

Abstract
Background:
Autosomal dominant polycystic kidney disease (ADPKD) is a progressive genetic disorder characterized by the development of numerous kidney cysts that result in kidney failure. Little is known regarding the key patient characteristics and utilization of healthcare resources for ADPKD patients along the continuum of disease progression. This observational study was designed to describe the characteristics of ADPKD patients and compare them with those of patients with other chronic kidney diseases.
Methods:
This retrospective cohort study involved patients with a claim for ADPKD or PKD unspecified from 1/1/2000–2/28/2013 and ≥6 months of previous continuous enrollment (baseline) within a large database of administrative claims in the USA. A random sample of chronic kidney disease (CKD) patients served as comparators. For a subset of ADPKD patients who had only a diagnosis code of unspecified PKD, abstraction of medical records was undertaken to estimate the proportion of patients who had medical chart-confirmed ADPKD. In patients with linked electronic laboratory data, the estimated glomerular filtration rate was calculated via serum creatinine values to determine CKD stage at baseline and during follow-up. Proportions of patients transitioning to another stage and the mean age at transition were calculated.
Results:
ADPKD patients were, in general, younger and had fewer physician visits, but had more specific comorbidities at observation start compared with CKD patients. ADPKD patients had a longer time in the milder stages and longer duration before recorded transition to a more severe stage compared with CKD patients. Patients with ADPKD at risk of rapid progression had a shorter time-to-end-stage renal disease than patients with CKD and ADPKD patients not at risk, but stage duration was similar between ADPKD patients at risk and those not at risk.
Conclusions:
These results suggest that distribution of patients by age at transition to next stage may be useful for identification of ADPKD patients at risk of rapid progression. The results also suggest that medical claims with diagnosis codes for “unspecified PKD”, in absence of a diagnosis code for autosomal recessive polycystic kidney disease, may be a good proxy for ADPKD.
Keywords: autosomal dominant polycystic kidney disease, ADPKD, chronic kidney disease, serum creatinine, disease stage, end-stage renal disease
Introduction
Autosomal dominant polycystic kidney disease (ADPKD) is a progressive genetic disorder characterized by the development of numerous kidney cysts that result in kidney enlargement, which can lead to pain, hypertension, renal dysfunction and, ultimately, kidney failure [1,2]. ADPKD is the most common monogenic kidney disease in the USA and Europe [3,4]. Prevalence studies are outdated and have, in general, been conducted in small populations [5]. However, a recent landmark single-site study in the USA revealed the incidence of ADPKD to be ≈1:400 to 1:1000, depending on whether the estimation is based on symptomatic and family-screen cases or on autopsy data [6]. ADPKD is the leading inheritable cause of end-stage renal disease (ESRD) and one of the leading causes of ESRD overall [3,7]. ADPKD is also associated with a high financial burden. Total healthcare costs for ADPKD have been reported to be $51,048 per patient-year [8]. Also, predicted four-year healthcare costs by chronic kidney disease (CKD) stage have been estimated to be $40,164 (stage I), $33,397 (stage II), $42,686 (stage III), $148,402 (stage IV), and $207,548 (stage V) [9].
Hallmarks of ADPKD include development of numerous fluid-filled renal cysts that result in kidney enlargement, renal insufficiency, renal failure and, ultimately ESRD [10,11]. Progression of ADPKD is marked by declining kidney function along with increasing frequency and intensity of acute hospitalizations. Additional renal complications with ADPKD include urinary-tract infections (UTIs) and renal stones [1,2]. ADPKD is a systemic disease with complications arising from cysts developing in organs beyond the kidneys (most frequently in the liver and pancreas) as well as other extrarenal manifestations such as intracranial aneurysms, vascular events, cardiac events, and hematuria [1,2,12,13]. Hypertension in ADPKD patients is an early identifiable marker of renal disease and cardiovascular disease [1,12,14].
Little is known about the key characteristics and utilization of healthcare resources for patients with ADPKD along the continuum of disease progression. Increased understanding of the natural history of ADPKD may help identify patients at risk of rapid progression of, or the complications associated with, ADPKD, who could benefit from early treatment to potentially delay ESRD. The goal of ADPKD treatments for non-ESRD patients is control of blood pressure [15] and pain relief [16]. The goal of ADPKD treatments for ESRD patients is dialysis or transplantation. However, there remains an unmet need for treatments that slow or reverse ADPKD progression. Pharmacologic agents are being tested [17] and combination therapies have been suggested [18], but as yet there is no pharmacologic therapy indicated for ADPKD in the USA.
This observational study was designed to describe the characteristics of ADPKD patients and compare them with those of patients with other chronic kidney diseases. The primary objective was to describe the natural history of ADPKD in a real-world setting. The present study focused on: characterization of disease progression based on data from administrative claims; comparison of the characteristics among ADPKD patients vs. other CKD patients; characterization of a subset of ADPKD patients potentially at risk of rapid disease progression using validation of medical-chart reviews.
Methods
Study design and data sources
This was a retrospective observational cohort study using data on eligibility, pharmacy, medical claims, and electronic laboratory results from the Optum Research Database* (ORD). The ORD represents patients from a large health plan in the USA. The ORD is a proprietary research database containing medical and pharmacy claims with linked enrollment information from 1993 to the present. For 2012, data relating to ≈12.6 million individuals with medical and pharmacy benefit coverage were available. Laboratory data were available for a subpopulation of patients.
Study population
The study population comprised members of commercial health plans in the ORD who had a medical claim associated with a diagnosis of ADPKD or CKD between 1 January 2000 and 28 February 2013, the timeframe available for analysis. Overall requirements for inclusion were: continuous enrollment in the health plan with medical coverage and pharmacy benefits for ≥6 months before the earliest qualifying diagnosis of ADPKD or CKD (index date); continued complete medical coverage and pharmacy benefits throughout the period of observation within the study period.
For inclusion in the ADPKD cohort, patients had to have at least two claims for either of the following ICD-9 codes on separate dates: ICD-9-CM 753.12 (PKD, unspecified) or 753.13 (ADPKD), excluding claims for laboratories and diagnostic centers. Patients were excluded from the ADPKD cohort if they had a claim with a diagnosis code for autosomal recessive polycystic kidney disease (ARPKD, ICD-9-CM 753.14) at any time during the study baseline or follow-up period, or any evidence of kidney transplantation prior to the first diagnosis of ADPKD.
A subset of ADPKD patients at potential risk of rapid progression during the study baseline or follow-up period was also identified based on one or more of the following: hypertension before the age of 35 years; hematuria before the age of 30 years; albuminuria at any time; advanced-stage kidney disease according to age (stage II by age 30 years, stage III by age 50 years, and stage IV/V, ESRD or transplantation by age 55 years). These criteria were identified based on a review of the literature on ADPKD [2,5,11–14,19,20].
Abstraction of medical records was undertaken for a subset of ADPKD patients who had only an ICD-9-CM diagnosis code of 753.12 (PKD, unspecified). Charts were used to validate the claims-based PKD diagnosis and to estimate the proportion of patients who had chart-confirmed ADPKD in the absence of a corresponding ICD-9-CM diagnosis.
For inclusion in the CKD cohort, patients had to have at least two claims for CKD (ICD-9-CM 585.1–585.9) on separate dates, excluding claims from laboratories or diagnostic centers. Patients with a claim for ADPKD (ICD-9-CM 753.13), PKD unspecified (753.12), or ARPKD (ICD-9-CM 753.14) at any time during the study period inclusive of the baseline period, as well as those with any evidence of kidney transplantation prior to the first diagnosis of CKD, were excluded. Patients meeting eligibility criteria for the CKD cohort were selected randomly so that the CKD cohort was threefold larger than the ADPKD cohort.
Measurement
Progression to a more severe stage of kidney disease, and the age at progression during the follow-up period, were the primary outcomes of the study. In a subgroup of patients with linked electronic data for laboratory results, identification of serum creatinine (SCr) results and calculation of estimated glomerular filtration rate (eGFR) was undertaken to determine the stage of kidney disease at baseline and during follow-up using the following quadratic equation with age, sex, and SCr [21]:
Stage of kidney disease was defined based on the following classification of eGFR (in mL/min): stage I, ≥90; II, 60–89; III, 30–59; IV, 15–29; V, <15 (or on dialysis). Duration for stage transition was defined as the time interval between two consecutive stages, and was used to describe the time to the next stage at transition. Patients were assessed for forward transition(s); due to fluctuations in SCr, patients could appear to have a “backward” transition, but such transitions were not included in the assessment. Claims data were used to calculate time-to-ESRD overall and stratified by baseline stage of kidney disease starting from the day after index. Duration of follow-up for each cohort member extended from the day after cohort entry until either the first occurrence of a study event (e.g., ESRD), discontinuation from the health plan, or the end of the study period, whichever occurred first. Similar analyses were conducted in a subset of patients at risk of rapid progression. Covariates were determined on the basis of healthcare claims during the 6-month baseline period for healthcare utilization; clinical characteristics and demographics were ascertained on entry date for the cohort.
Statistical analyses
Baseline descriptive characteristics were tabulated and stratified by cohort and stage. Number and percentage were calculated for categorical variables. Mean/standard deviation or median/interquartile range was calculated for continuous variables. The number of patients who transitioned to the next stage was tabulated to describe stage transition. Mean and standard deviation of age at each stage and the median and interquartile range of stage duration were described accordingly. Median survival times, incidence rates, and associated 95% confidence intervals for ESRD were estimated by study cohort and baseline stage. Kaplan–Meier plots were generated to depict the cumulative probability of event-free time among those with ADPKD, CKD, and those at risk of rapid progression.
Results
Characteristics of patients and the study
A total of 7,617 patients with ADPKD and 22,851 patients with CKD were identified, of which 4,101 ADPKD patients and 11,862 CKD patients had ≥1 SCr laboratory result during the baseline or follow-up periods (Figure 1). There were 3,415 patients with ADPKD who were identified as being at risk for rapid progression; 2,097 of these patients had ≥1 SCr laboratory result during baseline or follow-up. Charts for 146 patients with an unspecified diagnosis of PKD were procured, and 132 were determined by clinical reviewers to have sufficient information for ADPKD determination. Of these 132 patients, 125 were chart-confirmed to have ADPKD by a single clinical adjudicator, representing a positive predictive value of 94.7%. Demographic characteristics of ADPKD patients and CKD patients are described in Table 1.
Figure 1.

Flow chart for the study.
ADPKD, autosomal dominant polycystic kidney disease; ARPKD, autosomal recessive polycystic kidney disease; CKD, chronic kidney disease.
Table 1.
Demographic characteristics of ADPKD patients and CKD patients by disease stage at cohort entry: Optum Research Database, January 1, 2000–February 28, 2013.
| (n) | Overall (N=30,468) | Stage I (n=1,561) | Stage II (n=2,001) | Stage III (n=2,265) | Stage IV (n=743) | Stage V (n=2,697) | Unknown (n=22,201) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||||||||
| ADPKD (7,617) | CKD (22,851) | ADPKD (627) | CKD (934) | ADPKD (343) | CKD (1,658) | ADPKD (328) | CKD (1,937) | ADPKD (166) | CKD (577) | ADPKD (498) | CKD (2,199) | ADPKD (5,655) | CKD (15,546) | ||
| Age, n (%) | |||||||||||||||
| <20 | 401 (5.3) | 250 (1.1) | 45 (7.2) | 26 (2.8) | 0 | 7 (0.4) | 0 | 9 (0.5) | 0 | 1 (0.2) | 4 (0.8) | 40 (1.8) | 352 (6.2) | 167 (1.1) | |
| 20–29 | 613 (8.1) | 492 (2.2) | 104 (16.6) | 52 (5.6) | 9 (2.6) | 17 (1.0) | 3 (0.9) | 16 (0.8) | 0 | 17 (3.0) | 4 (0.8) | 98 (4.5) | 493 (8.7) | 292 (1.9) | |
| 30–39 | 1,469 (19.3) | 1,292 (5.7) | 193 (30.8) | 114 (12.2) | 48 (14.0) | 77 (4.6) | 33 (10.1) | 78 (4.0) | 13 (7.8) | 56 (9.7) | 28 (5.6) | 202 (9.2) | 1,154 (20.4) | 765 (4.9) | |
| 40–49 | 2,097 (27.5) | 2,990 (13.1) | 165 (26.3) | 226 (24.2) | 109 (31.8) | 279 (16.8) | 101 (30.8) | 237 (12.2) | 57 (34.3) | 97 (16.8) | 143 (28.7) | 456 (20.7) | 1,522 (26.9) | 1,695 (10.9) | |
| 50–59 | 1,933 (25.4) | 5,922 (25.9) | 83 (13.2) | 314 (33.6) | 133 (38.8) | 637 (38.4) | 120 (36.6) | 664 (34.3) | 72 (43.4) | 177 (30.7) | 197 (39.6) | 651 (29.6) | 1,328 (23.5) | 3,479 (22.4) | |
| 60–69 | 868 (11.4) | 6,299 (27.6) | 36 (5.7) | 191 (20.5) | 44 (12.8) | 565 (34.1) | 66 (20.1) | 780 (40.3) | 20 (12.1) | 173 (30.0) | 95 (19.1) | 510 (23.2) | 607 (10.7) | 4,080 (26.2) | |
| ≥70 | 236 (3.1) | 5,606 (24.5) | 1 (0.2) | 11 (1.2) | 0 | 76 (4.6) | 5 (1.5) | 153 (7.9) | 4 (2.4) | 56 (9.7) | 27 (5.4) | 242 (11.0) | 199 (3.5) | 5,068 (32.6) | |
|
| |||||||||||||||
| Sex, n (%) | |||||||||||||||
| Male | 3,622 (47.6) | 13,614 (59.6) | 230 (36.7) | 461 (49.4) | 154 (44.9) | 910 (54.9) | 197 (60.1) | 1,251 (64.6) | 97 (58.4) | 343 (59.5) | 255 (51.2) | 1,325 (60.3) | 2,689 (47.6) | 9,324 (60.0) | |
| Female | 3,995 (52.5) | 9,237 (40.4) | 397 (63.3) | 473 (50.6) | 189 (55.1) | 748 (45.1) | 131 (39.9) | 686 (35.4) | 69 (41.6) | 234 (40.6) | 243 (48.8) | 874 (39.8) | 2,966 (52.5) | 6,222 (40.0) | |
|
| |||||||||||||||
| Geographic area, n (%) | |||||||||||||||
| Northeast | 771 (10.1) | 1,888 (8.3) | 71 (11.3) | 67 (7.2) | 22 (6.4) | 100 (6.0) | 25 (7.6) | 148 (7.6) | 21 (12.7) | 47 (8.2) | 50 (10.0) | 170 (7.7) | 582 (10.3) | 1,356 (8.7) | |
| South | 3,648 (47.9) | 12,116 (53.0) | 413 (65.9) | 671 (71.8) | 216 (63.0) | 1,124 (67.8) | 199 (60.7) | 1,341 (69.2) | 113 (68.1) | 419 (72.6) | 273 (54.8) | 1,260 (57.3) | 2,434 (43.0) | 7,301 (47.0) | |
| Midwest | 1,921 (25.2) | 5,298 (23.2) | 67 (10.7) | 81 (8.7) | 47 (13.7) | 167 (10.1) | 47 (14.3) | 218 (11.3) | 17 (10.2) | 71 (12.3) | 110 (22.1) | 502 (22.8) | 1,633 (28.9) | 4,259 (27.4) | |
| West | 1,277 (16.8) | 3,549 (15.5) | 76 (12.1) | 115 (12.3) | 58 (16.9) | 267 (16.1) | 57 (17.4) | 230 (11.9) | 15 (9.0) | 40 (6.9) | 65 (13.1) | 267 (12.1) | 1,006 (17.8) | 2,630 (16.9) | |
|
| |||||||||||||||
| Cohort entry year, n (%) | |||||||||||||||
| 2000–2001 | 839 (11.0) | 1,031 (4.5) | 27 (4.3) | 3 (0.3) | 19 (5.5) | 11 (0.7) | 15 (4.6) | 31 (1.6) | 11 (6.6) | 25 (4.3) | 59 (11.8) | 333 (15.1) | 708 (12.5) | 628 (4.0) | |
| 2002–2003 | 1,115 (14.6) | 1,550 (6.8) | 84 (13.4) | 41 (4.4) | 52 (15.2) | 37 (2.2) | 42 (12.8) | 120 (6.2) | 23 (13.9) | 65 (11.3) | 68 (13.7) | 281 (12.8) | 846 (15.0) | 1,006 (6.5) | |
| 2004–2005 | 1,237 (16.2) | 2,233 (9.8) | 104 (16.6) | 55 (5.9) | 47 (13.7) | 76 (4.6) | 58 (17.7) | 190 (9.8) | 30 (18.1) | 93 (16.1) | 86 (17.3) | 351 (16.0) | 912 (16.1) | 1,468 (9.4) | |
| 2006–2007 | 1,367 (17.9) | 5,177 (22.7) | 98 (15.6) | 169 (18.1) | 61 (17.8) | 247 (14.9) | 62 (18.9) | 442 (22.8) | 42 (25.3) | 164 (28.4) | 80 (16.1) | 444 (20.2) | 1,024 (18.1) | 3,711 (23.9) | |
| 2008–2009 | 1,393 (18.3) | 5,363 (23.5) | 149 (23.8) | 252 (27.0) | 69 (20.1) | 476 (28.7) | 60 (18.3) | 455 (23.5) | 19 (11.4) | 98 (17.0) | 100 (20.0) | 357 (16.2) | 996 (17.6) | 3,725 (24.0) | |
| 2010–2011 | 1,176 (15.4) | 5,420 (23.7) | 115 (18.3) | 288 (30.8) | 69 (20.1) | 636 (38.4) | 67 (20.4) | 517 (26.7) | 29 (17.5) | 100 (17.3) | 69 (13.9) | 296 (13.5) | 827 (14.6) | 3,583 (23.0) | |
| 2012–2013 | 490 (6.4) | 2,077 (9.1) | 50 (8.0) | 126 (13.5) | 26 (7.6) | 175 (10.6) | 24 (7.3) | 182 (9.4) | 12 (7.2) | 32 (5.5) | 36 (7.2) | 137 (6.2) | 342 (6.0) | 1,425 (9.2) | |
|
| |||||||||||||||
| Healthcare utilization during 6-month baseline period, median (IQR) | |||||||||||||||
| Number of physician visits | 4 (2–6) | 6 (3–9) | 4 (3–7) | 6 (4–10) | 4 (3–7) | 6 (3–9) | 4 (3–7) | 6 (4–10) | 5 (3–8) | 7 (4–11) | 5 (2–8) | 5 (2–10) | 3 (2–6) | 5 (3–9) | |
| Number of hospitalizations | 0 |(0–0) | 0 (0–1) | 0 (0–0) | 0 (0–1) | 0 (0–0) | 0 (0–0) | 0 (0–0) | 0 (0–1) | 0 (0–0) | 0 (0–1) | 1 (0–1) | 1 (0–1) | 0 (0–0) | 0 (0–1) | |
ADPKD, autosomal dominant polycystic kidney disease; CKD, chronic kidney disease; IQR, interquartile range.
Among patients with known stage at baseline, the highest proportion of patients with ADPKD were in stage I, and the highest proportion of patients with CKD were in stage V. Cohort entry of patients with ADPKD was, in general, distributed evenly across the study period. However, a slightly higher percentage of CKD patients entered the cohort during the latter half of the study period (2006–2011). In addition, ADPKD patients were, in general, younger than CKD patients; 60.1% of patients in the ADPKD cohort were <50 years of age compared with 22.0% of patients in the CKD cohort. ADPKD patients also had a lower median number of visits to the physician compared with CKD patients.
ADPKD patients tended to have a lower comorbidity score than CKD patients: 60.1% of ADPKD patients had a score of 0 compared with 34.9% of CKD patients (Table 2). ADPKD patients had a lower proportion of general comorbid conditions but were more likely to have UTIs, kidney stones, hepatic conditions, flank/abdominal pain, and kidney pain.
Table 2.
Baseline clinical characteristics of ADPKD patients and CKD patients identified on the basis of healthcare claims, stratified by disease stage: Optum Research Database, January 1, 2000–February 28, 2013.
| (n) | Overall (N=30,468) | Stage I (N=1,561) | Stage II (N=2,001) | Stage III (N=2,265) | Stage IV (N=743) | Stage V (N=2,697) | Unknown (N=22,201) | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
| ||||||||||||||
| ADPKD (7,617) | CKD (22,851) | ADPKD (627) | CKD (934) | ADPKD (343) | CKD (1,658) | ADPKD (328) | CKD (1,937) | ADPKD (166) | CKD (577) | ADPKD (498) | CKD (2,199) | ADPKD (5,655) | CKD (15,546) | |
| Quan–Charlson Comorbidity Index score, n (%) | ||||||||||||||
| 0 | 4,580 (60.1) | 7,967 (34.9) | 436 (69.5) | 363 (38.9) | 196 (57.1) | 744 (44.9) | 129 (39.3) | 610 (31.5) | 37 (22.3) | 128 (22.2) | 36 (7.2) | 201 (9.1) | 3,746 (66.2) | 5,921 (38.1) |
| 1–2 | 2,461 (32.3) | 9,338 (40.9) | 162 (25.8) | 392 (42.0) | 125 (36.4) | 641 (38.7) | 157 (47.9) | 866 (44.7) | 113 (68.1) | 303 (52.5) | 299 (60.0) | 1,022 (46.5) | 1,605 (28.4) | 6,114 (39.3) |
| 3–4 | 464 (6.1) | 3,806 (16.7) | 22 (3.5) | 102 (10.9) | 15 (4.4) | 179 (10.8) | 36 (11.0) | 343 (17.7) | 15 (9.0) | 94 (16.3) | 123 (24.7) | 639 (29.1) | 253 (4.5) | 2,449 (15.8) |
| 5+ | 112 (1.5) | 1,740 (7.6) | 7 (1.1) | 77 (8.2) | 7 (2.0) | 94 (5.7) | 6 (1.8) | 118 (6.1) | 1 (0.6) | 52 (9.0) | 40 (8.0) | 337 (15.3) | 51 (0.9) | 1,062 (6.8) |
|
| ||||||||||||||
| Quan–Charlson Comorbidity Index score (continuous, mean ± SD) | 0.7±1.1 | 1.6±1.9 | 0.6±1.1 | 1.5±2 | 0.7±1.2 | 1.3±1.7 | 1±1.2 | 1.6±1.7 | 1.1±1 | 1.9±2 | 2±1.7 | 2.6±2.1 | 0.6±1 | 1.5±1.8 |
|
| ||||||||||||||
| General comorbid conditions, n (%) | ||||||||||||||
| Genitourinary congenital anomalies | 7,617 (100.0) | 551 (2.4) | 627 (100.0) | 20 (2.1) | 343 (100.0) | 42 (2.5) | 328 (100.0) | 52 (2.7) | 166 (100.0) | 15 (2.6) | 498 (100.0) | 39 (1.8) | 5,655 (100.0) | 383 (2.5) |
| Diseases of the urinary system | 5,062 (66.5) | 22,851 (100.0) | 403 (64.3) | 934 (100.0) | 229 (66.8) | 1,658 (100.0) | 278 (84.8) | 1,937 (100.0) | 156 (94.0) | 577 (100.0) | 492 (98.8) | 2,199 (100.0) | 3,504 (62.0) | 15,546 (100.0) |
| Hypertension | 5,021 (65.9) | 18,797 (82.3) | 334 (53.3) | 686 (73.4) | 262 (76.4) | 1,341 (80.9) | 297 (90.5) | 1,725 (89.1) | 152 (91.6) | 521 (90.3) | 453 (91.0) | 1,732 (78.8) | 3,523 (62.3) | 12,792 (82.3) |
| Disorders of lipid metabolism | 2,211 (29.0) | 12,312 (53.9) | 183 (29.2) | 547 (58.6) | 141 (41.1) | 1,129 (68.1) | 159 (48.5) | 1,329 (68.6) | 96 (57.8) | 362 (62.7) | 188 (37.8) | 749 (34.1) | 1,444 (25.5) | 8,196 (52.7) |
| Heart diseases | 1,731 (22.7) | 11,897 (52.1) | 107 (17.1) | 378 (40.5) | 79 (23.0) | 667 (40.2) | 79 (24.1) | 923 (47.7) | 44 (26.5) | 294 (51.0) | 263 (52.8) | 1,394 (63.4) | 1,159 (20.5) | 8,241 (53.0) |
| Respiratory infections | 1,594 (20.9) | 5,397 (23.6) | 174 (27.8) | 239 (25.6) | 71 (20.7) | 385 (23.2) | 67 (20.4) | 445 (23.0) | 36 (21.7) | 127 (22.0) | 86 (17.3) | 613 (27.9) | 1,160 (20.5) | 3,588 (23.1) |
| Other diseases of the lower respiratory tract | 1,123 (14.7) | 7,830 (34.3) | 91 (14.5) | 279 (29.9) | 50 (14.6) | 446 (26.9) | 49 (14.9) | 573 (29.6) | 30 (18.1) | 170 (29.5) | 179 (35.9) | 1,030 (46.8) | 724 (12.8) | 5,332 (34.3) |
| Diseases of arteries, arterioles, and capillaries | 925 (12.1) | 6,645 (29.1) | 52 (8.3) | 221 (23.7) | 40 (11.7) | 379 (22.9) | 51 (15.5) | 527 (27.2) | 22 (13.3) | 149 (25.8) | 158 (31.7) | 900 (40.9) | 602 (10.6) | 4,469 (28.7) |
|
| ||||||||||||||
| Specific comorbid conditions, n (%) | ||||||||||||||
| Urinary-tract infection | 1,208 (15.9) | 3,333 (14.6) | 123 (19.6) | 177 (19.0) | 64 (18.7) | 235 (14.2) | 54 (16.5) | 283 (14.6) | 26 (15.7) | 94 (16.3) | 75 (15.1) | 358 (16.3) | 866 (15.3) | 2,186 (14.1) |
| Kidney stone | 639 (8.4) | 1,038 (4.5) | 84 (13.4) | 52 (5.6) | 35 (10.2) | 95 (5.7) | 24 (7.3) | 113 (5.8) | 8 (4.8) | 27 (4.7) | 17 (3.4) | 65 (3.0) | 471 (8.3) | 686 (4.4) |
| Pancreatic conditions | 99 (1.3) | 521 (2.3) | 8 (1.3) | 20 (2.1) | 4 (1.2) | 28 (1.7) | 7 (2.1) | 38 (2.0) | 1 (0.6) | 14 (2.4) | 14 (2.8) | 99 (4.5) | 65 (1.2) | 322 (2.1) |
| Hepatic conditions | 932 (12.2) | 2,184 (9.6) | 98 (15.6) | 127 (13.6) | 55 (16.0) | 172 (10.4) | 43 (13.1) | 186 (9.6) | 14 (8.4) | 51 (8.8) | 100 (20.1) | 409 (18.6) | 622 (11.0) | 1,239 (8.0) |
| Flank/abdominal pain | 1,775 (23.3) | 4,024 (17.6) | 178 (28.4) | 202 (21.6) | 82 (23.9) | 290 (17.5) | 55 (16.8) | 303 (15.6) | 31 (18.7) | 96 (16.6) | 108 (21.7) | 561 (25.5) | 1,321 (23.4) | 2,572 (16.5) |
| Kidney pain | 139 (1.8) | 143 (0.6) | 15 (2.4) | 13 (1.4) | 7 (2.0) | 14 (0.8) | 3 (0.9) | 10 (0.5) | 6 (3.6) | 6 (1.0) | 5 (1.0) | 10 (0.5) | 103 (1.8) | 90 (0.6) |
| Renal ultrasound | 1,266 (16.6) | 4,257 (18.6) | 144 (23.0) | 179 (19.2) | 54 (15.7) | 301 (18.2) | 72 (22.0) | 393 (20.3) | 31 (18.7) | 150 (26.0) | 66 (13.3) | 565 (25.7) | 899 (15.9) | 2,669 (17.2) |
| Dialysis | 376 (4.9) | 1,979 (8.7) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 376 (75.5) | 1,979 (90.0) | 0 | 0 |
ADPKD, autosomal dominant polycystic kidney disease; CKD, chronic kidney disease; SD, standard deviation.
Distributions of demographic features and baseline clinical characteristics of ADPKD patients at risk of rapid progression (data not shown) were similar to those shown in Table 2 for the overall ADPKD group.
Stage transition
In both patients for whom the baseline stage was known (Table 3a) and those for whom initial stage was defined during follow-up (Table 3b), ADPKD patients were, in general, younger than CKD patients within the same stage and at transition to a more advanced stage based on recorded laboratory values. Among patients for whom the baseline stage was known and who started in stage I, median duration to stage II was 10.0 months for patients with ADPKD and 5.1 months for patients with CKD; median duration to stage III was 57.2 months and 7.3 months; to stage IV was 81.8 months and 15.0 months; and to stage V was 55.6 months and 2.4 months for patients with ADPKD and CKD, respectively. Among patients for whom the first stage was identified during follow-up and who started in stage I, median duration to stage II was 11.2 months for patients with ADPKD and 5.9 months for patients with CKD; to stage III was 35.3 months and 13.6 months; to stage IV was 78.2 months and 23.2 months, and to stage V was 50.6 months and 18.4 months for patients with ADPKD and CKD, respectively. Overall follow-up for these patients was 1.4–4.7 years for the ADPKD group and 1.0–2.7 years for the CKD group. Numbers of patients with no transition, missing a second SCr value, and those who had an apparent backward transition (potentially an artifact of recorded SCr levels) are also presented in Tables 3a and 3b, and the proportions were not substantially different between ADPKD and CKD groups.
Compared with overall ADPKD patients, similar median durations to transition to a more severe stage were observed among patients at risk of rapid progression in patients for whom the baseline stage was known (Table 4a) as well as those for whom stage was defined during follow-up (Table 4b). However, a younger age of stage transition was observed in all stages for patients at risk of rapid progression compared with overall ADPKD patients. Overall follow-up for these patients was 1.3–4.7 years.
Table 4.
Stage transition obtained from laboratory results among patients at risk of rapid progression: Optum Research Database, January 1, 2000–February 28, 2013; (a) patients for whom the baseline stage was known, and (b) patients for whom the first stage was defined during follow-up.
| (a) | Transition to next stage during follow-upa | |||||
|---|---|---|---|---|---|---|
|
| ||||||
| Baseline stageb | No transition/backward/missing | Stage II | Stage III | Stage IV | Stage V | |
| Stage I, n | 262 | 145/0/65 | 42 | 10 | 4 | 6 |
| Mean age, y±SD | 33.3±11.0 | 40.2±9.5 | 43.7±9.0 | 45.8±9.0 | 43.3±4.8 | |
| Median stage duration, mo (IQR) | 10.1 (4.8–24.8) | 56.9 (23.6–61.6) | 81.8 (51.9–90.1) | 56.1 (48.2–58.1) | ||
|
| ||||||
| Stage II, n | 132 | 34/18/22 | 56 | 20 | 10 | |
| Mean age, y±SD | 44.6±9.3 | 46.9±8.5 | 47.6±7.5 | 47.6±8.3 | ||
| Median stage duration, mo (IQR) | 9.0 (3.1–23.4) | 39.0 (20.9–70.2) | 53.2 (21.7–63.9) | |||
|
| ||||||
| Stage III, n | 174 | 42/12/25 | 89 | 46 | ||
| Mean age, y±SD | 47.5±9.1 | 49.6±8.2 | 51.2±9.1 | |||
| Median stage duration, mo (IQR) | 16.1 (5.6–34.2) | 43.2 (28.6–60.2) | ||||
|
| ||||||
| Stage IV, n | 110 | 22/9/8 | 71 | |||
| Mean age, y±SD | 48.1±7.6 | 48.8±7.9 | ||||
| Median stage duration, mo (IQR) | 8.9 (3.8–20.7) | |||||
|
| ||||||
| Stage V, n | 321 | 294/15/12 | ||||
| Mean age, y±SD | 47.8±8.4 | |||||
| (b) | Transition to next stage during follow-upa | |||||
|---|---|---|---|---|---|---|
|
| ||||||
| First stage during follow-upb | No transition/backward/missing | Stage II | Stage III | Stage IV | Stage V | |
| Stage I, n | 355 | 178/0/127 | 43 | 11 | 2 | 5 |
| Mean age, y±SD | 31.2±10.9 | 43.2±10.1 | 43.8±5.2 | 48.0±5.7 | 39.6±17.3 | |
| Median stage duration, mo (IQR) | 11.2 (3.6–26.4) | 35.3 (11.0–75.7) | 95.6 (78.2–113.0) | 82.8 (18.4–89.4) | ||
|
| ||||||
| Stage II, n | 150 | 53/28/19 | 47 | 14 | 4 | |
| Mean age, y±SD | 41.8±10.0 | 46.7±8.7 | 47.2±10.4 | 48.5±7.7 | ||
| Median stage duration, mo (IQR) | 21.1 (9.0–39.2) | 65.2 (32.3–80.1) | 83.5 (57.0–103.3) | |||
|
| ||||||
| Stage III, n | 170 | 52/18/23 | 68 | 35 | ||
| Mean age, y±SD | 46.1±11.5 | 48.7±11.2 | 51.1±9.2 | |||
| Median stage duration, mo (IQR) | 15.4 (7.0–28.4) | 45.8 (29.4–54.4) | ||||
|
| ||||||
| Stage IV, n | 109 | 33/8/11 | 57 | |||
| Mean age, y±SD | 47.0±8.5 | 48.8±7.1 | ||||
| Median stage duration, mo (IQR) | 13.3 (6.2–24.0) | |||||
|
| ||||||
| Stage V, n | 314 | 67/8/239 | ||||
| Mean age, y±SD | 47.4±9.0 | |||||
Patients with more than one transition were counted more than once.
Median follow-up was 1.3–2.7 years.
Median follow-up was 2.3–4.7 years.
ADPKD, autosomal dominant polycystic kidney disease; CKD, chronic kidney disease; IQR, interquartile range; mo, months; SD, standard deviation; y, years.
Time-to-ESRD was shortest among patients with ADPKD at risk of rapid progression, followed by CKD patients, and then ADPKD patients not at risk of rapid progression (Figure 2). In addition, progression to ESRD was most pronounced among those patients in advanced stages during the baseline period (Figure 3a–d).
Figure 2.

Time-to-ESRD among ADPKD patients, CKD patients, and ADPKD patients at risk of rapid progression, overalla.
aFor the overall cohort, P<0.0001.
ADPKD, autosomal dominant polycystic kidney disease; CKD, chronic kidney disease; ESRD, end-stage renal disease.
Figure 3.
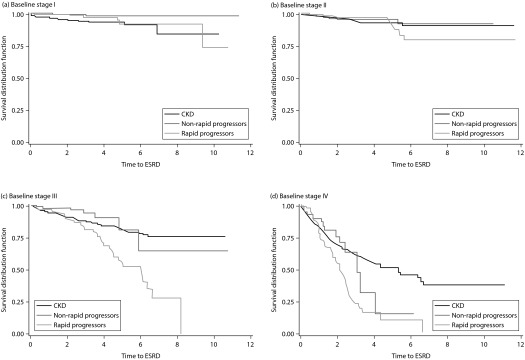
Time-to-ESRD among ADPKD patients, CKD patients, and ADPKD patients at risk of rapid progression: baseline stage I (a), baseline stage II (b), baseline stage III (c), and baseline stage IV (d)a.
aBaseline stage I (a), P=0.0002; baseline stage II (b), P=0.7808; baseline stage III (c), P<0.0001; baseline stage IV (d), P<0.0001. ADPKD, autosomal dominant polycystic kidney disease; ESRD, end-stage renal disease; CKD, chronic kidney disease.
Discussion
Understanding the natural history of ADPKD may help in the identification of patients who could benefit from early treatment and could potentially delay progression. We described the natural history of ADPKD in a large, geographically diverse commercially insured population in the USA over a long duration in a real-world setting.
In general, ADPKD patients were younger with a shorter median duration of health-plan membership and fewer physician visits, but had more specific comorbidities (e.g., UTIs, kidney stones, flank/abdominal pain, kidney pain) at the start of observation compared with CKD patients. Furthermore, in results unadjusted for differences between the two study populations, ADPKD patients had a longer time in the milder stages and longer duration before recorded transition to a more severe stage compared with CKD patients. Implications of these observations could be useful for determination of the transplantation eligibility of patients with ADPKD compared with the move to dialysis.
We also investigated a subgroup of patients considered to be at risk of rapid progression. These patients were younger at each stage, but had a similar recorded time to transition to a more severe stage compared with the overall ADPKD cohort, among patients with observed transitions to more severe stages. Criteria for entry into this cohort were based on reaching milestones at specific ages. Nevertheless, results suggested that age at transition to next stage may be useful for identification of ADPKD patients at risk of rapid progression, in combination with other measures (e.g., blood pressure, kidney stones, kidney size, number of kidney cysts). These results could be because the number of patients at risk of rapid progression transitioning to a more advanced stage was sparse, and because the ADPKD group included patients with ADPKD and PKD. However, the proportion of patients with PKD unspecified that had medical chart-specified ADPKD resulted in a positive predictive value of 94.7%.
Unadjusted time-to-event analyses suggested that ADPKD patients at risk of rapid progression (particularly those in stage III) had a shorter time-to-ESRD than patients with CKD and ADPKD patients not at risk. This phenomenon is most likely because cystogenesis is a constant event; therefore, if cysts are more plentiful and develop earlier, the disease progresses earlier, without increasing the time spent in each stage [22]. Time-to-event analyses also showed that progression to ESRD was most pronounced among patients in advanced stages during the baseline period.
Recent studies have begun to focus on the progression of ADPKD as well as the characteristics of ADPKD patients, highlighting the importance of studies that help to identify patients who may benefit from early treatment. Analyses of ADPKD progression between 1978 and 2012 in a natural-history setting found that a difference in eGFR decline can be predicted at least 10 years before the onset of ESRD [23]. In two recent studies by Higashihara et al., total kidney volume and eGFR were investigated for 39.7 months in 64 patients [24] and up to 13.9 years in 225 patients [25]. Results confirmed a declining rate of eGFR and increased total kidney volume as clinically meaningful markers in ADPKD.
Characteristics of ADPKD patients and treatment patterns over time were investigated in two recent studies. One study compared patients with ADPKD with non-ADPKD controls and investigated initiation of renal replacement therapy (RRT) [26]. The other study compared patients with ADPKD diagnosed between 1961 and 1990 with a cohort diagnosed between 1991 and 2011 [27]. The study investigating RRT found that neither change in age nor alteration in the male:female ratio among patients who started RRT changed over time [26]. It was speculated this observation was due to the disease impact of unmodifiable genetic factors in the absence of a specific treatment. The study comparing a diagnosis in an earlier timeframe versus in a later timeframe found an earlier age at the diagnosis, a lower mean blood pressure, better eGFR, and a longer timeframe from birth to ESRD and birth to death in the patients diagnosed in the later timeframe [27].
The studies described above underscore the importance of investigating progression of ADPKD as well as the characteristics of ADPKD patients. Unique characteristics of the present study are that disease progression and patient characteristics were described in a large retrospective cohort of ADPKD patients with a comparison cohort of CKD patients, both with a widespread geographic distribution. This comparison helps to place the data on ADPKD progression in context. In addition, the investigation of a cohort of patients at risk of rapid progression is helpful for obtaining more in-depth understanding of ADPKD patients.
A better overall knowledge of ADPKD that includes patient characteristics, disease progression, and utilization of healthcare resources is necessary to help identify treatments that may delay progression, increase the quality of life of patients, and reduce the utilization of healthcare resources associated with this disease.
Results of the present study increase our understanding of ADPKD, but study limitations should be noted. As mentioned above, the ADPKD cohort included PKD unspecified patients, which could have decreased the specificity of the ADPKD cohort. However, results from our sub-study on validation of PKD charts suggested a positive predictive value of 94.7% for classifying this group of patients as ADPKD patients. In addition, only results from laboratories reporting data to the ORD were available for stage analyses, so only ≈25% of patients had SCr results available at baseline to determine disease stage. First stage was identified during follow-up in an additional 28% of patients. Moreover, for each of these groups, 25% of patients were in stage V or ESRD, and had already achieved maximum progression of disease. Finally, some misclassification of disease stage and timing of progression could occur due to (i) assignment of disease stage based on the earliest record of an eGFR (calculated based on SCr laboratory results) that met threshold criteria for a more advanced stage; (ii) variability in the timing or frequency of laboratory testing in clinical practice; and (iii) misclassification due to missing or inaccurate electronic laboratory results, serum inaccuracies, or lack of information on ethnicity which impacts calculation of eGFR from SCr.
The present study suggests that distribution of patients by age at transition to next stage may be useful for the identification of ADPKD patients at risk of rapid progression. It also suggests that medical claims with diagnosis codes for “unspecified PKD” in the absence of diagnosis codes for ARPKD or ADPKD may serve as a good proxy for ADPKD. Inclusion of these criteria in claims-based algorithms for ADPKD could improve the capture (or identification) of this population in future studies. Taking into account the inherent limitations, electronic laboratory data can be used to describe disease progression. Future large, long-term cohort studies should continue to explore the genetic, clinical, and imaging characteristics of patients with ADPKD at risk of rapid progression who may demonstrate the greatest benefit from therapeutic interventions.
Table 3a.
Stage transition obtained from laboratory results among ADPKD patients and CKD patients: Optum Research Database, January 1, 2000–February 28, 2013; patients for whom the baseline stage was known.
| Baseline stageb | No transition/backward/missing |
Transition to next stage during follow-upa
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Stage II | Stage III | Stage IV | Stage V | |||||||||
|
| ||||||||||||
| ADPKD | CKD | ADPKD | CKD | ADPKD | CKD | ADPKD | CKD | ADPKD | CKD | ADPKD | CKD | |
| Stage I, n | 627 | 934 | 341/0/175 | 434/0/196 | 99 | 237 | 19 | 76 | 4 | 24 | 7 | 40 |
| Mean age, y±SD | 38.4±12.8 | 49.1±12.6 | 46.6±11.1 | 52.4±10.9 | 46.6±9.2 | 52.8±11.8 | 45.8±9.0 | 50.7±10.9 | 45.1±6.5 | 46.6±15.6 | ||
| Median stage duration, mo (IQR) | 10.0 (3.6–23.7) | 5.1 (1.4–10.8) | 57.2 (12.8–75.0) | 7.3 (1.8–18.4) | 81.8 (51.9–90.1) | 15.0 (9.1–27.7) | 55.6 (36.0–58.1) | 2.4 (0.2–17.1) | ||||
|
| ||||||||||||
| Stage II, n | 343 | 1,658 | 107/64/64 | 518/336/282 | 104 | 465 | 24 | 78 | 14 | 61 | ||
| Mean age, y±SD | 49.1±9.2 | 55.6±9.9 | 50.4±8.7 | 57.3±9.5 | 48.1±7.7 | 58.0±9.6 | 49.3±8.9 | 54.4±12.6 | ||||
| Median stage duration, mo (IQR) | 10.1 (4.7–23.9) | 5.3 (1.5–13.0) | 39.0 (17.9–61.0) | 8.9 (2.5–26.2) | 46.4 (17.5–63.2) | 7.4 (1.2–22.9) | ||||||
|
| ||||||||||||
| Stage III, n | 328 | 1,937 | 104/34/56 | 509/654/297 | 125 | 385 | 54 | 155 | ||||
| Mean age, y±SD | 51.3±9.2 | 57.7±10.2 | 52.1±8.9 | 57.4±10.8 | 52.5±9.1 | 58.3±11.0 | ||||||
| Median stage duration, mo (IQR) | 16.2 (6.8–31.4) | 6.2 (2.4–16.6) | 41.8 (29.1–58.6) | 13.8 (4.3–28.5) | ||||||||
|
| ||||||||||||
| Stage IV, n | 166 | 577 | 38/21/18 | 98/175/82 | 89 | 222 | ||||||
| Mean age, y±SD | 51.4±8.6 | 55.0±12.4 | 50.9±8.5 | 54.0±12.7 | ||||||||
| Median stage duration, mo (IQR) | 9.0 (3.9–18.5) | 7.8 (2.4–17.8) | ||||||||||
|
| ||||||||||||
| Stage V, n | 498 | 2,199 | 451/23/24 | 1,815/230/154 | ||||||||
| Mean age, y±SD | 52.9±10.7 | 53.2±14.2 | ||||||||||
Table 3b.
Stage transition obtained from laboratory results among ADPKD patients and CKD patients: Optum Research Database, January 1, 2000–February 28, 2013; patients for whom the first stage was defined during follow-up.
| First stage during follow-upc | No transition/backward/missing |
Transition to next stage during follow-upa
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Stage II | Stage III | Stage IV | Stage V | |||||||||
|
| ||||||||||||
| ADPKD | CKD | ADPKD | CKD | ADPKD | CKD | ADPKD | CKD | ADPKD | CKD | ADPKD | CKD | |
| Stage I, n | 816 | 616 | 411/0/302 | 283/0/213 | 94 | 103 | 13 | 24 | 3 | 6 | 6 | 8 |
| Mean age, y±SD | 37.4±12.8 | 47.2±14.0 | 48.1±9.9 | 53.4±11.4 | 44.8±6.0 | 53.1±13.8 | 52.0±8.0 | 54.2±11.4 | 38.3±15.8 | 46.5±19.3 | ||
| Median stage duration, mo (IQR) | 11.2 (4.3–26.4) | 5.9 (2.3–11.5) | 35.3 (17.5–63.4) | 13.6 (5.5–27.5) | 78.2 (2.3–113.0) | 23.2 (8.9–40.4) | 50.6 (4.8–89.4) | 18.4 (7.3–27.6) | ||||
|
| ||||||||||||
| Stage II, n | 378 | 957 | 133/56/85 | 372/120/211 | 101 | 235 | 18 | 24 | 5 | 12 | ||
| Mean age, y±SD | 47.5±10.2 | 57.8±10.7 | 51.4±9.2 | 60.8±9.9 | 50.0±10.8 | 57.7±13.3 | 50.0±7.4 | 57.0±10.9 | ||||
| Median stage duration, mo (IQR) | 19.5 (7.3–37.5) | 7.9 (3.5–16.8) | 60.6 (28.9–80.1) | 9.1 (5.2–20.1) | 79.9 (56.2–87.1) | 15.9 (7.6–34.8) | ||||||
|
| ||||||||||||
| Stage III, n | 321 | 1,069 | 130/38/52 | 423/226/216 | 89 | 175 | 45 | 58 | ||||
| Mean age, y±SD | 49.8±11.1 | 59.3±12.1 | 50.7±10.8 | 59.1±12.8 | 52.7±8.7 | 56.4±12.2 | ||||||
| Median stage duration, mo (IQR) | 15.1 (6.8–26.3) | 8.1 (3.4–18.5) | 40.0 (26.8–53.6) | 19.1 (7.4–39.5) | ||||||||
|
| ||||||||||||
| Stage IV, n | 186 | 406 | 66/15/28 | 126/84/68 | 77 | 128 | ||||||
| Mean age, y±SD | 50.7±9.4 | 56.5±12.8 | 51.8±8.4 | 54.9±11.8 | ||||||||
| Median stage duration, mo (IQR) | 15.2 (6.3–25.5) | 8.2 (3.2–17.0) | ||||||||||
|
| ||||||||||||
| Stage V, n | 438 | 1,509 | 96/12/330 | 163/93/1,253 | ||||||||
| Mean age, y±SD | 51.3±11.2 | 58.1±15.3 | ||||||||||
Patients with more than one transition were counted more than once.
Median follow-up was 1.4–2.7 years for the ADPKD group and 1.0–2.1 years for the CKD group.
Median follow-up was 2.3–4.7 years for the ADPKD group and 1.8–2.7 years for the CKD group.
ADPKD, autosomal dominant polycystic kidney disease; CKD, chronic kidney disease; IQR, interquartile range; mo, months; SD, standard deviation; y, years.
Abbreviations
- ADPKD
autosomal dominant polycystic kidney disease;
- ARPKD
autosomal recessive polycystic kidney disease;
- CKD
chronic kidney disease;
- eGFR
estimated glomerular filtration rate;
- ESRD
end-stage renal disease;
- ORD
Optum Research Database;
- RRT
renal replacement therapy;
- SCr
serum creatinine;
- UTIs
urinary-tract infections
Footnotes
Optum, Inc., USA
Contributions
Drs. Blanchette, Liang, Newsome, Rossetti, and Gutierrez contributed to the conception and design of this study. Supervisory contributions were made by Drs. Blanchette, Liang, Lubeck, Gutierrez, and Lin. Contributions to statistical analyses were made by Drs. Liang and Gu. All authors contributed to the writing, review, and revision of the manuscript. Drs Blanchette, Liang, Lubeck, Gu, Gutierrez, and Lin contributed to the analyses and interpretation of data.
Potential conflicts of interest
The International Committee of Medical Journal Editors’ (ICMJE) Potential Conflicts of Interests forms for the authors are available for download at: http://www.drugsincontext.com/wp-content/uploads/2015/04/dic.212275-COI.pdf
Sandro Rossetti is an employee of Otsuka America Pharmaceutical, Inc. Christopher M. Blanchette and Benjamin Gutierrez were employees of Otsuka Pharmaceutical Inc. during the study.
Caihua Liang, Nancy D. Lin, and Xiangmei Gu are employees of Optum Epidemiology and Deborah P. Lubeck is an employee of Outcomes Insights Inc.; both companies have received funds from Otsuka America Pharmaceutical, Inc., in connection with conduct of this study. Britt Newsome received funds from Otsuka America Pharmaceutical, Inc. in connection with conduct of this study.
Funding declaration
This study was sponsored by Otsuka America Pharmaceutical, Inc. (Princeton, NJ, USA). Medical writing and editorial support for preparation of this manuscript was provided by Scientific Connexions, Inc. (Lyndhurst, NJ, USA), an Ashfield Company, and funded by Otsuka America Pharmaceutical, Inc. None of the material in this manuscript has been presented previously.
References
- 1.Torres VE, Harris PC, Pirson Y. Autosomal dominant polycystic kidney disease. Lancet. 2007;369(9569):1287–1301. doi: 10.1016/S0140-6736(07)60601-1. http://dx.doi.org/10.1016/S0140-6736(07)60601-1. [DOI] [PubMed] [Google Scholar]
- 2.Grantham JJ. Clinical practice. Autosomal dominant polycystic kidney disease. N Engl J Med. 2008;359(14):1477–85. doi: 10.1056/NEJMcp0804458. http://dx.doi.org/10.1056/NEJMcp0804458. [DOI] [PubMed] [Google Scholar]
- 3.Chapman AB. Approaches to testing new treatments in autosomal dominant polycystic kidney disease: insights from the CRISP and HALT-PKD studies. Clin J Am Soc Nephrol. 2008;3(4):1197–1204. doi: 10.2215/CJN.00060108. http://dx.doi.org/10.2215/CJN.00060108. [DOI] [PubMed] [Google Scholar]
- 4.Hildebrant F. Genetic kidney disease. Lancet. 2010;375(9722):1287–95. doi: 10.1016/S0140-6736(10)60236-X. http://dx.doi.org/10.1016/S0140-6736(10)60236-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Blanchette CM, Matter S, Chawla A, et al. Burden of autosomal dominant polycystic kidney disease: systematic literature review. Am J Pharm Benefits. 2014 [Accepted for publication]. [Google Scholar]
- 6.Iglesias CG, Torres VE, Offord KP, Holley KE, Beard CM, Kurland LT. Epidemiology of adult polycystic kidney disease, Olmsted County, Minnesota. Am J Kidney Dis. 1983;2(6):630–9. doi: 10.1016/s0272-6386(83)80044-4. [DOI] [PubMed] [Google Scholar]
- 7.National Institutes of Health Fact Sheet – Autosomal Dominant Polycystic Kidney Disease. Available at: http://www2.niddk.nih.gov/ [Last accessed: July 10, 2014].
- 8.Brunelli SM, Blanchette CM, Claxton AJ, Roy D, Rossetti S, Gutierrez B. End-stage renal disease in autosomal dominant polycystic kidney disease: a comparison of dialysis-related utilization and costs with other chronic kidney diseases. Clinicoecon Outcomes Res. 2015;7:65–72. doi: 10.2147/CEOR.S76269. http://dx.doi.org/10.2147/CEOR.S76269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Blanchette CM, Lorga SR, Altan A, et al. Healthcare resource utilization and costs associated with autosomal dominant polycystic kidney disease. JHEOR. 2014;2:63–74. doi: 10.36469/9889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Grantham J, Cowley BJ, Torres VE. Progression of autosomal dominant polycystic kidney disease to renal failure. In: Seldin DW, Geibisch G, editors. The Kidney: Physiology and Pathophysiology. 3rd ed. Philadelphia: Lippincott Williams and Wilkins; 2000. pp. 2513–36. [Google Scholar]
- 11.Grantham JJ, Mulamalla S, Swenson-Fields KI. Why kidneys fail in autosomal dominant polycystic kidney disease. Nat Rev Nephrol. 2011;7(10):556–66. doi: 10.1038/nrneph.2011.109. http://dx.doi.org/10.1038/nrneph.2011.109. [DOI] [PubMed] [Google Scholar]
- 12.Helal I, Reed B, Mettler P, et al. Prevalence of cardiovascular events in patients with autosomal dominant polycystic kidney disease. Am J Nephrol. 2012;36(4):362–70. doi: 10.1159/000343281. http://dx.doi.org/10.1159/000343281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Irazabal MV, Huston J, 3rd, Kubly V, et al. Extended follow-up of unruptured intracranial aneurysms detected by presymptomatic screening in patients with autosomal dominant polycystic kidney disease. Clin J Am Soc Nephrol. 2011;6(6):1274–85. doi: 10.2215/CJN.09731110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ecder T. Cardiovascular complications in autosomal dominant polycystic kidney disease. Curr Hypertens Rev. 2013;9(1):2–11. doi: 10.2174/1573402111309010002. http://dx.doi.org/10.2174/1573402111309010002. [DOI] [PubMed] [Google Scholar]
- 15.Rahbari-Oskoui F, Williams O, Chapman A. Mechanisms and management of hypertension in autosomal dominant polycystic kidney disease. Nephrol Dial Transplant. 2014;29(12):2194–201. doi: 10.1093/ndt/gft513. [DOI] [PubMed] [Google Scholar]
- 16.Davison SN, Koncicki H, Brennan F. Pain in chronic kidney disease: a scoping review. Semin Dial. 2014;27(2):188–20. doi: 10.1111/sdi.12196. http://dx.doi.org/10.1111/sdi.12196. [DOI] [PubMed] [Google Scholar]
- 17.Wüthrich RP, Mei C. Pharmacological management of polycystic kidney disease. Expert Opin Pharmacother. 2014;15(8):1085–95. doi: 10.1517/14656566.2014.903923. http://dx.doi.org/10.1517/14656566.2014.903923. [DOI] [PubMed] [Google Scholar]
- 18.Aguiari G, Catizone L, Del Senno L. Multidrug therapy for polycystic kidney disease: A review and perspective. Am J Nephrol. 2013;37(2):175–182. doi: 10.1159/000346812. http://dx.doi.org/10.1159/000346812. [DOI] [PubMed] [Google Scholar]
- 19.Johnson AM, Gabow PA. Identification of patients with autosomal dominant polycystic kidney disease at highest risk for end-stage renal disease. J Am Soc Nephrol. 1997;8:1560–7. doi: 10.1681/ASN.V8101560. [DOI] [PubMed] [Google Scholar]
- 20.Ecder T, Fick-Brosnahan G, Schrier RW. Polycystic Kidney Disease. In: Schrier RW, editor. Diseases of the Kidney and Urinary Tract. 8th ed. Vol. 2. Philadelphia: Lippincott, Williams & Wilkins; 2007. [Google Scholar]
- 21.Rule AD, Larson TS, Bergstralh EJ, Slezak JM, Jacobsen SJ, Cosio FG. Using serum creatinine to estimate glomerular filtration rate: Accuracy in good health and in chronic kidney disease. Ann Intern Med. 2004;141(12):929–37. doi: 10.7326/0003-4819-141-12-200412210-00009. http://dx.doi.org/10.7326/0003-4819-141-12-200412210-00009. [DOI] [PubMed] [Google Scholar]
- 22.Harris PC, Bae KT, Rossetti S, et al. Cyst number but not the rate of cystic growth is associated with the mutated gene in autosomal dominant polycystic kidney disease. J Am Soc Nephrol. 2006;17(11):3013–9. doi: 10.1681/ASN.2006080835. http://dx.doi.org/10.1681/ASN.2006080835. [DOI] [PubMed] [Google Scholar]
- 23.Thong KM, Ong AC. The natural history of autosomal dominant polycystic kidney disease: 30-year experience from a single centre. QJM. 2013;106(7):639–46. doi: 10.1093/qjmed/hct082. http://dx.doi.org/10.1093/qjmed/hct082. [DOI] [PubMed] [Google Scholar]
- 24.Higashihara E, Nutahara K, Okegawa T, et al. Kidney volume and function in autosomal dominant polycystic kidney disease. Clin Exp Nephrol. 2014;18(1):157–65. doi: 10.1007/s10157-013-0834-4. http://dx.doi.org/10.1007/s10157-013-0834-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Higashihara E, Horie S, Muto S, Mochizuki T, Nishio S, Nutahara K. Renal disease progression in autosomal dominant polycystic kidney disease. Clin Exp Nephrol. 2012;16(4):622–8. doi: 10.1007/s10157-012-0611-9. http://dx.doi.org/10.1007/s10157-012-0611-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Martinez V, Comas J, Arcos E, et al. Renal replacement therapy in ADPKD patients: a 25-year survey based on the Catalan registry. BMC Nephrology. 2013;14:186. doi: 10.1186/1471-2369-14-186. http://dx.doi.org/10.1186/1471-2369-14-186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Helal I, McFann K, Reed B, Yan XD, Schrier RW. Changing referral characteristics of patients with autosomal dominant polycystic kidney disease. Am J Med. 2013;126:832e7–11. doi: 10.1016/j.amjmed.2012.12.018. http://dx.doi.org/10.1016/j.amjmed.2012.12.018. [DOI] [PubMed] [Google Scholar]