

Abstract
Background
The metabolism of tyrosine kinase inhibitors (TKIs) is mainly mediated via hepatic route, but the mechanism responsible for their hepatocellular accumulation is still unknown. This study was designed to understand the contribution of organic anion transporting polypeptides (OATPs) in the hepatic uptake of selected TKIs – pazopanib, canertinib, erlotinib, vandetanib and nilotinib.
Methods
Michaelis-Menten (MM) kinetic parameters for TKIs were determined by concentration-dependent cellular accumulation of selected TKIs using Chinese hamster ovary cells – wild type as well as transfected with humanized OATP-1B1 and OATP-1B3 transporter proteins.
Results
The MM constant (Km) values of OATP-1B1 for nilotinib and vandetanib are 10.14±1.91 and 2.72±0.25 μM, respectively, and Vmax values of OATP-1B1 for nilotinib and vandetanib were 6.95±0.47 and 75.95±1.99 nmol/mg protein per minute, respectively. Likewise, Km values of OATP-1B3 for canertinib, nilotinib and vandetanib were 12.18±3.32, 7.84±1.43 and 4.37±0.79 μM, respectively and Vmax values of OATP-1B3 for canertinib, nilotinib and vandetanib were 15.34±1.59, 6.75±0.42 and 194.64±10.58 nmol/mg protein per minute, respectively. Canertinib did not exhibit any substrate specificity toward OATP-1B1. Also, erlotinib and pazopanib did not exhibit any substrate specificity toward OATP-1B1 and -1B3.
Conclusions
Because selected TKIs are the substrates of OATP-1B1 and -1B3 expressed in hepatic tissue, these compounds can be regarded as molecular targets for transporter-mediated drug-drug interactions (DDIs). Any alteration in the function of these hepatic OATPs might account for the pharmacokinetic variability of TKIs.
Keywords: drug interactions, hepatic disposition, metabolism, organic anion transporting polypeptide (OATP)-1B1, OATP-1B3, tyrosine kinase inhibitors
Introduction
Hepatic uptake of drugs is mediated via various members of membrane transporter families. Membrane transporters localized on the basolateral side of hepatic tissue are known to play an important role in the uptake of therapeutic agents/metabolites from blood into the hepatocytes. This uptake process is recognized as the first step in hepatocellular elimination and plays a vital role in hepatic drug disposition. Organic anion transporting polypeptides (OATPs) appear to play a critical role in bioavailability, distribution and excretion of numerous exogenous amphipathic organic anionic compounds including anionic oligopeptides, steroid conjugates, organic dyes, bile salts, thyroid hormones and many drugs such as pravastatin and rifampicin [1–4].
Tyrosine kinase inhibitors (TKIs) target the intracellular tyrosine kinase domain of various tyrosine kinase receptors, which are often overexpressed in cancer tissues. Human genome sequence data have indicated that deregulation of the protein kinase pathway is one of the main mechanisms underlying tumor growth. Over the past few years, attention has been focused toward the discovery of a targeted therapy that will act against defining characteristics of cancer resulting from abnormal function of protein kinases [5, 6]. In the past decade, the US Food and Drug Administration has approved several TKIs. Many of these compounds have been associated with low patient response along with unwanted effects of toxicity, which is unexpected and largely unexplained. Comprehensive data from Phase I studies of these TKIs establishes the optimized dose for Phase II. Even though TKIs offer theoretical advantages (selectively target/kill the cancer precursor cells and protect normal tissues) over traditional anticancer agents, these agents are still associated with unpredictable clinical effects because of interindividual pharmacokinetic variability and narrow therapeutic window [5, 7]. Substantial interindividual differences in the concentration-time profiles of TKIs range from 32% to 118% and is mostly unexplained. Many TKIs exhibit limited efficacy with significant degree of unexpected and unexplained toxicity [8]. Interindividual pharmacokinetic variation in TKIs can have both genetic and nongenetic origins. This pharmacokinetic variation can be due to many plausible sources, including interindividual differences in absorption, distribution, metabolism and excretion (ADME). TKIs are primarily metabolized in the liver by CYP enzymes (mainly CYP3A4) and are eliminated via the biliary excretion route in the feces as unchanged drug or metabolites [8]. Hepatic uptake of TKIs can be attributed as a major source of pharmacokinetic variability which is also recognized as one of the most crucial and complex steps in drug disposition [5]. Currently, various OATP family transporters such as OATP-1B1, -1B3 and -2B1 have been identified and characterized on the sinusoidal membrane of hepatic tissue [1]. However the mechanisms responsible for hepatocellular accumulation of TKIs prior to metabolism and biliary secretion are still largely unexplained. Previous investigations have reported that OATPs, namely, OATP-1B1 and -1B3, are responsible for uptake of TKIs into human liver cells [5]. Uptake of TKIs such as axitinib, lapatinib and sorafenib into human hepatocytes is regulated by OATP-1B1 and/or -1B3. Also, some of the TKIs, namely, pazopanib and lapatinib, are known to inhibit the functional capacity of OATP-1B1 and/or -1B3 transporter proteins [5]. However, there is still a need for a systematic approach to delineate the mechanism involved in hepatic uptake of these TKIs. Because the hepatic system possesses many transporters (both influx and efflux), it is difficult to delineate the affinity of individual transporters toward these TKIs. Hence, it is of utmost importance to estimate the relative contribution of OATP-1B1 and -1B3 in hepatic uptake of TKIs [1]. In the present study, we evaluated the interaction of TKIs (pazopanib, erlotinib, canertinib, nilotinib and vandetanib) with the human OATPs expressed on the sinusoidal membrane of the liver by employing an in vitro model system with wild-type (WT) and transfected Chinese hamster ovary (CHO) cells.
Materials and methods
Chemicals
Pazopanib, erlotinib, canertinib, nilotinib and vandetanib were purchased from LC Laboratories (Woburn, MA, USA). All other chemicals used were of high-performance liquid chromatography grade and were obtained from either Sigma Aldrich (MO, USA) or Fisher Scientific (NH.USA).
In vitro studies
Cell lines
CHO cells (passage number 17–50) were selected for all in vitro experiments. WT, OATP-1B1 and -1B3 CHO transfected cells were obtained as a gift from Dr. Bruno Stieger (Department of Clinical Pharmacology and Toxicology, University Hospital Zürich, Zürich, Switzerland). Cells were cultured in Dulbecco's modified Eagle's medium (DMEM), supplemented with 10% heat-inactivated fetal bovine serum, l-proline (50 μg/mL), 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), penicillin (100 μg/mL) and streptomycin (100 μg/mL), and maintained at 37°C with 5% CO2 under humidifying conditions. For OATP-1B1- and -1B3-expressing CHO cells, the medium was also supplemented with geneticin (100 μg/mL).
In vitro cellular accumulation studies
Cellular accumulation studies were conducted in 24-well polystyrene plates (Costar Corning, NY, USA). CHO cells (WT and transfected) were plated at a seeding density of 3×105 cells/well. The medium was changed every alternate day. Cells formed confluent monolayers in 3–4 days. Twenty-four hours before any experiment, the cells were exposed to 10 mM sodium butyrate to induce higher expression of the transfected transporter. On the day of the experiment, the medium was aspirated and cells were rinsed three times with cell assay buffer (116.4 mM NaCl, 5.3 mM KCl, 1 mM NaH2PO4, 0.8 mM MgSO4, 5.5 mM d-glucose and 20 mM HEPES/Tris; pH 7.4) prewarmed at 37°C. The uptake experiment was initiated by adding 0.5 mL of fresh serum-free medium containing 0.25 and 0.5 μM of TKIs (pazopanib, erlotinib, canertinib, nilotinib and vandetanib) in WT as well as OATP-1B type transfected cells. After the cells were incubated for 10 min with TKIs, the uptake solution was aspirated and the cells were washed twice with 2 mL of ice-cold uptake buffer. This resulted in removal of the nonspecifically bound substrate from the membrane as well as arrested further cellular accumulation. Finally, 0.5 mL of fresh DMEM was added to each well and cell lysis was carried out by storing the culture plates overnight at -80°C. On the following day, intracellular drug concentration was quantified using liquid chromatography-tandem mass spectrometry (LC/MS-MS) as described in previous publications from our group as well as others [9–13]. Based on the time points for uptake, the minimum concentrations observed were well beyond the detection limit. The amount of TKIs accumulated was normalized to the protein content in each well with Bradford's reagent (Bio-Rad, CA, USA). All stock solutions were prepared in dimethyl sulfoxide (DMSO) and diluted using medium such that the final DMSO concentration did not exceed 0.5% (v/v).
Estimation of Michaelis-Menten kinetics
To determine the kinetic basis for the differential uptake of OATP-1B1 and -1B3 transporter proteins, concentration-dependent uptake of TKIs was carried out. Using a concentrated stock solution of the TKIs, several working concentrations were prepared ranging from 0.01 to 50 μM in serum-free fresh medium. Uptake was carried out at different concentrations of TKIs in WT, OATP-1B1 and -1B3 transfected CHO cells.
Data analysis
Kinetic parameters of TKI uptake via hepatic OATP-1B1 and -1B3 were calculated with a nonlinear least-squares regression analysis program, KaleidaGraph version 3.5. The data were plotted and fitted to Michaelis-Menten (MM) equation (1), and the maximum transport rate (Vmax) and MM constant (Km) were calculated.
| (1) |
where v is the initial uptake rate, Vmax is the maximal velocity, Km is the MM constant, and [C] is the total concentration of TKIs.
Cytotoxicity studies
Cell Titer 96® Aqueous Non-Radioactive Cell Proliferation Assay Kit (Promega, Madison, WI, USA) was employed to carry out cytotoxicity assay. WT and OATP-1B1 and -1B3 transfected CHO cells were cultured in 96-well plates. Sterile drug solutions of the highest concentration (50 μM) of TKIs were prepared in the culture medium using 0.22-μm nylon sterile membrane filters. Aliquots of TKIs having a volume of 100 μL (previously made in culture medium) were added to each well and incubated for 24 h. Cell proliferation of the cells in the presence of TKIs was compared with a negative control (medium without TKIs) and a positive control (Triton X). Twenty microliters of dye solution was added to each well after 24 h of incubation with TKIs. Cells were then incubated for 4 h in order to complete the reaction of cells with dye. The ultraviolet absorbance of purple formazan formed was quantified at a wavelength of 590 nm with a 96-well microtiter plate reader (SpectraFluor Plus, Tecan, Maennedorf, Switzerland). The toxicity of TKIs in WT and OATP-1B-transfected CHO cells was estimated by the amount of formazan formed, which is directly proportional to the viable cells.
Results
In vitro cellular accumulation of TKIs
Initial in vitro uptake experiments were carried out to determine cellular accumulation of TKIs in WT, OATP-1B1 and -1B3 transfected CHO cells. Cellular accumulation was measured by exposing the WT and OATP-1B1 transfected CHO cells to two different concentrations (0.25 and 0.5 μM) of TKIs. In previously reported results, concentration ranges from 0.1 to 10 μM have been shown to be nonsaturating for OATP-1B1 and -1B3 mediated transport [5]. We performed our studies within these linear nonsaturable ranges and also at concentrations that were well within our detection limit. Also, while studying transporter-mediated uptakes, we always aim to use as low a concentration as possible so as to limit any toxicity. Hence, on the basis of these considerations, we chose 0.25 and 0.5 μM as our concentration ranges. Out of the selected TKIs, nilotinib and vandetanib showed significantly enhanced cellular accumulation in OATP-1B1-transfected cells relative to WT cells. The remaining three TKIs (can-ertinib, pazopanib and erlotinib) did not show any significant enhanced cellular accumulation in OATP-1B1 transfected cells compared to WT cells. Vandetanib (0.25 and 0.5 μM) showed the highest uptake, about 1.3-fold (p<0.01) in OATP-1B1 transfected cells compared to WT cells. Nilotinib (0.25 and 0.5 μM) also showed higher uptake, ∼1.3-fold (p<0.01) and 1.2-fold (p<0.05), respectively, in transfected cells relative to WT cells (Figure 1). It has been reported previously that OATP-1B3 shares 80% amino acid identity with OATP-1B1. Also, both the OATP isoforms share multiple overlapping substrates, such as rifampicin pravastatin, pitavastatin and docetaxel [14, 15]. In this study, we have also determined the cellular uptake of TKIs at two different concentrations (0.25 and 0.5 μM) in WT and OATP-1B3-transfected cells. Canertinib, nilotinib and vandetanib at both concentrations showed significantly enhanced cellular accumulation in OATP-1B3 transfected cells compared to WT cells (p<0.01). No difference was observed in cellular accumulation of pazopanib and erlotinib in OATP-1B3 transfected cells compared to WT cells (Figure 2). The highest uptake of canertinib (0.25 and 0.5 μM) was observed, about two (p<0.01) and five (p<0.01) times, respectively, in OATP-1B3 transfected cells than the WT cells. A significantly higher uptake of vandetanib and nilotinib was also evident in OATP-1B3 cells than WT (Figure 2). No statistically significant changes in cellular accumulation of pazopanib and erlotinib were found between WT and OATP-1B3 transfected cells (Figure 2). Nilotinib and vandetanib showed overlapping substrate specificity toward OATP-1B1 and -1B3, whereas canertinib only showed affinity toward OATP-1B3.
Figure 1.
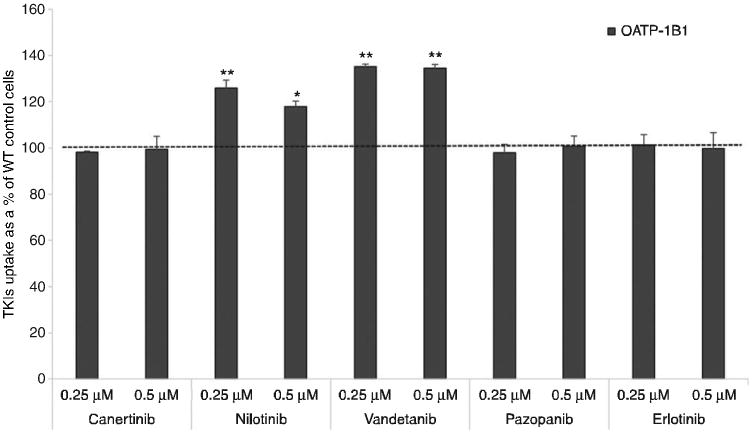
Cellular accumulation of TKIs at two concentrations (0.25 and 0.5 μM) by OATP-1B1 transporter.
TKIs were incubated with WT and CHO-OATP-1B1 transfected CHO cells for 10 min. Intracellular drug concentration was quantified using LC/MS-MS. Data represent the mean±SD, n=4 (*p<0.05, **p<0.01).
Figure 2.
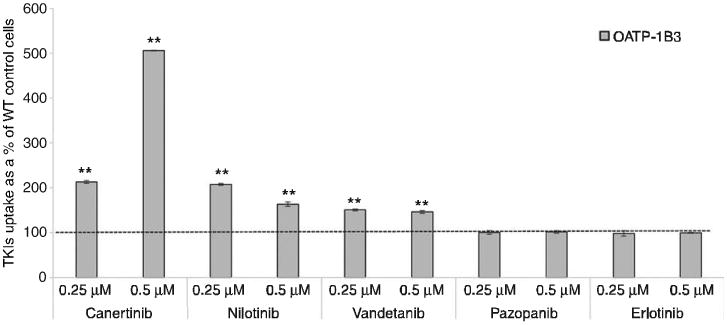
Cellular accumulation of TKIs at two concentrations (0.25 and 0.5 μM) by OATP-1B3 transporter.
TKIs were incubated with WT and OATP-1B3 transfected CHO cells for 10 min. Intracellular drug concentration was quantified using LC/MS-MS. Data represent the mean±SD, n=4 (*p<0.05, **p<0.01).
Estimation of MM kinetics
For estimation of MM kinetics, WT and OATP-1B1- and -1B3 transfected CHO cells were incubated with various concentrations (0.01–50 μM) of TKIs. Previous studies have shown time-dependent uptake on similar cell lines to be linear up to 15 min [16–18]. We incubated our cells for 10 min as it falls within the linear range of uptake as well as gives us concentration that lie well within the detectable range. Figures 3 and 4 clearly demonstrate that carrier-mediated uptake of TKIs via OATP-1B1 and OATP-1B3 is concentration dependent and saturable at higher concentrations. This is the first study where MM kinetic parameters of OATP-1B1 and OATP-1B3 have been evaluated for selected TKIs (canertinib, pazopanib, nilotinib, vandetanib and erlotinib). The values obtained for kinetic parameters have been summarized in Table 1. Despite no significant changes in cellular accumulation of canertinib and nilotinib at lower concentrations (0.01–0.075 μM), these drugs showed greater accumulation at higher concentrations (0.1–50 μM) in OATP-1B1 and/or -1B3 cells in comparison to CHO-WT cells. For vandetanib, kinetic parameter were evaluated in the concentration range of 0.01–50 μM. Intracellular accumulation of nilotinib and vandetanib was mediated via OATP-1B1 transporter protein. Km value of 2.72±0.25 μM for vandetanib showed higher affinity toward OATP-1B1 transporter than nilotinib (Km =10.14±1.91 μM). Three TKIs (vandetanib, nilotinib and canertinib) appeared to have affinity toward OATP-1B3. The affinity of these TKIs toward OATP-1B3 transporter was in the following order: vandetanib>nilotinib>canertinib (Km =4.37±0.79, 7.84±1.43 and 19.87±2.20 μM, respectively). Also, vandetanib showed greater affinity toward OATP-1B1 than OATP-1B3 transporter protein, whereas nilotinib showed higher affinity for OATP-1B3 than OATP-1B1. Canertinib exhibited its affinity only toward OATP-1B3. Vmax values of both the hepatic uptake transporters for TKIs were calculated and summarized in Table 1. No statistically significant cellular accumulation was observed for pazopanib and erlotinib in OATP-1B1- and -1B3-transfected cells in comparison to WT cells.
Figure 3.
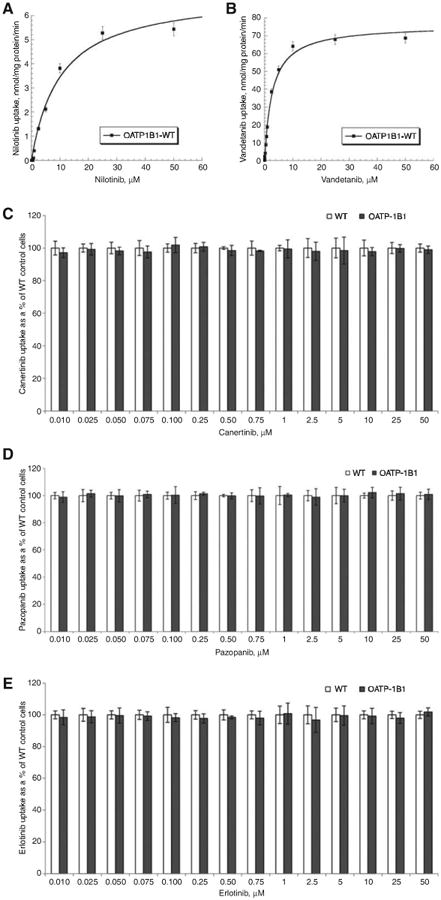
Concentration-dependent uptake of TKIs in OATP-1B1 transfected CHO cells.
(A) Concentration-dependent uptake of nilotinib in OATP-1B1 transfected CHO cells. OATP-1B1 mediated nilotinib transport determined as the difference in uptake in OATP-1B1 and WT CHO cells at each substrate concentration. (B) Concentration-dependent uptake of vandetanib in OATP-1B1 transfected CHO cells. OATP-1B1 mediated vandetanib transport determined as the difference in uptake in OATP-1B1 and WT CHO cells at each substrate concentration. (C) Concentration-dependent uptake of canertinib in WT and OATP-1B1 transfected CHO cells. (D) Concentration-dependent uptake of pazopanib in WT and OATP-1B1 transfected CHO cells. (E) Concentration-dependent uptake of erlotinib in WT and OATP-1B1 transfected CHO cells. Each data point is expressed as mean±SD, n=4.
Figure 4.
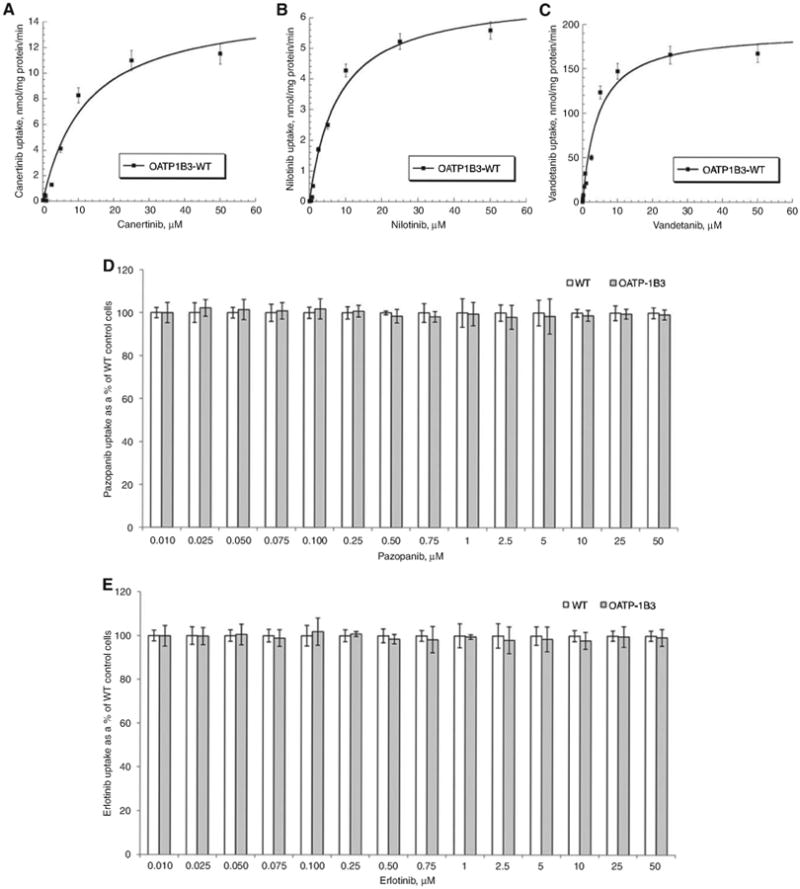
Concentration-dependent uptake of TKIs in OATP-1B3 transfected CHO cells.
(A) Concentration-dependent uptake of canertinib in OATP-1B3 transfected CHO cells. OATP-1B3 mediated canertinib transport determined as the difference in uptake in OATP-1B3 and WT CHO cells at each substrate concentration. (B) Concentration-dependent uptake of nilotinib in OATP-1B3 transfected CHO cells. OATP-1B3 mediated nilotinib transport determined as the difference in uptake in OATP-1B3 and WT CHO cells at each substrate concentration. (C) Concentration-dependent uptake of vandetanib in OATP-1B3 transfected CHO cells. OATP-1B3 mediated vandetanib transport determined as the difference in uptake in OATP-1B3 and WTCHO cells at each substrate concentration. (D) Concentration-dependent uptake of pazopanib in WT and OATP-1B3 transfected CHO cells. (E) Concentration-dependent uptake of erlotinib in WT and OATP-1B3 transfected CHO cells. Each data point is expressed as mean±SD, n=4.
Table 1.
Michaelis-Menten kinetic parameters (Vmax and Km) and catalytic efficiency (Vmax/Km) estimated for tested TKIs for OATP-1B1 and/or OATP-1B3 transporter proteins.
| TKIs | OATP-1B1 | OATP-1B3 | ||||
|---|---|---|---|---|---|---|
|
|
|
|||||
| Km | Vmax | Vmax/Km | Km | Vmax | Vmax/Km | |
| Canertinib | – | – | – | 12.18±3.32 | 15.34±1.59 | 1.25 |
| Nilotinib | 10.14±1.91 | 6.95±0.47 | 0.68 | 7.84±1.43 | 6.75±0.42 | 0.86 |
| Vandetanib | 2.72±0.25 | 75.95±1.99 | 27.92 | 4.37±0.79 | 194.64±10.58 | 44.31 |
| Erlotinib | – | – | – | – | – | – |
| Pazopanib | – | – | – | – | – | – |
Units: Vmax, nmol/mg protein per minute; Km, μM; Vmax/Km, mL/mg protein per minute.
Cytotoxicity studies
To evaluate the cytotoxic effect of the selected TKIs, a cell proliferation assay was performed on cell monolayers of WT and transfected CHO cells for a period of 24 h. No cytotoxic effects of pazopanib, erlotinib, canertinib, vandetanib and nilotinib at a concentration of 50 μM were observed with WT and transfected cells in comparison to positive control (Triton X). The findings from this study clearly demonstrate that the selected TKIs are noncyto-toxic at a concentration of 50 μM (Figure 5).
Figure 5.
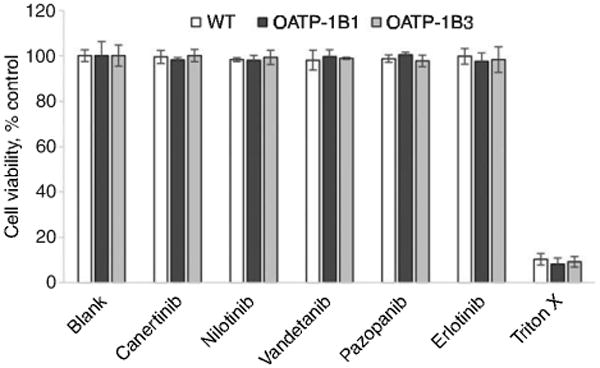
Cytotoxicity in the presence of TKIs at the highest studied concentration (50 μM) on CHO-WT and OATP-1B1- and -1B3-transfected cells.
Data represent the mean±SD, n=4.
Discussion
In the present study, we investigated the role of OATP family transporters in the hepatic uptake process of TKIs. This work also estimated the contribution of OATP-1B1 and -1B3 in hepatocellular accumulation of TKIs with in vitro model-based systems. For uptake transporter analysis, we utilized CHO cells, WT as well as transfected with specifically expressing OATP transport protein. CHO cells were originally selected for the transfection because of the lack of expression of this family of proteins in the parent cell line, resulting in minimal background activity. Also, these cells can be maintained in culture for sustained periods and can be ready for use in a specific experiment within a few days. Our findings are in partial agreement with the study done by Zimmerman et al. [5], where the authors reported the uptake of nilotinib (only at 0.1 μM) in HEK293 cells expressing OATP-1B1 and -1B3 and uptake of vandetanib (only at 0.1 μM) via OATP-1B3 transporter. Similar results were observed in the in vitro cellular accumulation studies performed, demonstrating the uptake of nilotinib and vandetanib via OATP-1B1 and -1B3 transporter proteins. Zimmermann et al. [5] also reported the uptake of pazopanib in cells expressing OATP-1B1 and -1B3 at a concentration of 0.1 μM and no statistically significant uptake of vandetanib (0.1 μM) in cells expressing OATP-1B1 transporter protein. Conversely, our findings demonstrate that there is no active involvement of OATP-1B1 and -1B3 in intracellular accumulation of pazopanib, whereas uptake of vandetanib in hepatic tissue is mediated via OATP-1B1 and -1B3. These contrasting results suggest that utilization of different concentration of TKIs and in vitro model-based systems may affect the cellular accumulation of TKIs via OATP and can lead to misinterpretation of the role of OATPs in cellular accumulation of TKIs. The current concentrations used in this article (0.25 and 0.5 μM) were selected on the basis of previously published results for in vitro uptake studies of TKIs and their toxicity for CHO cells. The concentration ranges used were close to clinically observed Cmax values but may not exactly mimic tissue concentration in the clinic. These studies act as a proof of concept, but further studies may be needed to make the data more clinically relevant. It has been already reported that the TKIs are substrates of efflux transporter proteins such as P-glyco-protein (P-gp), multidrug resistance-associated protein (MRP) and breast cancer resistance protein (BCRP) [10,11, 19–22]. These efflux transporters are also expressed in the bile canalicular membrane and are responsible for excretion of drug and their metabolites from blood into bile. The membrane transporters (OATPs) along with efflux transporters and metabolizing enzymes play a vital role in hepatic disposition of drugs. Seithel et al. reported that coadministration of pravastatin together with macrolides (clarithromycin and roxithromycin) results in increased plasma concentration of pravastatin. It is a substrate for OATP-1B1, -1B3 and CYP3A4, whereas clarithromycin and roxithromycin are inhibitors of OATP-1B1, -1B3 and CYP3A4. Because pravastatin is not extensively metabolized and is excreted almost unchanged into bile and urine, these DDI studies suggest the major involvement of hepatic uptake transporters than metabolizing enzymes for macrolide-induced altered plasma concentration of pravastatin [23]. Similarly, Backman et al. demonstrated that coadministration of cerivastatin and simvastatin (OATP-1B1 and CYP2C8 substrate) together with gemifibrozil resulted in a sixfold rise in the cerivastatin and simvastatin exposure. Such increase in plasma concentration of cerivastatin and simvastatin was due to the inhibitory potency of gemifibrozil on OATP-1B1 [24, 25]. Uptake of drugs mediated via OATP can be considered as an important additional mechanism essential for DDIs [26]. Unlike statins, information on the role of hepatic uptake transporters (OATP-1B1 and -1B3) for TKIs is very sparse. Thus, it is of utmost importance to understand the role of OATPs in hepatocellular accumulation of TKIs. The results obtained from in vitro cellular accumulation studies suggest for the very first time the involvement of OATP-1B1 and/or OATP-1B3 in hepatocellular accumulation of TKIs (canertinib, nilotinib and vandetanib). This article is the first to report the affinity of selected TKIs for OATP-1B1 and/or -1B3 by estimating the MM kinetic parameters. These findings suggest that OATP-1B1 exhibits greater affinity toward vandetanib than nilotinib. The ratio of these kinetic parameters (Vmax/Km) provides an estimate of the catalytic efficiency of OATP-1B1 transporter. The transport efficiency (Vmax/Km) of OATP-1B1 transporter was observed for nilotinib and vandetanib as 0.68 and 27.92 mL/mg protein per minute, respectively. While comparing the efficiency values, OATP-1B1 transport efficiency was found to be higher for vandetanib than nilotinib. No significant changes in uptake of canertinib, pazopanib and erlotinib were observed in WT and OATP-1B1 transfected cells. Similarly, for OATP-1B3 transporter protein, the ratio of Vmax/Km provides an estimate of the catalytic efficiency of OATP-1B3 transporter. Such transport efficiency (Vmax/Km) of OATP-1B3 transporter observed for canertinib, nilotinib and vandetanib was 1.25, 0.86 and 44.31 mL/mg protein per minute, respectively. OATP-1B3 transporter exhibits the highest transport rate for vandetanib than nilotinib and canertinib. In pazopanib and erlotinib, no significant difference was observed in uptake values of WT and OATP-1B3, suggesting that OATP-1B3 transporter does not play a major role in their hepatic accumulation. On comparing the Km values of OATP-1B1 and -1B3 transporter for nilotinib, it is apparent that nilotinib exerts higher affinity toward OATP-1B3 relative to OATP-1B1, whereas, Vmax values of OATP-1B1 and -1B3 are comparable to nilotinib. These MM kinetic parameters play an important role in determining the differential transport efficiency of OATP-1B1 and -1B3 in hepatic uptake of nilotinib. A distinction in Km values of OATP-1B1 and -1B3 for nilotinib can be attributed as the primary driving force in determining its transport efficiency, as no difference was observed in Vmax values of both the OATP-1B type transporters. The difference in the transport efficiencies (Vmax/Km) of OATP-1B1 and -1B3 was not very large, suggesting equal involvement of both the OATPs in the hepatic uptake of nilotinib. On comparing Km values, vandetanib exhib-its higher affinity toward OATP-1B1 than -1B3. Higher Vmax values were observed for OATP-1B3 (∼2.6-fold) than -1B1. Large differences in Vmax have a significant contribution in determining the substrate specificity of vandetanib for OATP-1B1 and -1B3. The transport efficiency (Vmax/Km) of OATP-1B3 for vandetanib showed approximately twofold difference than -1B1, showing greater contribution of OATP-1B3 than -1B1 in hepatic uptake of vandetanib. Therefore, hepatic uptake of vandetanib and nilotinib is tightly regulated by OATP-1B1 and -1B3, which act as two gatekeepers localized on the liver. Hence, OATP-1B1 and -1B3 can be considered as important factors in determining pharmacokinetics or DDIs of vandetanib and nilotinib. For canertinib, only OATP-1B3 was observed to be responsible for its hepatic uptake and it can act as a key determinant in the bioavailability of canertinib. These results though act mainly as a proof of concept, and the actual rate constants in humans may vary based on various physiological and pathological conditions resulting in altered expression of these transporters within the liver.
Interindividual pharmacokinetic variability can arise at many stages of ADME. For individual therapy, dosing strategies can be based on pharmacokinetic properties. Decisive tailoring of individual dosing to a patient necessitates minimizing these interindividual pharmacokinetic differences and reducing the risks of both toxicity and subtherapeutic dosing [7]. Till now, most of the studies performed with TKIs have not considered the affinity of hepatic uptake transporters as a determinant of pharmacokinetic profiles of TKIs. Interindividual pharmacokinetic variability has been associated with OATP-1B1 and -1B3 genetic polymorphisms in patients taking statins and irinotecan [18, 27]. Expression and involvement of these OATP-1B-type isoforms in liver has important implications for better understanding of the factors governing ADME of TKIs. Any compromise in the activity of OATP-1B-type transporter proteins will result in suboptimal treatment or high toxicity considering the wide interindividual pharmacokinetic variability of the tested TKIs. As vandetanib and nilotinib exhibit substrate specificity toward OATP-1B1 and -1B3, these results led us to the hypothesis that an inactive phenotypic variant of OATP-1B type transporter may determine the clinical pharmacokinetics of these TKIs.
Previously published reports have only considered the role of efflux transporters and metabolizing enzymes in determining the pharmacokinetic profile of TKIs [10, 11, 14, 28–31]. In this study, we have observed that a carrier-mediated uptake process via OATP-1B1 transporter is involved in cellular accumulation of nilotinib and vandetanib in hepatic tissue. Also, OATP-1B3 is responsible for hepatocellular accumulation of canertinib, vandetanib and nilotinib. OATP-1B1 and/or -1B3 regulate the initial step of hepatic elimination of TKIs by carrying out the uptake of selected TKIs into the hepatic tissue, exposing the molecules to CYP enzyme-mediated metabolism and elimination via biliary secretion. These OATPs expressed on the basolateral membrane of hepatocytes will induce uptake of TKIs and can be regarded as one of the determinants of the overall metabolic rate of TKIs in the liver [4]. An efficient directional movement of therapeutic agents across hepatic tissues requires the manifestation and synchronized activity of hepatic uptake, metabolizing enzymes and efflux transporters [32]. Duckett and Cameron [33] reported that coadministration of ketoconazole (CYP3A4 and P-gp inhibitor) with nilotinib resulted in threefold increase in the plasma concentration of nilotinib. Ketoconazole is also a well-known inhibitor of OATP-1B type transporters [34], and we have shown that OATP-1B1 and -1B3 transporters are responsible for the hepatic uptake of nilotinib. Therefore, the threefold increase in the plasma concentration of nilotinib may not be just due to the inhibition of efflux transporter and metabolizing enzyme but also could be due to inhibition of OATP-1B type transporter. OATP-1B1 and -1B3 transporters also play a vital role in the hepatic uptake of nilotinib, making it vulnerable to metabolism by CYP3A4 and ultimately causing its elimination by biliary secretion via P-gp. As a major fraction of nilotinib is eliminated in the feces in unchanged form, suggesting that the transmembrane localization of nilotinib by OATPs in the liver has a major impact on its pharmacokinetics, Minocha et al. [11] reported that the coadministration of vandetanib and everolimus resulted in increased plasma concentration of vandetanib. Everolimus increases the plasma concentration of vandetanib by inhibiting efflux transporters (P-gp and BCRP), which are also localized on the bile canalicular membrane. Everolimus is also a well-known inhibitor of OATP-1B1 and -1B3 transporters, which are responsible for hepatocellular accumulation of therapeutic agents. In this article, we have reported the involvement of OATP-1B1 and -1B3 in the hepatic uptake of vandetanib. Taken together, it is reasonable to assume that elevation in plasma concentration of vandetanib is not only due to the inhibition of P-gp and BCRP but also due to inhibition of OATP-1B1 and -1B3 by everolimus. Complex interplay of OATPs (localized on the basolateral membrane of hepatic tissues) and efflux transporters (expressed on the bile canalicular membrane) may be responsible for the hepatobiliary excretion of TKIs. Inhibition of TKI uptake via hepatic OATPs by coadministration of drugs that are also substrates or inhibitors of these hepatic uptake transporters is a plausible explanation for several in vivo observed DDIs. Inhibition of metabolizing enzymes and/or efflux transporter could not be the only cause of the experimental effects. Drug-induced alteration of OATP-1B1 and -1B3 transporter function is, therefore, an essential auxiliary mechanism underlying DDIs [26].
OATP-mediated DDIs have the potential to completely influence drug efficacy and toxicity. Therefore, coadministration of canertinib, vandetanib and nilotinib (OATP-1B1 and/or -1B3 substrates) along with other hepatic OATP substrates/inhibitors (paclitaxel, cyclosporine, protease inhibitors, rifampicin, statins, telmisartan, valsartan, mTOR inhibitors, antibiotics etc.) may result in altered pharmacokinetics and pharmacodynamics of TKIs. However, induction of the expression of hepatic OATPs can also result in increased detoxification and elimination of numerous OATP-1B1 and -1B3 substrates from the body [14, 35–37]. The pharmacokinetic profile of TKIs is likely to be modified in subjects with hepatic impairment, as TKIs are primarily eliminated via hepatic metabolism and biliary excretion. Downregulation of OATP-1B-type transporter protein in liver disease has been reported by Keitel et al. and Oswald et al. [38, 39]. Also, reduced expression of OATP-1B1 and -1B3 in liver cancer compared to noncancerous liver tissues has been reported by various investigators [40–42]. For the hepatically impaired population, dose reduction of nilotinib has been proposed along with recommendation of lower starting dose and monitoring of any liver function abnormalities [43, 44]. Also, the dose of vandetanib in patients with moderate and severe hepatic impairment has not been recommended as its safety and efficacy has not been established [43, 44]. In vitro accumulation studies along with estimation of MM kinetic profiles confirms the role of OATP-1B1 and -1B3 in hepatic uptake of nilotinib and vandetanib. On the basis of our findings along with already published reports mentioned above, we can hypothesize that higher plasma concentration of nilotinib or other TKIs in patients with hepatic impairment can be attributed to compromised/downregu-lated expression of OATP-1B1 and -1B3 transporters, which can be responsible for any alteration in the pharmacokinetic profile of nilotinib or other TKIs. Similar findings have been reported by Zimmerman et al., showing the involvement of OATP-1B1 and-1B3 in hepatic elimination of sorafenib and longer systemic accumulation due to compromised activity of OATP-1B-type transporter protein [5].
In conclusion, we have shown that OATP-1B1 and OATP-1B3 are responsible for hepatocellular accumulation of nilotinib and vandetanib, whereas only OATP-1B3 is responsible for the carrier-mediated uptake of canerti-nib in hepatic tissue. These findings delineate the involvement of OATP-1B1 and/or -1B3 in hepatic uptake of tested TKIs and confirms the affinity of these hepatic uptake transporters as a determinant of the pharmacokinetic profile of TKIs. As coadministration of TKIs with other therapeutic agents is becoming common in multidrug therapy, hepatic uptake transporters OATP-1B1 and -1B3 can be regarded as important molecular targets for potential DDIs. Thus, hepatic uptake mediated by OATP-1B1 and -1B3 for selected TKIs should be dynamically scrutinized in order to circumvent DDIs. These transporters, in conjunction with the efflux proteins, may eventually decide on the overall flux of the TKIs within the hepatic tissue. These studies act as a proof of concept substantiating the need for further clinical studies investigating the OATP-based DDI potential of TKIs. Further in vivo studies are required for better understanding of the contribution of OATP-1B1 and/or -1B3 transporter proteins in the hepatic disposition of TKIs and for predicting any adverse drug reactions associated with DDIs.
Acknowledgments
This work was supported by National Institutes of Health grant 1R01 AI071199. The authors highly appreciate Dr. Bruno Stieger for the generous gift of CHO-WT and OATP-1B type transporter protein transfected cell lines.
Research funding: None declared.
Footnotes
Conflict of interest statement: Authors' conflict of interest disclosure: The authors stated that there are no conflicts of interest regarding the publication of this article. Research support played no role in the study design; in the collection, analysis, and interpretation of data; in the writing of the report; or in the decision to submit the report for publication.
Employment or leadership: None declared.
Honorarium: None declared.
Contributor Information
Varun Khurana, Division of Pharmaceutical Sciences, School of Pharmacy, University of Missouri-Kansas City, Kansas City, MO 64108, USA.
Mukul Minocha, Division of Pharmaceutical Sciences, School of Pharmacy, University of Missouri-Kansas City, Kansas City, MO 64108, USA; and Center for Translational Medicine, School of Pharmacy, University of Maryland, Baltimore, MD, USA.
Dhananjay Pal, Division of Pharmaceutical Sciences, School of Pharmacy, University of Missouri-Kansas City, Kansas City, MO 64108, USA.
References
- 1.Shimizu M, Fuse K, Okudaira K, Nishigaki R, Maeda K, Kusuhara H, et al. Contribution of OATP (organic anion-transporting polypeptide) family transporters to the hepatic uptake of fexofenadine in humans. Drug Metab Dispos. 2005;33:1477–81. doi: 10.1124/dmd.105.004622. [DOI] [PubMed] [Google Scholar]
- 2.Hagenbuch B, Meier PJ. Organic anion transporting polypeptides of the OATP/SLC21 family: phylogenetic classification as OATP/SLCO superfamily, new nomenclature and molecular/functional properties. Pflugers Arch. 2004;447:653–65. doi: 10.1007/s00424-003-1168-y. [DOI] [PubMed] [Google Scholar]
- 3.Barton HA, Lai Y, Goosen TC, Jones HM, El-Kattan AF, Gosset JR, et al. Model-based approaches to predict drug-drug interactions associated with hepatic uptake transporters: preclinical, clinical and beyond. Expert Opin Drug Metab Toxicol. 2013;9:459–72. doi: 10.1517/17425255.2013.759210. [DOI] [PubMed] [Google Scholar]
- 4.Shitara Y, Maeda K, Ikejiri K, Yoshida K, Horie T, Sugiyama Y. Clinical significance of organic anion transporting polypeptides (OATPs) in drug disposition: their roles in hepatic clearance and intestinal absorption. Biopharm Drug Dispos. 2013;34:45–78. doi: 10.1002/bdd.1823. [DOI] [PubMed] [Google Scholar]
- 5.Zimmerman EI, Hu S, Roberts JL, Gibson AA, Orwick SJ, Li L, et al. Contribution of OATP1B1 and OATP1B3 to the disposition of sorafenib and sorafenib-glucuronide. Clin Cancer Res. 2013;19:1458–66. doi: 10.1158/1078-0432.CCR-12-3306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pearson MA, Fabbro D. Targeting protein kinases in cancer therapy: a success? Expert Rev Anticancer Ther. 2004;4:1113–24. doi: 10.1586/14737140.4.6.1113. [DOI] [PubMed] [Google Scholar]
- 7.Undevia SD, Gomez-Abuin G, Ratain MJ. Pharmacokinetic variability of anticancer agents. Nat Rev Cancer. 2005;5:447–58. doi: 10.1038/nrc1629. [DOI] [PubMed] [Google Scholar]
- 8.van Erp NP, Gelderblom H, Guchelaar HJ. Clinical pharmacokinetics of tyrosine kinase inhibitors. Cancer Treat Rev. 2009;35:692–706. doi: 10.1016/j.ctrv.2009.08.004. [DOI] [PubMed] [Google Scholar]
- 9.Minocha M, Khurana V, Mitra AK. Determination of pazopanib (GW-786034) in mouse plasma and brain tissue by liquid chromatography-tandem mass spectrometry (LC/MS-MS) J Chromatogr B Analyt Technol Biomed Life Sci. 2012;901:85–92. doi: 10.1016/j.jchromb.2012.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Minocha M, Khurana V, Qin B, Pal D, Mitra AK. Enhanced brain accumulation of pazopanib by modulating P-gp and Bcrpl mediated efflux with canertinib or erlotinib. Int J Pharm. 2012;436:127–34. doi: 10.1016/j.ijpharm.2012.05.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Minocha M, Khurana V, Qin B, Pal D, Mitra AK. Co-administration strategy to enhance brain accumulation of vandetanib by modulating P-glycoprotein (P-gp/Abcb1) and breast cancer resistance protein (Bcrp1/Abcg2) mediated efflux with m-TOR inhibitors. Int J Pharm. 2012;434:306–14. doi: 10.1016/j.ijpharm.2012.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gotze L, Hegele A, Metzelder SK, Renz H, Nockher WA. Development and clinical application of a LC-MS/MS method for simultaneous determination of various tyrosine kinase inhibitors in human plasma. Clin Chim Acta. 2012;413:143–9. doi: 10.1016/j.cca.2011.09.012. [DOI] [PubMed] [Google Scholar]
- 13.Lankheet NA, Hillebrand MJ, Rosing H, Schellens JH, Beijnen JH, Huitema AD. Method development and validation for the quantification of dasatinib, erlotinib, gefitinib, imatinib, lapatinib, nilotinib, sorafenib and sunitinib in human plasma by liquid chromatography coupled with tandem mass spectrometry. Biomed Chromatogr. 2013;27:466–76. doi: 10.1002/bmc.2814. [DOI] [PubMed] [Google Scholar]
- 14.Kalliokoski A, Niemi M. Impact of OATP transporters on pharmacokinetics. Br J Pharmacol. 2009;158:693–705. doi: 10.1111/j.1476-5381.2009.00430.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Konig J, Cui Y, Nies AT, Keppler D. Localization and genomic organization of a new hepatocellular organic anion transporting polypeptide. J Biol Chem. 2000;275:23161–8. doi: 10.1074/jbc.M001448200. [DOI] [PubMed] [Google Scholar]
- 16.De Bruyn T, van Westen GJ, Ijzerman AP, Stieger B, de Witte P, Augustijns PF, et al. Structure-based identification of OATP1B1/3 inhibitors. Mol Pharmacol. 2013;83:1257–67. doi: 10.1124/mol.112.084152. [DOI] [PubMed] [Google Scholar]
- 17.Gui C, Obaidat A, Chaguturu R, Hagenbuch B. Development of a cell-based high-throughput assay to screen for inhibitors of organic anion transporting polypeptides 1B1 and 1B3. Curr Chem Genomics. 2010;4:1–8. doi: 10.2174/1875397301004010001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nozawa T, Minami H, Sugiura S, Tsuji A, Tamai I. Role of organic anion transporter OATP1B1 (OATP-C) in hepatic uptake of irinotecan and its active metabolite, 7-ethyl-10-hydroxyca-mptothecin: in vitro evidence and effect of single nucleotide polymorphisms. Drug Metab Dispos. 2005;33:434–9. doi: 10.1124/dmd.104.001909. [DOI] [PubMed] [Google Scholar]
- 19.Agarwal S, Mittapalli RK, Zellmer DM, Gallardo JL, Donelson R, Seiler C, et al. Active efflux of Dasatinib from the brain limits efficacy against murine glioblastoma: broad implications for the clinical use of molecularly targeted agents. Mol Cancer Ther. 2012;11:2183–92. doi: 10.1158/1535-7163.MCT-12-0552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Agarwal S, Elmquist WF. Insight into the cooperation of P-glycoprotein (ABCB1) and breast cancer resistance protein (ABCG2) at the blood-brain barrier: a case study examining sorafenib efflux clearance. Mol Pharm. 2012;9:678–84. doi: 10.1021/mp200465c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang T, Agarwal S, Elmquist WF. Brain distribution of cediranib is limited by active efflux at the blood-brain barrier. J Pharmacol Exp Ther. 2012;341:386–95. doi: 10.1124/jpet.111.190488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Agarwal S, Sane R, Gallardo JL, Ohlfest JR, Elmquist WF. Distribution of gefitinib to the brain is limited by P-glycoprotein (ABCB1) and breast cancer resistance protein (ABCG2)-mediated active efflux. J Pharmacol Exp Ther. 2010;334:147–55. doi: 10.1124/jpet.110.167601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Seithel A, Eberl S, Singer K, Auge D, Heinkele G, Wolf NB, et al. The influence of macrolide antibiotics on the uptake of organic anions and drugs mediated by OATP1B1 and OATP1B3. Drug Metab Dispos. 2007;35:779–86. doi: 10.1124/dmd.106.014407. [DOI] [PubMed] [Google Scholar]
- 24.Backman JT, Kyrklund C, Neuvonen M, Neuvonen PJ. Gemfibrozil greatly increases plasma concentrations of cerivastatin. Clin Pharmacol Ther. 2002;72:685–91. doi: 10.1067/mcp.2002.128469. [DOI] [PubMed] [Google Scholar]
- 25.Backman JT, Kyrklund C, Kivisto KT, Wang JS, Neuvonen PJ. Plasma concentrations of active simvastatin acid are increased by gemfibrozil. Clin Pharmacol Ther. 2000;68:122–9. doi: 10.1067/mcp.2000.108507. [DOI] [PubMed] [Google Scholar]
- 26.Fahrmayr C, Fromm MF, Konig J. Hepatic OATP and OCT uptake transporters: their role for drug-drug interactions and pharmacogenetic aspects. Drug Metab Rev. 2010;42:380–401. doi: 10.3109/03602530903491683. [DOI] [PubMed] [Google Scholar]
- 27.Romaine SP, Bailey KM, Hall AS, Balmforth AJ. The influence of SLC01B1 (OATP1B1) gene polymorphisms on response to statin therapy. Pharmacogenomics J. 2010;10:1–11. doi: 10.1038/tpj.2009.54. [DOI] [PubMed] [Google Scholar]
- 28.Hu S, Franke RM, Filipski KK, Hu C, Orwick SJ, de Bruijn EA, et al. Interaction of imatinib with human organic ion carriers. Clin Cancer Res. 2008;14:3141–8. doi: 10.1158/1078-0432.CCR-07-4913. [DOI] [PubMed] [Google Scholar]
- 29.Burger H, van Tol H, Brok M, Wiemer EA, de Bruijn EA, Guetens G, et al. Chronic imatinib mesylate exposure leads to reduced intracellular drug accumulation by induction of the ABCG2 (BCRP) and ABCB1 (MDR1) drug transport pumps. Cancer Biol Ther. 2005;4:747–52. doi: 10.4161/cbt.4.7.1826. [DOI] [PubMed] [Google Scholar]
- 30.Burger H, van Tol H, Boersma AW, Brok M, Wiemer EA, Stoter G, et al. Imatinib mesylate (STI571) is a substrate for the breast cancer resistance protein (BCRP)/ABCG2 drug pump. Blood. 2004;104:2940–2. doi: 10.1182/blood-2004-04-1398. [DOI] [PubMed] [Google Scholar]
- 31.Hamada A, Miyano H, Watanabe H, Saito H. Interaction of imatinib mesilate with human P-glycoprotein. J Pharmacol Exp Ther. 2003;307:824–8. doi: 10.1124/jpet.103.055574. [DOI] [PubMed] [Google Scholar]
- 32.Kim RB. Organic anion-transporting polypeptide (OATP) transporter family and drug disposition. Eur J Clin Invest. 2003;33(Suppl 1):21–5. doi: 10.1046/j.1365-2362.33.s2.5.x. [DOI] [PubMed] [Google Scholar]
- 33.Duckett DR, Cameron MD. Metabolism considerations for kinase inhibitors in cancer treatment. Expert Opin Drug Metab Toxicol. 2010;6:1175–93. doi: 10.1517/17425255.2010.506873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Choi MK, Jin QR, Choi YL, Ahn SH, Bae MA, Song IS. Inhibitory effects of ketoconazole and rifampin on OAT1 and OATP1B1 transport activities: considerations on drug-drug interactions. Biopharm Drug Dispos. 2011;32:175–84. doi: 10.1002/bdd.749. [DOI] [PubMed] [Google Scholar]
- 35.Koenen A, Kroemer HK, Grube M, Meyer zu Schwabedissen HE. Current understanding of hepatic and intestinal OATP-mediated drug-drug interactions. Expert Rev Clin Pharmacol. 2011 Nov 2;4:729–42. doi: 10.1586/ecp.11.58. [DOI] [PubMed] [Google Scholar]
- 36.Muller F, Fromm MF. Transporter-mediated drug-drug interactions. Pharmacogenomics. 2011;12:1017–37. doi: 10.2217/pgs.11.44. [DOI] [PubMed] [Google Scholar]
- 37.Niemi M. Role of OATP transporters in the disposition of drugs. Pharmacogenomics. 2007;8:787–802. doi: 10.2217/14622416.8.7.787. [DOI] [PubMed] [Google Scholar]
- 38.Keitel V, Burdelski M, Warskulat U, Kuhlkamp T, Keppler D, Haussinger D, et al. Expression and localization of hepatobiliary transport proteins in progressive familial intrahepatic cholestasis. Hepatology. 2005;41:1160–72. doi: 10.1002/hep.20682. [DOI] [PubMed] [Google Scholar]
- 39.Oswald M, Kullak-Ublick GA, Paumgartner G, Beuers U. Expression of hepatic transporters OATP-C and MRP2 in primary sclerosing cholangitis. Liver. 2001;21:247–53. doi: 10.1034/j.1600-0676.2001.021004247.x. [DOI] [PubMed] [Google Scholar]
- 40.Wlcek K, Svoboda M, Riha J, Zakaria S, Olszewski U, Dvorak Z, et al. The analysis of organic anion transporting polypeptide (OATP) mRNA and protein patterns in primary and metastatic liver cancer. Cancer Biol Ther. 2011;11:801–11. doi: 10.4161/cbt.11.9.15176. [DOI] [PubMed] [Google Scholar]
- 41.Vavricka SR, Jung D, Fried M, Grutzner U, Meier PJ, Kullak-Ublick GA. The human organic anion transporting polypeptide 8 (SLC01B3) gene is transcriptionally repressed by hepatocyte nuclear factor 3beta in hepatocellular carcinoma. J Hepatol. 2004;40:212–8. doi: 10.1016/j.jhep.2003.10.008. [DOI] [PubMed] [Google Scholar]
- 42.Cui Y, Konig J, Nies AT, Pfannschmidt M, Hergt M, Franke WW, et al. Detection of the human organic anion transporters SLC21A6 (OATP2) and SLC21A8 (OATP8) in liver and hepatocellular carcinoma. Lab Invest. 2003;83:527–38. doi: 10.1097/01.lab.0000065015.02412.48. [DOI] [PubMed] [Google Scholar]
- 43.Di Gion P, Kanefendt F, Lindauer A, Scheffler M, Doroshyenko O, Fuhr U, et al. Clinical pharmacokinetics of tyrosine kinase inhibitors: focus on pyrimidines, pyridines and pyrroles. Clin Pharmacokinet. 2011;50:551–603. doi: 10.2165/11593320-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 44.Feng B, Xu JJ, Bi YA, Mireles R, Davidson R, Duignan DB, et al. Role of hepatic transporters in the disposition and hepatotoxicity of a HER2 tyrosine kinase inhibitor CP-724,714. Toxicol Sci. 2009;108:492–500. doi: 10.1093/toxsci/kfp033. [DOI] [PubMed] [Google Scholar]