nm in alginate-chitosan hydrogel for the treatment of IBD via mouse model [79]. The hydrogel collapsed in the colon and NPs quickly released the drug on or within the closed area of colonocytes. KPV-loaded NPs also exhibited significant anti-inflammatory efficacy in mice. Moreover, KPV-loaded NPs generated similar therapeutic efficacy with 12,000-fold lower concentration in the formulation than that of KPV solution. These observations clearly indicate the potential of drug-loaded NPs as an effective therapeutic approach for the treatment of IBD.
Nasal Delivery
Nasal route is commonly explored for non-invasive protein and peptide delivery. Recent advancement in biotechnology, inhalation devices and targeting motifs has considerably raised research interest in protein and peptide delivery via this route. It offers advantages including large surface area, highly vascularized mucosa, porous endothelial membrane, lower enzymatic activity relative to GIT and avoidance of first-pass metabolism [101, 102]. However, pattern of deposition and size distribution through delivery device and nasal clearance mechanisms might pose a significant challenge to protein delivery [103]. Following intranasal administration, proteins can be absorbed directly into systemic circulation, central nervous system (CNS) or across GIT [104]. Miacalcin®, DDAVP®, Synarel®, Fortical® and Syntocinon® are few of the marketed proteins and peptides for nasal administration [9].
Absorption through nasal mucosa is highly dependent on nasal physiology, physicochemical properties of permeants and effectiveness of delivery devices [105]. About 1.5 to 21 mL of mucus generated by serous and seromucous glands forms a double layer of 2-5 μm in thickness [106]. Mucus acts as a first-line defense by filtering and entrapping foreign particles. Entrapped particles are expelled by cilia in the epithelial surface. This process might significantly impair protein absorption through nasal mucosa. Moreover, mucociliary apparatus poses a formidable barrier for nasal absorption. Cilia along with mucus layer generate a fluctuating movement to clear drug in approximately 12-15 min from nasal cavity [107]. In addition to physiological factors, physicochemical properties of permeants play a considerable role in altering absorption across nasal mucosa. Proteins of size greater than 1 kDa have demonstrated overall bioavailability of 0.5 to 5% [108]. Moreover, particles of size 1 μm have been reported to reach blood circulation after intranasal administration [108]. Importantly, lipophilicity and partition coefficient may alter protein and peptide absorption across nasal mucosal surface. In addition, the particle size and distribution pattern may also determine nasal protein absorption. Particles of size 10 μm or higher have been demonstrated to accumulate in the upper respiratory region. On the other hand, particles of less than 0.5 μm in size are carried by the inhaled air to the lungs. Importantly, particles of the size in the range of 5-7 μm significantly accumulate in the nasal cavity [109].
Various polymeric NPs have been examined for their efficacy in improving protein and peptide absorption across nasal mucosa. Chitosan-N-acetyl-L-cysteine (CS-NAC) NPs have been investigated for nasal delivery of insulin [110]. The particle size ranged from 140-210 nm with a zeta potential of 119.5 to 131.7 mV. Particles were spherical in shape and had loading efficiency of 13-42%. CS-NAC NPs displayed an excellent swelling behavior and generated an initial burst followed by slow release of insulin, in vitro. In vivo studies revealed that CS-NAC NPs were more promising in reducing blood glucose levels relative to CS NPs and insulin control solution. Intranasal administration of insulin-loaded CS-NAC and CS particles diminished glucose level to 59% and 74% at 30 and 60 min post dosing, respectively. Total reduction in plasma glucose levels observed with insulin-loaded CS-NAC and CS NPs were 16.2% and 8.3% within 5 h, respectively. These results clearly signify the potential of thiolated CS in improving nasal absorption of proteins such as insulin. Hybrid poly-oligosaccharide NPs comprising of CS and cyclodextrins have been developed and investigated for nasal insulin delivery [111]. These NPs demonstrated high efficacy in reducing transepithelial resistance of nasal membrane reversibly. Moreover, intranasal administration of insulin-loaded hybrid NPs generated more than 35% reduction in glucose levels at 1 h post dosing in conscious rabbits. In contrast, insulin solution resulted in 14% reduction in plasma glucose levels after 30 min of dose administration.
CS and alginate have been employed commonly for formulating NPs due to their excellent biodegradability, biocompatibility, and mucoadhesive properties. Such high bioadhesiveness is beneficial as it will prolong the contact time of NPs with biological membrane. Moreover, CS has been known to enhance absorption across epithelium by causing transient loosening of tight junctions. Recently, chitosan/alginate blended NPs have been developed by Goycoolea et al. [112]. Particle size of insulin-loaded NPs ranged from 273 to 396 nm. In vitro release studies demonstrated nearly 80% insulin release within 20 min. The insulin-loaded NPs generated 35% reduction in plasma glucose level at 45 min post intranasal administration. PEGylated trimethyl CS nanocomplexes have demonstrated high potential in improving nasal insulin absorption [113]. These nanocomplexes have generated approximately 34-37% reduction in plasma glucose levels in rats. Thiolated CS-thioglycolic acid (CS-TGA) NPs have been developed by Shahnaz et al. as a promising drug carrier for leuprolide. Mean particle size of these NPs was observed to be 252 ± 82 nm. Leuprolide was released slowly over a period of 6 h. Thiolated CS NPs generated 5.2-fold higher leuprolide transport across porcine nasal mucosa relative to leuprolide solution. These NPs displayed 6.9 and 3.8- fold higher area under the curve (AUC) and maximum plasma concentration relative to leuprolide solution, respectively.
Nose to Brain Delivery
Protein and peptide brain delivery via nasal route has gained considerable attention as it can bypass blood-brain barrier (BBB) and first-pass metabolism and provides rapid brain absorption due to high blood flow and porous endothelial membrane structures [114]. BBB is considered as the most formidable barrier for drug delivery into the brain from systemic circulation [115]. Hence, direct brain delivery through intranasal route offers a more promising and viable strategy to improve absorption of therapeutic agents which have short plasma half-life. Nasal cavity can be distinguished into nasal vestibular, respiratory and olfactory regions. Olfactory region has been proposed as the major route for brain transport following intranasal administration (Fig. 3) [116-124]. Several polymeric NPs have been investigated for direct and sustained release of proteins and peptides into the brain following intranasal administration.
Figure 3.
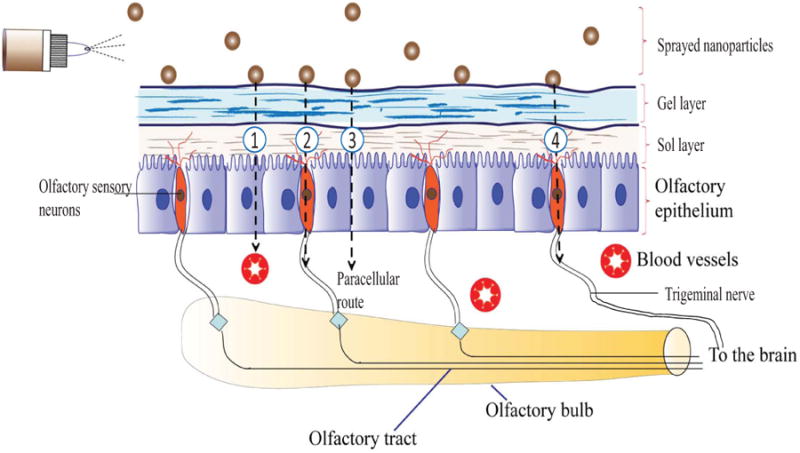
Various transport mechanisms to brain following intranasal administration: (1) Nasal vasculature into the systemic circulation (2) Olfactory tract into the cerebrospinal fluid (CSF) and brain (3) Paravascular route into the brain parenchyma and (4) Trigeminal nerve pathways to the brain stem.
Cheng et al. demonstrated brain delivery of neurotoxin-I with polylactic acid NPs (NT-I-NPs) following intranasal administration [125]. Intranasal administration of NT-I-NPs demonstrated shorter time to reach maximum concentration (Tmax) relative to intravenous (i.v.) administration of NT-I-NPs and NT-I solution. Tmax values observed for intranasally and i.v. administered NT-I-NPs was 65 and 95 min, respectively. Tmax value displayed by NT-I solution was 145 min. Moreover, AUC generated by intranasal administration of NT-I-NPs was significantly higher relative to i.v. administration of NT-I-NPs and NT-I solution. Absolute bioavailability produced by intranasal administration was about 160 and 196% higher relative to i.v. administration of NT-I-NPs and NT-I solution. These results indicate that the brain delivery of peptide loaded NPs can be significantly improved by intranasal administration relative to i.v. administration. Xia et al. have demonstrated the efficacy of low-molecular-weight protamine (LMWP)-modified PEG-PLA NPs to deliver therapeutic agents to CNS via olfactory and trigeminal nerve pathways [126]. Such nanoparticulate formulation could be employed for improving brain protein or peptide absorption.
PLGA NPs loaded with thyrotropin-releasing hormone (TRH) of size 108 ± 12 nm have been developed by Veronesi et al. [127]. Intranasal administration of TRH-loaded NPs significantly raised the stimulations required to reach stage V seizures in rats. Moreover, seizure after duration discharge was observed to be significantly reduced in rats treated with TRH-loaded NPs intranasally. These observations clearly indicate the neuroprotective potential of intranasally administered TRH-loaded NPs. A recent report discussed brain concentrations of neurotoxin-I (NT-I) peptide after intranasal administration as solution and PLA NPs coated with polysorbate-80 [128]. Investigators observed an approximately 3-fold higher accumulation of NT-I with NPs compared to solution. Concentrations of NT-I observed with NPs and solutions were 18.23 ± 3.30 ng/mL and 6.26 ± 0.23 ng/mL, respectively. Moreover, NPs generated about 2-fold higher brain concentration relative to i.v. administration (8.56 ± 0.33 ng/mL). The same group of investigators developed neurotoxin-II (NT-II)-loaded PLA NPs coated with polysorbate-80 [129]. Brain concentrations of NT-II observed after intranasal administration of solution and NPs were 0.26 ±0.03 and 8.66 ± 0.30 ng/g, respectively. NPs generated more than 30-fold higher brain concentrations of NT-II relative to solution. Moreover, NPs generated about 3-fold higher brain concentration relative to i.v. administration (2.50 ±0.21 ng/g). These observations clearly indicate that PLA NPs coated with polysorbate-80 have significant potential to enhance the antinociceptive activity of these peptides after intranasal delivery. Recently, Liu et al. demonstrated the transport of wheat germ agglutinin-modified PEG-PLA NPs into the brain following intranasal administration [130]. Ex vivo imaging analysis clearly displayed that olfactory and trigeminal nerve pathways were primarily responsible for transport of NPs to brain. Investigators also proposed that the brain transport was possibly due to extracellular transport along the nerve fibers.
Lactoferrin receptor has been reported to be overly expressed on the luminal surface of respiratory epithelium, brain capillary endothelial cells and neurons [131]. Moreover, it has been observed to be highly expressed in CNS neurodegenerative conditions such as Alzheimer's, Parkinson's and Huntington's disease. Such high expression could be useful in generating lactoferrin receptor targeted delivery of polymeric NPs to promote absorption of proteins and peptides in these tissues. Lactoferrin functionalized PEG-co-PCL NPs exhibited significantly higher brain accumulation of coumarin-6 relative to unmodified NPs [131]. AUC0-8h of coumarin-6 generated by lactoferrin-modified NPs in rat cerebrum, cerebellum, olfactory tract, olfactory bulb and hippocampus were 1.36, 1.53, 1.70, 1.57 and 1.23-times higher relative to unmodified NPs. Moreover, these functionalized NPs encapsulating a neuroprotective peptide -NAPVSIPQ produced remarkable memory improvement effects at relatively lower dose than unmodified NPs. These observations demonstrate the utilization of nanoparticulate based targeted approach to improve brain delivery and efficacy of proteins and peptides. Recently, Mistry et al. demonstrated that polystyrene or polysorbate-coated polystyrene NPs of size 100 nm were primarily located in olfactory epithelial cells but were absent in olfactory bulb in mice [132]. Hence, investigators hypothesized that the average nanoparticulate size required for optimal axonal transport may be less than 100 nm.
Pulmonary Delivery
Pulmonary route is one of the most commonly investigated non-invasive routes to improve absorption of proteins and peptides. This route provides numerous advantages including enormous absorptive surface area (100 m2), high vascularization, thin alveolar epithelial membrane (0.1-0.2 μm) and low enzymatic activity [109]. Despite these advantages, several factors may regulate pulmonary protein and peptide absorption. Central airway epithelium is largely constituted of ciliated columnar cells which expresses tight intercellular junctions. These tight junctions may significantly limit paracellular transport of proteins and peptides. Diffusion rate in this region is directly proportional to concentration gradient and lipid solubility. However, diffusion is also observed to be influenced by molecular size and ionization capabilities of the proteins and peptides [109]. Mucus binding and subsequent clearance via mucociliary apparatus may also reduce concentrations available for absorption. Particle wettability, aggregation, crystallinity, polymorphism and susceptibility towards enzymatic degradation may also determine absorption rate of proteins and peptides. Importantly, the deposition in the respiratory tract significantly depends on the aerodynamic properties of inhaled particles and breathing patterns [133-135]. Particles with aerodynamic diameter between 5 to 9 μm are deposited by impaction in bronchial airways. Fast breathing patterns may result in deposition of 3 to 6 μm particles in similar mechanism as above. Gravitational sedimentation results in deposition of 1 to 5 μm particles in smaller airways. Importantly, particles with aerodynamic diameter less than 3 μm might be deposited in respiratory bronchioles. Slow breathing patterns can promote absorption of particles in these ranges. Particles with aerodynamic diameter of less than 500 nm can easily be deposited in alveoli through Brownian diffusion however; there are chances that small particles can be exhaled significantly. Despite these challenges, omalizumab, α1-Antitrypsin, cyclosporine A, IFN (α, β and γ), IL-2 and IL-4 mutein are some of the inhaled (pulmonary delivery) proteins that are in ongoing clinical trials [136].
Several polymeric NPs have been previously explored to improve systemic absorption of proteins and peptides following pulmonary administration. Alpha 1-antitrypsin-loaded PLGA NPs have been anticipated as a promising formulation for the treatment of respiratory diseases [137]. NPs were spherical in shape with an average size of 100-1000 nm and entrapment efficiency of around 90%. PLGA (50:50) NPs released 60% of alpha 1-antitrypsin in 8 h however; an initial burst release of about 30% was observed. Mannitol based dry powder of insulin-loaded CS NPs has been suggested as an excellent formulation for local and systemic delivery of proteins and peptides [138]. Glycol and thiolated glycol CS NPs significantly enhanced the pharmacological availability of calcitonin following pulmonary administration [16]. Calcitonin-loaded thiolated and non-thiolated CS NPs generated availability of 40% and 27%, respectively. A marked hypocalcemic activity was observed for a period of 24 and 12 h with thiolated and unmodified CS NPs. Particles were of size 230 to 330 nm and demonstrated high calcitonin entrapment efficiency. Following intratracheal administration, thiolated CS NPs displayed 2-fold higher mucoadhesion relative to glycol CS NPs. Pulmonary delivery of modified gelatin based NPs has been proposed as an excellent delivery system to enhance insulin absorption [139]. Recently, Wanakule et al. suggested an enzyme responsive NPs entrapped microgel system as a promising formulation for pulmonary delivery of proteins and peptides [140]. This delivery system was proposed to overcome challenges such as alveolar macrophage uptake, non-specificity, low respirable fractions and poor deposition in deeper lung regions. Microgel system was formulated with a new Michael addition during w/o emulsion method and displayed a highly porous structure and optimal aerodynamic properties for deposition in alveolar region.
Mechanofusion™ is an excellent technique to impart remarkable inhalation properties to lyophilized nanoparticulate systems [141]. Nanocomposites were developed by loading salmon calcitonin adsorbed PLGA nanospheres on inhalable lactose (Pharmatose325M™) in Mechanofusion apparatus at 372 rpm for 30 min. This process improved the inhalation efficiency of lyophilized powder probably due to excellent powder flow ability and de-agglomeration. The respirable fraction of NP composites and nanospheres were 34.9 ± 1.5 and 12.4 ± 13.1, respectively. Importantly, nanocomposites generated superior hypocalcemic effects relative to salmon calcitonin solution and nanospheres. Following intratracheal administration of nanocomposites, approximately 50% (w/w) of nanospheres were observed to be deposited in alveoli region. However, nearly 60% of nanospheres were eliminated within 1 h of nanocomposite administration. To overcome this problem, CS-modified PLGA nanocomposites were developed by the same group of investigators [142]. Modification of PLGA with CS was performed to improve mucoadhesive properties to the nanospheres. Salmon calcitonin nanocomposites were prepared with spray drying fluidized bed granulation (Agglomaster™) and dry powder coating (Mechanofusion™) techniques. Nanocomposites developed by these processes demonstrated excellent inhalation efficiency with 50% deposition in alveoli regions following intratracheal administration in rats. Interestingly, nanocomposites developed using Agglomaster™ technique exhibited excellent redispersibility, retention time and hypocalcemic effects relative to Mechanofusion™. Moreover, the hypocalcemic effect generated by nanocomposites developed using Agglomaster™ technique were more significant relative to non-modified nanocomposites and nanocomposites developed using Mechanofusion™. These observations clearly indicate the suitability of applying nanocomposite systems developed with Agglomaster™ technique for pulmonary protein and peptide delivery.
Parenteral Delivery
Parenteral route has attracted considerable attention for nanoparticulate delivery as it can avoid first-pass metabolism, provide highest bioavailability and active targeting for drug delivery [143]. However, NPs properties such as size, charge, dissolution rate and surface coating can significantly alter the in vivo fate [143, 144]. Moreover, NPs with size greater than 200 nm or slow dissolution rate can be rapidly cleared by mononuclear phagocytic system [145]. This process might significantly enhance nanoparticulate uptake in liver and spleen. PEGylation has been proposed as a promising approach to evade reticuloendothelial system uptake [146-148]. In addition, the use of poly (N-vinyl-2-pyrrolidone) as a coating material to improve blood circulation time of particulate carriers has also been reported [149]. Interestingly, NPs with size between 100-300 nm can highly accumulate in tumors due to enhanced permeability and retention effect [143, 150]. For systemic to brain administration, BBB poses a formidable barrier for brain transport of proteins and peptides (Fig. 4). Brain capillary endothelial cells (BCECs) express tight intracellular junctions and occlude paracellular spaces. Moreover, BCECs are also encircled by astrocytic end feet and pericytes which further forms an implausible barrier for protein and peptide transport. Hence, there is an imperative need for the development of drug delivery systems capable of overcoming BBB following systemic administration.
Figure 4.
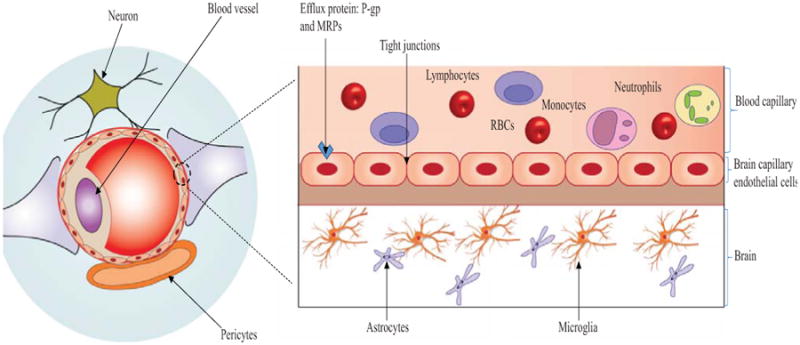
A schematic representation of BBB. Note: Paracellular diffusion is highly restricted for proteins and peptides due to expression of tight junction. Carrier mediated transport and receptor/adsorptive mediated endocytosis are the major pathways for transport of proteins and peptides across BBB.
In recent years, several polymeric NPs have been investigated to target poorly permeable tissues such as brain and lung following systemic administration. Lactoferrin-modified PEG-PLGA NPs have been investigated for their efficacy to treat Parkinson's disease [151]. Urocortin-loaded NPs were spherical in shape and exhibited size of 120 nm. In vivo studies clearly demonstrated that i.v. injection of urocortin-loaded NPs significantly reduced striatum lesions caused by 6-hydroxydopamine. Angiopep-2 peptide and EGFP-EGF1 protein functionalized PEG-PCL NPs have been developed by Huile et al. [152]. These functionalized NPs demonstrated remarkable potential in penetrating BBB (angiopep-2) and binding to neuroglial cells (EGFP-EGF1). Moreover, these dual functionalized NPs generated significantly higher brain accumulation relative to unmodified NPs. These targeting potential of angiopep-2 peptide and EGFP-EGF1 protein represents a novel promising formulation for the treatment of neuroglial related diseases following systemic administration. A novel CS based NPs loaded with caspase inhibitor N-Benzyloxycarbonyl-Asp(OMe)-Glu(OMe)-Val-Asp(OMe)-fluoromethyl ketone (Z-DEVD-FMK) or basic fibroblast growth factor have been observed to rapid transport across BBB following systemic circulation [153, 154].
Recently, Bao et al. developed OX26 functionalized hyperbranched polyglycerol-conjugated PLGA to improve brain delivery of endomorphins [155]. NPs were spherical in shape with an average size of 170 ± 20 nm. Drug loading and entrapment efficiency observed were 8.65 ± 1.27% and 83.96 ± 2.60 %, respectively. Nearly, 65% of endomorphin was released in first 72 h. Moreover, OX26-modified NPs generated significantly higher analgesic activity in chronic constriction injured rats relative to unmodified NPs and endomorphin itself, following i.v. injection. Xia et al. demonstrated penetratin (CPP with low cationic amino acid content) modified polyethylene glycol)-poly(lactic acid) NPs as an excellent brain drug delivery system compared to protamine (high arginine content) modified NPs [156]. Such systems can be utilized for improving brain protein or peptide delivery following systemic administration. RVG29- and CS-conjugated pluronic-based nanocarrier has been proposed as a promising formulation for improving brain delivery of proteins such as β-galactosidase [157]. B6 peptide (CGHKAKGPRK) functionalized PEG-PLA NPs have been demonstrated as an excellent delivery system for the brain delivery of neuroprotective peptide-NAPVSIPQ (NAP) [158]. NAP-loaded B6 NPs were 118.3 ± 7.8 nm in size with entrapment and loading efficiency of 0.65 ± 0.021% and 0.57 ± 0.023%, respectively. NAP-loaded B6 NPs demonstrated remarkable improvement in learning impairment recovery, loss of hippocampal neurons, and cholinergic disruption at significantly lower doses.
The shape of NPs has been observed to affect the hydrodynamic properties and interactions with target tissues significantly [159]. Moreover, size, surface charge and modification (targeting ligand) can significantly influence the uptake mechanism at BBB [160]. This process in turn affects the endocytotic pathway mechanisms and therefore, intracellular fate of NPs. Such information might be valuable for brain specific delivery of NPs following i.v. administration.
Transdermal Delivery
Transdermal delivery offers several advantages such as enormous surface area, avoidance of hepatic first-pass, continuous and non-invasive administration and patient compliance. Moreover, skin is easily accessible and delivery can be controlled and terminated whenever required. However, transdermal protein and peptide delivery is limited by penetrative and enzymatic barriers [161]. Large size and hydrophilic nature forbid the passive diffusion of proteins across the skin. Several endopeptidases and exopeptidases are also expressed in epidermal and dermal layers of the skin which may significantly degrade proteins and peptides. Several formulation and permeation enhancing approaches have been studied for improving transdermal delivery. Among formulation approaches, encapsulation of a protein or peptide in polymeric NPs seems to be an attractive approach for enhancing transdermal delivery. However, intact polymeric NPs are only able to penetrate into the superficial layers of the stratum corneum [162]. Hence, for efficient delivery of polymeric NPs, permeation enhancers, iontophoresis or sonophoresis may be needed to facilitate permeation through stratum corneum. In a recent study, Rastogi et al. have studied the potential of transdermal electroporation of insulin-loaded nanocarriers for insulin delivery [163]. Insulin-loaded PCL-PEG-PCL NPs were prepared by w/o/w double emulsion solvent evaporation method. Insulin entrapment efficiency was observed to be 32.9 ± 7.6% with an average NPs diameter of about 85 ± 9.4 nm. The efficacy of electroporation of insulin as solution and NPs was compared both in vitro and in vivo. Electroporation of NPs resulted in 4-fold higher insulin deposition in rat skin relative to solution. In vivo efficacy was evaluated in streptozotocin-diabetic male Wistar rats. This studies demonstrated 77 ± 5% (87.2 ± 6.4 mIU/mL, t = 2 h) and 85 ± 8% (37.8 ± 10.2 mIU/mL, t = 4 h) reduction in blood glucose levels with therapeutic levels maintained for 24 and 36 h for solution and NPs, respectively. These results demonstrate the efficiency of electroporation of nanosystems as an alternative to injectable insulin administration.
Ocular Delivery
Ocular route has been utilized for both systemic and localized delivery of proteins and peptides. For systemic delivery, ocular route provides advantages such as ease of administration, avoidance of first-pass metabolism and comparatively rapid rate of absorption (over oral administration) [164]. Systemic absorption of several proteins and peptides including insulin, calcitonin, and enkephalins has been studied following ocular administration [165-167]. Currently, ocular route is mainly used for the delivery of proteins and peptides for the treatment of local ocular disorders such as age related macular degeneration, dry eye disease, or proliferative diabetic retinopathy. Lucentis® and Eylea® are the recently marketed proteins intended for the treatment of ocular diseases [9, 168]. The physiological and anatomical barriers and enzymatic degradation within the ocular environment limit the efficacy of proteins and peptides administered by ocular route [169]. Polymeric NPs have been successfully designed to overcome these barriers and improve ocular bioavailability of proteins and peptides. Polymeric NPs can improve localized ocular delivery by providing sustain release, protection from enzymatic degradation and enhancing precorneal residence time compared to aqueous eye drops. However, NPs may be eliminated rapidly from precorneal pocket. Hence, for topical administration it is highly desirable to formulate NPs with mucoadhesive properties to increase their retention time in the cul-de-sac. PEG, carbopol and hyaluronic acid have been utilized to improve NPs precorneal residence time [170-172].
The prolonged residence time allows optimal contact between the formulation and mucosa and thereby sufficient drug concentration in external ocular tissues can be achieved. In a recent study, coating of cyclosporine A (CsA)-loaded poly caprolactone (PCL)/benzalkonium chloride (BKC) nanospheres with hyaluronic acid resulted in high concentrations of cyclosporine A into the cornea compared with non-coated NPs [172]. Cationic polymers such as Eudragit® have also been employed to increase precorneal residence time. It was observed that Eudragit® can prolong the residence time of NPs by interacting with the anionic mucins present in the mucus layer at the eye surface. Aksungur et al. evaluated efficiency of Eudragit® in improving effectiveness of CsA NPs formulations against inflammation of the eye surface [171]. The NPs were prepared using either PLGA alone or a mixture of Eudragit® RL with PLGA or were coated with Carbopol®. Tear kinetic parameters were determined following topical application of CsA-loaded NPs suspension and RestasisA® (drug-loaded emulsion) in rabbit eye (Table 4). Cellular uptake, tear film concentration of the drug and AUC0→24h were significantly higher for PLGA: Eudragit® RL (75:25)-CsA NPs (cationic NPs) and Carbopol® coated PLGA-CsA NPs (adhesive) formulations compared to Restasis A®. hese results clearly demonstrate the effect of surface characteristics of NPs in the improvement of ocular retention and bioavailability.
Table 4. Tear pharmacokinetic parameters following topical application of CsA-loaded NPs suspension and RestasisA® (Reproduced with permission from reference [171]).
| Fornulation code | a AUC0→24h (ng h/g) | b C max (ng/g) |
|---|---|---|
| P-CsA | 490.42 | 126.12 |
| P:E-CsA (75:25) | 972.59 | 366.30 |
| P:C-CsA | 776.57 | 211.08 |
| Restasis A® | 514.24 | 299.02 |
Area under the concentration-time curve between 0 and 24 h.
Peak drug concentration (ng CsA/g tear).
Even though topical administration is the major route for the anterior segment conditions, it is difficult to deliver proteins and peptides to posterior ocular tissues via topical route. Currently, intravitreal injection is the most commonly employed technique to treat posterior ocular diseases. However, short vitreal half-life of proteins and peptides and chronic nature of most of the posterior ocular diseases lead to requirement of frequent intravitreal injections. These administrations are usually associated with potential undesired side effects such as increased risk of cataract development, vitreous hemorrhage, retinal detachment, and endophthalmitis leading to poor patient acceptance. Several research studies have demonstrated suitability of intravitreally injected NPs in the treatment of posterior segment ocular diseases. In a recent study, Kim et al. have developed polylactic acid/polylactic acid-polyethylene oxide NPs (PLA/PLA-PEO) for the delivery of C16Y (an integrin-antagonist peptide) to the sub-retinal space for the treatment of choroidal neovascularization (CNV) [173]. Efficacy of C16Y-NPs was evaluated in laser-induced CNV in rats. A single intravitreal administration of both C16Y peptide and C16Y-NPs inhibited CNV at 5 and 9 days post laser photocoagulation. However, CNV inhibition was significantly higher for C16Y-NPs than the C16Y peptide solution on day 5. Because of short intravitreal half-life of C16Y peptide, inhibition of the experimental CNV was less with the peptide solution as compared to C16Y-NPs. On the contrary, the prolonged C16Y peptide release from C16Y-NPs aided to more CNV inhibition following intravitreal injection. These results demonstrate the potential of intravitreally injected biodegradable polymer-based NPs for treating choroidal neovascularization related to age-related macular degeneration.
Conclusion
Proteins and peptides have attracted considerable attention in the treatment of various chronic diseases due to their high potency and specificity. However, physicochemical properties of proteins and peptides and complex physiology of the non-invasive routes pose significant challenges for site specific delivery of these macromolecules. In recent years, polymeric NPs have demonstrated considerable potential in promoting absorption of macromolecules via non-invasive routes. An ideal polymeric nanoparticulate system should be capable of generating high loading and entrapment efficiency, protecting protein integrity until it reaches the target site and releasing the encapsulated protein or peptide in a sustained manner to avoid frequent administration. Recent advancements in biotechnology and newly emerging targeting ligands have significantly boosted interest in developing novel polymeric NPs system for systemic and local delivery of proteins and peptides. Importantly, surface conjugation of polymeric NPs with targeting ligands such as antibodies and cell penetrating peptides may be further explored to improve the effectiveness of formulation. Surface modification such as coating with mucoadhesive polymers or co-administration of protease inhibitors may significantly enhance the efficacy of polymeric NPs for nasal and pulmonary delivery. Development of novel polymeric NPs to enhance bioavailability of proteins and peptides remains an active field of research. Further studies are still required to develop novel targeted NPs formulation for site specific and sustained release of proteins and peptides in a non-invasive patient complaint manner.
Acknowledgments
This work was supported by NIH grant R01EY09171 and RO1EY10659.
Abbreviations
- PASP
Poly(L-aspartic acid)
- BBB
Blood-brain barrier
- BCECs
Brain capillary endothelial cells
- BSA
Bovine serum albumin
- CKS9
CKSTHPLSC targeting peptide
- CNV
Choroidal neovascularization
- CPP
Cell-penetrating peptide
- CS
Chitosan
- CsA
Cyclosporine A
- CSK
CSKSSDYQC cell targeting peptide
- CS-TGA
Thiolated Chitosan -thioglycolic acid
- DS
Dextran sulfate
- DTPA
Diethylene triamine pentaacetic acid
- FAE
Follicle-associated epithelium
- GIT
Gastrointestinal tract
- HIP
Hydrophobic ion-pairing
- IBD
Inflammatory bowel disease
- KPV
Tripeptide Lys-Pro-Val
- NT-I
Neurotoxin-I
- LMWP
Low-molecular-weight protamine
- OVA
Ovalbumin
- PAA
Polyacrylic acid
- PCL
Polycaprolactone
- PD
Pharmacodynamic
- PK
Pharmacokinetic
- PLA
Poly (lactic acid)
- PLA-PEO
Polylactic acid-polyethylene oxide
- PLGA
Poly (DL-lactide co-glycolide)
- PMMA
Poly(methyl methacrylate)
- PNIPPAm
Poly-N-isopropylacrylamide
- SOD
Superoxide dismutase
- TMC
Trimethylchitosan
- TMC-Cys
Trimethyl chitosan-cysteine conjugate
- TPP
Tripolyphosphate
Footnotes
Conflict of interest: The authors confirm that this article content has no conflict of interest.
References
- 1.Tan ML, Choong PF, Dass CR. Recent developments in liposomes, microparticles and nanoparticles for protein and peptide drug delivery. Peptides. 2010;31(1):184–93. doi: 10.1016/j.peptides.2009.10.002. [DOI] [PubMed] [Google Scholar]
- 2.Patel A, Cholkar K, Mitra AK. Recent Developments in Protein and Peptide Parenteral Delivery Approaches. Ther Delivery. 2014;5(3):337–65. doi: 10.4155/tde.14.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kumar TR, Soppimath K, Nachaegari SK. Novel delivery technologies for protein and peptide therapeutics. Curr Pharm Biotechnol. 2006;7(4):261–76. doi: 10.2174/138920106777950852. [DOI] [PubMed] [Google Scholar]
- 4.Jitendra, Sharma PK, Bansal S, Banik A. Noninvasive routes of proteins and peptides drug delivery. Indian J Pharm Sci. 2011;73(4):367–75. doi: 10.4103/0250-474X.95608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lee C, Choi JS, Kim I, Oh KT, Lee ES, Park ES, Lee KC, Youn YS. Long-acting inhalable chitosan-coated poly(lactic-co-glycolic acid) nanoparticles containing hydrophobically modified exendin-4 for treating type 2 diabetes. Int J Nanomedicine. 2013;8:2975–83. doi: 10.2147/IJN.S48197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pinto Reis C, Neufeld RJ, Ribeiro J, Veiga F. Nanoencapsulation II. Biomedical applications and current status of peptide and protein nanoparticulate delivery systems. Nanomedicine. 2006;2(2):53–65. doi: 10.1016/j.nano.2006.04.009. [DOI] [PubMed] [Google Scholar]
- 7.des Rieux A, Fievez V, Garinot M, Schneider YJ, Préat V. Nanoparticles as potential oral delivery systems of proteins and vaccines: a mechanistic approach. J Control Release. 2006;116(1):1–27. doi: 10.1016/j.jconrel.2006.08.013. [DOI] [PubMed] [Google Scholar]
- 8.Shukla RS, Chen Z, Cheng K. Advanced Drug Delivery. Wiley; 2013. Strategies of drug targeting; p. 105. [Google Scholar]
- 9.labels.fda.gov.
- 10.Vrignaud S, Benoit JP, Saulnier P. Strategies for the nanoen-capsulation of hydrophilic molecules in polymer-based nanoparticles. Biomaterials. 2011;32(33):8593–604. doi: 10.1016/j.biomaterials.2011.07.057. [DOI] [PubMed] [Google Scholar]
- 11.Patel A, Cholkar K, Vibhuti A, Mitra AK. Ocular drug delivery systems: An overview. World J Pharmacol. 2013;2(2):47–64. doi: 10.5497/wjp.v2.i2.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sung HW, Sonaje K, Liao ZX, Hsu LW, Chuang EY. pH-responsive nanoparticles shelled with chitosan for oral delivery of insulin: from mechanism to therapeutic applications. Acc Chem Res. 2012;45(4):619–29. doi: 10.1021/ar200234q. [DOI] [PubMed] [Google Scholar]
- 13.Iqbal J, Vigl C, Moser G, Gasteiger M, Perera G, Bernkop-Schnürch A. Development and in vivo evaluation of a new oral nanoparticulate dosage form for leuprolide based on polyacrylic acid. Drug Deliv. 2011;18(6):432–40. doi: 10.3109/10717544.2011.577108. [DOI] [PubMed] [Google Scholar]
- 14.Mahapatro A, Singh DK. Biodegradable nanoparticles are excellent vehicle for site directed in-vivo delivery of drugs and vaccines. J Nanobiotechnol. 2011;9:55. doi: 10.1186/1477-3155-9-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cuña M, Alonso-Sandel M, Remuñán-López C, Pivel JP, Alonso-Lebrero JL, Alonso MJ. Development of phosphorylated glucomannan-coated chitosan nanoparticles as nanocarriers for protein delivery. J Nanosci Nanotechnol. 2006;6(9-10):2887–95. doi: 10.1166/jnn.2006.435. [DOI] [PubMed] [Google Scholar]
- 16.Makhlof A, Werle M, Tozuka Y, Takeuchi H. Nanoparticles of glycol chitosan and its thiolated derivative significantly improved the pulmonary delivery of calcitonin. Int J Pharm. 2010;397(1-2):92–5. doi: 10.1016/j.ijpharm.2010.07.001. [DOI] [PubMed] [Google Scholar]
- 17.Sinsuebpol C, Chatchawalsaisin J, Kulvanich P. Preparation and in vivo absorption evaluation of spray dried powders containing salmon calcitonin loaded chitosan nanoparticles for pulmonary delivery. Drug Des Devel Ther. 2013;7:861–73. doi: 10.2147/DDDT.S47681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yadav SC, Kumari A, Yadav R. Development of peptide and protein nanotherapeutics by nanoencapsulation and nanobioconjugation. Peptides. 2011;32(1):173–87. doi: 10.1016/j.peptides.2010.10.003. [DOI] [PubMed] [Google Scholar]
- 19.Kafka AP, McLeod BJ, Rades T, McDowell A. Release and bioactivity of PACA nanoparticles containing D-Lys(6)-GnRH for brushtail possum fertility control. J Control Release. 2011;149(3):307–13. doi: 10.1016/j.jconrel.2010.10.029. [DOI] [PubMed] [Google Scholar]
- 20.Zhang Q, Shen Z, Nagai T. Prolonged hypoglycemic effect of insulin-loaded polybutylcyanoacrylate nanoparticles after pulmonary administration to normal rats. Int J Pharm. 2001;218(1-2):75–80. doi: 10.1016/s0378-5173(01)00614-7. [DOI] [PubMed] [Google Scholar]
- 21.Mukherjee B, Santra K, Pattnaik G, Ghosh S. Preparation, characterization and in-vitro evaluation of sustained release protein-loaded nanoparticles based on biodegradable polymers. Int J Nanomedicine. 2008;3(4):487–96. doi: 10.2147/ijn.s3938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Eley JG, Mathew P. Preparation and release characteristics of insulin and insulin-like growth factor-one from polymer nanoparticles. J Microencapsul. 2007;24(3):225–34. doi: 10.1080/02652040601162335. [DOI] [PubMed] [Google Scholar]
- 23.Santander-Ortega Manuel J, Csaba N, González L, Bastos-González D, Ortega-Vinuesa JL, Alonso Maria J. Protein-loaded PLGA–PEO blend nanoparticles: encapsulation, release and degradation characteristics. Colloid Polym Sci. 2010;288(2):141–150. [Google Scholar]
- 24.Bilati U, Allemann E, Doelker E. Nanoprecipitation versus emulsion-based techniques for the encapsulation of proteins into biodegradable nanoparticles and process-related stability issues. AAPS PharmSci Tech. 2005;6(4):E594–604. doi: 10.1208/pt060474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.van de Weert M, Hoechstetter J, Hennink WE, Crommelin DJ. The effect of a water/organic solvent interface on the structural stability of lysozyme. J Control Release. 2000;68(3):351–9. doi: 10.1016/s0168-3659(00)00277-7. [DOI] [PubMed] [Google Scholar]
- 26.Cleland JL, Powell MF, Shire SJ. The development of stable protein formulations: a close look at protein aggregation, deamidation, and oxidation. Crit Rev Ther Drug Carrier Syst. 1993;10(4):307–77. [PubMed] [Google Scholar]
- 27.Varshochian R, Jeddi-Tehrani M, Mahmoudi AR, Khoshayand MR, Atyabi F, Sabzevari A, Esfahani MR, Dinarvand R. The protective effect of albumin on bevacizumab activity and stability in PLGA nanoparticles intended for retinal and choroidal neovascularization treatments. Eur J Pharm Sci. 2013;50(3-4):341–52. doi: 10.1016/j.ejps.2013.07.014. [DOI] [PubMed] [Google Scholar]
- 28.Stanwick JC, Baumann MD, Shoichet MS. Enhanced neurotrophin-3 bioactivity and release from a nanoparticle-loaded composite hydrogel. J Control Release. 2012;160(3):666–75. doi: 10.1016/j.jconrel.2012.03.024. [DOI] [PubMed] [Google Scholar]
- 29.Son S, Lee WR, Joung YK, Kwon MH, Kim YS, Park KD. Optimized stability retention of a monoclonal antibody in the PLGA nanoparticles. Int J Pharm. 2009;368(1-2):178–85. doi: 10.1016/j.ijpharm.2008.09.061. [DOI] [PubMed] [Google Scholar]
- 30.Gaudana R, Khurana V, Parenky A, Mitra AK. Encapsulation of Protein-Polysaccharide HIP Complex in Polymeric Nanoparticles. J Drug Deliv. 2011;2011:458128. doi: 10.1155/2011/458128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Patel A, Gaudana R, Mitra AK. A novel approach to antibody nanocarriers development through hydrophobic ionpairing complexation. J Microencapsul. 2014;31(6):542–50. doi: 10.3109/02652048.2014.885606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rastogi R, Anand S, Koul V. Evaluation of pharmacological efficacy of ‘insulin-surfoplex’ encapsulated polymer vesicles. Int J Pharm. 2009;373(1-2):107–15. doi: 10.1016/j.ijpharm.2009.01.022. [DOI] [PubMed] [Google Scholar]
- 33.Bilati U, Allemann E, Doelker E. Development of a nanoprecipitation method intended for the entrapment of hydrophilic drugs into nanoparticles. Eur J Pharm Sci. 2005;24(1):67–75. doi: 10.1016/j.ejps.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 34.Gaudana R, Gokulgandhi M, Khurana V, Kwatra D, Mitra AK. Design and evaluation of a novel nanoparticulate-based formulation encapsulating a HIP complex of lysozyme. Pharm Dev Technol. 2013;18(3):752–9. doi: 10.3109/10837450.2012.737806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vandana M, Sahoo SK. Optimization of physicochemical parameters influencing the fabrication of protein-loaded chitosan nanoparticles. Nanomedicine (Lond) 2009;4(7):773–85. doi: 10.2217/nnm.09.54. [DOI] [PubMed] [Google Scholar]
- 36.Xu Y, Du Y. Effect of molecular structure of chitosan on protein delivery properties of chitosan nanoparticles. Int J Pharm. 2003;250(1):215–26. doi: 10.1016/s0378-5173(02)00548-3. [DOI] [PubMed] [Google Scholar]
- 37.Elvassore N, Bertucco A, Caliceti P. Production of insulinloaded poly(ethylene glycol)/poly(l-lactide) (PEG/PLA) nanoparticles by gas antisolvent techniques. J Pharm Sci. 2001;90(10):1628–36. doi: 10.1002/jps.1113. [DOI] [PubMed] [Google Scholar]
- 38.Marin E, Briceno MI, Caballero-George C. Critical evaluation of biodegradable polymers used in nanodrugs. Int J Nanomedicine. 2013;8:3071–90. doi: 10.2147/IJN.S47186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Goldberg M, Gomez-Orellana I. Challenges for the oral delivery of macromolecules. Nat Rev Drug Discov. 2003;2(4):289–95. doi: 10.1038/nrd1067. [DOI] [PubMed] [Google Scholar]
- 40.Salama NN, Eddington ND, Fasano A. Tight junction modulation and its relationship to drug delivery. Adv Drug Deliv Rev. 2006;58(1):15–28. doi: 10.1016/j.addr.2006.01.003. [DOI] [PubMed] [Google Scholar]
- 41.Lemmer HJ, Hamman JH. Paracellular drug absorption enhancement through tight junction modulation. Expert Opin Drug Deliv. 2013;10(1):103–14. doi: 10.1517/17425247.2013.745509. [DOI] [PubMed] [Google Scholar]
- 42.Antunes F, Andrade F, Ferreira D, Nielsen HM, Sarmento B. Models to predict intestinal absorption of therapeutic peptides and proteins. Curr Drug Metab. 2013;14(1):4–20. [PubMed] [Google Scholar]
- 43.Hamman JH, Enslin GM, Kotze AF. Oral delivery of peptide drugs: barriers and developments. BioDrugs. 2005;19(3):165–77. doi: 10.2165/00063030-200519030-00003. [DOI] [PubMed] [Google Scholar]
- 44.Herrero EP, Alonso MJ, Csaba N. Polymer-based oral peptide nanomedicines. Ther Deliv. 2012;3(5):657–68. doi: 10.4155/tde.12.40. [DOI] [PubMed] [Google Scholar]
- 45.Witt KA, Huber JD, Egleton RD, Roberts MJ, Bentley MD, Guo L, Wei H, Yamamura HI, Davis TP. Pharmacodynamic and pharmacokinetic characterization of poly(ethylene glycol) conjugation to met-enkephalin analog [D-Pen2, D-Pen5]-enkephalin (DPDPE) J Pharmacol Exp Ther. 2001;298(2):848–56. [PubMed] [Google Scholar]
- 46.Xu X, Li R, Ma M, Wang X, Wang Y, Zou H. Multidrug resistance protein P-glycoprotein does not recognize nanoparticle C60: experiment and modeling. Soft Matter. 2012;8(10):2915–2923. [Google Scholar]
- 47.Gupta S, Jain A, Chakraborty M, Sahni JK, Ali J, Dang S. Oral delivery of therapeutic proteins and peptides: a review on recent developments. Drug Deliv. 2013;20(6):237–46. doi: 10.3109/10717544.2013.819611. [DOI] [PubMed] [Google Scholar]
- 48.Brogden RN, Heel RC. Human insulin. A review of its biological activity, pharmacokinetics and therapeutic use. Drugs. 1987;34(3):350–71. doi: 10.2165/00003495-198734030-00003. [DOI] [PubMed] [Google Scholar]
- 49.Malik DK, Baboota S, Ahuja A, Hasan S, Ali J. Recent advances in protein and peptide drug delivery systems. Curr Drug Deliv. 2007;4(2):141–51. doi: 10.2174/156720107780362339. [DOI] [PubMed] [Google Scholar]
- 50.Shakweh M, Ponchel G, Fattal E. Particle uptake by Peyer's patches: a pathway for drug and vaccine delivery. Expert Opin Drug Deliv. 2004;1(1):141–63. doi: 10.1517/17425247.1.1.141. [DOI] [PubMed] [Google Scholar]
- 51.Desai MP, Labhasetwar V, Amidon GL, Levy RJ. Gastrointestinal uptake of biodegradable microparticles: effect of particle size. Pharm Res. 1996;13(12):1838–45. doi: 10.1023/a:1016085108889. [DOI] [PubMed] [Google Scholar]
- 52.Coppi G, Sala N, Bondi M, Sergi S, Iannuccelli V. Ex-vivo evaluation of alginate microparticles for Polymyxin B oral administration. J Drug Target. 2006;14(9):599–606. doi: 10.1080/10611860600864182. [DOI] [PubMed] [Google Scholar]
- 53.Giannasca PJ, Giannasca KT, Leichtner AM, Neutra MR. Human intestinal M cells display the sialyl Lewis A antigen. Infect Immun. 1999;67(2):946–53. doi: 10.1128/iai.67.2.946-953.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Prego C, García M, Torres D, Alonso MJ. Transmucosal macromolecular drug delivery. J Control Release. 2005;101(1-3):151–62. doi: 10.1016/j.jconrel.2004.07.030. [DOI] [PubMed] [Google Scholar]
- 55.Liu Y, Kong M, Feng C, Yang KK, Li Y, Su J, Cheng XJ, Park HJ, Chen XG. Biocompatibility, cellular uptake and biodistribution of the polymeric amphiphilic nanoparticles as oral drug carriers. Colloids Surf B Biointerfaces. 2013;103:345–53. doi: 10.1016/j.colsurfb.2012.11.012. [DOI] [PubMed] [Google Scholar]
- 56.Delie F, Blanco-Prieto MJ. Polymeric particulates to improve oral bioavailability of peptide drugs. Molecules. 2005;10(1):65–80. doi: 10.3390/10010065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.des Rieux A, Fievez V, Théate I, Mast J, Préat V, Schneider YJ. An improved in vitro model of human intestinal follicle-associated epithelium to study nanoparticle transport by M cells. Eur J Pharm Sci. 2007;30(5):380–91. doi: 10.1016/j.ejps.2006.12.006. [DOI] [PubMed] [Google Scholar]
- 58.des Rieux A, Ragnarsson EG, Gullberg E, Préat V, Schneider YJ, Artursson P. Transport of nanoparticles across an in vitro model of the human intestinal follicle associated epithelium. Eur J Pharm Sci. 2005;25(4-5):455–65. doi: 10.1016/j.ejps.2005.04.015. [DOI] [PubMed] [Google Scholar]
- 59.Jani P, Halbert GW, Langridge J, Florence AT. The uptake and translocation of latex nanospheres and microspheres after oral administration to rats. J Pharm Pharmacol. 1989;41(12):809–12. doi: 10.1111/j.2042-7158.1989.tb06377.x. [DOI] [PubMed] [Google Scholar]
- 60.Shakweh M, Besnard M, Nicolas V, Fattal E. Poly (lactide-co-glycolide) particles of different physicochemical properties and their uptake by peyer's patches in mice. Eur J Pharm Biopharm. 2005;61(1-2):1–13. doi: 10.1016/j.ejpb.2005.04.006. [DOI] [PubMed] [Google Scholar]
- 61.Tang BC, Dawson M, Lai SK, Wang YY, Suk JS, Yang M, Zeitlin P, Boyle MP, Fu J, Hanes J. Biodegradable polymer nanoparticles that rapidly penetrate the human mucus barrier. Proc Natl Acad Sci USA. 2009;106(46):19268–73. doi: 10.1073/pnas.0905998106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Florence AT. The oral absorption of micro- and nanoparticulates: neither exceptional nor unusual. Pharm Res. 1997;14(3):259–66. doi: 10.1023/a:1012029517394. [DOI] [PubMed] [Google Scholar]
- 63.Florence AT, Hillery AM, Hussain N, Jani PU. Factors affecting the oral uptake and translocation of polystyrene nanoparticles: histological and analytical evidence. J Drug Target. 1995;3(1):65–70. doi: 10.3109/10611869509015936. [DOI] [PubMed] [Google Scholar]
- 64.Reis CP, Veiga FJ, Ribeiro AJ, Neufeld RJ, Damgé C. Nanoparticulate biopolymers deliver insulin orally eliciting pharmacological response. J Pharm Sci. 2008;97(12):5290–305. doi: 10.1002/jps.21347. [DOI] [PubMed] [Google Scholar]
- 65.Cui F, Qian F, Zhao Z, Yin L, Tang C, Yin C. Preparation, characterization, and oral delivery of insulin loaded carboxylated chitosan grafted poly(methyl methacrylate) nanoparticles. Biomacromolecules. 2009;10(5):1253–8. doi: 10.1021/bm900035u. [DOI] [PubMed] [Google Scholar]
- 66.Gupta PN, Khatri K, Goyal AK, Mishra N, Vyas SP. M-cell targeted biodegradable PLGA nanoparticles for oral immunization against hepatitis B. J Drug Target. 2007;15(10):701–13. doi: 10.1080/10611860701637982. [DOI] [PubMed] [Google Scholar]
- 67.Yoo MK, Kang SK, Choi JH, Park IK, Na HS, Lee HC, Kim EB, Lee NK, Nah JW, Choi YJ, Cho CS. Targeted delivery of chitosan nanoparticles to Peyer's patch using M cell-homing peptide selected by phage display technique. Biomaterials. 2010;31(30):7738–47. doi: 10.1016/j.biomaterials.2010.06.059. [DOI] [PubMed] [Google Scholar]
- 68.Jin Y, Song Y, Zhu X, Zhou D, Chen C, Zhang Z, Huang Y. Goblet cell-targeting nanoparticles for oral insulin delivery and the influence of mucus on insulin transport. Biomaterials. 2012;33(5):1573–82. doi: 10.1016/j.biomaterials.2011.10.075. [DOI] [PubMed] [Google Scholar]
- 69.Zhang N, Ping Q, Huang G, Xu W, Cheng Y, Han X. Lectin-modified solid lipid nanoparticles as carriers for oral administration of insulin. Int J Pharm. 2006;327(1-2):153–9. doi: 10.1016/j.ijpharm.2006.07.026. [DOI] [PubMed] [Google Scholar]
- 70.Lochner N, Pittner F, Wirth M, Gabor F. Wheat germ agglutinin binds to the epidermal growth factor receptor of artificial Caco-2 membranes as detected by silver nanoparticle enhanced fluorescence. Pharm Res. 2003;20(5):833–9. doi: 10.1023/a:1023406224028. [DOI] [PubMed] [Google Scholar]
- 71.Lee GY, Kim JH, Oh GT, Lee BH, Kwon IC, Kim IS. Molecular targeting of atherosclerotic plaques by a stabilin-2-specific peptide ligand. J Control Release. 2011;155(2):211–7. doi: 10.1016/j.jconrel.2011.07.010. [DOI] [PubMed] [Google Scholar]
- 72.Akala EO, Elekwachi O, Chase V, Johnson H, Lazarre M, Scott K. Organic redox-initiated polymerization process for the fabrication of hydrogels for colon-specific drug delivery. Drug Dev Ind Pharm. 2003;29(4):375–86. doi: 10.1081/ddc-120018373. [DOI] [PubMed] [Google Scholar]
- 73.Chourasia MK, Jain SK. Pharmaceutical approaches to colon targeted drug delivery systems. J Pharm Pharm Sci. 2003;6(1):33–66. [PubMed] [Google Scholar]
- 74.Kompella UB, Lee VH. Delivery systems for penetration enhancement of peptide and protein drugs: design considerations. Adv Drug Deliv Rev. 2001;46(1-3):211–45. doi: 10.1016/s0169-409x(00)00137-x. [DOI] [PubMed] [Google Scholar]
- 75.Renukuntla J, Vadlapudi AD, Patel A, Boddu SH, Mitra AK. Approaches for enhancing oral bioavailability of peptides and proteins. Int J Pharm. 2013;447(1-2):75–93. doi: 10.1016/j.ijpharm.2013.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sinha V, Singh A, Kumar RV, Singh S, Kumria R, Bhinge J. Oral colon-specific drug delivery of protein and peptide drugs. Crit Rev Ther Drug Carrier Syst. 2007;24(1):63–92. doi: 10.1615/critrevtherdrugcarriersyst.v24.i1.30. [DOI] [PubMed] [Google Scholar]
- 77.Haupt S, Rubinstein A. The colon as a possible target for orally administered peptide and protein drugs. Crit Rev Ther Drug Carrier Syst. 2002;19(6):499–551. doi: 10.1615/critrevtherdrugcarriersyst.v19.i6.10. [DOI] [PubMed] [Google Scholar]
- 78.Coco R, Plapied L, Pourcelle V, Jérôme C, Brayden DJ, Schneider YJ, Préat V. Drug delivery to inflamed colon by nanoparticles: comparison of different strategies. Int J Pharm. 2013;440(1):3–12. doi: 10.1016/j.ijpharm.2012.07.017. [DOI] [PubMed] [Google Scholar]
- 79.Laroui H, Dalmasso G, Nguyen HT, Yan Y, Sitaraman SV, Merlin D. Drug-loaded nanoparticles targeted to the colon with polysaccharide hydrogel reduce colitis in a mouse model. Gastroenterology. 2010;138(3):843–53 e1-2. doi: 10.1053/j.gastro.2009.11.003. [DOI] [PubMed] [Google Scholar]
- 80.Sharma G, van der Walle CF, Ravi Kumar MN. Antacid coencapsulated polyester nanoparticles for peroral delivery of insulin: development, pharmacokinetics, biodistribution and pharmacodynamics. Int J Pharm. 2013;440(1):99–110. doi: 10.1016/j.ijpharm.2011.12.038. [DOI] [PubMed] [Google Scholar]
- 81.Zhang X, Sun M, Zheng A, Cao D, Bi Y, Sun J. Preparation and characterization of insulin-loaded bioadhesive PLGA nanoparticles for oral administration. Eur J Pharm Sci. 2012;45(5):632–8. doi: 10.1016/j.ejps.2012.01.002. [DOI] [PubMed] [Google Scholar]
- 82.Cetin M, Aktas MS, Vural I, Ozturk M. Salmon calcitonin-loaded Eudragit(R) and Eudragit(R)-PLGA nanoparticles: in vitro and in vivo evaluation. J Microencapsul. 2012;29(2):156–66. doi: 10.3109/02652048.2011.635426. [DOI] [PubMed] [Google Scholar]
- 83.Xiong XY, Li YP, Li ZL, Zhou CL, Tam KC, Liu ZY, Xie GX. Vesicles from Pluronic/poly(lactic acid) block copolymers as new carriers for oral insulin delivery. J Control Release. 2007;120(1-2):11–7. doi: 10.1016/j.jconrel.2007.04.004. [DOI] [PubMed] [Google Scholar]
- 84.Damgé C, Socha M, Ubrich N, Maincent P. Poly(epsilon-caprolactone)/eudragit nanoparticles for oral delivery of aspart-insulin in the treatment of diabetes. J Pharm Sci. 2010;99(2):879–89. doi: 10.1002/jps.21874. [DOI] [PubMed] [Google Scholar]
- 85.Thompson CJ, Tetley L, Uchegbu IF, Cheng WP. The complexation between novel comb shaped amphiphilic polyallylamine and insulin: towards oral insulin delivery. Int J Pharm. 2009;376(1-2):46–55. doi: 10.1016/j.ijpharm.2009.04.014. [DOI] [PubMed] [Google Scholar]
- 86.Deutel B, Greindl M, Thaurer M, Bernkop-Schnürch A. Novel insulin thiomer nanoparticles: in vivo evaluation of an oral drug delivery system. Biomacromolecules. 2008;9(1):278–85. doi: 10.1021/bm700916h. [DOI] [PubMed] [Google Scholar]
- 87.Prego C, Torres D, Alonso MJ. Chitosan nanocapsules as carriers for oral peptide delivery: effect of chitosan molecular weight and type of salt on the in vitro behaviour and in vivo effectiveness. J Nanosci Nanotechnol. 2006;6(9-10):2921–8. doi: 10.1166/jnn.2006.429. [DOI] [PubMed] [Google Scholar]
- 88.Sonaje K, Chen YJ, Chen HL, Wey SP, Juang JH, Nguyen HN, Hsu CW, Lin KJ, Sung HW. Enteric-coated capsules filled with freeze-dried chitosan/poly(gamma-glutamic acid) nanoparticles for oral insulin delivery. Biomaterials. 2010;31(12):3384–94. doi: 10.1016/j.biomaterials.2010.01.042. [DOI] [PubMed] [Google Scholar]
- 89.Sonaje K, Lin KJ, Wey SP, Lin CK, Yeh TH, Nguyen HN, Hsu CW, Yen TC, Juang JH, Sung HW. Biodistribution, pharmacodynamics and pharmacokinetics of insulin analogues in a rat model: Oral delivery using pH-responsive nanoparticles vs. subcutaneous injection. Biomaterials. 2010;31(26):6849–58. doi: 10.1016/j.biomaterials.2010.05.042. [DOI] [PubMed] [Google Scholar]
- 90.Makhlof A, Tozuka Y, Takeuchi H. Design and evaluation of novel pH-sensitive chitosan nanoparticles for oral insulin delivery. Eur J Pharm Sci. 2011;42(5):445–51. doi: 10.1016/j.ejps.2010.12.007. [DOI] [PubMed] [Google Scholar]
- 91.Su FY, Lin KJ, Sonaje K, Wey SP, Yen TC, Ho YC, Panda N, Chuang EY, Maiti B, Sung HW. Protease inhibition and absorption enhancement by functional nanoparticles for effective oral insulin delivery. Biomaterials. 2012;33(9):2801–11. doi: 10.1016/j.biomaterials.2011.12.038. [DOI] [PubMed] [Google Scholar]
- 92.Yin L, Ding J, He C, Cui L, Tang C, Yin C. Drug permeability and mucoadhesion properties of thiolated trimethyl chitosan nanoparticles in oral insulin delivery. Biomaterials. 2009;30(29):5691–700. doi: 10.1016/j.biomaterials.2009.06.055. [DOI] [PubMed] [Google Scholar]
- 93.Rekha MR, Sharma CP. Synthesis and evaluation of lauryl succinyl chitosan particles towards oral insulin delivery and absorption. J Control Release. 2009;135(2):144–51. doi: 10.1016/j.jconrel.2009.01.011. [DOI] [PubMed] [Google Scholar]
- 94.Woitiski CB, Neufeld RJ, Veiga F, Carvalho RA, Figueiredo IV. Pharmacological effect of orally delivered insulin facilitated by multilayered stable nanoparticles. Eur J Pharm Sci. 2010;41(3-4):556–63. doi: 10.1016/j.ejps.2010.08.009. [DOI] [PubMed] [Google Scholar]
- 95.Ribeiro C, Neto AP, das Neves J, Bahia MF, Sarmento B. Preparation of polyelectrolyte nanocomplexes containing recombinant human hepatocyte growth factor as potential oral carriers for liver regeneration. Methods Mol Biol. 2012;811:113–25. doi: 10.1007/978-1-61779-388-2_8. [DOI] [PubMed] [Google Scholar]
- 96.Xiong Y, Qi J, Yao P. Amphiphilic cholic-acid-modified dextran sulfate and its application for the controlled delivery of superoxide dismutase. Macromol Biosci. 2012;12(4):515–24. doi: 10.1002/mabi.201100367. [DOI] [PubMed] [Google Scholar]
- 97.Woitiski CB, Sarmento B, Carvalho RA, Neufeld RJ, Veiga F. Facilitated nanoscale delivery of insulin across intestinal membrane models. Int J Pharm. 2011;412(1-2):123–31. doi: 10.1016/j.ijpharm.2011.04.003. [DOI] [PubMed] [Google Scholar]
- 98.Sarmento B, Ribeiro AJ, Veiga F, Ferreira DC, Neufeld RJ. Insulin-loaded nanoparticles are prepared by alginate ionotropic pre-gelation followed by chitosan polyelectrolyte complexation. J Nanosci Nanotechnol. 2007;7(8):2833–41. doi: 10.1166/jnn.2007.609. [DOI] [PubMed] [Google Scholar]
- 99.Zhang Y, Wu X, Meng L, Zhang Y, Ai R, Qi N, He H, Xu H, Tang X. Thiolated Eudragit nanoparticles for oral insulin delivery: preparation, characterization and in vivo evaluation. Int J Pharm. 2012;436(1-2):341–50. doi: 10.1016/j.ijpharm.2012.06.054. [DOI] [PubMed] [Google Scholar]
- 100.Sakuma S, Suzuki N, Sudo R, Hiwatari K, Kishida A, Akashi M. Optimized chemical structure of nanoparticles as carriers for oral delivery of salmon calcitonin. Int J Pharm. 2002;239(1-2):185–95. doi: 10.1016/s0378-5173(02)00113-8. [DOI] [PubMed] [Google Scholar]
- 101.Turker S, Onur E, Ozer Y. Nasal route and drug delivery systems. Pharm World Sci. 2004;26(3):137–42. doi: 10.1023/b:phar.0000026823.82950.ff. [DOI] [PubMed] [Google Scholar]
- 102.Saindane NS, Pagar KP, Vavia PR. Nanosuspension based in situ gelling nasal spray of carvedilol: development, in vitro and in vivo characterization. AAPS PharmSciTech. 2013;14(1):89–99. doi: 10.1208/s12249-012-9896-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Smart JD. Nasal and buccal drug delivery: management forum conference. Ther Deliv. 2012;3(7):819–22. doi: 10.4155/tde.12.60. [DOI] [PubMed] [Google Scholar]
- 104.Rapoport A, Winner P. Nasal delivery of antimigraine drugs: clinical rationale and evidence base. Headache. 2006;46(Suppl 4):S192–201. doi: 10.1111/j.1526-4610.2006.00603.x. [DOI] [PubMed] [Google Scholar]
- 105.Ugwoke MI, Verbeke N, Kinget R. The biopharmaceutical aspects of nasal mucoadhesive drug delivery. J Pharm Pharmacol. 2001;53(1):3–21. doi: 10.1211/0022357011775145. [DOI] [PubMed] [Google Scholar]
- 106.Arora P, Sharma S, Garg S. Permeability issues in nasal drug delivery. Drug Discov Today. 2002;7(18):967–75. doi: 10.1016/s1359-6446(02)02452-2. [DOI] [PubMed] [Google Scholar]
- 107.Merkus FW, Verhoef JC, Schipper NG, Marttin E. Nasal mucociliary clearance as a factor in nasal drug delivery. Adv Drug Deliv Rev. 1998;29(1-2):13–38. doi: 10.1016/s0169-409x(97)00059-8. [DOI] [PubMed] [Google Scholar]
- 108.Huang Y, Donovan MD. Large molecule and particulate uptake in the nasal cavity: the effect of size on nasal absorption. Adv Drug Deliv Rev. 1998;29(1-2):147–155. doi: 10.1016/s0169-409x(97)00066-5. [DOI] [PubMed] [Google Scholar]
- 109.Paturi Durga, et al. Pulmonary Nanomedicine. Pan Stanford Publishing; 2012. Nasal and Pulmonary Delivery of Macromolecules to Treat Respiratory and Nonrespiratory Diseases; pp. 45–102. [Google Scholar]
- 110.Wang X, Zheng C, Wu Z, Teng D, Zhang X, Wang Z, Li C. Chitosan-NAC nanoparticles as a vehicle for nasal absorption enhancement of insulin. J Biomed Mater Res B Appl Biomater. 2009;88(1):150–61. doi: 10.1002/jbm.b.31161. [DOI] [PubMed] [Google Scholar]
- 111.Teijeiro-Osorio D, Remunan-Lopez C, Alonso MJ. New generation of hybrid poly/oligosaccharide nanoparticles as carriers for the nasal delivery of macromolecules. Biomacromolecules. 2009;10(2):243–9. doi: 10.1021/bm800975j. [DOI] [PubMed] [Google Scholar]
- 112.Goycoolea FM, Lollo G, Remuñán-López C, Quaglia F, Alonso MJ. Chitosan-alginate blended nanoparticles as carriers for the transmucosal delivery of macromolecules. Biomacromolecules. 2009;10(7):1736–43. doi: 10.1021/bm9001377. [DOI] [PubMed] [Google Scholar]
- 113.Jintapattanakit A, Peungvicha P, Sailasuta A, Kissel T, Junyaprasert VB. Nasal absorption and local tissue reaction of insulin nanocomplexes of trimethyl chitosan derivatives in rats. J Pharm Pharmacol. 2010;62(5):583–91. doi: 10.1211/jpp.62.05.0004. [DOI] [PubMed] [Google Scholar]
- 114.Alam MI, Beg S, Samad A, Baboota S, Kohli K, Ali J, Ahuja A, Akbar M. Strategy for effective brain drug delivery. Eur J Pharm Sci. 2010;40(5):385–403. doi: 10.1016/j.ejps.2010.05.003. [DOI] [PubMed] [Google Scholar]
- 115.Patel M, Vadlapatla RK, Pal D, Mitra AK. Molecular and functional characterization of riboflavin specific transport system in rat brain capillary endothelial cells. Brain Res. 2012;1468:1–10. doi: 10.1016/j.brainres.2012.05.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Wen MM. Olfactory targeting through intranasal delivery of biopharmaceutical drugs to the brain: current development. Discov Med. 2011;11(61):497–503. [PubMed] [Google Scholar]
- 117.Pardeshi CV, Belgamwar VS. Direct nose to brain drug delivery via integrated nerve pathways bypassing the blood-brain barrier: an excellent platform for brain targeting. Expert Opin Drug Deliv. 2013;10(7):957–72. doi: 10.1517/17425247.2013.790887. [DOI] [PubMed] [Google Scholar]
- 118.Bahadur S, Pathak K. Physicochemical and physiological considerations for efficient nose-to-brain targeting. Expert Opin Drug Deliv. 2012;9(1):19–31. doi: 10.1517/17425247.2012.636801. [DOI] [PubMed] [Google Scholar]
- 119.Mistry A, Stolnik S, Illum L. Nanoparticles for direct nose-to-brain delivery of drugs. Int J Pharm. 2009;379(1):146–57. doi: 10.1016/j.ijpharm.2009.06.019. [DOI] [PubMed] [Google Scholar]
- 120.Illum L. Transport of drugs from the nasal cavity to the central nervous system. Eur J Pharm Sci. 2000;11(1):1–18. doi: 10.1016/s0928-0987(00)00087-7. [DOI] [PubMed] [Google Scholar]
- 121.Thorne RG, Frey WH., 2nd Delivery of neurotrophic factors to the central nervous system: pharmacokinetic considerations. Clin Pharmacokinet. 2001;40(12):907–46. doi: 10.2165/00003088-200140120-00003. [DOI] [PubMed] [Google Scholar]
- 122.Chen XQ, Fawcett JR, Rahman YE, Ala TA, Frey WH., II Delivery of Nerve Growth Factor to the Brain via the Olfactory Pathway. J Alzheimers Dis. 1998;1(1):35–44. doi: 10.3233/jad-1998-1102. [DOI] [PubMed] [Google Scholar]
- 123.Thorne RG, Pronk GJ, Padmanabhan V, Frey WH., 2nd Delivery of insulin-like growth factor-I to the rat brain and spinal cord along olfactory and trigeminal pathways following intranasal administration. Neuroscience. 2004;127(2):481–96. doi: 10.1016/j.neuroscience.2004.05.029. [DOI] [PubMed] [Google Scholar]
- 124.Shi CG, Wang LM, Wu Y, Wang P, Gan ZJ, Lin K, Jiang LX, Xu ZQ, Fan M. Intranasal administration of nerve growth factor produces antidepressant-like effects in animals. Neurochem Res. 2010;35(9):1302–14. doi: 10.1007/s11064-010-0183-6. [DOI] [PubMed] [Google Scholar]
- 125.Cheng Q, Feng J, Chen J, Zhu X, Li F. Brain transport of neurotoxin-I with PLA nanoparticles through intranasal administration in rats: a microdialysis study. Biopharm Drug Dispos. 2008;29(8):431–9. doi: 10.1002/bdd.621. [DOI] [PubMed] [Google Scholar]
- 126.Xia H, Gao X, Gu G, Liu Z, Zeng N, Hu Q, Song Q, Yao L, Pang Z, Jiang X, Chen J, Chen H. Low molecular weight protamine-functionalized nanoparticles for drug delivery to the brain after intranasal administration. Biomaterials. 2011;32(36):9888–98. doi: 10.1016/j.biomaterials.2011.09.004. [DOI] [PubMed] [Google Scholar]
- 127.Veronesi MC, Aldouby Y, Domb AJ, Kubek MJ. Thyrotropin-releasing hormone d,l polylactide nanoparticles (TRH-NPs) protect against glutamate toxicity in vitro and kindling development in vivo. Brain Res. 2009;1303:151–60. doi: 10.1016/j.brainres.2009.09.039. [DOI] [PubMed] [Google Scholar]
- 128.Ruan Y, Yao L, Zhang B, Zhang S, Guo J. Antinociceptive properties of nasal delivery of neurotoxin-loaded nanoparticles coated with polysorbate-80. Peptides. 2011;32(7):1526–9. doi: 10.1016/j.peptides.2011.05.002. [DOI] [PubMed] [Google Scholar]
- 129.Ruan Y, Yao L, Zhang B, Zhang S, Guo J. Nanoparticle-mediated delivery of neurotoxin-II to the brain with intranasal administration: an effective strategy to improve antinociceptive activity of neurotoxin. Drug Dev Ind Pharm. 2012;38(1):123–8. doi: 10.3109/03639045.2011.592533. [DOI] [PubMed] [Google Scholar]
- 130.Liu Q, Shen Y, Chen J, Gao X, Feng C, Wang L, Zhang Q, Jiang X. Nose-to-brain transport pathways of wheat germ agglutinin conjugated PEG-PLA nanoparticles. Pharm Res. 2012;29(2):546–58. doi: 10.1007/s11095-011-0641-0. [DOI] [PubMed] [Google Scholar]
- 131.Liu Z, Jiang M, Kang T, Miao D, Gu G, Song Q, Yao L, Hu Q, Tu Y, Pang Z, Chen H, Jiang X, Gao X, Chen J. Lactoferrin-modified PEG-co-PCL nanoparticles for enhanced brain delivery of NAP peptide following intranasal administration. Biomaterials. 2013;34(15):3870–81. doi: 10.1016/j.biomaterials.2013.02.003. [DOI] [PubMed] [Google Scholar]
- 132.Mistry A, Glud SZ, Kjems J, Randel J, Howard KA, Stolnik S, Illum L. Effect of physicochemical properties on intranasal nanoparticle transit into murine olfactory epithelium. J Drug Target. 2009;17(7):543–52. doi: 10.1080/10611860903055470. [DOI] [PubMed] [Google Scholar]
- 133.Sturm R. Theoretical models of carcinogenic particle deposition and clearance in children's lungs. J Thorac Dis. 2012;4(4):368–76. doi: 10.3978/j.issn.2072-1439.2012.08.03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Slomkowski S, Gosecki M. Progress in nanoparticulate systems for peptide, proteins and nucleic acid drug delivery. Curr Pharm Biotechnol. 2011;12(11):1823–39. doi: 10.2174/138920111798377003. [DOI] [PubMed] [Google Scholar]
- 135.Heyder J. Particle transport onto human airway surfaces. Eur J Respir Dis Suppl. 1982;119:29–50. [PubMed] [Google Scholar]
- 136.Depreter F, Pilcer G, Amighi K. Inhaled proteins: challenges and perspectives. Int J Pharm. 2013;447(1-2):251–80. doi: 10.1016/j.ijpharm.2013.02.031. [DOI] [PubMed] [Google Scholar]
- 137.Pirooznia N, Hasannia S, Lotfi AS, Ghanei M. Encapsulation of alpha-1 antitrypsin in PLGA nanoparticles: in vitro characterization as an effective aerosol formulation in pulmonary diseases. J Nanobiotechnol. 2012;10:20. doi: 10.1186/1477-3155-10-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Al-Qadi S, Grenha A, Carrión-Recio D, Seijo B, Remuñán-López C. Microencapsulated chitosan nanoparticles for pulmonary protein delivery: in vivo evaluation of insulin-loaded formulations. J Control Release. 2012;157(3):383–90. doi: 10.1016/j.jconrel.2011.08.008. [DOI] [PubMed] [Google Scholar]
- 139.Zhao YZ, Li X, Lu CT, Xu YY, Lv HF, Dai DD, Zhang L, Sun CZ, Yang W, Li XK, Zhao YP, Fu HX, Cai L, Lin M, Chen LJ, Zhang M. Experiment on the feasibility of using modified gelatin nanoparticles as insulin pulmonary administration system for diabetes therapy. Acta Diabetol. 2012;49(4):315–25. doi: 10.1007/s00592-011-0356-z. [DOI] [PubMed] [Google Scholar]
- 140.Wanakule P, Liu GW, Fleury AT, Roy K. Nano-inside-micro: Disease-responsive microgels with encapsulated nanoparticles for intracellular drug delivery to the deep lung. J Control Release. 2012;162(2):429–37. doi: 10.1016/j.jconrel.2012.07.026. [DOI] [PubMed] [Google Scholar]
- 141.Yang M, Yamamoto H, Kurashima H, Takeuchi H, Yokoyama T, Tsujimoto H, Kawashima Y. Design and evaluation of poly(DL-lactic-co-glycolic acid) nanocomposite particles containing salmon calcitonin for inhalation. Eur J Pharm Sci. 2012;46(5):374–80. doi: 10.1016/j.ejps.2012.02.024. [DOI] [PubMed] [Google Scholar]
- 142.Yang M, Yamamoto H, Kurashima H, Takeuchi H, Yokoyama T, Tsujimoto H, Kawashima Y. Design and evaluation of inhalable chitosan-modified poly (DL-lactic-co-glycolic acid) nanocomposite particles. Eur J Pharm Sci. 2012;47(1):235–43. doi: 10.1016/j.ejps.2012.05.016. [DOI] [PubMed] [Google Scholar]
- 143.Tiana X, Lib H, Zhanga D, Shena J, Jiaa L, Zhenga D, Liua G, Haoa L, Shenc Y, Zhangd Q. Parenteral nanosuspension of a novel synthesized antitumor candidate: Investigation of tissue bio-distributions and plasma pharmacokinetics. Colloids Surf A Phys-icochem Eng Asp. 2013;436(0):868–872. [Google Scholar]
- 144.Sun B, Yeo Y. Nanocrystals for the parenteral delivery of poorly water-soluble drugs. Curr Opin Solid State Mater Sci. 2012;16(6):295–301. doi: 10.1016/j.cossms.2012.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Sonavane G, Tomoda K, Makino K. Biodistribution of colloidal gold nanoparticles after intravenous administration: effect of particle size. Colloids Surf B Bio interfaces. 2008;66(2):274–80. doi: 10.1016/j.colsurfb.2008.07.004. [DOI] [PubMed] [Google Scholar]
- 146.Li SD, Huang L. Nanoparticles evading the reticuloendothelial system: role of the supported bilayer. Biochim Biophys Acta. 2009;1788(10):2259–66. doi: 10.1016/j.bbamem.2009.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Li SD, Huang L. Stealth nanoparticles: high density but sheddable PEG is a key for tumor targeting. J Control Release. 2010;145(3):178–81. doi: 10.1016/j.jconrel.2010.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Dufort S, Sancey L, Coll JL. Physico-chemical parameters that govern nanoparticles fate also dictate rules for their molecular evolution. Adv Drug Deliv Rev. 2012;64(2):179–89. doi: 10.1016/j.addr.2011.09.009. [DOI] [PubMed] [Google Scholar]
- 149.Ishihara T, Maeda T, Sakamoto H, Takasaki N, Shigyo M, Ishida T, Kiwada H, Mizushima Y, Mizushima T. Evasion of the accelerated blood clearance phenomenon by coating of nanoparticles with various hydrophilic polymers. Biomacromolecules. 2010;11(10):2700–6. doi: 10.1021/bm100754e. [DOI] [PubMed] [Google Scholar]
- 150.Wong J, Brugger A, Khare A, Chaubal M, Papadopoulos P, Rabinow B, Kipp J, Ning J. Suspensions for intravenous (IV) injection: a review of development, preclinical and clinical aspects. Adv Drug Deliv Rev. 2008;60(8):939–54. doi: 10.1016/j.addr.2007.11.008. [DOI] [PubMed] [Google Scholar]
- 151.Hu K, Shi Y, Jiang W, Han J, Huang S, Jiang X. Lactoferrin conjugated PEG-PLGA nanoparticles for brain delivery: preparation, characterization and efficacy in Parkinson's disease. Int J Pharm. 2011;415(1-2):273–83. doi: 10.1016/j.ijpharm.2011.05.062. [DOI] [PubMed] [Google Scholar]
- 152.Huile G, Shuaiqi P, Zhi Y, Shijie C, Chen C, Xinguo J, Shun S, Zhiqing P, Yu H. A cascade targeting strategy for brain neuroglial cells employing nanoparticles modified with angiopep-2 peptide and EGFP-EGF1 protein. Biomaterials. 2011;32(33):8669–75. doi: 10.1016/j.biomaterials.2011.07.069. [DOI] [PubMed] [Google Scholar]
- 153.Yemisci M, Gürsoy-Özdemir Y, Caban S, Bodur E, Capan Y, Dalkara T. Transport of a caspase inhibitor across the blood-brain barrier by chitosan nanoparticles. Methods Enzymol. 2012;508:253–69. doi: 10.1016/B978-0-12-391860-4.00013-6. [DOI] [PubMed] [Google Scholar]
- 154.Caban S, Capan Y, Couvreur P, Dalkara T. Preparation and characterization of biocompatible chitosan nanoparticles for targeted brain delivery of peptides. Methods Mol Biol. 2012;846:321–32. doi: 10.1007/978-1-61779-536-7_27. [DOI] [PubMed] [Google Scholar]
- 155.Bao H, Jin X, Li L, Lv F, Liu T. OX26 modified hyperbranched polyglycerol-conjugated poly(lactic-co-glycolic acid) nanoparticles: synthesis, characterization and evaluation of its brain delivery ability. J Mater Sci Mater Med. 2012;23(8):1891–901. doi: 10.1007/s10856-012-4658-7. [DOI] [PubMed] [Google Scholar]
- 156.Xia H, Gao X, Gu G, Liu Z, Hu Q, Tu Y, Song Q, Yao L, Pang Z, Jiang X, Chen J, Chen H. Penetratin-functionalized PEG-PLA nanoparticles for brain drug delivery. Int J Pharm. 2012;436(1-2):840–50. doi: 10.1016/j.ijpharm.2012.07.029. [DOI] [PubMed] [Google Scholar]
- 157.Kim JY, Choi WI, Kim YH, Tae G. Brain-targeted delivery of protein using chitosan- and RVG peptide-conjugated, pluronicbased nano-carrier. Biomaterials. 2013;34(4):1170–8. doi: 10.1016/j.biomaterials.2012.09.047. [DOI] [PubMed] [Google Scholar]
- 158.Liu Z, Gao X, Kang T, Jiang M, Miao D, Gu G, Hu Q, Song Q, Yao L, Tu Y, Chen H, Jiang X, Chen J. B6 peptide-modified PEG-PLA nanoparticles for enhanced brain delivery of neuroprotective peptide. Bioconjug Chem. 2013;24(6):997–1007. doi: 10.1021/bc400055h. [DOI] [PubMed] [Google Scholar]
- 159.Kolhar P, Anselmo AC, Gupta V, Pant K, Prabhakarpandian B, Ruoslahti E, Mitragotri S. Using shape effects to target antibody-coated nanoparticles to lung and brain endothelium. Proc Natl Acad Sci USA. 2013;110(26):10753–8. doi: 10.1073/pnas.1308345110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Georgieva JV, Kalicharan D, Couraud PO, Romero IA, Weksler B, Hoekstra D, Zuhorn IS. Surface characteristics of nanoparticles determine their intracellular fate in and processing by human blood-brain barrier endothelial cells in vitro. Mol Ther. 2011;19(2):318–25. doi: 10.1038/mt.2010.236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Herwadkar A, Banga AK. Peptide and Protein Delivery. Elsevier Inc; 2011. Transdermal Delivery of Peptides and Proteins; p. 380. [Google Scholar]
- 162.Desai P, Patlolla RR, Singh M. Interaction of nanoparticles and cell-penetrating peptides with skin for transdermal drug delivery. Mol Membr Biol. 2010;27(7):247–59. doi: 10.3109/09687688.2010.522203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Rastogi R, Anand S, Koul V. Electroporation of polymeric nanoparticles: an alternative technique for transdermal delivery of insulin. Drug Dev Ind Pharm. 2010;36(11):1303–11. doi: 10.3109/03639041003786193. [DOI] [PubMed] [Google Scholar]
- 164.Vyas SP, Paliwal R, Paliwal SR. Ocular Delivery of Peptides and Proteins. In: Walle CVD, editor. Peptide and Protein Delivery. Elsevier Inc; 2011. pp. 87–103. [Google Scholar]
- 165.Xuan B, McClellan DA, Moore R, Chiou GC. Alternative delivery of insulin via eye drops. Diabetes Technol Ther. 2005;7(5):695–8. doi: 10.1089/dia.2005.7.695. [DOI] [PubMed] [Google Scholar]
- 166.Ahsan F, Arnold J, Meezan E, Pillion DJ. Enhanced bioavailability of calcitonin formulated with alkylglycosides following nasal and ocular administration in rats. Pharm Res. 2001;18(12):1742–6. doi: 10.1023/a:1013330815253. [DOI] [PubMed] [Google Scholar]
- 167.Chiou GC, Shen ZF, Zheng YQ, Li BH. Enhancement of systemic delivery of met-enkephalin and leu-enkephalin eyedrops with permeation enhancers. Methods Find Exp Clin Pharmacol. 1992;14(5):361–6. [PubMed] [Google Scholar]
- 168.Vadlapudi AD, Patel A, Cholkar K, Mitra AK. Recent Patents on Emerging Therapeutics for the Treatment of Glaucoma, Age Related Macular Degeneration and Uveitis. Recent Pat Biomed Eng. 2012;5(1):83–101. doi: 10.2174/1874764711205010083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Lee VH, Carson LW, Kashi SD, Stratford R., Jr Metabolic and permeation barriers to the ocular absorption of topically applied enkephalins in albino rabbits. J Ocul Pharmacol. 1986;2(4):345–52. doi: 10.1089/jop.1986.2.345. [DOI] [PubMed] [Google Scholar]
- 170.Musumeci T, Bucolo C, Carbone C, Pignatello R, Drago F, Puglisi G. Polymeric nanoparticles augment the ocular hypotensive effect of melatonin in rabbits. Int J Pharm. 2013;440(2):135–40. doi: 10.1016/j.ijpharm.2012.10.014. [DOI] [PubMed] [Google Scholar]
- 171.Aksungur P, Demirbilek M, Denkbas EB, Vandervoort J, Ludwig A, Unlü N. Development and characterization of Cyclosporin A loaded nanoparticles for ocular drug delivery: Cellular toxicity, uptake, and kinetic studies. J Control Release. 2011;151(3):286–94. doi: 10.1016/j.jconrel.2011.01.010. [DOI] [PubMed] [Google Scholar]
- 172.Yenice I, Mocan MC, Palaska E, Bochot A, Bilensoy E, Vural I, Irkeç M, Hincal AA. Hyaluronic acid coated poly-epsilon-caprolactone nanospheres deliver high concentrations of cyclosporine A into the cornea. Exp Eye Res. 2008;87(3):162–7. doi: 10.1016/j.exer.2008.04.002. [DOI] [PubMed] [Google Scholar]
- 173.Kim H, Csaky KG. Nanoparticle-integrin antagonist C16Y peptide treatment of choroidal neovascularization in rats. J Control Release. 2010;142(2):286–93. doi: 10.1016/j.jconrel.2009.10.031. [DOI] [PubMed] [Google Scholar]