

Abstract
The bile salt export pump (BSEP) plays an important role in bile acid excretion. Impaired BSEP function may result in liver injury. Bile acids also undergo basolateral efflux, but the relative contributions of biliary (CLBile) versus basolateral efflux (CLBL) clearance to hepatocellular bile acid excretion have not been determined. In the present study, taurocholic acid (TCA; a model bile acid) disposition was characterized in human and rat sandwich-cultured hepatocytes (SCH) combined with pharmacokinetic modeling. In human SCH, biliary excretion of TCA predominated (CLBile = 0.14 ± 0.04 ml/min per g liver; CLBL = 0.042 ± 0.019 ml/min per g liver), whereas CLBile and CLBL contributed approximately equally to TCA hepatocellular excretion in rat SCH (CLBile = 0.34 ± 0.07 ml/min per g liver; CLBL = 0.26 ± 0.07 ml/min per g liver). Troglitazone decreased TCA uptake, CLBile, and CLBL; membrane vesicle assays revealed for the first time that the major metabolite, troglitazone sulfate, was a noncompetitive inhibitor of multidrug resistance–associated protein 4, a basolateral bile acid efflux transporter. Simulations revealed that decreased CLBile led to a greater increase in hepatic TCA exposure in human than in rat SCH. A decrease in both excretory pathways (CLBile and CLBL) exponentially increased hepatic TCA in both species, suggesting that 1) drugs that inhibit both pathways may have a greater risk for hepatotoxicity, and 2) impaired function of an alternate excretory pathway may predispose patients to hepatotoxicity when drugs that inhibit one pathway are administered. Simulations confirmed the protective role of uptake inhibition, suggesting that a drug’s inhibitory effects on bile acid uptake also should be considered when evaluating hepatotoxic potential. Overall, the current study precisely characterized basolateral efflux of TCA, revealed species differences in hepatocellular TCA efflux pathways, and provided insights about altered hepatic bile acid exposure when multiple transport pathways are impaired.
Introduction
Bile acids are important endogenous molecules that are involved in the digestion and absorption of fats and regulation of lipid and glucose homeostasis (Hofmann, 1999a; Nguyen and Bouscarel, 2008). However, bile acids can exert toxic effects at supraphysiologic concentrations through disruption of mitochondrial ATP synthesis, necrosis, and apoptosis (Perez and Briz, 2009; Maillette de Buy Wenniger and Beuers, 2010); thus, defects in excretion may lead to hepatic accumulation of bile acids and subsequent hepatotoxicity.
Hepatic transport proteins play important roles in vectorial transport of bile acids. Sodium-taurocholate cotransporting polypeptide (NTCP) and organic anion transporting polypeptides are responsible for sodium-dependent and -independent uptake of bile acids from sinusoidal blood into hepatocytes, respectively. Bile acids in hepatocytes are excreted into bile across the canalicular membrane, predominantly via the bile salt export pump (BSEP). Consistent with the important role of BSEP in bile acid excretion, impaired BSEP function due to genetic polymorphisms has been shown to induce liver injury (e.g., progressive familial intrahepatic cholestasis type II) (Jansen et al., 1999). Also, increasing evidence suggests that inhibition of BSEP by drugs is associated with cholestatic/mixed-type drug-induced liver injury (DILI) (Morgan et al., 2010, 2013; Dawson et al., 2012; Pedersen et al., 2013).
In addition to BSEP-mediated canalicular excretion, bile acids also are transported from the hepatocyte into sinusoidal blood via basolateral efflux transporters, including multidrug resistance–associated protein (MRP) 3, MRP4, and organic solute transporter α (OSTα)–OSTβ. Expression levels of MRP3 and MRP4 are upregulated under cholestatic conditions, suggesting that they function as a compensatory route of bile acid excretion and thereby serve as an important part of adaptive response (Akita et al., 2001; Trauner et al., 2005; Zollner et al., 2007). OSTα–OSTβ mediates basolateral efflux of bile acids from enterocytes into the portal circulation by facilitated diffusion (Ballatori et al., 2009), but the role of OSTα–OSTβ in hepatic bile acid efflux remains to be further characterized. Recently, Morgan et al. (2013) reported that prediction of DILI was improved by considering the inhibitory effect of a drug on MRP2, MRP3, and MRP4, compared with BSEP inhibition alone. In addition, studies from our laboratory demonstrated that MRP4 inhibition was associated with cholestatic/mixed DILI among BSEP noninhibitors, emphasizing the role of MRP4 in DILI (Köck et al., 2014).
DILI is one of the primary reasons for withdrawal of approved drugs from the market and a major concern during drug development (Watkins and Seeff, 2006). One prominent example is troglitazone (TGZ), the first of the thiazolidinedione class of antidiabetic drugs that was withdrawn from worldwide markets due to severe DILI. Although mechanisms of TGZ-mediated hepatotoxicity remain unclear, in vitro vesicular transport assays demonstrated that TGZ and its major metabolite, TGZ sulfate (TS), are potent BSEP inhibitors, suggesting a cholestatic component in TGZ-induced hepatotoxicity (Funk et al., 2001). TGZ also inhibits NTCP, MRP3, and MRP4 (Marion et al., 2007; Morgan et al., 2013); although TS accumulates extensively in hepatocytes (Funk et al., 2001; Lee et al., 2010), the effect of TS on basolateral efflux transporters has not been investigated.
Due to extensive biliary excretion, it generally has been accepted that the contribution of basolateral efflux to hepatocellular bile acid excretion is minimal under normal conditions. However, as proposed in the “hepatocyte hopping” theory of bilirubin glucuronides (Iusuf et al., 2012), it is plausible that bile acids may undergo extensive basolateral efflux (through MRP3 and/or MRP4) and reuptake into downstream hepatocytes (through NTCP and/or organic anion-transporting polypeptide). This would prevent saturation of biliary transporters in upstream hepatocytes and transfer bile acids to downstream hepatocytes, protecting hepatocytes from bile acid toxicity. To our knowledge, the contribution of basolateral efflux versus biliary excretion to hepatocellular bile acid disposition has not been precisely characterized.
The purpose of the present studies was to characterize taurocholic acid (TCA) hepatobiliary disposition (basolateral uptake, basolateral efflux, biliary excretion, flux from canalicular networks) in human and rat sandwich-cultured hepatocytes (SCH) using a novel uptake and efflux protocol developed by our laboratory combined with pharmacokinetic modeling (Pfeifer et al., 2013). Results from the current investigation revealed that species differences exist in cellular TCA efflux pathways in human versus rat SCH; simulations suggested differential hepatobiliary TCA disposition in human and rat SCH due to inhibitors of canalicular excretion and/or basolateral efflux. This novel finding might explain, in part, the underlying mechanisms of species differences in hepatotoxicity mediated by BSEP inhibitors. This study also investigated the effects of TGZ and its metabolites on TCA disposition in human and rat SCH, and is the first to report that TS inhibits MRP4, a basolateral bile acid efflux transporter. Last, simulations based on the constructed mechanistic models provided insights regarding altered hepatic bile acid exposure when multiple bile acid transport pathways are impaired.
Materials and Methods
All chemicals were purchased from Sigma-Aldrich (St. Louis, MO) unless otherwise stated. TGZ [5-(4-[(6-hydroxy-2,5,7,8-tetramethylchroman-2-yl)methoxy]benzyl)thiazolidine-2,4-dione] was purchased from Cayman Chemical Company (Ann Arbor, MI). TS (5-[[4-[[3,4-dihydro-2,5,7,8-tetramethyl-6-(sulfooxy)-2H-1-benzopyran-2-yl]methoxy]phenyl]methyl]-2,4-thiazolidinedione) was kindly provided by Daiichi-Sankyo Co., Ltd. (Tokyo, Japan). TS was also synthesized from TGZ in-house as described by Saha et al. (2010). [3H]TCA (5 Ci/mmol) and [3H]dehydroepiandrosterone sulfate (DHEAS; 79.5 Ci/mmol) were purchased from PerkinElmer (Waltham, MA). Dimethylsulfoxide was obtained from Fisher Scientific (Fairlawn, NJ). GIBCO brand fetal bovine serum, recombinant human insulin, and Dulbecco’s modified Eagle’s medium (DMEM) were purchased from Life Technologies (Carlsbad, CA). Insulin/transferrin/selenium culture supplement, BioCoat culture plates, and Matrigel extracellular matrix were purchased from BD Biosciences Discovery Labware (Bedford, MA).
Sandwich-Cultured Hepatocytes.
Rat hepatocytes were isolated from male Wistar rats (234–245 g; Charles River Laboratories, Inc., Wilmington, MA) using a two-step collagenase perfusion method as previously described (LeCluyse et al., 1996). Animals had free access to water and food before surgery and were allowed to acclimate for at least 5 days. All animal procedures complied with the guidelines of the Institutional Animal Care and Use Committee (University of North Carolina, Chapel Hill, NC). Rat hepatocytes were seeded onto six-well BioCoat culture plates at a density of 1.75 × 106 cells/well in seeding medium (DMEM containing 5% fetal bovine serum, 10 µM insulin, 1 µM dexamethasone, 2 mM l-glutamine, 1% minimum essential medium nonessential amino acids, 100 units/ml of penicillin G sodium, and 100 µg/ml of streptomycin) as described previously (Swift et al., 2010). Hepatocytes were incubated for 2 hours at 37°C in a humidified incubator (95% O2, 5% CO2) and allowed to attach to the collagen substratum, after which time the medium was aspirated to remove unattached cells and replaced with fresh medium. The next day, cells were overlaid with BD Matrigel at a concentration of 0.25 mg/ml in ice-cold feeding medium (DMEM supplemented with 0.1 µM dexamethasone, 2 mM l-glutamine, 1% minimum essential medium nonessential amino acids, 100 units/ml of penicillin G sodium, 100 µg/ml of streptomycin, and 1% insulin/transferrin/selenium). The culture medium was changed daily until experiments were performed on day 4. Fresh human SCH, seeded onto 24-well BioCoat culture plates and overlaid with Matrigel, were purchased from Triangle Research Laboratories (Research Triangle Park, NC). Fresh human hepatocytes were obtained from two Caucasian females [31 years old, body mass index (BMI) 29.1 kg/m2; 56 years old, BMI 22.3 kg/m2) and one African American female (48 years old, BMI 24.9 kg/m2). The culture medium (the same feeding medium used for rat SCH) was changed daily until experiments were performed on day 7.
Uptake and Efflux Studies in SCH.
Uptake and efflux studies of TCA were performed in human and rat SCH as previously described (Pfeifer et al., 2013). In brief, on day 4 (rat) or day 7 (human) of culture, SCH were preincubated for 10 minutes in 1.5 ml/well (rat) or 0.3 ml/well (human) standard (Ca2+-containing) or Ca2+-free (Ca2+/Mg2+-free buffer containing EGTA) Hanks’ balanced salt solution (HBSS). Incubating SCH in Ca2+-free HBSS disrupts the tight junctions that form the bile canalicular networks (B-CLEAR technology; Qualyst Transporter Solutions, Research Triangle Park, NC). For uptake and efflux studies with TCA, SCH were treated with 1 μM [3H]TCA (400 nCi/ml) in 1.5 ml/well (rat) or 0.3 ml/well (human) standard HBSS for 20 minutes at 37°C. After the 20-minute uptake phase, buffers containing TCA were removed, cells were washed twice with 1.5 ml/well (rat) or 0.3 ml/well (human) standard or Ca2+-free HBSS buffer at 37°C, and the third application of buffer was added to SCH for the 15-minute (rat) or 10-minute (human) efflux phase (Fig. 1). For determination of TGZ effects on TCA disposition, SCH were preincubated with 10 µM TGZ for 30 minutes in 1.5 ml/well (rat) or 0.3 ml/well (human) standard HBSS before 10-minute application of standard or Ca2+-free HBSS. The rest of the experiment (uptake and efflux) was performed as described earlier. Preincubation was selected to minimize the inhibitory effects of TGZ and its metabolites on TCA uptake, and to allow enough time for the formation of TS, a potent BSEP inhibitor. TCA accumulation in cells + bile (standard HBSS) and cells (Ca2+-free HBSS) during uptake (2, 5, 10, and 20 minutes in human SCH; 2, 5, 10, 15, and 20 minutes in rat SCH) and efflux (2, 3.5, 5, and 10 minutes in human SCH; 2, 3.5, 5, 10, and 15 minutes in rat SCH) phases was determined by terminal sampling of n = 3 wells at each time point. During the efflux phase, incubation buffer (standard HBSS or Ca2+-free HBSS) also was collected at the end of each incubation period. Cells were washed twice in ice-cold HBSS, and were solubilized in 0.3 ml (24-well; human SCH) or 1 ml (six-well; rat SCH) 0.5% Triton X-100. Radioactivity in cell lysates and buffer samples was quantified by liquid scintillation counting (Packard TriCarb; PerkinElmer).
Fig. 1.

Schemes depicting the uptake and efflux protocol and the mechanistic model of [3H]TCA disposition in SCH. (A) Uptake and efflux studies were conducted in the presence of standard (+Ca2+) Hanks’ balanced salt solution (Std HBSS). Tight junctions remained sealed throughout the study period. (B) Tight junctions remained open throughout the study period by preincubating with Ca2+-free HBSS, then performing an uptake phase in standard HBSS to provide relief from the removal of Ca2+, followed by a brief wash and efflux in Ca2+-free HBSS. In the uptake and efflux protocols, the dashed box represents preincubation with 10 µM TGZ in standard HBSS for TGZ-treated groups. Gray shading represents inclusion of the substrate, 1 µM [3H]TCA, in standard HBSS during the uptake phase. Black shading represents 1-minute wash followed by 10- (human SCH) or 15-minute (rat SCH) efflux phase in standard or Ca2+-free HBSS. In the model schemes, X, V, and C denote the mass of TCA, compartmental volume, and TCA concentration, respectively. Subscripts on mass, volume, and concentration terms denote the corresponding compartment in the model scheme. Superscripts represent the presence (+, intact tight junctions; cells + bile) and absence (−, modulated tight junctions; cells) of Ca2+ in the preincubation and efflux buffer. CLuptake, CLBL, and CLBile represent clearance values for uptake from buffer into hepatocytes, efflux from hepatocytes into buffer, and canalicular excretion from hepatocytes, respectively. KFlux represents the first-order rate constant for flux from bile networks into buffer.
Pharmacokinetic Modeling.
Pharmacokinetic modeling was used to evaluate the hepatobiliary disposition of TCA (control), and to determine the effects of TGZ on TCA disposition (+TGZ) in human and rat SCH. A model scheme incorporating linear parameters governing TCA disposition (Fig. 1) was fit to mass versus time data from individual SCH experiments (Fig. 2). The model fitting was performed with Phoenix WinNonlin, version 6.1 (Certara, St. Louis, MO) using the stiff estimation method and a power model to account for residual error. The following differential equations, which were developed based on the model scheme depicted in Fig. 1, were fit simultaneously to data generated in SCH in the presence of intact and disrupted bile canaliculi for each condition (human and rat; control and +TGZ):
Fig. 2.

[3H]TCA mass versus time data in rat and human SCH in the absence (control) or presence of 10 µM TGZ preincubation. Closed symbols/solid lines represent [3H]TCA in cells + bile (top) or standard HBSS (bottom), and open symbols/dashed lines represent [3H]TCA in cells (top) or Ca2+-free HBSS (bottom). The simulated mass-time profiles were generated from the relevant equations based on the model scheme depicted in Fig. 1, and the final parameter estimates are reported in Table 1. Data (picomoles per milligram protein) represent the mean ± S.E.M. (n = 3 SCH preparations in triplicate per group).
Mass in standard HBSS buffer:
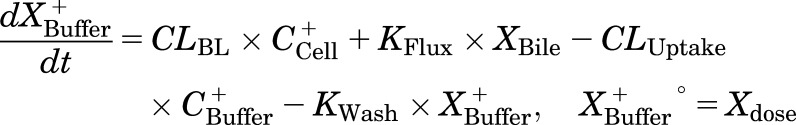 |
Mass in Ca2+-free HBSS buffer:
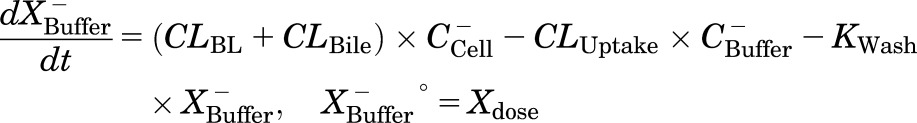 |
Mass in cells:
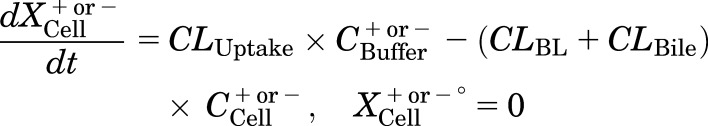 |
Mass in bile (standard HBSS):
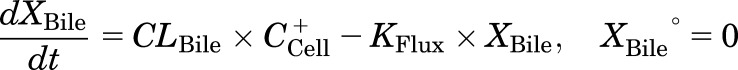 |
Mass in cells+bile (standard HBSS):
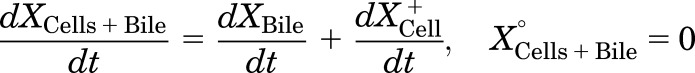 |
where CLBile is the biliary clearance, CLBL is the basolateral efflux clearance, CLUptake is the uptake clearance, and variables and parameters are defined as in Fig. 1, and Kwash was activated for 1 minute at the end of the 20-minute uptake phase and fixed at 1 × 104 min−1 based on simulations to eliminate the TCA dose from the buffer compartment and represent the wash step. CCell represents the intracellular concentration, calculated as XCell/VCell, where cellular volume (VCell) was estimated based on the protein content of each preparation, using a value of 7.4 μl/mg protein (Lee and Brouwer, 2010). CBuffer represents the buffer concentration, calculated as XBuffer/VBuffer, where the buffer volume (VBuffer) was constant (1.5 ml for rat SCH and 0.3 ml for human SCH). Initial parameter estimates were obtained from noncompartmental analysis of SCH data, where CLUptake was estimated from the initial (2 minutes) uptake data as follows: CLUptake = (dXcells+bile/dt)/CBuffer. CLBL and CLBile were estimated from efflux phase data under Ca2+-free conditions, where (CLBL + CLBile) = X−Buffer,0-15min/AUCcells,0-15min (area under the cellular TCA concentration versus time curve from 0 to 15 minutes, obtained using the linear trapezoidal rule). KFlux, which represents the flux of substrate out of bile networks in standard HBSS conditions, was estimated initially from simulations using Berkeley-Madonna. The impact of impaired function of canalicular and/or basolateral efflux transporters on hepatic TCA exposure in human and rat SCH was simulated using the TCA model and parameter estimates (Fig. 1; Table 1); parameters representing transport-mediated efflux (CLBL and CLBile) were decreased by 10-fold in isolation, or in combination, in human and rat SCH; the resulting changes in predicted cellular TCA concentrations are plotted in Fig. 4. To determine the net effect of impaired function of uptake and/or efflux (basolateral and canalicular) transporters on hepatic TCA exposure in human SCH, simulations were performed by decreasing CLUptake and CLEfflux (sum of biliary and basolateral efflux clearances; CLBL + CLBile) gradually by 10- to 100-fold in combination; it was assumed that both efflux pathways (CLBL and CLBile) were impaired to the same extent. Simulated cellular TCA concentrations are presented in Fig. 5. All simulations were performed using Berkeley-Madonna version 8.3.11.
TABLE 1.
Summary of recovered parameter estimates based on the model scheme depicted in Fig. 1 describing TCA disposition in human and rat SCH without (control) or with 10 µM troglitazone (+TGZ) preincubation
Human and rat SCH were treated with 1 µM [3H]TCA (see Fig. 1 for details of incubation conditions), and the model was fit simultaneously to all data from each preparation. Data are presented as the mean ± S.D. of individual fits from n = 3 SCH preparations.
| Conditions |
CLUptake |
CLBile |
CLBL |
KFlux |
|---|---|---|---|---|
| ml/min per g liver | min−1 | |||
| Human SCH | ||||
| Control | 2.2 ± 0.4 | 0.14 ± 0.04 | 0.042 ± 0.019 | 0.043 ± 0.015 |
| +TGZ | 0.23 ± 0.04* | 0.084 ± 0.069 | 0.022 ± 0.018 | 0.070 ± 0.036 |
| Rat SCH | ||||
| Control | 1.2 ± 0.5 | 0.34 ± 0.07 | 0.26 ± 0.07 | 0.053 ± 0.015 |
| +TGZ | 0.20 ± 0.04 | 0.18 ± 0.05* | 0.22 ± 0.06 | 0.077 ± 0.038 |
Significantly different from control (P < 0.05).
Fig. 4.

Simulations depicting the impact of impaired function of canalicular and/or basolateral efflux transporters on hepatic TCA exposure. Cellular TCA concentrations in human and rat SCH were simulated based on the TCA model scheme depicted in Fig. 1 and parameter estimates (Table 1) for human SCH (A), rat SCH (B), and rat SCH where CLUptake was 5× the value listed in Table 1 (C). Parameters representing transport-mediated efflux (CLBL and CLBile) were decreased by 10-fold in isolation, or in combination, to represent impaired function of canalicular efflux transporters (solid line with open circle), basolateral efflux transporters (dashed line), and both pathways (dashed line with closed circle). Simulations were performed for 200 minutes to obtain steady-state intracellular concentrations; the time to reach steady state was longer when efflux pathways were impaired compared with control (solid line).
Fig. 5.

Net effects of inhibition of uptake and efflux transporters on hepatic TCA exposure. Cellular TCA concentrations in human SCH were simulated as a function of decreased (10- to 100-fold) CLUptake and CLEfflux (defined as CLBL + CLBile); both efflux pathways (CLBL and CLBile) were assumed to be impaired to the same extent. The z-axis represents the fold change in steady-state hepatic TCA concentrations: 10- to 100-fold (red), 5- to 10-fold (orange), 1- to 5-fold (light green), 0.1- to 1-fold (dark green), and 0.001- to 0.1-fold (blue).
Membrane Vesicles.
Human MRP4 plasmid [pcDNA3.1(−)-MRP4] was provided by Dr. Dietrich Keppler (German Cancer Research Center, Heidelberg, Germany). Human embryonic kidney 293T cell lines stably transfected with pcDNA3.1(−)-MRP4 or an empty plasmid vector (control) were established as previously described (Köck et al., 2014). Membrane vesicles were prepared from these cell lines, and transport experiments were carried out by a rapid filtration assay as described previously (Ghibellini et al., 2008). In brief, membrane vesicles (5 μg of protein) were incubated at 37°C in Tris-sucrose buffer (TSB; 50 mM Tris-HCl/250 mM sucrose) containing 10 mM MgCl2, 10 mM creatine phosphate, 100 μg/ml creatine kinase, 4 mM ATP or AMP, and [3H]DHEAS (0.7 μCi/ml) in the absence and presence of TS, in a volume of 50 μl. After incubation for 2 minutes, the reaction was stopped by the addition of 0.8 ml of ice-cold TSB and immediately applied to a glass fiber filter (type A/E; Pall Corp., Port Washington, NY) and washed twice with 2 ml of ice-cold TSB. Filters were mixed by vortexing in 5 ml of scintillation fluid, and radioactivity was quantified by liquid scintillation counting (Packard TriCarb). The ATP-dependent uptake of substrate was calculated by subtracting substrate uptake in the presence of AMP from substrate uptake in the presence of ATP. The MRP4-dependent uptake of substrate was calculated by subtracting ATP-dependent uptake in MRP4-overexpressing vesicles from that in control vesicles. Initially, the inhibitory effect of TS (10 μM) on MRP4-dependent transport of [3H]DHEAS (2 μM) was evaluated in the presence or absence of 3 mM glutathione (GSH). Further studies were performed using concentration ranges of [3H]DHEAS (0.5–20 μM) and TS (5–50 μM) in the absence of GSH to determine the inhibition constant (Ki). Initial estimates of Ki values and the type of inhibition were derived from Dixon plots of TS concentrations versus 1/velocity data. Then the kinetic parameters (Km, Vmax, and Ki) and type of inhibition were determined by fitting competitive, noncompetitive, and uncompetitive models to the untransformed data by nonlinear regression analysis using Phoenix WinNonlin, version 6.1. Equations used for each inhibition model are as follows:
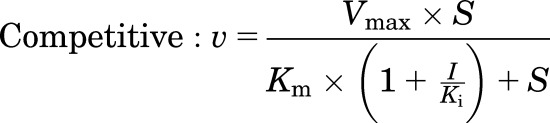 |
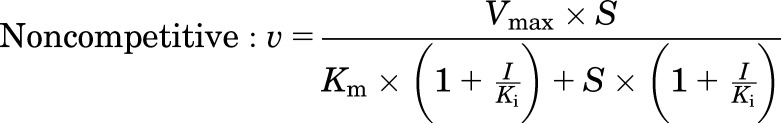 |
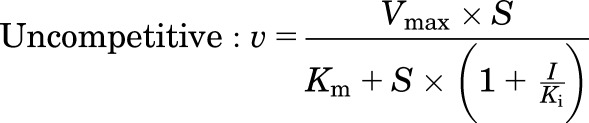 |
where S represents the concentration of [3H]DHEAS, I represents the concentration of TS, and V denotes the rate of [3H]DHEAS transport. The best-fit model was assessed from visual inspection of the observed versus predicted data and Akaike Information Criterion (AIC). Representative data from n = 2 independent experiments in triplicate are presented in Fig. 3.
Fig. 3.

Inhibition of MRP4-mediated transport of [3H]DHEAS by TS in membrane vesicles from MRP4-overexpressing and control human embryonic kidney 293T cells. (A) Effect of GSH (3 mM) on MRP4-mediated transport of 2 μM [3H]DHEAS and inhibition by 10 μM TS. (B) Effect of increasing concentrations of TS (0, 5, 10, and 50 μM) on MRP4-mediated [3H]DHEAS (2 minutes, 0.5–20 μM) transport in the absence of GSH. Each point represents the mean ± S.D. A noncompetitive inhibition model best described the data. Lines represent model fits based on the noncompetitive inhibition model. Representative plots from n = 2 independent studies are presented. The estimated Ki values based on a noncompetitive inhibition model were 8.0 ± 0.7 and 8.5 ± 1.2 µM from each study.
Data Analysis.
TCA accumulation was corrected for nonspecific binding to the BioCoat plate without cells, and normalized to protein concentration measured by the BCA protein assay (Pierce Chemical, Rockford, IL). The intracellular concentration of TCA was obtained by dividing TCA accumulation (picomoles per milligram protein) by the previously reported hepatocyte volume (7.4 µl/mg protein) (Lee and Brouwer, 2010). Apparent (CLBile,app) and intrinsic (CLBile,int) biliary clearance values were calculated using B-CLEAR technology (Qualyst Transporter Solutions) based on the following equations:
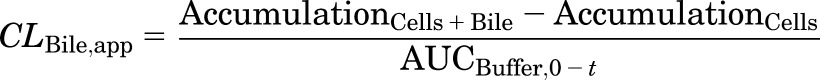 |
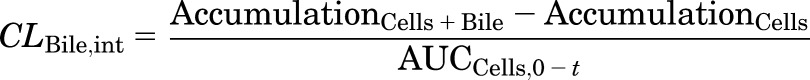 |
where 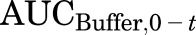 is the area under the TCA buffer concentration versus time curve, which is the product of the initial TCA buffer concentration (1 µM) and the incubation time (t), assuming that sink conditions of TCA in the buffer are maintained (concentration changes <10% during the uptake phase).
is the area under the TCA buffer concentration versus time curve, which is the product of the initial TCA buffer concentration (1 µM) and the incubation time (t), assuming that sink conditions of TCA in the buffer are maintained (concentration changes <10% during the uptake phase). 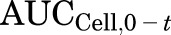 is the area under the TCA cellular concentration versus time curve, which was obtained using the linear trapezoidal rule. Clearance units (microliters per minute per milligram protein) were converted to milliliters per minute per gram liver based on the protein content in liver tissue (90 and 112 mg protein/g liver for human and rat, respectively) (Sohlenius-Sternbeck, 2006). The paired Student’s t test was used to compare parameters in the presence or absence of TGZ preincubation. In all cases, P < 0.05 was considered statistically significant. All statistical analyses were performed using SigmaStat 3.5 (Systat Software, San Jose, CA).
is the area under the TCA cellular concentration versus time curve, which was obtained using the linear trapezoidal rule. Clearance units (microliters per minute per milligram protein) were converted to milliliters per minute per gram liver based on the protein content in liver tissue (90 and 112 mg protein/g liver for human and rat, respectively) (Sohlenius-Sternbeck, 2006). The paired Student’s t test was used to compare parameters in the presence or absence of TGZ preincubation. In all cases, P < 0.05 was considered statistically significant. All statistical analyses were performed using SigmaStat 3.5 (Systat Software, San Jose, CA).
Results
TCA Disposition in Human and Rat SCH with and without TGZ Preincubation.
TCA uptake and efflux studies were conducted as described in Fig. 1. The mass-time profiles of TCA in cells + bile and cells (during the uptake and efflux phases) and buffer (during the efflux phase) in human and rat SCH in the absence (control) and presence (+TGZ) of TGZ preincubation are presented in Fig. 2. Under all conditions, TCA accumulation in cells + bile and cells increased during the uptake phase and decreased during the efflux phase. Appearance of TCA in the standard and Ca2+-free HBSS buffer increased during the efflux phase. TGZ preincubation decreased TCA accumulation in cells + bile, cells, and the efflux into buffers in both human and rat SCH. CLBile,app of TCA during the uptake phase was also significantly decreased after preincubation with TGZ compared with the control group, indicating that TGZ decreased uptake and/or biliary excretion of TCA; in human SCH, CLBile,app values after 10-minute uptake (standard B-CLEAR method) in control and +TGZ groups were 1.1 ± 0.3 and 0.10 ± 0.02 ml/min per g liver, respectively (P = 0.035). The corresponding values in rat SCH were 0.48 ± 0.08 and 0.07 ± 0.01 ml/min per g liver, respectively (P = 0.008). TCA CLBile,int also was significantly decreased after TGZ preincubation, suggesting that TGZ decreased TCA biliary excretion; in human SCH, CLBile,int values after 10-minute uptake in control and +TGZ groups were 0.31 ± 0.07 and 0.15 ± 0.09 ml/min per g liver, respectively (P = 0.004). The corresponding values in rat SCH were 0.62 ± 0.11 and 0.22 ± 0.08 ml/min per g liver, respectively (P = 0.049).
Parameter estimates recovered from fitting differential equations (see Materials and Methods) based on the model scheme in Fig. 1 to TCA accumulation data from independent SCH preparations are presented in Table 1. In the absence of TGZ preincubation (control), human SCH showed greater CLUptake, slightly lower CLBile, and notably lower CLBL relative to rat SCH. This is consistent with greater cellular accumulation of TCA observed in human SCH (Fig. 2). Interestingly, CLBile was about 3.3-fold greater than CLBL in human SCH, whereas CLBile and CLBL showed a similar contribution to the total cellular efflux of TCA in rat SCH in the absence of TGZ. In human SCH, TGZ preincubation significantly decreased CLUptake (P = 0.017); there were trends toward decreased CLBL and CLBile after TGZ preincubation compared with the control groups. In rat SCH, CLBile was significantly decreased after TGZ preincubation (P = 0.017); there were trends toward decreased CLUptake and CLBL after TGZ preincubation compared with the control groups. However, these differences failed to reach statistical significance due to large variability in mean differences.
Inhibitory Effects of TS on MRP4-Mediated [3H]DHEAS Transport in Membrane Vesicles.
The inhibitory effects of TS on MRP4, a basolateral bile acid efflux transporter, were evaluated using membrane vesicles prepared from human embryonic kidney 293T cells overexpressing MPR4 or control cells. TS (10 μM) inhibited MRP4-mediated transport of [3H]DHEAS (2 µM) by 78 and 72% in the absence and presence of GSH, respectively (Fig. 3A). Inhibition of MRP4-mediated [3H]DHEAS transport by TS was determined in two independent membrane vesicle studies over a range of substrate concentrations (DHEAS, 0.5–20 µM) and inhibitor concentrations (TS, 5–50 µM). In both studies, the noncompetitive inhibition model best described the inhibition data visually and generated the lowest AIC value; AIC values for competitive, noncompetitive, and uncompetitive inhibition models were 632.4, 628.9, and 664.6, respectively, in the first study, and were 740.0, 738.3, and 786.0, respectively, in the second study. Ki values based on a noncompetitive inhibition model were 8.0 ± 0.70 and 8.5 ± 1.2 µM in the first and the second studies, respectively. A representative fit of the noncompetitive inhibition model to the data is presented in Fig. 3B.
Impact of Impaired Function of Canalicular versus Basolateral Efflux Transporters on Hepatic TCA Exposure.
The altered hepatobiliary disposition of TCA due to impaired function of bile acid efflux transporters was simulated based on the TCA model described in this report (Fig. 1; Table 1). Simulated hepatic TCA concentrations, up to and including steady state (CH,ss) in human and rat SCH, are shown in Fig. 4, A and B, respectively. CLUptake of TCA in rat SCH might have been underestimated compared with rats in vivo because it has been reported that TCA uptake clearance was decreased by 5-fold in rat SCH on day 4 compared with day 0 due to decreased Ntcp protein expression, whereas TCA uptake clearance remained unchanged over time in human SCH (Liu et al., 1998; Kotani et al., 2011; Tchaparian et al., 2011). To account for decreased Ntcp function over days of culture, simulations were also performed with a 5-fold higher CLUptake in rat SCH (Fig. 4C). TCA CH,ss was higher in human SCH (11.1 µM) compared with rat SCH (1.9 µM) (Fig. 4, A and B); this is consistent with the observed higher cellular TCA accumulation during uptake and efflux studies compared with rat SCH (Fig. 2). However, TCA CH,ss in rat SCH with 5-fold greater CLUptake (9.1 µM) is comparable to that in human SCH (Fig. 4, A and C). Simulations revealed that in human SCH, a 10-fold decrease in CLBile increased TCA CH,ss by 2.9-fold compared with control, whereas a 1.3-fold increase in TCA CH,ss occurred relative to control when CLBL was decreased by 10-fold (Fig. 4A). Interestingly, a 10-fold decrease in both CLBile and CLBL increased TCA CH,ss by 7.0-fold compared with control, which is a greater than proportional increase compared with inhibiting either pathway in isolation. Simulations in rat SCH revealed that TCA CH,ss was increased by 2.0- and 1.6-fold when CLBile and CLBL were decreased by 10-fold, respectively, relative to control (Fig. 4B). TCA CH,ss increased by 9.3-fold relative to control when both CLBile and CLBL were decreased by 10-fold; similar to human SCH simulations, the increase in CH,ss is greater than proportional compared with conditions in which either pathway is impaired in isolation. The same trends were observed in rat SCH when 5-fold greater CLUptake was simulated (Fig. 4C); the fold increase in TCA CH,ss was 1.9, 1.6, and 7.3 relative to control when CLBile, CLBL, and both CLBile and CLBL were decreased 10-fold, respectively.
Impact of Impaired Function of Uptake versus Efflux Transporters on Hepatic TCA Exposure in Human SCH.
Hepatic bile acid concentrations are determined by both hepatic uptake and efflux processes; drugs that inhibit efflux transporters often also inhibit uptake transporters. To understand the net effects of impaired function of uptake and efflux transporters on hepatic TCA exposure, TCA CH,ss in human SCH was simulated based on various values of CLUptake and CLEfflux (defined as CLBile + CLBL) (Fig. 5). When CLUptake remained unchanged (for example, fractional inhibition of CLUptake = 0), TCA CH,ss increased exponentially as the fractional inhibition of CLEfflux increased. On the other hand, CH,ss decreased proportionally as the fractional inhibition of CLUptake increased, when CLEfflux remained unchanged (for example, fractional inhibition of CLEfflux = 0). When the fractional inhibition of CLUptake and CLEfflux was the same, TCA CH,ss remained unchanged (fold change = 1). If the fractional inhibition of CLUptake was greater than the fractional inhibition of CLEfflux, then TCA CH,ss was decreased (fold change < 1). If the fractional inhibition of CLUptake was less than the fractional inhibition of CLEfflux, then the fold change in TCA CH,ss was greater than 1; TCA CH,ss increased exponentially with increasing fractional inhibition of CLEfflux, but the fold change in TCA CH,ss decreased with increasing fractional inhibition of CLUptake. Notably, a greater than 10-fold increase in TCA CH,ss was observed only when the fractional inhibition of CLUptake was less than 0.6.
Discussion
The present study determined the hepatobiliary disposition of TCA in human and rat SCH using a novel uptake and efflux protocol recently developed in our laboratory combined with pharmacokinetic modeling (Pfeifer et al., 2013). The results demonstrated that species differences exist in the hepatocellular excretion of TCA; in human SCH, biliary excretion predominated, whereas biliary excretion and basolateral efflux contributed approximately equally to hepatocellular TCA excretion in rat SCH (Table 1). Jemnitz et al. (2010) reported that basolateral and biliary excretion contribute equally to TCA efflux in human SCH, whereas basolateral efflux was the dominant cellular efflux pathway of TCA in rat SCH. The likely reason for these discrepancies is that these investigators did not account for the TCA “flux” from the canalicular spaces into the buffer, which results from regular “pulsing” of the bile canaliculi in SCH (KFlux in Fig. 1) (Pfeifer et al., 2014). Regular, ordered contraction of bile canaliculi has been reported previously in isolated couplets and cultured hepatocytes (Oshio and Phillips, 1981; Phillips et al., 1982), and has been shown to facilitate bile flow in vivo in rat liver (Watanabe et al., 1991). In the study by Jemnitz et al. (2010), basolateral efflux was evaluated by measuring TCA in standard buffer during the efflux phase. However, the amount of TCA that appeared in the buffer during the efflux phase was actually the sum of basolateral efflux and flux from the bile canalicular spaces, which led to an overestimation of basolateral efflux. To circumvent these issues and accurately estimate the relative contributions of CLBL, CLBile, and KFlux, pharmacokinetic modeling was used in the current study.
Troglitazone, a known hepatotoxic compound, was selected in the current study because disposition of TGZ and its derived metabolites was well characterized in human and rat SCH (Lee et al., 2010). After preincubation with TGZ, CLBile was significantly decreased (rat) or tended to decrease (human) compared with control (Table 1), consistent with reported inhibitory effects of TGZ and TS on BSEP (Funk et al., 2001; Dawson et al., 2012). Interestingly, CLBL tended to decrease after TGZ preincubation compared with control, suggesting that TGZ and/or TS also might inhibit basolateral efflux of TCA. TGZ has been reported to inhibit the basolateral efflux transporters MRP3 and MRP4 (Morgan et al., 2013), but hepatic TGZ concentrations are minimal, whereas TS accumulates in hepatocytes due to extensive hepatic metabolism of TGZ (Funk et al., 2001; Lee et al., 2010). Thus, inhibitory effects of TS on MRP4-mediated transport were investigated. MRP4 was selected because TCA is transported by human MRP4, but not by human MRP3 (Akita et al., 2002; Rius et al., 2006). Since GSH is cotransported with bile acids by MRP4 (Rius et al., 2006), the inhibitory effect of TS at 10 µM was tested initially in the absence and presence of GSH. TS inhibited MRP4-mediated transport of [3H]DHEAS to a similar extent regardless of GSH, suggesting that the inhibitory effects of TS on MRP4 are independent of GSH (Fig. 3A). Further studies were performed in the absence of GSH, and revealed that TS inhibited MRP4-mediated [3H]DHEAS transport by noncompetitive inhibition, with a Ki value of 8 µM (Fig. 3B).
In addition to inhibition of efflux, CLUptake of TCA was significantly decreased (human) or showed trends toward a decrease (rat) compared with control after TGZ preincubation (Table 1). Although TGZ is a potent inhibitor of NTCP/Ntcp-mediated bile acid uptake (Marion et al., 2007), TGZ concentrations in the buffer were minimal during the uptake phase because TGZ-containing buffer was removed and replaced with TGZ-free buffer during the 10-minute preincubation (standard or Ca2+-free buffers) as well as the 20-minute uptake phase. These data suggest that TGZ might inhibit NTCP/Ntcp by mechanisms other than direct inhibition; further studies are needed to characterize the precise mechanism(s) of inhibition.
Preclinical animals often are less sensitive to bile acid–mediated DILI compared with humans, and thus, do not reliably predict human hepatotoxicity. Potential reasons include species differences in toxic bile acid composition, substrate and/or inhibitor specificity of bile acid transporters, and metabolism/detoxification pathways of drugs as well as bile acids (Setchell et al., 1997; Hofmann, 2004; Leslie et al., 2007; Chiang, 2009). In addition, differential inhibition of hepatocellular excretion pathways, as demonstrated in the current study, may contribute to species differences in bile acid–mediated hepatotoxicity. Simulations revealed that impaired function of canalicular and/or basolateral efflux transporters led to differential hepatobiliary disposition of TCA in human and rat SCH. In human SCH, hepatic TCA concentrations, which are relevant to hepatotoxicity, were increased 2.9-fold relative to control when canalicular transporter function was impaired, whereas impaired function of basolateral efflux transporters minimally increased hepatic TCA concentration (1.3-fold) (Fig. 4A). This was expected due to the predominant role of biliary excretion and the minor contribution of basolateral efflux to the overall hepatocellular excretion of TCA in human SCH. Interestingly, impaired function of both canalicular and basolateral efflux transporters further increased hepatic TCA concentrations by 7-fold compared with control (Fig. 4A), suggesting that basolateral efflux, despite serving as a minor route of hepatic excretion under normal conditions, plays an important role as a compensatory efflux pathway when canalicular excretion is impaired in human hepatocytes.
Expression and/or function of Ntcp has been reported to decrease over days of culture in rat SCH, whereas NTCP expression remains constant in human SCH; in rat SCH, TCA uptake clearance was decreased 5-fold on day 4 compared with day 0 (Liu et al., 1998; Kotani et al., 2011; Tchaparian et al., 2011). Thus, the CLUptake of TCA is likely underestimated in rat SCH, but not in human SCH. To account for the decreased function of Ntcp in day 4 rat SCH, simulations were performed in rat SCH with an CLUptake estimate obtained in day 4 rat SCH (1× CLUptake) as well as a 5-fold greater CLUptake (5× CLUptake). Although robust functional or quantitative proteomics data for BSEP, MRP3, and MRP4 in SCH over time do not exist, available data suggest that Bsep protein expression in rat SCH and MRP3/Mrp3 and MRP4/Mrp4 in rat and human SCH remain relatively unchanged over days of culture under our culture conditions (Swift et al., 2010; Tchaparian et al., 2011).
In both human and rat SCH, an exponential increase in hepatic TCA concentrations was only observed when the function of both efflux pathways was decreased (Fig. 5). These results are consistent with the mathematical relationship that governs fold change in cellular exposure: 1/(1 − fe), where fe is the total fraction excreted by all pathways (biliary or basolateral) (Zamek-Gliszczynski et al., 2009). Zamek-Gliszczynski et al. demonstrated that, if multiple excretion pathways exist, minor changes in exposure (<2-fold) are expected when a transport pathway that contributes to no more than 50% of total excretion is impaired, as noted when biliary excretion (rat) or basolateral efflux (human and rat) pathways alone are decreased in the current study. However, hepatic exposure increases exponentially in response to loss of function of transport pathways that contribute to >50% of total excretion, as noted in the current study when both biliary excretion and basolateral efflux transporters are impaired.
Bile acids undergo efficient enterohepatic recirculation; only ∼5% of the bile acid pool is synthesized in hepatocytes, whereas the remaining 95% is reabsorbed from the intestinal lumen after biliary excretion and taken up into hepatocytes (Hofmann, 1999b). Therefore, in addition to canalicular and basolateral efflux transporters, hepatic bile acid exposure is also regulated by hepatic uptake transporters. Inhibition of bile acid efflux transporters by drugs is reported to be associated with cholestatic/mixed-type DILI, but often, these drugs also inhibit uptake transporters, which may exert protective effects (Leslie et al., 2007); the net effect will be determined by the relative extent (potency) of uptake inhibition versus efflux inhibition. As might be expected, simulations suggest that hepatic TCA exposure increases only when the extent of efflux inhibition exceeds that of uptake inhibition (Fig. 5). Notably, fractional inhibition of CLUptake > 0.6 prevents hepatic TCA exposure from increasing by more than 10-fold, thereby confirming the protective effects of uptake inhibition. Simulations in the current study were performed using a constant fractional inhibition of uptake and efflux transporters throughout the simulation, assuming steady-state drug (inhibitor) concentrations in the medium and in the cell. In reality, drug concentrations change over time. Thus, dynamic changes in inhibitor concentrations should be considered by incorporating drug disposition into the model to more accurately predict altered bile acid disposition by drugs.
In the current study, species differences in hepatic excretion of TCA in human and rat SCH were identified. In human SCH, biliary excretion predominated, whereas biliary excretion and basolateral efflux contributed approximately equally to TCA efflux in rat SCH. As a result, the hepatic accumulation of TCA in rat SCH due to inhibition of BSEP alone might not be as extensive as that observed in human SCH. In human and rat SCH, inhibition of both excretion pathways led to exponential increases in hepatic TCA exposure, suggesting that inhibition of both excretion pathways might increase DILI liability. Alternatively, administration of a drug that inhibits one excretion pathway may predispose individuals with impaired transport function (due to disease or genetic polymorphisms) in the alternate pathway to hepatic bile acid accumulation and subsequent DILI. Simulations confirmed that uptake inhibition plays a protective role by helping minimize hepatic bile acid accumulation. This work emphasizes that the inhibitory effects of a drug on bile acid transporters mediating uptake as well as multiple efflux pathways should be considered when evaluating the hepatotoxic potential of drugs.
Acknowledgments
Phoenix WinNonlin software was generously provided to the Division of Pharmacotherapy and Experimental Therapeutics, UNC Eshelman School of Pharmacy, by Certara as a member of the Pharsight Academic Center of Excellence Program. The authors thank Daiichi-Sankyo Co., Ltd. for providing TS. The authors also thank Dr. Goto (UNC Eshelman School of Pharmacy) for synthesis of TS, and Dr. Keppler (German Cancer Research Center, Heidelberg, Germany) for providing the MRP4 expression vector [pcDNA3.1(+)-MRP4].
Abbreviations
- AIC
Akaike Information Criterion
- BSEP
bile salt export pump
- BMI
body mass index
- DHEAS
dehydroepiandrosterone sulfate
- DILI
drug-induced liver injury
- DMEM
Dulbecco’s modified Eagle’s medium
- GSH
glutathione
- HBSS
Hanks’ balanced salt solution
- MRP
multidrug resistance–associated protein
- NTCP
sodium-taurocholate cotransporting polypeptide
- OST
organic solute transporter
- SCH
sandwich-cultured hepatocytes
- TCA
taurocholic acid
- TGZ
troglitazone/5-(4-[(6-hydroxy-2,5,7,8-tetramethylchroman-2-yl)methoxy]benzyl)thiazolidine-2,4-dione
- TS
TGZ sulfate/5-[[4-[[3,4-dihydro-2,5,7,8-tetramethyl-6-(sulfooxy)-2H-1-benzopyran-2-yl]methoxy]phenyl]methyl]-2,4-thiazolidinedione
- TSB
Tris-sucrose buffer
Authorship Contributions
Participated in research design: Yang, Pfeifer, Köck, Brouwer.
Conducted experiments: Yang, Pfeifer, Köck.
Performed data analysis: Yang, Pfeifer, Köck.
Wrote or contributed to the writing of the manuscript: Yang, Pfeifer, Köck, Brouwer.
Footnotes
This work was supported by the National Institutes of Health National Institute of General Medical Sciences [Grant R01-GM041935 to K.L.R.B.] and by Deutsche Forschungsgemeinschaft [Grant Ko4186/1-1 to K.K.]. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This work was presented, in part, as a poster at the following meeting: Yang K and Brouwer KLR (2012) Pharmacokinetic modeling and simulation study to predict the impact of troglitazone (TGZ) on the hepatobiliary disposition of taurocholate (TC) in rat sandwich-cultured hepatocytes (SCH). 2012 AAPS Annual Meeting and Exposition; 2012 Oct 14–18; Chicago, IL.
References
- Akita H, Suzuki H, Hirohashi T, Takikawa H, Sugiyama Y. (2002) Transport activity of human MRP3 expressed in Sf9 cells: comparative studies with rat MRP3. Pharm Res 19:34–41. [DOI] [PubMed] [Google Scholar]
- Akita H, Suzuki H, Sugiyama Y. (2001) Sinusoidal efflux of taurocholate is enhanced in Mrp2-deficient rat liver. Pharm Res 18:1119–1125. [DOI] [PubMed] [Google Scholar]
- Ballatori N, Li N, Fang F, Boyer JL, Christian WV, Hammond CL. (2009) OST alpha-OST beta: a key membrane transporter of bile acids and conjugated steroids. Front Biosci (Landmark Ed) 14:2829–2844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang JY. (2009) Bile acids: regulation of synthesis. J Lipid Res 50:1955–1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson S, Stahl S, Paul N, Barber J, Kenna JG. (2012) In vitro inhibition of the bile salt export pump correlates with risk of cholestatic drug-induced liver injury in humans. Drug Metab Dispos 40:130–138. [DOI] [PubMed] [Google Scholar]
- Funk C, Ponelle C, Scheuermann G, Pantze M. (2001) Cholestatic potential of troglitazone as a possible factor contributing to troglitazone-induced hepatotoxicity: in vivo and in vitro interaction at the canalicular bile salt export pump (Bsep) in the rat. Mol Pharmacol 59:627–635. [PubMed] [Google Scholar]
- Ghibellini G, Leslie EM, Pollack GM, Brouwer KLR. (2008) Use of Tc-99m mebrofenin as a clinical probe to assess altered hepatobiliary transport: integration of in vitro, pharmacokinetic modeling, and simulation studies. Pharm Res 25:1851–1860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann AF. (1999a) Bile Acids: The good, the bad, and the ugly. News Physiol Sci 14:24–29. [DOI] [PubMed] [Google Scholar]
- Hofmann AF. (1999b) The continuing importance of bile acids in liver and intestinal disease. Arch Intern Med 159:2647–2658. [DOI] [PubMed] [Google Scholar]
- Hofmann AF. (2004) Detoxification of lithocholic acid, a toxic bile acid: relevance to drug hepatotoxicity. Drug Metab Rev 36:703–722. [DOI] [PubMed] [Google Scholar]
- Iusuf D, van de Steeg E, Schinkel AH. (2012) Hepatocyte hopping of OATP1B substrates contributes to efficient hepatic detoxification. Clin Pharmacol Ther 92:559–562. [DOI] [PubMed] [Google Scholar]
- Jansen PL, Strautnieks SS, Jacquemin E, Hadchouel M, Sokal EM, Hooiveld GJ, Koning JH, De Jager-Krikken A, Kuipers F, Stellaard F, et al. (1999) Hepatocanalicular bile salt export pump deficiency in patients with progressive familial intrahepatic cholestasis. Gastroenterology 117:1370–1379. [DOI] [PubMed] [Google Scholar]
- Jemnitz K, Veres Z, Vereczkey L. (2010) Contribution of high basolateral bile salt efflux to the lack of hepatotoxicity in rat in response to drugs inducing cholestasis in human. Toxicol Sci 115:80–88. [DOI] [PubMed] [Google Scholar]
- Köck K, Ferslew BC, Netterberg I, Yang K, Urban TJ, Swaan PW, Stewart PW, Brouwer KLR. (2014) Risk factors for development of cholestatic drug-induced liver injury: inhibition of hepatic basolateral bile acid transporters multidrug resistance-associated proteins 3 and 4. Drug Metab Dispos 42:665–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotani N, Maeda K, Watanabe T, Hiramatsu M, Gong LK, Bi YA, Takezawa T, Kusuhara H, Sugiyama Y. (2011) Culture period-dependent changes in the uptake of transporter substrates in sandwich-cultured rat and human hepatocytes. Drug Metab Dispos 39:1503–1510. [DOI] [PubMed] [Google Scholar]
- LeCluyse EL, Bullock PL, Parkinson A, Hochman JH. (1996) Cultured rat hepatocytes. Pharm Biotechnol 8:121–159. [DOI] [PubMed] [Google Scholar]
- Lee JK, Brouwer KR. (2010) Determination of intracellular volume of rat and human sandwich-cultured hepatocytes (Abstract ID 1595). The Toxicologist. Supplement to Toxicological Sciences 114:339. [Google Scholar]
- Lee JK, Marion TL, Abe K, Lim C, Pollack GM, Brouwer KLR. (2010) Hepatobiliary disposition of troglitazone and metabolites in rat and human sandwich-cultured hepatocytes: use of Monte Carlo simulations to assess the impact of changes in biliary excretion on troglitazone sulfate accumulation. J Pharmacol Exp Ther 332:26–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leslie EM, Watkins PB, Kim RB, Brouwer KLR. (2007) Differential inhibition of rat and human Na+-dependent taurocholate cotransporting polypeptide (NTCP/SLC10A1) by bosentan: a mechanism for species differences in hepatotoxicity. J Pharmacol Exp Ther 321:1170–1178. [DOI] [PubMed] [Google Scholar]
- Liu X, Brouwer KLR, Gan LS, Brouwer KR, Stieger B, Meier PJ, Audus KL, LeCluyse EL. (1998) Partial maintenance of taurocholate uptake by adult rat hepatocytes cultured in a collagen sandwich configuration. Pharm Res 15:1533–1539. [DOI] [PubMed] [Google Scholar]
- Maillette de Buy Wenniger L, Beuers U. (2010) Bile salts and cholestasis. Dig Liver Dis 42:409–418. [DOI] [PubMed] [Google Scholar]
- Marion TL, Leslie EM, Brouwer KLR. (2007) Use of sandwich-cultured hepatocytes to evaluate impaired bile acid transport as a mechanism of drug-induced hepatotoxicity. Mol Pharm 4:911–918. [DOI] [PubMed] [Google Scholar]
- Morgan RE, Trauner M, van Staden CJ, Lee PH, Ramachandran B, Eschenberg M, Afshari CA, Qualls CW, Jr, Lightfoot-Dunn R, Hamadeh HK. (2010) Interference with bile salt export pump function is a susceptibility factor for human liver injury in drug development. Toxicol Sci 118:485–500. [DOI] [PubMed] [Google Scholar]
- Morgan RE, van Staden CJ, Chen Y, Kalyanaraman N, Kalanzi J, Dunn RT, 2nd, Afshari CA, Hamadeh HK. (2013) A multifactorial approach to hepatobiliary transporter assessment enables improved therapeutic compound development. Toxicol Sci 136:216–241. [DOI] [PubMed] [Google Scholar]
- Nguyen A, Bouscarel B. (2008) Bile acids and signal transduction: role in glucose homeostasis. Cell Signal 20:2180–2197. [DOI] [PubMed] [Google Scholar]
- Oshio C, Phillips MJ. (1981) Contractility of bile canaliculi: implications for liver function. Science 212:1041–1042. [DOI] [PubMed] [Google Scholar]
- Pedersen JM, Matsson P, Bergström CA, Hoogstraate J, Norén A, LeCluyse EL, Artursson P. (2013) Early identification of clinically relevant drug interactions with the human bile salt export pump (BSEP/ABCB11). Toxicol Sci 136:328–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez MJ, Briz O. (2009) Bile-acid-induced cell injury and protection. World J Gastroenterol 15:1677–1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeifer ND, Hardwick RN, Brouwer KLR. (2014) Role of hepatic efflux transporters in regulating systemic and hepatocyte exposure to xenobiotics. Annu Rev Pharmacol Toxicol 54:509–535. [DOI] [PubMed] [Google Scholar]
- Pfeifer ND, Yang K, Brouwer KLR. (2013) Hepatic basolateral efflux contributes significantly to rosuvastatin disposition I: characterization of basolateral versus biliary clearance using a novel protocol in sandwich-cultured hepatocytes. J Pharmacol Exp Ther 347:727–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips MJ, Oshio C, Miyairi M, Katz H, Smith CR. (1982) A study of bile canalicular contractions in isolated hepatocytes. Hepatology 2:763–768. [DOI] [PubMed] [Google Scholar]
- Rius M, Hummel-Eisenbeiss J, Hofmann AF, Keppler D. (2006) Substrate specificity of human ABCC4 (MRP4)-mediated cotransport of bile acids and reduced glutathione. Am J Physiol Gastrointest Liver Physiol 290:G640–G649. [DOI] [PubMed] [Google Scholar]
- Saha S, New LS, Ho HK, Chui WK, Chan EC. (2010) Direct toxicity effects of sulfo-conjugated troglitazone on human hepatocytes. Toxicol Lett 195:135–141. [DOI] [PubMed] [Google Scholar]
- Setchell KD, Rodrigues CM, Clerici C, Solinas A, Morelli A, Gartung C, Boyer J. (1997) Bile acid concentrations in human and rat liver tissue and in hepatocyte nuclei. Gastroenterology 112:226–235. [DOI] [PubMed] [Google Scholar]
- Sohlenius-Sternbeck AK. (2006) Determination of the hepatocellularity number for human, dog, rabbit, rat and mouse livers from protein concentration measurements. Toxicol In Vitro 20:1582–1586. [DOI] [PubMed] [Google Scholar]
- Swift B, Pfeifer ND, Brouwer KLR. (2010) Sandwich-cultured hepatocytes: an in vitro model to evaluate hepatobiliary transporter-based drug interactions and hepatotoxicity. Drug Metab Rev 42:446–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tchaparian EH, Houghton JS, Uyeda C, Grillo MP, Jin L. (2011) Effect of culture time on the basal expression levels of drug transporters in sandwich-cultured primary rat hepatocytes. Drug Metab Dispos 39:2387–2394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trauner M, Wagner M, Fickert P, Zollner G. (2005) Molecular regulation of hepatobiliary transport systems: clinical implications for understanding and treating cholestasis. J Clin Gastroenterol 39(4, Suppl 2):S111–S124. [DOI] [PubMed] [Google Scholar]
- Watanabe N, Tsukada N, Smith CR, Phillips MJ. (1991) Motility of bile canaliculi in the living animal: implications for bile flow. J Cell Biol 113:1069–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkins PB, Seeff LB. (2006) Drug-induced liver injury: summary of a single topic clinical research conference. Hepatology 43:618–631. [DOI] [PubMed] [Google Scholar]
- Zamek-Gliszczynski MJ, Kalvass JC, Pollack GM, Brouwer KLR. (2009) Relationship between drug/metabolite exposure and impairment of excretory transport function. Drug Metab Dispos 37:386–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zollner G, Wagner M, Fickert P, Silbert D, Gumhold J, Zatloukal K, Denk H, Trauner M. (2007) Expression of bile acid synthesis and detoxification enzymes and the alternative bile acid efflux pump MRP4 in patients with primary biliary cirrhosis. Liver Int 27:920–929. [DOI] [PubMed] [Google Scholar]