

Abstract
There is an urgent need in oncology to link molecular aberrations in tumors with therapeutics that can be administered in a personalized fashion. One approach identifies synthetic-lethal genetic interactions or dependencies that cancer cells acquire in the presence of specific mutations. Using engineered isogenic cells, we generated a systematic and quantitative chemical-genetic interaction map that charts the influence of 51 aberrant cancer genes on 90 drug responses. The dataset strongly predicts drug responses found in cancer cell line collections, indicating that isogenic cells can model complex cellular contexts. Applied to triple-negative breast cancer, we report clinically actionable interactions with the MYC oncogene including resistance to AKT/PI3K pathway inhibitors and an unexpected sensitivity to dasatinib through LYN inhibition in a synthetic-lethal manner, providing new drug and biomarker pairs for clinical investigation. This scalable approach enables the prediction of drug responses from patient data and can accelerate the development of new genotype-directed therapies.
Keywords: systems biology, synthetic lethal, genetic interactions, networks
INTRODUCTION
Recent advances in sequencing technology have led to a dramatic increase in the discovery of altered genes in patient tumors. This rapid accumulation of genetic information has led to a bottleneck at the level of understanding of the functional and therapeutic implications of aberrant gene activities in cancer (1). The pressing clinical need to identify therapeutic biomarkers has spurred several large-scale screening efforts using genomically characterized cancer cell line collections to identify molecular correlates of drug responses (2–4). While these collections reflect the diversity of mutations found in human tumors, each cell line carries mutational ‘baggage’ in the form of hundreds to thousands of different genomic alterations. This makes it difficult to link drug responses with the presence of a single causal mutation. In addition, rare mutations that occur with low frequency may not be adequately represented in cancer cell line collections. Therefore, new sensitive and scalable approaches to model genetic aberrations are required to address these emerging challenges in oncology.
Another challenge for the development of personal cancer therapies is the lack of direct therapeutic approaches for many oncogenes, such as transcription factors or other non-kinase targets. In these cases an especially useful alternative method to identify potential therapeutic liabilities is through a synthetic lethal approach. This strategy identifies interactions between mutant genes and inhibition of alternative pathways using functional genomics (5, 6). This framework exploits mutational changes in cells that result in a dependence on pathways that are otherwise non-essential. In lower organisms, systematic genetic interaction maps have transformed our understanding of basic biological processes and drug responses (7, 8). In mammals, synthetic lethal screens using RNAi or small molecules have identified several vulnerabilities in RAS-mutated cell lines (9–15). Previous work has shown that isogenic cell lines can be used to explore therapeutic responses for candidate inhibitors (16–19). However, this approach has not yet been applied in a systematic and quantitative fashion that is able to measure both resistance and sensitivity. Here we apply a systematic approach to determine the degree to which isogenic lines can serve as a starting point to map chemical-genetic interactions and identify novel therapeutic strategies in oncology.
Breast cancer has served as a prime example for biomarker-driven therapy. Several targeted therapies are now given as standard-of-care for patients who present with the overexpression of the HER2 (human epidermal growth factor 2/ERBB2) receptor or the estrogen and progesterone receptors. However, no biomarker-driven therapy is available to treat the most aggressive and challenging receptor triple-negative breast cancer (TNBC) subtype. Previous studies have shown that the transcription factor MYC is a breast cancer oncogene and plays an important functional role in TNBC (20–22). In the breast TCGA study, MYC was found to be focally amplified in 40% of TNBCs and a MYC transcriptional signature was significantly upregulated in these tumors (23). Several early transgenic mouse models have shown that specific expression of MYC in the mammary gland by itself (24) or with cooperating oncogenes induces mammary tumor formation (25, 26). A conditional mouse model system subsequently demonstrated that MYC is a true driver of mammary tumorigenesis by showing that tumor formation could be regressed completely upon MYC withdrawal (27). More recent studies have shown that inhibition of endogenous MYC by a dominant negative MYC mutant can attenuate tumor formation in lung and pancreatic cancer mouse models driven by other oncogenes (28, 29). In an osteogenic sarcoma MYC-driven mouse model, even transient inactivation of MYC induced sustained tumor regression, indicating the potential efficacy for MYC-inhibitory therapies (30). These studies together clearly demonstrate that MYC is an important therapeutic target for cancer tumorigenesis. However, despite this enthusiasm, specific small molecule inhibitors of MYC have yet to be translated into clinically viable therapies for patients. Recently, efforts to target upstream regulation of MYC by BET bromodomain inhibitors have shown dramatic effects in some MYC-driven hematopoietic cancers (31). However, whether MYC is the key target of such inhibitors in solid tumors is still unclear (32). Hence, there is a great need to target MYC indirectly and several studies have employed synthetic lethal strategies to exploit MYC overexpression in breast, lung and liver cancers (17, 33–36). These studies have led to the identification of a diverse set of candidates including Cyclin dependent kinases (CDK1), Aurora kinases, SUMO-activating enzymes (SAE1/2), and casein kinase (CSNK1E) which could point to a dependency on DNA repair and cell-cycle checkpoints in cells that harbor high MYC (37). While CDK1 and Aurora kinases may be pharmacologically tractable targets, currently no inhibitors of these molecules have been approved for use in TNBC.
To aid in the development of new synthetic lethal strategies, we have developed an interaction mapping strategy using isogenic cell lines to measure direct relationships between expression of cancer-associated genes and the proliferative response to clinically relevant compounds. We show that this dataset is highly complementary to drug responses found by profiling tumor cell line collections that are an order of magnitude larger. Furthermore, we demonstrate ways in which these data could aid in the design of new personalized clinical trials. In particular, these data identify a novel synthetic lethal relationship between expression of the MYC proto-oncogene and sensitivity to the multi-kinase inhibitor dasatinib, providing a novel application for an already FDA-approved drug and an associated biomarker for clinical interrogation.
RESULTS
Creation of a quantitative chemical-genetic interaction map
We developed a chemical-genetic interaction mapping strategy to uncover the impact of expression of specific genes on proliferative responses to a panel of emerging and established therapeutics (Fig. 1A). To study the impact of aberrant gene activity in isolation, we developed an isogenic model of triple-negative breast cancer (TNBC) using the receptor negative, non-tumorigenic cell line MCF10A. This epithelial cell line is derived from healthy breast tissue and is diploid and largely devoid of somatic alterations (38). Importantly, MCF10A cells are amenable to transformation by a wide-variety of oncogenes, making them an appropriate cell type to study diverse oncogene signaling pathways (38). We created 51 stable cell lines by ectopic expression of wild-type and mutant genes that are common in breast and other cancers to model the impact of recurrent gene mutation, amplification, and overexpression (Supplementary Table S1, Supplementary Fig. S1A). Gene expression was confirmed via immunoblot and some of the genes tested were able to promote growth factor independence and anchorage-independent growth indicating the capacity for transformation (Supplementary Table S1, Supplementary Fig. S1B,C).
Figure 1. An isogenic cell line screen reveals genomic drivers of drug response.

(A) Overview of screening approach in MCF10A isogenic cell lines. For each isogenic line, relative drug responses are comparing empty vector expressing MCF10A cells exposed to the same drugs. (B) Pie chart depicting FDA approval status of 90 compounds in this study. (C) Distribution of drugs targeting distinct cancer pathways and particular kinase targets (inset). (D) Volcano plot comparing magnitude and significance score of altered drug responses as compared against control MCF10A parental cells for 4,541 chemical-genetic interactions interrogated in this study. Maximum FDR rates of score ranges are indicated (see methods). Data points reflecting resistance compared to control of G12V mutant H/K/N-RAS MCF10A cells to EGFR inhibitors erlotinib and vandetanib are highlighted. (E) The 51 genes analyzed in this study sorted based on the number of high scoring chemical interactions (number of interactions with |S|>4 or |S|>2).
The majority of current cancer drugs have not been linked to specific genomic alterations that could be used as biomarkers to specify their selective therapeutic efficacy. To measure the impact of gene activation on cellular responses systematically, we screened our isogenic panel against a library of 90 anti-cancer therapeutics spanning multiple stages of clinical development. Seventy-nine percent of these drugs have already been through at least one clinical trial, and 25% are already FDA-approved compounds (Supplementary Table S2, Fig. 1B). Together, they target a broad variety of canonical cancer pathways and targets (Fig. 1C). We developed a robust screening method to quantitatively assess the impact of gene expression on drug responses. In this screen, isogenic cells expressing control vector or a gene of interest are plated separately and their relative proliferation after 72 hours of drug treatment is compared by high-content microscopy. Cell numbers from each line and treatment are compared and the effect size is determined by the fold-change in cell number at the IC50 as compared to control, averaged over replicates (Fig. 1D, supplementary methods). Next, the p-value of significance is converted to a signed chemical-genetic interaction score (S). Positive S values indicate that the expression of the gene drove drug resistance and negative S values indicate that the gene caused drug sensitivity as compared to vector controls. The screen displayed a high correlation of scores across replicates (r=0.618, Supplementary Fig. S2A,B) and an empirical false-discovery rate (FDR) of 1% and 10% corresponding to score cutoffs of approximately S=±4 and S=±2, respectively (Supplementary Fig. S2C). Notably, these numbers compare favorably to similar screens performed in yeast (39). Altogether, we determined quantitative scores for 4,541 gene-drug interactions and identified 174 resistance interactions and 97 sensitivity interactions at S=±2, corresponding to a 10% FDR (Supplementary Table S3).
As a control, we examined the impact of activating G12V mutations in the RAS family of oncogenes (HRAS, KRAS, and NRAS) on drug responses that drove resistance to multiple EGFR inhibitors, including erlotinib and vandetanib (Fig. 1D). It is well-established that KRAS operates downstream of EGFR and our results are consistent with this known relationship. In addition, our results confirm findings from other cancer cell line drug screens and clinical observations that indicate KRAS mutations can drive acquired resistance to EGFR inhibitors in patients (2, 40). A number of other activated oncogenes also induced resistance to erlotinib, including the TPR-MET fusion (S=4.3), IGF1R (S=3.4), BRAF V600E (S=2.1) and constitutively active MEK (MEKDD) (S=4.4), delineating several routes of resistance to EGFR inhibitor therapy, most of which have been observed in the clinic (41–43) (Supplementary Fig. S2D). These results were largely consistent with other EGFR inhibitors including BIBW-2992 (Supplementary Fig. S2E). We also observed that cells expressing a common activating mutation in PIK3CA (H1047R) were resistant to MEK inhibitors AZD-6244 (S=2.1) and CI-1040 (S=3.1), reflecting known redundancy between PI3K and MEK pathways. As MEK inhibitor clinical trials are ongoing, these observations support emerging data that patients with PIK3CA activating mutations are not likely to respond to this therapeutic approach (44) and predicts that PIK3CA mutation may drive acquired resistance to MEK inhibitors. In addition, expected drug sensitivities between kinases and drugs that directly target them were identified, including expression of EGFR which led to sensitivity to the EGFR inhibitor gefitinib (S=−2.8) and activation of the AKT pathway by myristoylated AKT (MYR-AKT) led to sensitivity to the PI3K/mTOR inhibitor BEZ-235 (S=−3.5). We also identified the PLK inhibitor BI-3536 as the top synthetic lethal hit with RAS genes (mean S=−2.0), confirming a previous synthetic lethal RNAi screen that identified PLK1 dependency and mitotic stress as a hallmark of the RAS oncogenic state (10). Among the 51 genes in this study, RAS-family oncogenes altered the most drug responses. This highlights their importance in the selection of drug-treatment regimes, especially since they are among the most mutated genes in human cancer (Fig. 1E). Analysis of the mutational spectrum of breast cancers also revealed that many less frequently altered genes can modulate the response to a large number of compounds, providing rationale for their consideration as cancer targets and modifiers of clinical responses (Supplementary Fig. S2F). Thus, the resulting map highlights known drug responses driven by gene activation and provides a roadmap for the exploration of novel molecular drivers of therapeutic responses.
Prediction of cancer cell line responses and drug similarities
The ability of isogenic cell line screens to recapitulate known clinical and cellular drug responses raises the possibility that they could complement cancer cell line screens of therapeutics, an established paradigm for biomarker identification. Recent screens have used regression techniques to identify molecular markers correlated with drug responses (2, 4). Comparison of the 21 genes and 40 drugs in common with the Cancer Genome Project (CGP) study (4) revealed a strong degree of overlap between drug responses using isogenic lines and responses found to be significantly correlated with genotypes in the CGP. Reflecting the quantitative nature of our dataset, this overlap was related to the S-score cutoff used to define hits (over 50% at |S|>4.5, Fig. 2A) and was strongly significant at a variety of cutoffs (p=1.4×10−5 at |S|>2.5, Supplementary Fig. S3). Thus, our approach complements cancer cell line screening and provides a quantitative basis for the prediction of genotype-specific dependencies that can be explored in other established model systems.
Figure 2. Global analysis of the chemical-genetic interaction map.

(A) Comparison of chemical-genetic interactions from this study at a variety of significance cutoffs with 489 drug-gene associations spanning 21 genes and 40 drugs identified in the CGP through regression analysis (p-value of 0.05) (4). The score cutoff reflects the absolute value of the S-score, and therefore encapsulates both resistance and sensitivity. Dotted line represents background probability of overlap. (B) A genetic interaction profile for each drug is calculated across 51 cell lines. Using a sliding cutoff based on correlation of profiles, the similarity of genetic interaction profiles for two drugs is plotted against the fraction of these drugs that have the same annotated molecular target. (C) Hierarchical clustering of drug profile similarities for compounds targeting multiple distinct biological pathways.
Existing drugs target a limited number of pathways and can have unexpected but significant off-target effects that dominate their biological activities. To identify the degree to which off-target effects dominate the chemical-genetic interaction map, we asked whether independent small molecules targeting the same pathway have a similar spectrum of genetic interactions. We used the profile of interactions for a given drug across the isogenic panel to provide a sensitive phenotypic signature and evaluated the degree to which this profile was shared between drugs. We found that independent drugs with the same annotated molecular target had a highly correlated profile that was predictive of the probability that they targeted the same pathway (Fig. 2B). Furthermore, drugs targeting the same pathways had highly similar profiles that were distinct from other classes of inhibitors (Fig. 2C) suggesting that their cellular effects are primarily through inhibition of the intended molecular target. These data indicate that the interaction map has the ability to link novel compounds to existing classes of drugs and serve as a platform for exploring drug mechanism of action.
New pharmacologically tractable dependencies for the MYC oncogene
A powerful use of the chemical-genetic interaction map is to identify synthetic lethal relationships involving oncogenes for which no specific small molecule inhibitors exist and are thus considered undruggable. One such oncogene, the transcription factor MYC, is among the most frequently amplified genes in breast cancer and associated with the basal molecular or TNBC pathologic subtype, the most aggressive form of breast cancer (23, 33). Previous synthetic lethal approaches centered on MYC have identified several new genes that have not yet been easily targeted pharmacologically (34, 35, 45, 46). Therefore, we interrogated the chemical-genetic interaction map to identify existing, clinically-relevant small molecules that can modulate the response of cells over-expressing MYC. We uncovered that MYC drove resistance to 6 distinct AKT/PI3K/mTOR pathway inhibitors, most strongly with the AKT inhibitor MK-2206 (S=4.5) (Fig. 3A). In validation studies all 6 inhibitors significantly inhibited the relative proliferation of control MCF10APURO cells while leaving MCF10AMYC cells unaffected (Fig. 3B). Mining previously published gene expression and drug-response data, we found that increased MYC expression could significantly predict resistance to MK-2206 in a panel of 20 breast cancer cell lines (p=0.01), further indicating strong corroboration between isogenic and cancer cell line responses (Fig. 3C). These data are consistent with prior reports of MYC-driven resistance to other PI3K pathway inhibitors in cell lines (18) and mouse models (47). Together, these results shed light on previous data suggesting that AKT/PI3K inhibitors are not effective in the basal breast cancer molecular subtype (48), where MYC expression is known to be high (23, 33). Since a number of similar compounds are approved or under investigation in breast cancer, we hypothesize that MYC status may be a useful criterion for exclusion of patients from trials involving these inhibitors.
Figure 3. Validation of MYC-driven drug responses.
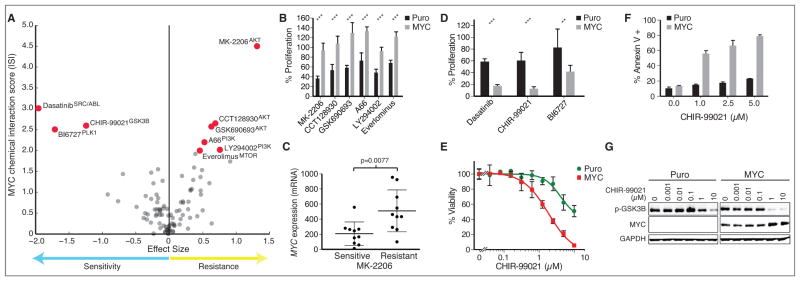
(A) Volcano plot of MYC driven drug responses identified in MCF10AMYC cells versus control. Drug responses with S≥2 are highlighted. (B) Validation of relative growth rates of drug treated MCF10APURO and MCF10AMYC cells compared to DMSO control in the presence of AKT/PI3K/MTOR pathway inhibitors as indicated in panel A. (C) Sensitivity to AKT inhibitor MK-2206 compared with MYC expression across a panel of 20 breast cancer cell lines separated into two equally sized groups to define sensitive and resistant lines with RNAseq data from (54). (D) Validation of relative growth rates of drug treated MCF10A lines with synthetic lethal hits in A. (E) Concentration-response of viability of isogenic cell lines to GSK3B inhibitor CHIR-99021. (F) Fraction of total cell population undergoing apoptosis in response to drug treatment for 24 hours as measured by Annexin V staining. (G) Levels of p-GSK3B Ser9 and total MYC after treatment of MCF10A cells for 18 hours. GAPDH is used as loading control. Unless otherwise noted, drug concentrations are the near IC50 listed in Table S2. *** = p<0.001, **= p<0.01.
Several strong synthetic lethal interactions pointed us toward new therapies that might be effective against tumors harboring high levels of MYC (Fig. 3A). Detailed analysis of three top candidates, BI-6727 (Polo-like kinase inhibitor), CHIR-99021 (GSK3β), and dasatinib (ABL and SRC-family kinase), revealed that all drugs were significantly more effective in a MYC-dependent manner in MCF10A cells (Fig. 3D). Sensitivity to BI-6727 (S=−2.5), a PLK inhibitor that targets the mitotic machinery, is consistent with previous reports that inhibitors of related mitotic kinases have been shown to have preferential activity in MYC-high cancers (17, 36). Likewise, the CDK inhibitor flavopiridol (S=−1.4), the kinesin inhibitor SB743921 (S=−1.55), as well as a structurally distinct PLK inhibitor BI2536 (S=−1.47) scored negatively with MYC, indicating that MYC expression leads to an increased dependence on multiple mitotic processes.
An RNAi screen previously identified depletion of GSK3β as synthetic lethal with MYC (35), but a small molecule that can phenocopy knockdown of GSK3β has not yet been identified. We found that MYC expression resulted in cellular sensitivity to CHIR-99021 through a reduction in cell viability (Fig. 3E), and induction of apoptosis in a MYC-dependent manner (Fig. 3F), confirming a synthetic lethal relationship. CHIR-99021 (S=−2.6) targets GSK3β, which phosphorylates MYC to promote its degradation (49). Indicating an on-target effect, the cellular response to CHIR-99021 resulted in potent phospho-GSK3β kinase inhibition and an increase in MYC protein, consistent with an increase in stability due to loss of GSK3β activity (Fig. 3G). Aberrant activation of MYC has been shown to induce apoptosis in a variety of model systems and therefore it is plausible that CHIR-99021 induces apoptosis through an increase in MYC activity (50). While more work is required to further explore its utility in pre-clinical systems, we hypothesize that CHIR-99021 or other GSK3β inhibitors that are currently in trials for neurodegenerative disorders (51) could potentially be repurposed for use in MYC-driven cancers.
Dasatinib treatment is synthetic lethal with MYC expression in TNBC model systems
Mapping of synthetic lethal interactions with already FDA-approved inhibitors can lead to the discovery of previously unknown connections and can ultimately accelerate new clinical trials by repurposing clinically-viable drugs. The strongest MYC synthetic lethal interaction was with dasatinib (Sprycel, S=−3.0), a tyrosine kinase inhibitor that is approved for use in BCR-ABL+ CML and GIST with known off-target activities including inhibition of the SRC-family kinases and ephrin kinases. Analysis across a range of concentrations revealed specific inhibition of cell number in MCF10AMYC cells compared to controls after dasatinib treatment for 3 days (Fig. 4A). We also confirmed MYC-specific sensitivity using an orthogonal FACS based competition assay where MCF10APURO cells outcompeted their MYC counterparts over a range of dasatinib concentrations (Supplementary Fig. S4A). The parental MCF10A cells contain a copy-number gain of the MYC locus presumably acquired during the immortalization process (38, 52). To model a more MYC-naïve state, we used a model system based on primary human mammary epithelial cells (HMEC) that are derived from healthy breast tissue, have a limited lifespan, and low MYC expression (53). We created a derivative of this cell line that constitutively expresses an inactive MYC-ER fusion protein that is activated in the presence of 4-hydroxy tamoxifen (4-OHT). HMEC cells in the presence of 4-OHT were 5-fold more sensitive to dasatinib (IC50 ~ 200nM) as compared to vehicle-treated controls (IC50 ~ 1uM) (Fig. 4B). The sensitivity was matched by a significant 6-fold induction of apoptosis in cells with activated MYC versus controls (p = 1×10−7) (Fig. 4C). Apoptosis was evidenced by PARP cleavage, attenuated expression of mitochondrial anti-apoptotic markers MCL1 and BCL-xL, and induction of the pro-apoptotic BIM protein (Fig. 4D, Supplementary Fig. S4B). Thus, cellular sensitivity in isogenic cell line model systems indicates that expression of MYC can drive a cytotoxic response to dasatinib in breast epithelial cells.
Figure 4. MYC is synthetic lethal with dasatinib in engineered cell lines.

(A) Relative proliferation of MCF10APURO versus MCF10AMYC cell lines to a range of dasatinib concentrations. (B) Relative viability of HMEC MYC-ER cells treated with vehicle or 4-OHT to activate MYC in response to dasatinib for 48 hours. (C) Fraction of total cell population undergoing apoptosis in HMEC MYC-ER cells treated with vehicle or 4-OHT in the presence of dasatinib. (D) Response to dasatinib (250nM) in HMEC MYC-ER cells through measurement of molecular correlates of apoptosis including PARP cleavage, MCL1, BCL-xL and BIM. Actin is loading control.
Dasatinib has preferential activity in MYC expressing cancer cell lines
Synthetic lethal interactions found in isogenic cell lines provide a basis for exploration in cancer cell lines, which more closely mimic the complex genotypes and biology present in patient tumors. Indeed, a global comparison of results from our screen and those found through cancer cell line screens indicated substantial overlap (Fig. 2A). Therefore, we tested the hypothesis that MYC is a predictive biomarker for cancer cell lines that are sensitive to dasatinib. We performed high-throughput cell line sensitivity screening of dasatinib against a panel of 664 cancer cell lines spanning a variety of tumor types (Supplementary Table S4, see methods). As controls, we verified that CML cell lines harboring BCR-ABL fusions were specifically sensitive to dasatinib (p=2×10−9) (Supplementary Fig. S5A,B). Integration of these data with previously published gene expression data (2) revealed that sensitive cell lines had higher MYC expression at the mRNA level (p=5×10−4) (Fig. 5A). In contrast, cell lines with low levels of MYC expression (relative expression <10) were >90% likely to be drug resistant, suggesting that at least a basal level of MYC expression is required for sensitivity (9 sensitive versus 83 resistant, Fig. 5A). However, this relationship was highly dependent on the tissue of origin (Supplementary Fig. S5C) and we therefore sought to investigate this link specifically in breast cancer. Integration of drug sensitivity with focused molecular annotations of breast cancer cell lines (54) revealed a significant relationship between sensitivity and MYC mRNA and protein levels (p=0.0089 and p=0.001, respectively) (Fig. 5B,C). Next, we selected three MYChigh cancer cell lines (SUM149, MDAMB231 and HCC1143) and two MYClow lines (T47D and HCC1428) for further interrogation, confirming their MYC levels (Fig. 5D) and MYC dependence as assessed through siRNA mediated knockdown (Supplementary Fig. S6A). We found increased sensitivity to dasatinib in MYChigh cancer cell lines (IC50>100nM for MYClow and <100nM for MYChigh, Fig. 5E). To investigate if dasatinib can inhibit breast tumor growth in vivo, xenografts of MDAMB231 and HCC1428 were generated in nude mice and treated daily with dasatinib or vehicle administered orally for 15 days. Tumor volume was significantly reduced in MYChigh, MDAMB231 xenografts (p=0.01) but not in the MYClow, HCC1428 derived tumors (Fig. 5F). These data corroborate isogenic cell line responses and show that MYC levels predict dasatinib sensitivity in cancer cell lines in vitro and in vivo.
Figure 5. MYC is correlated with dasatinib sensitivity in cancer cell lines and in vivo.

(A) Relationship between dasatinib sensitivity as determined in this study and published MYC gene expression data across 664 cancer cell lines (2). An AUC<3 is used to define sensitive cell lines. (B-C) Relationship between dasatinib sensitivity and MYC expression as assessed through RNAseq (B) and reverse-phase protein array (RPPA) (C) data from breast cancer cell lines published in (54). (D) MYC protein levels assessed by western blot across established breast cancer cell lines. (E) Relative viability of breast cancer cell lines across a range of concentrations of dasatinib. (F) Percent change in tumor volume of human cell lines xenografted into mice and treated daily with the indicated concentration of dasatinib via oral gavage. A minimum of 5 mice were used in each group. n.s. = not significant.
Dasatinib synthetic lethality is through LYN inhibition in MYChigh breast cancers
We next sought to understand the mechanisms by which breast cancer cells with high MYC expression respond to dasatinib. Dasatinib has been shown to bind up to 38 kinases with high affinity (55) and we reasoned that the molecular target of dasatinib might be selectively up-regulated in a MYC-dependent manner. To elucidate upregulated drug targets, we used a proteomic approach wherein immobilized dasatinib is used to affinity purify proteins that bind the drug that are subsequently identified using quantitative mass spectrometry (56). Using this approach, we identified multiple unique peptides for the SRC-family tyrosine kinase LYN which were selectively bound and enriched in MCF10AMYC cells compared to control cells (Fig. 6A, Supplementary Table S5). LYN is a direct target of dasatinib (55) and is important for B-cell activation and has been shown to be active in prostate and breast cancer (57). Immunoblot confirmed that LYN is upregulated, LYN activated by auto-phosphorylation of Y416 is increased in a MYC-dependent manner, and that LYN activation is inhibited upon drug treatment (Fig. 6B). Mirroring the changes found in isogenic cells, both total and phospho-LYN were strongly linked to MYC levels across our breast cancer cell lines (Fig. 6C). Interestingly, SRC, a canonical target of dasatinib and known oncogene, was found to be expressed at higher levels in cell lines that were drug resistant and MYClow (T47D and HCC1428) suggesting that it does not play a role in the response to dasatinib in breast cancer (Fig. 6C). We hypothesized that in MYChigh breast cancer cell lines LYN is necessary for cell viability and its inhibition is the basis for dasatinib sensitivity. Indeed, siRNA-mediated knockdown of LYN significantly inhibited the proliferation of all three MYChigh but not MYClow cell lines (Fig. 6D, Supplementary Fig. S6B,C) and expression of a dasatinib-resistant gatekeeper mutant of LYN (T319I) significantly rescued viability of all three MYChigh lines when treated with dasatinib compared to GFP control (Fig. 6E, Supplementary Fig. S6D) (58). Together, these data indicate that MYChigh breast cancer cells require LYN and their sensitivity to dasatinib is mediated by a LYN-dependent mechanism.
Figure 6. Dependence on LYN mediates synthetic lethality between MYC and dasatinib.

(A) Peptides enriched through a dasatinib-bead based affinity purification followed by quantitative mass spectrometry to identify bound peptides. Peptides representing kinases 2-fold more abundant in MCF10AMYC cells are highlighted. (B) Validation via western blot of LYN, p-LYN (Y416) and MYC levels in MCF10APURO cells and MCF10AMYC cells treated with dasatinib for 18 hours at indicated concentrations (nM). (C) Western blot measurement of MYC, LYN, pLYN, SRC levels in five characterized breast cancer cell lines. (D) Relative viability after siRNA mediated knockdown of LYN compared to non-targeting (NT), scrambled control in five breast cancer cell lines. (E) Relative viability of dasatinib sensitive breast cancer cell lines over-expressing GFP control, LYN and LYN T319I constructs after treatment with 1uM dasatinib compared to DMSO.
MYC and LYN are strongly linked and have interdependent clinical outcomes
This synthetic lethal interaction suggests that patient tumors harboring high levels of MYC may respond to dasatinib through inhibition of LYN. Indeed, expression of MYC and LYN transcripts were tightly linked across a panel of 807 cancer cell lines from diverse origins (Fig. 7A), and correlated in breast cancer cell lines (r=0.53, p=2×10−4) (Fig. S7). In patients, both MYC and LYN were significantly co-expressed across 919 patients in the Breast TCGA (r=0.23, p=1×10−12) and highest in the basal subtype and TNBC patient population (Fig. 7B,C) (23). To interrogate whether the combined activity of MYC and LYN could influence clinical outcomes, we investigated data from the I-SPY breast cancer clinical trial (n=149 patients) where we stratified patients based on their tertile of expression of MYC and LYN. In patients with MYChigh tumors, those with higher expression of LYN were more likely to relapse and had a decreased survival (Fig. 7D, p=0.017, log rank test). By contrast, high expression of LYN did not correlate with outcome in MYClow patients (Fig. 7E). These data suggest MYC hyper-activation leads to an increased dependency on LYN in human breast cancers.
Figure 7. MYC and LYN are co-expressed and have interdependent clinical outcomes.
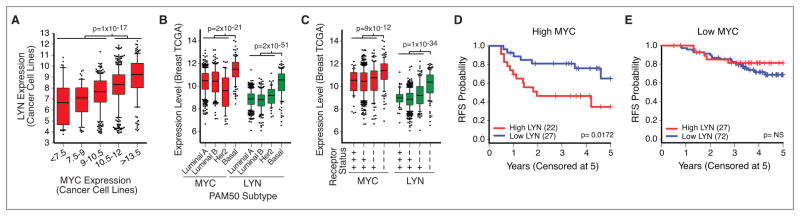
(A) Coexpression of MYC and LYN across 789 cancer cell lines, data from (2). Groups of cell lines are binned by MYC expression. Expression of MYC and LYN across patients in the Breast TCGA (23) separated based on patient PAM50 subtype (B) and the number of positively stained molecular receptors (ER, PR or HER2) (C). Whiskers span the 10–90th percentiles. (D–E) Kaplan Meier relapse-free survival (RFS) curves of I-SPY 1 patients stratified by LYN expression levels (D) patient subset with highest tertile of MYC expression levels (n=50), (E) patient subset with lowest tertile of MYC expression levels (n=99).
DISCUSSION
We present a quantitative platform and dataset for mapping genotype-specific responses to clinically relevant inhibitors using an isogenic panel of cell lines harboring distinct genetic events. We envision that this dataset can help shape systems pharmacology-based approaches for cancer therapy. As opposed to prior barcoded-based approaches that were unable to capture both resistance and sensitivity (10, 11, 18), the quantitative nature of our dataset allowed us to perform several key comparisons. We uncovered a strong overlap of drug response biomarkers through comparison with previous large-scale cancer cell line screening efforts as well as focused comparison with the AKT inhibitor MK-2206 and dasatinib. Our dataset is strongly predictive of cancer cell line drug sensitivities and indicates that engineered isogenic cell lines can accurately model the biology of mutations present in genetically complex tumor samples. The proposed platform has several distinct advantages over correlative screening approaches in cancer cell lines. While cancer cell lines represent the natural heterogeneity of clinical cancer cases, effective screening requires a panel of cell lines harboring each mutation of interest. For rare mutations, gathering sufficient lines may be prohibitive or impossible. In addition, the presence of many mutations in any single cell line makes statistical association difficult. Lastly, a known limitation of current synthetic lethal screening platforms using cancer cell line collections is the inability to accurately model cellular contexts specific to particular disease types (6). While we have focused on breast cancer, future work may develop an expanded and tailored isogenic cell line encyclopedia that encompasses the majority of recurrent oncogenic mutations, amplifications, and deletions found in a particular tumor type. Integrative analysis of drug responses, pathway alterations, and emerging dependencies in these lines will likely illuminate previously unexplored therapeutic avenues.
This chemical-genetic interaction map revealed a number of novel connections and provides a valuable dataset for the exploration of therapeutic responses for a variety of cancer genes. As proof-of-concept that the interaction map can predict biologically relevant and clinically actionable responses, we investigated dependencies induced by MYC. Analysis indicated that MYC could drive resistance to multiple PI3K/AKT/mTOR pathway inhibitors. As many of these inhibitors are being explored in the clinic, this finding provides a possible route to both innate as well as acquired resistance to these drugs in patients. The top synthetic lethal hit with MYC was dasatinib, which has previously been found to be effective in basal breast cancer cell lines in vitro (59), a subtype often expressing high MYC (33). Here we show it can also be effective against breast tumor xenografts harboring high MYC in vivo. In addition, our results indicate that MYC-driven dasatinib sensitivity is likely through LYN inhibition. This connection is intriguing since MYC has previously been suggested to operate both upstream and downstream of SRC-family kinases, including LYN, in other tumor types (60, 61). Like MYC, LYN has also been shown to be preferentially active in basal breast cancers (62, 63). Interestingly, dasatinib was also found to be synthetic lethal with CCND3 (S=−2.6), a component of the CDK4/6 complex. Since CDKs have been shown to be synthetic lethal with MYC activation (17, 33, 64), one possibility is that dasatinib may be more effective in cells with enhanced cell cycle progression through either CCND3 or MYC. Further studies will be necessary to determine the exact molecular mechanisms by which MYC-expressing cells become dependent on LYN. However, the fact that MYC and LYN are highly co-expressed in patients and combine to influence outcomes provides strong evidence of their functional relationship.
Limited therapeutic options currently exist for patients with TNBC. This work indicates that the approved drug dasatinib may be an immediately applicable and efficacious treatment for this challenging subset of breast cancer patients. Previous trials of dasatinib in TNBC patient populations have had limited response rates (65, 66) that may be enhanced in the future by employing MYC and LYN as biomarkers for patient selection. As dasatinib is FDA-approved, it provides an example of how chemical-genetic interaction maps can provide valuable insights that can ultimately be used to repurpose existing drugs for new clinical trials, thus accelerating therapeutic development. The ability to systematically map molecular drivers of drug responses revealed a plethora of unexpected but actionable connections and provides a blueprint for new systems approaches for precision medicine.
METHODS
MCF10A cell line generation and screening
MFC10A parental cell lines were grown according to published protocols (38). Derivative isogenic cell lines were generated though stable infection using viral infection of cell pools using the indicated vectors (Supplementary Table S1). Control MCF10A cell lines were generated by expressing empty vectors conferring puromycin, or blasticidin gene resistance as appropriate. Proliferation was measured by staining with Hoescht nuclear dye and cell (nuclear) number counted using a Thermo CellInsight high content microscope. The parental cell line was first screened against all 90 compounds (Selleckchem, Houston, TX) to determine concentration-response curves and approximate IC50 concentrations (Supplementary Table S2). The maximum concentration assayed for any drug was approximately 20μM. Each line was independently screened by plating 1,000 cells/well in 384-well plates for 24 hours then exposed to each drug at IC50 concentration for 72 hours using a minimum of 8 replicates. Statistical scoring is described in detail in the supplemental methods.
Viability and apoptosis assays
Cell viability was determined using the CellTiter-Glo cell viability assay per manufacturer instructions (Promega). Apoptosis was measured through cell fixation and staining with early-apoptosis marker Annexin V and quantified through FACS using standard protocols.
High throughput cancer cell line screening
Cancer cell lines were obtained from the Broad Institute’s Biological Samples Platform and are a subset of the Cancer Cell Line Encyclopedia’s human cancer cell lines (2). Cell lines were grown in their specified medium at 37C/5% CO2 and plated into duplicate 1536-well assay plates at a density of 500 cells per well in 6 ul of total volume. The cells were incubated overnight and then treated with dasatinib in a 16-pt, 2-fold concentration range for 72 hours. ATP levels were measured using CellTiter-Glo as a surrogate for cell viability. Cellular responses to compounds were based on a normalized area under the dose-response curve (AUC) as described previously (2). Sensitive cell lines are annotated as those with AUC < 3.
Cell Culture, siRNA Transfection, and Immunoblotting
MDAMB231, cells were obtained from the American Type Culture Collection (ATCC) and were propagated in DMEM containing 10% FBS. T47D, HCC1143 and HCC1428 cells were obtained from ATCC and propagated in RPMI1640 containing 10% FBS. SUM149 cells were obtained from the lab of Dr. Joe Gray and propagated in F-12 with 5%FBS, insulin and hydrocortizone. No additional cell line authentication was conducted by the authors. The following antibodies were used for immunoblot analyses: MYC and MCL-1 (Abcam), β, actin and BCL-xl (Santa Cruz Biotechnology, Inc.), PARP, SRC, LYN and p-LYN (Cell Signaling Technology) and BIM (Assay Designs).
Xenograft analysis
Animal work was conducted in accordance with protocols approved by the Institutional Care and Use Committee for animal research at the University of California, San Francisco. Nude mice (BALB/c nude/nude) were subcutaneously injected with 1.5×106 MDAMB231 cells or 6×106 HCC1428 cells mixed 1:1 with Basement Membrane Matrix (BD Biosciences). Initial tumor dimensions were monitored three times weekly and the treatment was initiated when tumor volume reached about 80mm3. Once animals reached indicated tumor volume, they were randomly placed into control or treatment groups. Animals were treated with 50mg/kg crushed Dasatinb (Sprycel) tablets from the UCSF pharmacy dissolved in water daily for 14 days via oral gavage. Tumor volume was calculated daily from two diameter measurements using calipers, one along the anterior-posterior axis and the other along the lateral-medial axis. Percent change for tumor growth is based on volumes calculated from size on day 1 of treatment compared to day 15.
Statistical parameters
All p-values are based on a two-tailed Student’s t-test unless otherwise noted. All error bars are standard deviation unless otherwise noted.
Supplementary Material
STATEMENT OF SIGNIFICANCE.
Determining how the plethora of genomic abnormalities that exist within a given tumor cell impacts drug responses remains a major challenge in oncology. Here, we develop a new mapping approach to connect cancer genotypes to drug responses using engineered isogenic cell lines and demonstrate how the resulting dataset can guide clinical interrogation.
Acknowledgments
This work was supported by Martha and Bruce Atwater (SB, AG), UCSF Breast Oncology SPORE development award (SB, AG), NCI U01CA168370 (SB, FM), NIGMS R01GM107671 (SB, NK), NIH T32 Postdoctoral Training Award 5T32CA108462-10 (AZ), NCI 5R01CA136717 (AG), Leukemia and Lymphoma Scholar Award (AG) and CDMRP Award W81XWH-12-1-0272 (AG).
We acknowledge members of the McCormick lab for advice and reagents, Mercedes Joaquin for assistance with mouse experiments, and Susan Samson, UCSF Breast Oncology Program Advocacy Core, for helpful discussions.
Footnotes
There are no potential conflicts of interest to disclose.
AUTHORS’ CONTRIBUTIONS
Conception and design: S. Bandyopadhyay. Development of methodology: M.M. Martins. Acquisition of data: M.M. Martins, A.Y. Zhou, A. Corella, T. Rakshandehroo, J.D. Gordan, R.S. Levin, J. Jascur, J. Cheah. Analysis and interpretation of data: M.M. Martins, A.Y. Zhou, D. Horiuchi, C. Yau, J. Johnson, P.A. Clemons, A. Shamji, S. Bandyopadhyay. Writing, review, and/or revision of the manuscript: A.Y. Zhou, A. Goga, S. Bandyopadhyay with input from all authors. Administrative, technical, or material support: M. Shales, A. Sorrentino, S. Schreiber, N.J. Krogan, K.M. Shokat, F. McCormick. Study supervision: A. Goga, S. Bandyopadhyay
References
- 1.Yaffe MB. The scientific drunk and the lamppost: massive sequencing efforts in cancer discovery and treatment. Science signaling. 2013;6:pe13. doi: 10.1126/scisignal.2003684. [DOI] [PubMed] [Google Scholar]
- 2.Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483:603–7. doi: 10.1038/nature11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Basu A, Bodycombe NE, Cheah JH, Price EV, Liu K, Schaefer GI, et al. An interactive resource to identify cancer genetic and lineage dependencies targeted by small molecules. Cell. 2013;154:1151–61. doi: 10.1016/j.cell.2013.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Garnett MJ, Edelman EJ, Heidorn SJ, Greenman CD, Dastur A, Lau KW, et al. Systematic identification of genomic markers of drug sensitivity in cancer cells. Nature. 2012;483:570–5. doi: 10.1038/nature11005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ashworth A, Lord CJ, Reis-Filho JS. Genetic interactions in cancer progression and treatment. Cell. 2011;145:30–8. doi: 10.1016/j.cell.2011.03.020. [DOI] [PubMed] [Google Scholar]
- 6.Nijman SM, Friend SH. Cancer. Potential of the synthetic lethality principle. Science. 2013;342:809–11. doi: 10.1126/science.1244669. [DOI] [PubMed] [Google Scholar]
- 7.Bandyopadhyay S, Mehta M, Kuo D, Sung MK, Chuang R, Jaehnig EJ, et al. Rewiring of genetic networks in response to DNA damage. Science. 2010;330:1385–9. doi: 10.1126/science.1195618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Costanzo M, Baryshnikova A, Bellay J, Kim Y, Spear ED, Sevier CS, et al. The genetic landscape of a cell. Science. 2010;327:425–31. doi: 10.1126/science.1180823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Corcoran RB, Cheng KA, Hata AN, Faber AC, Ebi H, Coffee EM, et al. Synthetic lethal interaction of combined BCL-XL and MEK inhibition promotes tumor regressions in KRAS mutant cancer models. Cancer Cell. 2013;23:121–8. doi: 10.1016/j.ccr.2012.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Luo J, Emanuele MJ, Li D, Creighton CJ, Schlabach MR, Westbrook TF, et al. A genome-wide RNAi screen identifies multiple synthetic lethal interactions with the Ras oncogene. Cell. 2009;137:835–48. doi: 10.1016/j.cell.2009.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Scholl C, Frohling S, Dunn IF, Schinzel AC, Barbie DA, Kim SY, et al. Synthetic lethal interaction between oncogenic KRAS dependency and STK33 suppression in human cancer cells. Cell. 2009;137:821–34. doi: 10.1016/j.cell.2009.03.017. [DOI] [PubMed] [Google Scholar]
- 12.Steckel M, Molina-Arcas M, Weigelt B, Marani M, Warne PH, Kuznetsov H, et al. Determination of synthetic lethal interactions in KRAS oncogene-dependent cancer cells reveals novel therapeutic targeting strategies. Cell research. 2012;22:1227–45. doi: 10.1038/cr.2012.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang WS, Stockwell BR. Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chemistry & biology. 2008;15:234–45. doi: 10.1016/j.chembiol.2008.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang Y, Ngo VN, Marani M, Yang Y, Wright G, Staudt LM, et al. Critical role for transcriptional repressor Snail2 in transformation by oncogenic RAS in colorectal carcinoma cells. Oncogene. 2010;29:4658–70. doi: 10.1038/onc.2010.218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Barbie DA, Tamayo P, Boehm JS, Kim SY, Moody SE, Dunn IF, et al. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature. 2009;462:108–12. doi: 10.1038/nature08460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Beaver JA, Gustin JP, Yi KH, Rajpurohit A, Thomas M, Gilbert SF, et al. PIK3CA and AKT1 mutations have distinct effects on sensitivity to targeted pathway inhibitors in an isogenic luminal breast cancer model system. Clinical cancer research: an official journal of the American Association for Cancer Research. 2013;19:5413–22. doi: 10.1158/1078-0432.CCR-13-0884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Goga A, Yang D, Tward AD, Morgan DO, Bishop JM. Inhibition of CDK1 as a potential therapy for tumors over-expressing MYC. Nat Med. 2007;13:820–7. doi: 10.1038/nm1606. [DOI] [PubMed] [Google Scholar]
- 18.Muellner MK, Uras IZ, Gapp BV, Kerzendorfer C, Smida M, Lechtermann H, et al. A chemical-genetic screen reveals a mechanism of resistance to PI3K inhibitors in cancer. Nature chemical biology. 2011;7:787–93. doi: 10.1038/nchembio.695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zecchin D, Boscaro V, Medico E, Barault L, Martini M, Arena S, et al. BRAF V600E is a determinant of sensitivity to proteasome inhibitors. Molecular cancer therapeutics. 2013;12:2950–61. doi: 10.1158/1535-7163.MCT-13-0243. [DOI] [PubMed] [Google Scholar]
- 20.Alles MC, Gardiner-Garden M, Nott DJ, Wang Y, Foekens JA, Sutherland RL, et al. Meta-analysis and gene set enrichment relative to er status reveal elevated activity of MYC and E2F in the “basal” breast cancer subgroup. PloS one. 2009;4:e4710. doi: 10.1371/journal.pone.0004710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chandriani S, Frengen E, Cowling VH, Pendergrass SA, Perou CM, Whitfield ML, et al. A core MYC gene expression signature is prominent in basal-like breast cancer but only partially overlaps the core serum response. PloS one. 2009;4:e6693. doi: 10.1371/journal.pone.0006693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gatza ML, Lucas JE, Barry WT, Kim JW, Wang Q, Crawford MD, et al. A pathway-based classification of human breast cancer. Proc Natl Acad Sci U S A. 2010;107:6994–9. doi: 10.1073/pnas.0912708107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cancer Genome Atlas N. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stewart TA, Pattengale PK, Leder P. Spontaneous mammary adenocarcinomas in transgenic mice that carry and express MTV/myc fusion genes. Cell. 1984;38:627–37. doi: 10.1016/0092-8674(84)90257-5. [DOI] [PubMed] [Google Scholar]
- 25.Sinn E, Muller W, Pattengale P, Tepler I, Wallace R, Leder P. Coexpression of MMTV/v-Ha-ras and MMTV/c-myc genes in transgenic mice: synergistic action of oncogenes in vivo. Cell. 1987;49:465–75. doi: 10.1016/0092-8674(87)90449-1. [DOI] [PubMed] [Google Scholar]
- 26.Podsypanina K, Politi K, Beverly LJ, Varmus HE. Oncogene cooperation in tumor maintenance and tumor recurrence in mouse mammary tumors induced by Myc and mutant Kras. Proc Natl Acad Sci U S A. 2008;105:5242–7. doi: 10.1073/pnas.0801197105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.D’Cruz CM, Gunther EJ, Boxer RB, Hartman JL, Sintasath L, Moody SE, et al. c-MYC induces mammary tumorigenesis by means of a preferred pathway involving spontaneous Kras2 mutations. Nat Med. 2001;7:235–9. doi: 10.1038/84691. [DOI] [PubMed] [Google Scholar]
- 28.Soucek L, Helmer-Citterich M, Sacco A, Jucker R, Cesareni G, Nasi S. Design and properties of a Myc derivative that efficiently homodimerizes. Oncogene. 1998;17:2463–72. doi: 10.1038/sj.onc.1202199. [DOI] [PubMed] [Google Scholar]
- 29.Soucek L, Jucker R, Panacchia L, Ricordy R, Tato F, Nasi S. Omomyc, a potential Myc dominant negative, enhances Myc-induced apoptosis. Cancer research. 2002;62:3507–10. [PubMed] [Google Scholar]
- 30.Jain M, Arvanitis C, Chu K, Dewey W, Leonhardt E, Trinh M, et al. Sustained loss of a neoplastic phenotype by brief inactivation of MYC. Science. 2002;297:102–4. doi: 10.1126/science.1071489. [DOI] [PubMed] [Google Scholar]
- 31.Delmore JE, Issa GC, Lemieux ME, Rahl PB, Shi J, Jacobs HM, et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell. 2011;146:904–17. doi: 10.1016/j.cell.2011.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lockwood WW, Zejnullahu K, Bradner JE, Varmus H. Sensitivity of human lung adenocarcinoma cell lines to targeted inhibition of BET epigenetic signaling proteins. Proc Natl Acad Sci U S A. 2012;109:19408–13. doi: 10.1073/pnas.1216363109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Horiuchi D, Kusdra L, Huskey NE, Chandriani S, Lenburg ME, Gonzalez-Angulo AM, et al. MYC pathway activation in triple-negative breast cancer is synthetic lethal with CDK inhibition. J Exp Med. 2012;209:679–96. doi: 10.1084/jem.20111512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kessler JD, Kahle KT, Sun T, Meerbrey KL, Schlabach MR, Schmitt EM, et al. A SUMOylation-dependent transcriptional subprogram is required for Myc-driven tumorigenesis. Science. 2012;335:348–53. doi: 10.1126/science.1212728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Toyoshima M, Howie HL, Imakura M, Walsh RM, Annis JE, Chang AN, et al. Functional genomics identifies therapeutic targets for MYC-driven cancer. Proc Natl Acad Sci U S A. 2012;109:9545–50. doi: 10.1073/pnas.1121119109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yang D, Liu H, Goga A, Kim S, Yuneva M, Bishop JM. Therapeutic potential of a synthetic lethal interaction between the MYC proto-oncogene and inhibition of aurora-B kinase. Proc Natl Acad Sci U S A. 2010;107:13836–41. doi: 10.1073/pnas.1008366107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cermelli S, Jang IS, Bernard B, Grandori C. Synthetic lethal screens as a means to understand and treat MYC-driven cancers. Cold Spring Harbor perspectives in medicine. 2014:4. doi: 10.1101/cshperspect.a014209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Debnath J, Mills KR, Collins NL, Reginato MJ, Muthuswamy SK, Brugge JS. The role of apoptosis in creating and maintaining luminal space within normal and oncogene-expressing mammary acini. Cell. 2002;111:29–40. doi: 10.1016/s0092-8674(02)01001-2. [DOI] [PubMed] [Google Scholar]
- 39.Collins SR, Roguev A, Krogan NJ. Quantitative genetic interaction mapping using the E-MAP approach. Methods in enzymology. 2010;470:205–31. doi: 10.1016/S0076-6879(10)70009-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Misale S, Yaeger R, Hobor S, Scala E, Janakiraman M, Liska D, et al. Emergence of KRAS mutations and acquired resistance to anti-EGFR therapy in colorectal cancer. Nature. 2012;486:532–6. doi: 10.1038/nature11156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007;316:1039–43. doi: 10.1126/science.1141478. [DOI] [PubMed] [Google Scholar]
- 42.Vazquez-Martin A, Cufi S, Oliveras-Ferraros C, Torres-Garcia VZ, Corominas-Faja B, Cuyas E, et al. IGF-1R/epithelial-to-mesenchymal transition (EMT) crosstalk suppresses the erlotinib-sensitizing effect of EGFR exon 19 deletion mutations. Scientific reports. 2013;3:2560. doi: 10.1038/srep02560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ohashi K, Sequist LV, Arcila ME, Moran T, Chmielecki J, Lin YL, et al. Lung cancers with acquired resistance to EGFR inhibitors occasionally harbor BRAF gene mutations but lack mutations in KRAS, NRAS, or MEK1. Proc Natl Acad Sci U S A. 2012;109:E2127–33. doi: 10.1073/pnas.1203530109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wee S, Jagani Z, Xiang KX, Loo A, Dorsch M, Yao YM, et al. PI3K pathway activation mediates resistance to MEK inhibitors in KRAS mutant cancers. Cancer Res. 2009;69:4286–93. doi: 10.1158/0008-5472.CAN-08-4765. [DOI] [PubMed] [Google Scholar]
- 45.Lin CJ, Nasr Z, Premsrirut PK, Porco JA, Jr, Hippo Y, Lowe SW, et al. Targeting synthetic lethal interactions between Myc and the eIF4F complex impedes tumorigenesis. Cell reports. 2012;1:325–33. doi: 10.1016/j.celrep.2012.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu L, Ulbrich J, Muller J, Wustefeld T, Aeberhard L, Kress TR, et al. Deregulated MYC expression induces dependence upon AMPK-related kinase 5. Nature. 2012;483:608–12. doi: 10.1038/nature10927. [DOI] [PubMed] [Google Scholar]
- 47.Liu P, Cheng H, Santiago S, Raeder M, Zhang F, Isabella A, et al. Oncogenic PIK3CA-driven mammary tumors frequently recur via PI3K pathway-dependent and PI3K pathway-independent mechanisms. Nature medicine. 2011;17:1116–20. doi: 10.1038/nm.2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Heiser LM, Sadanandam A, Kuo WL, Benz SC, Goldstein TC, Ng S, et al. Subtype and pathway specific responses to anticancer compounds in breast cancer. Proc Natl Acad Sci U S A. 2012;109:2724–9. doi: 10.1073/pnas.1018854108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gregory MA, Qi Y, Hann SR. Phosphorylation by glycogen synthase kinase-3 controls c-myc proteolysis and subnuclear localization. The Journal of biological chemistry. 2003;278:51606–12. doi: 10.1074/jbc.M310722200. [DOI] [PubMed] [Google Scholar]
- 50.Evan GI, Wyllie AH, Gilbert CS, Littlewood TD, Land H, Brooks M, et al. Induction of apoptosis in fibroblasts by c-myc protein. Cell. 1992;69:119–28. doi: 10.1016/0092-8674(92)90123-t. [DOI] [PubMed] [Google Scholar]
- 51.Hooper C, Killick R, Lovestone S. The GSK3 hypothesis of Alzheimer’s disease. Journal of neurochemistry. 2008;104:1433–9. doi: 10.1111/j.1471-4159.2007.05194.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kadota M, Yang HH, Gomez B, Sato M, Clifford RJ, Meerzaman D, et al. Delineating genetic alterations for tumor progression in the MCF10A series of breast cancer cell lines. PloS one. 2010;5:e9201. doi: 10.1371/journal.pone.0009201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yaswen P, Stampfer MR. Molecular changes accompanying senescence and immortalization of cultured human mammary epithelial cells. The international journal of biochemistry & cell biology. 2002;34:1382–94. doi: 10.1016/s1357-2725(02)00047-x. [DOI] [PubMed] [Google Scholar]
- 54.Daemen A, Griffith OL, Heiser LM, Wang NJ, Enache OM, Sanborn Z, et al. Modeling precision treatment of breast cancer. Genome biology. 2013;14:R110. doi: 10.1186/gb-2013-14-10-r110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Davis MI, Hunt JP, Herrgard S, Ciceri P, Wodicka LM, Pallares G, et al. Comprehensive analysis of kinase inhibitor selectivity. Nature biotechnology. 2011;29:1046–51. doi: 10.1038/nbt.1990. [DOI] [PubMed] [Google Scholar]
- 56.Bantscheff M, Eberhard D, Abraham Y, Bastuck S, Boesche M, Hobson S, et al. Quantitative chemical proteomics reveals mechanisms of action of clinical ABL kinase inhibitors. Nature biotechnology. 2007;25:1035–44. doi: 10.1038/nbt1328. [DOI] [PubMed] [Google Scholar]
- 57.Ingley E. Functions of the Lyn tyrosine kinase in health and disease. Cell communication and signaling: CCS. 2012;10:21. doi: 10.1186/1478-811X-10-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Azam M, Seeliger MA, Gray NS, Kuriyan J, Daley GQ. Activation of tyrosine kinases by mutation of the gatekeeper threonine. Nature structural & molecular biology. 2008;15:1109–18. doi: 10.1038/nsmb.1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Finn RS, Dering J, Ginther C, Wilson CA, Glaspy P, Tchekmedyian N, et al. Dasatinib, an orally active small molecule inhibitor of both the src and abl kinases, selectively inhibits growth of basal-type/”triple-negative” breast cancer cell lines growing in vitro. Breast cancer research and treatment. 2007;105:319–26. doi: 10.1007/s10549-006-9463-x. [DOI] [PubMed] [Google Scholar]
- 60.Bowman T, Broome MA, Sinibaldi D, Wharton W, Pledger WJ, Sedivy JM, et al. Stat3-mediated Myc expression is required for Src transformation and PDGF-induced mitogenesis. Proc Natl Acad Sci U S A. 2001;98:7319–24. doi: 10.1073/pnas.131568898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Seitz V, Butzhammer P, Hirsch B, Hecht J, Gutgemann I, Ehlers A, et al. Deep sequencing of MYC DNA-binding sites in Burkitt lymphoma. PloS one. 2011;6:e26837. doi: 10.1371/journal.pone.0026837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Croucher DR, Hochgrafe F, Zhang L, Liu L, Lyons RJ, Rickwood D, et al. Involvement of Lyn and the atypical kinase SgK269/PEAK1 in a basal breast cancer signaling pathway. Cancer research. 2013;73:1969–80. doi: 10.1158/0008-5472.CAN-12-1472. [DOI] [PubMed] [Google Scholar]
- 63.Hochgrafe F, Zhang L, O’Toole SA, Browne BC, Pinese M, Porta Cubas A, et al. Tyrosine phosphorylation profiling reveals the signaling network characteristics of Basal breast cancer cells. Cancer research. 2010;70:9391–401. doi: 10.1158/0008-5472.CAN-10-0911. [DOI] [PubMed] [Google Scholar]
- 64.Horiuchi D, Huskey NE, Kusdra L, Wohlbold L, Merrick KA, Zhang C, et al. Chemical-genetic analysis of cyclin dependent kinase 2 function reveals an important role in cellular transformation by multiple oncogenic pathways. Proc Natl Acad Sci U S A. 2012;109:E1019–27. doi: 10.1073/pnas.1111317109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Finn RS, Bengala C, Ibrahim N, Roche H, Sparano J, Strauss LC, et al. Dasatinib as a single agent in triple-negative breast cancer: results of an open-label phase 2 study. Clinical cancer research: an official journal of the American Association for Cancer Research. 2011;17:6905–13. doi: 10.1158/1078-0432.CCR-11-0288. [DOI] [PubMed] [Google Scholar]
- 66.Moulder S, Yan K, Huang F, Hess KR, Liedtke C, Lin F, et al. Development of candidate genomic markers to select breast cancer patients for dasatinib therapy. Molecular cancer therapeutics. 2010;9:1120–7. doi: 10.1158/1535-7163.MCT-09-1117. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.