

Abstract
Synaptic GABAA receptors respond to synaptically released GABA and are considered to be unaffected by the low levels of ambient transmitter in the brain. We show that synaptic-type α1β2γ2L GABAA receptors expressed in HEK293 cells respond with large steady-state currents to combinations of a low concentration (0.5 μM) of GABA and clinically used GABAergic modulators propofol, etomidate, or pentobarbital or the steroid alphaxalone. At a maximally effective concentration of modulator, the current levels at the end of 2-minute applications of drug combinations were >10% of the peak response to saturating GABA. In the absence of modulators, 0.5 μM GABA generated a steady-state response of 1% of the peak response to saturating GABA. The concentration-response curves for enhancement of steady-state currents by propofol, etomidate, pentobarbital, or alphaxalone were at similar or lower drug concentrations compared with concentration-response relationships for enhancement of peak responses. We propose that modulation of tonically activated synaptic-type GABAA receptors contributes to the clinical actions of sedative drugs.
Introduction
GABAA receptors in the brain are exposed to phasic high (near millimolar) concentrations of GABA during synaptic transmission and low ambient (<1 μM) concentrations of GABA, which is continuously present in the cerebrospinal fluid (reviewed in Farrant and Nusser, 2005). Ambient GABA activates extrasynaptic GABAA receptors, whose major identifying characteristics are high affinity to the transmitter and weak desensitization in the persistent presence of an agonist. The targets of phasic GABA are synaptic GABAA receptors, which are characterized by relatively low affinity to GABA and high maximal peak open probability. The conventional view is that synaptic-type receptors are minimally affected by the presence of low ambient GABA. Indeed, the concentration of extracellular GABA (Houston et al., 2012) is several tens of folds lower than the apparent affinity to GABA of synaptic subtypes of GABAA receptors (Li et al., 2006; Picton and Fisher, 2007).
Under physiologic conditions, GABAA receptors, including synaptic receptors, are exposed to endogenous modulators such as neuroactive steroids (Carver and Reddy, 2013). Under clinical conditions, GABAA receptors can be exposed to GABAergic sedative drugs such as propofol, etomidate, or barbiturates. Although these drugs are ineffective at modulating peak responses of synaptic events due to saturation of GABA binding and a high maximal open probability of GABA-activated receptors, there remains a possibility that GABAergic modulators potentiate the low-level activity from synaptic-type receptors exposed to ambient GABA.
To test this hypothesis, we exposed heterologously expressed rat α1β2γ2L receptors to a low concentration (0.5 μM) of GABA, intended to mimic steady-state GABA, in the absence and presence of several common modulators of the GABAA receptor. The data show that a combination of a low concentration of GABA with clinical concentrations of propofol, etomidate, pentobarbital, or the steroid alphaxalone enhances the level of quasi–steady-state current by ∼10-fold. Concentration-response measurements indicate that the EC50s for potentiation of steady-state currents are at similar or lower concentrations than those measured for potentiation of peak responses. Our data raise the possibility of a role for tonically activated synaptic-type GABAA receptors in the clinical actions of some anesthetic drugs.
Materials and Methods
Experiments were carried out on rat wild-type α1β2γ2L GABAA receptors. Receptors were transiently expressed in HEK293 cells, using a modified calcium phosphate precipitation–based transient transfection technique (Akk, 2002). The amino terminus of the α1 subunit was tagged with the FLAG epitope (Ueno et al., 1996), and bead-binding with immunobeads against the FLAG epitope was used to identify cells expressing high levels of GABAA receptors.
Electrophysiological recordings were done on lifted cells using the standard whole-cell voltage-clamp approach. Cells were clamped at −60 mV. Recordings were conducted at room temperature. The bath solution contained the following (in mM): NaCl, 140; KCl, 5; MgCl2, 1; CaCl2, 2; d-glucose, 10; and HEPES, 10 (pH 7.4). The pipette solution contained the following (in mM): CsCl, 140; NaCl, 4; MgCl2, 4; CaCl2, 0.5; EGTA, 5; and HEPES, 10 (pH 7.4). Agonist (GABA) and modulators (propofol, etomidate, pentobarbital, or alphaxalone) were applied onto cells through bath using an SF-77B fast perfusion stepper system (Warner Instruments, Hamden, CT). We have previously found that the solution exchange time with this system is 10–20 milliseconds with a typical-size HEK cell (Li and Akk, 2008). Durations of drug applications were 2 minutes in most recordings. Consecutive drug applications were separated by 2- to 4-minute washouts in bath solution. The currents were recorded using an Axopatch 200B amplifier (Molecular Devices, Sunnyvale, CA), low-pass filtered at 2 kHz and digitized with a Digidata 1320 series interface (Molecular Devices) at 10 kHz. The analysis of current traces was conducted using pClamp 9.0 software (Molecular Devices). Current traces were analyzed with respect to the amplitudes of peak and residual currents at the end of a 2-minute drug application. No visually noticeable current decay was observed at the end of 2-minute applications when 0.5 μM GABA was coapplied with modulators. Each cell was also exposed to 50 μM GABA to obtain a reference point for maximal peak response. This concentration, at ∼5 times the macroscopic EC50 value (Li et al., 2006), produces a near-maximal response. Amplitudes of responses to low GABA and combinations of low GABA and modulators are expressed as percentage of the peak response to 50 μM GABA from the same cell. The data are reported as mean ± S.E.M.
Concentration-response curves were fitted, individually for each cell, with the following equation:
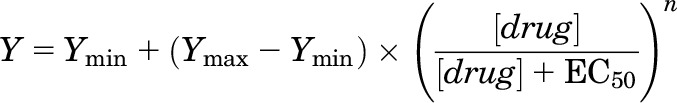 |
where EC50 is the concentration of drug producing a half-maximal effect, n describes the slope of relationship, and Ymin and Ymax are the low- and high-concentration asymptotes, respectively. Fitting was conducted using the NFIT software (University of Texas Medical Branch at Galveston, Galveston, TX). Parameters of the fit are reported as mean ± S.E.M.
The potentiating effect of alphaxalone on steady-state current elicited by GABA is given as response ratio, calculated as response in the presence of steroid divided by response in the absence of steroid. Statistical analysis was performed using Excel (Microsoft, Redmond, WA). Statistical significance was determined by comparing the response ratio to 1, i.e., no effect, using a two-tailed paired t test. This test is equivalent to a one-sample t test with a hypothetical value of 1, and is designed to verify whether the effect of the drug is statistically significant.
GABA, inorganic salts used in buffers, pentobarbital, and alphaxalone were bought from Sigma-Aldrich (St. Louis, MO). Propofol was from MP Biomedicals (Solon, OH). Etomidate was purchased from R&D Systems (Minneapolis, MN).
Results
Rat α1β2γ2L GABAA receptors expressed in HEK293 cells have a whole-cell GABA EC50 of ∼10 μM (Li et al., 2006). Activation of α1β2γ2L receptors by 50 μM GABA resulted in fast-rising large inward currents that desensitized to 14 ± 1% (mean ± S.E.M.; 14 cells) of the peak response by the end of a 2-minute drug application. Exposure of same set of cells to 0.5 μM GABA elicited a peak response that was 3 ± 1% and a steady-state current that was 1 ± 1% of the peak response to 50 μM GABA. Sample currents are shown in Fig. 1, A and B.
Fig. 1.

Activation of rat α1β2γ2L GABAA receptors by GABA. Currents were elicited by 2-minute applications of 50 μM GABA (A) or 0.5 μM GABA (B). In (B), the framed inset shows the same current trace at a higher current resolution. Traces in (A) and (B) are from the same cell.
The anesthetic compound propofol potentiates currents from α1β2γ2L receptors activated by submaximal concentrations of GABA (Ruesch et al., 2012). We found that coapplication of 1 μM propofol with 0.5 μM GABA potentiated the peak response by 7- ± 2-fold (14 cells). Propofol alone at this concentration elicited a negligible response (0.3 ± 0.1% of the peak response to 50 μM GABA). The current at the end of a 2-minute drug application was enhanced by 14- ± 5-fold when 1 μM propofol was coapplied with 0.5 μM GABA. Relative to the peak response to 50 μM GABA, the steady-state current at the end of a 2-minute application of 0.5 μM GABA + 1 μM propofol was 9 ± 2% (14 cells).
To investigate the concentration-response relationship of propofol-elicited enhancement of steady-state currents, we tested, in a separate set of five cells, the effect of 0.1–3 μM propofol on steady-state currents elicited by low GABA. Compared with the peak response to 50 μM GABA in the same cell, the mean current level at the end of a 2-minute drug application was 2 ± 1%, 5 ± 1%, 8 ± 2%, or 8 ± 1% in the presence of 0.5 μM GABA and 0.1, 0.3, 1, and 3 μM propofol, respectively. Curve-fitting the data from each cell individually yielded an EC50 of 0.44 ± 0.06 μM and a Hill coefficient of 2.4 ± 0.6 (mean ± S.E.M.; Fig. 2B). The fit maximum was 9 ± 1% (five cells).
Fig. 2.

Exposure to propofol (PRO) enhances steady-state currents from α1β2γ2L GABAA receptors. (A) Sample current responses from receptors activated by 50 μM GABA (red line), 0.5 μM GABA (blue line), or 0.5 μM GABA + 0.1, 0.3, 1, or 3 μM propofol (black lines). For clarity, only the first 10 seconds of the 50 μM GABA trace are shown. The inset shows the final 30 seconds of the traces. All traces are from the same cell. (B) A concentration-response relationship for propofol-induced enhancement of steady-state currents. The steady-state currents were normalized to the peak response to 50 μM GABA from the same cells. Data show mean ± S.E.M. from a set of five cells. The curve shows the prediction of the equation Y = Ymin + (Ymax − Ymin) × ([propofol]/([propofol] + EC50))n, where EC50 is the concentration of propofol producing a half-maximal effect, n is the slope of the curve, and Ymin and Ymax are the low- and high-concentration asymptotes, respectively. The curve was generated with the overall mean parameters from the five cells. The EC50 was 0.44 ± 0.06 μM (mean ± S.E.M.), n was 2.4 ± 0.6, and Ymax was 9 ± 1%. The value for Ymin was held at the value obtained from the relative steady-state current from the same five cells in the presence of 0.5 μM GABA alone (1 ± 0.3%).
To determine whether other common GABAA receptor potentiators modulate steady-state currents from α1β2γ2L receptors, we examined the effects of etomidate and pentobarbital on currents elicited by 0.5 μM GABA. Coapplication of 0.1–10 μM etomidate with 0.5 μM GABA enhanced steady-state current. The relative (compared with peak in the presence of 50 μM GABA) steady-state current levels were 1.3 ± 0.4%, 3 ± 1%, 6 ± 1%, 11 ± 1%, or 11 ± 1% when receptors were activated by 0.5 μM GABA in the presence of 0.1, 0.3, 1, 3, or 10 μM etomidate, respectively (a set of seven cells). Curve-fitting yielded an EC50 of 0.9 ± 0.4 μM, a Hill coefficient of 2.2 ± 0.4, and a fitted maximal response of 11 ± 1%. Sample traces and the concentration-response plot are shown in Fig. 3A.
Fig. 3.

Exposure to etomidate, pentobarbital, or alphaxalone enhances steady-state currents from α1β2γ2L GABAA receptors. (A) Sample current responses from receptors activated by 50 μM GABA (red line), 0.5 μM GABA (blue line), or 0.5 μM GABA + 10 μM etomidate (ETO; black line). For clarity, only the first 10 seconds of the 50 μM GABA trace are shown. All traces are from the same cell. The lower portion of (A) shows the concentration-response relationship for etomidate-induced enhancement of steady-state current levels. The steady-state currents were normalized to the peak response to 50 μM GABA from the same cells. Data show mean ± S.E.M. from seven cells exposed to 0.5 μM GABA and 0.1–10 μM etomidate. The curve was generated as in Fig. 2 with the following overall parameters: EC50 = 0.9 ± 0.4 μM, n = 2.2 ± 0.4, Ymin = 0.6 ± 0.2%, Ymax = 11 ± 1%. (B) Sample current traces from receptors activated by 50 μM GABA (red line), 0.5 μM GABA (blue line), or 0.5 μM GABA + 100 μM pentobarbital (PEB; black line). The lower portion of (B) shows the concentration-response relationship for pentobarbital-induced enhancement of steady-state current levels. Data show mean ± S.E.M. from five cells exposed to 0.5 μM GABA and 3–100 μM pentobarbital. The curve was generated as in Fig. 2 with the following overall parameters: EC50 = 12 ± 2 μM, n = 2.6 ± 0.4, Ymin = 0.7 ± 0.1%, Ymax = 13 ± 2%. (C) Sample current traces from receptors activated by 50 μM GABA (red line), 0.5 μM GABA (blue line), 0.5 μM GABA + 1 μM alphaxalone (ALF; black line), or 0.5 μM GABA + 1 μM alphaxalone + 1 μM propofol (PRO; gray line). The lower portion of (C) shows the effect of 10, 30, 100, 300, or 1000 nM alphaxalone on steady-state responses to 0.5 μM GABA. The data give mean ± S.E.M. for the response ratio (1 = no effect) from 4–11 cells at each steroid concentration. All data points were obtained from individual cells. Statistical analysis was conducted by comparing the response ratio to 1 using two-tailed paired t test (Excel). *P < 0.05; **P < 0.01; †P > 0.05.
Steady-state currents elicited by 0.5 μM GABA were also enhanced in the presence of pentobarbital. Coapplication of 3, 10, 30, or 100 μM pentobarbital with GABA resulted in relative current levels of 2 ± 1%, 6 ± 1%, 13 ± 1%, or 12 ± 2% (five cells at each condition), respectively. Curve-fitting yielded an EC50 of 12 ± 2 μM, a Hill coefficient of 2.6 ± 0.4, and a maximal current level of 13 ± 2% (Fig. 3B).
We previously showed that the application of a neuroactive steroid enhances peak responses from α1β2γ2L GABAA receptors activated by allosteric activators or combinations of low GABA and an allosteric activator (Li et al., 2014). To test whether a steroid can enhance steady-state currents from receptors activated by GABA and an allosteric drug, we coapplied 10–1000 nM alphaxalone with 0.5 μM GABA + 1 μM propofol. Due to numerous controls and comparisons in this experimental protocol, the effects of increasing concentrations of steroid were tested on separate cells. The steady-state current level at the end of a 2-minute application was 8 ± 1% (10 nM alphaxalone; three cells), 15 ± 2% (30 nM; three cells), 11 ± 2% (100 nM; three cells), 10 ± 2% (300 nM; four cells), and 16 ± 6% (1000 nM; three cells) of the peak response to 50 μM GABA. These values were not significantly different (two-sample t test; Excel) from the mean steady-state response observed for 0.5 μM GABA + 1 μM propofol in the absence of alphaxalone (9 ± 2% of the peak response to 50 μM GABA; see above). Sample traces are shown in Fig. 3C.
To verify that alphaxalone is capable of potentiating steady-state responses to GABA alone, we evaluated data obtained as part of the experimental protocol in the preceding experiment where each cell had additionally been challenged with 0.5 μM GABA + alphaxalone. In six cells exposed to 0.5 μM GABA + 1000 nM alphaxalone, the steady-state response at the end of a 2-minute drug application was 9 ± 2% of the peak response to 50 μM GABA. This represented an 11- ± 2-fold increase in the current level compared with steady-state response to 0.5 μM GABA alone in the same set of cells. Note that the number of cells here is greater than in the preceding experiment because not all cells yielded data for all drug conditions. Analogous data evaluation for other steroid concentrations showed that steady-state currents from receptors activated by 0.5 μM GABA were potentiated by 2.4- ± 0.3-fold (11 cells) and 4.1- ± 0.4-fold (4 cells) in the presence of 100 and 300 nM alphaxalone, respectively. Coapplication of 10 or 30 nM alphaxalone with 0.5 μM GABA had a statistically insignificant effect on steady-state response. We infer that alphaxalone concentration-dependently enhances steady-state response to low GABA. Sample current traces and the concentration-response relationship are given in Fig. 3C.
Discussion
The potentiating effects of GABAA receptor modulators are typically studied with regard to effect on the peak response. Here, we demonstrate that some common, clinically used GABAA receptor potentiators (propofol, etomidate, pentobarbital, and the steroid alphaxalone) potentiate quasi–steady-state currents from α1β2γ2L receptors activated by 0.5 μM GABA. The steady-state response under control conditions was 1% of the peak response to saturating GABA. At a maximally effective concentration of modulator, the amplitude of steady-state current was potentiated by ∼10-fold.
GABAA receptors containing α1β2γ2L receptors have a high (>0.8) maximal open probability in the presence of GABA (Steinbach and Akk, 2001). This predicts that the steady-state open probability of α1β2γ2L receptors in the presence of 0.5 μM GABA and propofol, etomidate, pentobarbital, or alphaxalone reaches ∼0.1. Although the latter value may be overestimated if rapid desensitization reduces the maximal peak estimate, we believe that the error is insignificant, for the following reasons. First, a more rapid solution exchange when 1 mM, instead of 50 μM, GABA is used to activate receptors has only minor effect on the peak response. Second, the peak response to saturating GABA is minimally affected by potentiators, indicative of high open probability during peak current. Finally, our relative steady-state responses to saturating GABA are generally similar to those obtained from rapid drug applications to excised patches (e.g., Haas and Macdonald, 1999).
The EC50s for potentiation of steady-state currents from α1β2γ2L were similar or lower than the EC50s from available data for potentiation of peak responses. For example, we estimate that the half-maximal concentration of etomidate producing an increase of steady-state currents is 0.9 μM (Fig. 3A), whereas our recent study on the effect of etomidate on peak currents from α1β2γ2L receptors revealed an EC50 of 2 μM (Li et al., 2014). Similarly, while the EC50 for potentiation of steady-state currents by propofol was 0.4 μM (Fig. 2B), estimates for EC50 for potentiation of peak currents from α1β2γ2 receptors range from 2 (Krasowski et al., 2001) to 25 μM (Lam and Reynolds, 1998). The differences in EC50 estimates may be due to state dependence of potentiation or simply reflect greater occupancy of the transmitter site in steady-state recordings.
The concentration of free propofol in plasma of surgical patients is ∼1.3 μM (Rehberg and Duch, 1999). Thus, potentiation of steady-state currents from α1β2γ2L receptors may contribute to the sedative effect of propofol. The concentration of steady-state GABA to which the receptors are exposed under physiologic conditions is estimated at or below 0.5 μM (multiple references in Houston et al., 2012). Although exposure of α1β2γ2L to 0.5 μM GABA elicited an almost negligible response (1% of the response to saturating GABA), the combination of 0.5 μM GABA and 1 μM propofol is expected to generate a steady level of ∼10% of maximal activity from these receptors.
Maximal steady-state current levels were similar for other potentiators. Coapplication of 100 μM pentobarbital with 0.5 μM GABA produced steady-state activity that was 12% of the peak response to 50 μM GABA. The mean steady-state current was 11% of the maximal peak response in the presence of 0.5 μM GABA and 10 μM etomidate, and 9% in the presence of 0.5 μM GABA and 1000 nM alphaxalone. Interestingly, the addition of 10–1000 nM alphaxalone to the combination of 0.5 μM GABA + 1 μM propofol did not lead to further enhancement of steady-state current. This is in contrast to our recent finding that neuroactive steroids can potentiate peak currents elicited by a combination of GABA and an allosteric activator (Li et al., 2014).
The ambient GABA concentration is likely dynamic, showing greater fluctuations at or near the synapse where the high-millimolar concentration of GABA during a synaptic event rapidly drops due to uptake and diffusion. A recent study found that the ability of GABAA receptor potentiators to enhance tonic currents depends on ambient GABA concentration (Houston et al., 2012). This introduces yet another variable capable of affecting the final level of tonic inhibition in the presence of sedative drugs.
Two major questions are raised by these observations. The first is the mechanism by which the steady-state response is enhanced. One possibility is that desensitization is reduced. This notion is supported by prior work that showed slower desensitization or potentiation of steady-state currents elicited by saturating GABA in the presence of some anesthetic drugs (Feng and Macdonald, 2004; Feng et al., 2004; Hall et al., 2004). Potentiation of steady-state currents from receptors activated by low concentrations of GABA additionally includes an effect on channel gating. Our data obtained with 0.5 μM GABA demonstrate a major contribution from gating given that the peak response, for which desensitization-based effect is minimal, was enhanced by 7-fold, whereas the steady-state response, sensitive to changes in both desensitization and gating, was enhanced by 14-fold during coapplication with 1 μM propofol. In future single-channel work, it will be interesting to determine the precise kinetic makeup of potentiation of currents elicited by ambient GABA.
The second question relates to the physiologic consequences of the additional, tonic current. Receptors containing the α1 and γ2 subunits constitute the majority of GABAA receptors in the brain (Whiting et al., 1999). If it is assumed that these receptors are present at about 10-fold–higher numbers than the classic tonic receptors (largely α4/δ or α5/γ2 subtypes), then the steady-state contribution we have demonstrated would be a considerable additional tonic inhibitory influence on the cells. This rough calculation will require much more refinement as information on the relative numbers of different receptor types is gathered, as well as further studies on the abilities of these modulators to enhance responses to endogenous levels of GABA. There is evidence that α1/γ2 receptors can be located outside the immediate postsynaptic membrane (Thomas et al., 2005; Mortensen and Smart, 2006; Kasugai et al., 2010). This pool of extrasynaptically located α1/γ2 GABAA receptors is expected to be minimally active in the presence of steady-state GABA but respond with an ∼10-fold increase in current amplitude during exposure to anesthetics. Unlike synaptic α1/γ2 receptors that undergo periodic activation and desensitization by high-millimolar concentrations of GABA, extrasynaptic α1/γ2 receptors would be ideally suited to provide steady inhibitory current until exposure to the anesthetic is terminated.
To recapitulate, the data show that synaptic-type α1β2γ2L GABAA receptors exposed to a low, steady-state concentration of GABA and clinical concentrations of several anesthetic drugs produce persistent currents that have an amplitude of ∼10% of the maximal peak response. This type of potentiation of inhibitory tone is distinct from the anesthetic-mediated prolongation of synaptic currents or enhancement of tonic inhibition mediated by classic α4/6- or α5-containing extrasynaptic receptors. We propose that these persistent, nondesensitizing currents from α1βγ2 GABAA receptors, in addition to previously identified anesthetic targets or modes of potentiation, contribute to the clinical effects of anesthetic drugs.
Acknowledgments
The authors thank Dr. J. H. Steinbach for many fruitful discussions during the course of the study.
Authorship Contributions
Participated in research design: Akk.
Conducted experiments: Li.
Performed data analysis: Li, Akk.
Wrote or contributed to the writing of the manuscript: Li, Akk.
Footnotes
This work was supported by the National Institutes of Health National Institute of General Medical Sciences [Grants P01-GM047969 and R01-GM108580].
References
- Akk G. (2002) Contributions of the non-alpha subunit residues (loop D) to agonist binding and channel gating in the muscle nicotinic acetylcholine receptor. J Physiol 544:695–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carver CM, Reddy DS. (2013) Neurosteroid interactions with synaptic and extrasynaptic GABA(A) receptors: regulation of subunit plasticity, phasic and tonic inhibition, and neuronal network excitability. Psychopharmacology (Berl) 230:151–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrant M, Nusser Z. (2005) Variations on an inhibitory theme: phasic and tonic activation of GABA(A) receptors. Nat Rev Neurosci 6:215–229. [DOI] [PubMed] [Google Scholar]
- Feng HJ, Bianchi MT, Macdonald RL. (2004) Pentobarbital differentially modulates alpha1beta3delta and alpha1beta3gamma2L GABAA receptor currents. Mol Pharmacol 66:988–1003. [DOI] [PubMed] [Google Scholar]
- Feng HJ, Macdonald RL. (2004) Multiple actions of propofol on alphabetagamma and alphabetadelta GABAA receptors. Mol Pharmacol 66:1517–1524. [DOI] [PubMed] [Google Scholar]
- Haas KF, Macdonald RL. (1999) GABAA receptor subunit gamma2 and delta subtypes confer unique kinetic properties on recombinant GABAA receptor currents in mouse fibroblasts. J Physiol 514:27–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall AC, Rowan KC, Stevens RJN, Kelley JC, Harrison NL. (2004) The effects of isoflurane on desensitized wild-type and alpha 1(S270H) gamma-aminobutyric acid type A receptors. Anesth Analg 98:1297–1304. [DOI] [PubMed] [Google Scholar]
- Houston CM, McGee TP, Mackenzie G, Troyano-Cuturi K, Rodriguez PM, Kutsarova E, Diamanti E, Hosie AM, Franks NP, Brickley SG. (2012) Are extrasynaptic GABAA receptors important targets for sedative/hypnotic drugs? J Neurosci 32:3887–3897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasugai Y, Swinny JD, Roberts JDB, Dalezios Y, Fukazawa Y, Sieghart W, Shigemoto R, Somogyi P. (2010) Quantitative localisation of synaptic and extrasynaptic GABAA receptor subunits on hippocampal pyramidal cells by freeze-fracture replica immunolabelling. Eur J Neurosci 32:1868–1888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krasowski MD, Jenkins A, Flood P, Kung AY, Hopfinger AJ, Harrison NL. (2001) General anesthetic potencies of a series of propofol analogs correlate with potency for potentiation of gamma-aminobutyric acid (GABA) current at the GABA(A) receptor but not with lipid solubility. J Pharmacol Exp Ther 297:338–351. [PubMed] [Google Scholar]
- Lam DW, Reynolds JN. (1998) Modulatory and direct effects of propofol on recombinant GABAA receptors expressed in xenopus oocytes: influence of alpha- and gamma2-subunits. Brain Res 784:179–187. [DOI] [PubMed] [Google Scholar]
- Li P, Akk G. (2008) The insecticide fipronil and its metabolite fipronil sulphone inhibit the rat alpha1beta2gamma2L GABA(A) receptor. Br J Pharmacol 155:783–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P, Bracamontes JR, Manion BD, Mennerick S, Steinbach JH, Evers AS, Akk G. (2014) The neurosteroid 5β-pregnan-3α-ol-20-one enhances actions of etomidate as a positive allosteric modulator of α1β2γL GABAA receptors. Br J Pharmacol 171:5446–5457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P, Covey DF, Steinbach JH, Akk G. (2006) Dual potentiating and inhibitory actions of a benz[e]indene neurosteroid analog on recombinant alpha1beta2gamma2 GABAA receptors. Mol Pharmacol 69:2015–2026. [DOI] [PubMed] [Google Scholar]
- Mortensen M, Smart TG. (2006) Extrasynaptic alphabeta subunit GABAA receptors on rat hippocampal pyramidal neurons. J Physiol 577:841–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picton AJ, Fisher JL. (2007) Effect of the alpha subunit subtype on the macroscopic kinetic properties of recombinant GABA(A) receptors. Brain Res 1165:40–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehberg B, Duch DS. (1999) Suppression of central nervous system sodium channels by propofol. Anesthesiology 91:512–520. [DOI] [PubMed] [Google Scholar]
- Ruesch D, Neumann E, Wulf H, Forman SA. (2012) An allosteric coagonist model for propofol effects on α1β2γ2L γ-aminobutyric acid type A receptors. Anesthesiology 116:47–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinbach JH, Akk G. (2001) Modulation of GABA(A) receptor channel gating by pentobarbital. J Physiol 537:715–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas P, Mortensen M, Hosie AM, Smart TG. (2005) Dynamic mobility of functional GABAA receptors at inhibitory synapses. Nat Neurosci 8:889–897. [DOI] [PubMed] [Google Scholar]
- Ueno S, Zorumski C, Bracamontes J, Steinbach JH. (1996) Endogenous subunits can cause ambiguities in the pharmacology of exogenous gamma-aminobutyric acidA receptors expressed in human embryonic kidney 293 cells. Mol Pharmacol 50:931–938. [PubMed] [Google Scholar]
- Whiting PJ, Bonnert TP, McKernan RM, Farrar S, Le Bourdellès B, Heavens RP, Smith DW, Hewson L, Rigby MR, Sirinathsinghji DJ, et al. (1999) Molecular and functional diversity of the expanding GABA-A receptor gene family. Ann N Y Acad Sci 868:645–653. [DOI] [PubMed] [Google Scholar]