

Summary
Staphylococcus aureus skin colonization is universal in atopic dermatitis and common in cancer patients treated with epidermal growth factor receptor inhibitors. However, the causal relationship of dysbiosis and eczema has yet to be clarified. Herein, we demonstrate that Adam17fl/flSox9-Cre mice, generated to model ADAM17-deficiency in human, developed eczematous dermatitis with naturally occurring dysbiosis, similar to that observed in atopic dermatitis. Corynebacterium mastitidis, S. aureus, and Corynebacterium bovis sequentially emerged during the onset of eczematous dermatitis, and antibiotic specific for these bacterial species almost completely reversed dysbiosis and eliminated skin inflammation. Whereas S. aureus prominently drove eczema formation, C. bovis induced robust T helper 2 cell responses. Langerhans cells were required for eliciting immune responses against S. aureus inoculation. These results characterize differential contributions of dysbiotic flora during eczema formation, and highlight the microbiota-host immunity axis as a possible target for future therapeutics in eczematous dermatitis.
Introduction
Microbiome analyses in recent years have revealed that mammalian body surfaces are colonized by vast numbers of bacterial communities (Grice et al., 2009), providing impetus to explore the roles of the microbiota in normal and diseased skin. Staphylococcus aureus has long been known to heavily colonize skin of patients with atopic dermatitis (AD) (Leyden et al., 1974), a chronic inflammatory skin disease that develops in individuals with barrier-disrupted skin, and that leads to the progressive development of asthma and food allergy referred to as the atopic march (Spergel and Paller, 2003). Microbiome analysis has revealed temporal dybiosis dominated by S. aureus during AD flares (Kong et al., 2012), suggesting an important role of S. aureus and/or dysbiosis during active atopic inflammation.
A major cause for stratum corneum barrier disruption in AD is attributed to loss-of-function mutations in FLG that encodes filaggrin, a structural protein in keratinocytes/corneocytes, thereby allowing outside-in penetration of foreign antigens and subsequent sensitization (Irvine et al., 2011). Other rare genetic diseases also manifest eczematous dermatitis that are clinically nearly identical to AD, and that could represent prototypic eczema with defects in relatively well-characterized genes (Kubo et al., 2012). Among such diseases are Netherton syndrome and hyper IgE syndrome, resulting from mutations in SPINK5 and STAT3, respectively (Chavanas et al., 2000; Minegishi et al., 2007). S. aureus is commonly isolated from skin in both of these diseases, and microbiome analysis has revealed dysbiosis with overrepresentation of S. aureus and Corynebacterium species in inflammatory skin lesions of hyper IgE syndrome (Oh et al., 2013).
Further supporting the possible involvement of dysbiosis in eczema pathophysiology, dysbiosis is implicated in other inflammatory skin conditions. Cancer patients treated with epidermal growth factor receptor (EGFR) inhibitors commonly develop eczematous rashes and pustules (Lacouture, 2006), from which S. aureus can be isolated (Eilers et al., 2010; Lichtenberger et al., 2013). Despite its apparent association with eczematous inflammation in AD, genetic diseases and EGFR inhibitor usage, the cause-or-effect relationship of dysbiosis and AD or eczematous dermatitis has not been determined to date. To address this issue would require a mouse model in which the microbiota alterations recapitulate human conditions.
A disintegrin and metalloproteinase 17 (ADAM17) is a transmembrane protease that cleaves a variety of membrane-bound proteins to release soluble forms (Blobel, 2005). ADAM17 plays a major role in TNFα and EGFR signaling pathways, in which pro-TNFα and pro-EGFR ligands are shed by ADAM17 from cell surfaces into active forms (Blobel, 2005). Genomic ablation of Adam17 in mice leads to perinatal lethality (Peschon et al., 1998). ADAM17-deficiency has been recently identified in humans (Blaydon et al., 2011), defining a new genetic disorder. Importantly, described patients exhibited eczematous dermatitis and pustular lesions, and were prone to S. aureus infections.
Herein, we show that mice lacking Adam17 in Sox9-expressing tissues including epidermis (Adam17fl/flSox9-Cre mice) manifested phenotypes similar to their human counterparts, exhibiting eczematous skin with disrupted barriers, elevated serum IgE and mixed T helper cell responses. Microbiome analysis of eczematous lesions in these mice revealed prominent dysbiosis that recapitulated that in human AD and eczematous dermatitis. We also demonstrate that eczematous inflammation in Adam17fl/flSox9-Cre mice was driven by dysbiosis, in which S. aureus and C. bovis to contribute differentially;, that dysbiosis could be induced by targeted ablation of EGFR signaling;, and that epidermal Langerhans cells mediate early immune responses to S. aureus.
Results
Adam17fl/flSox9-Cre mice exhibit eczematous inflammation
We generated mice that lack Adam17 in Sox9-expressing tissues including epidermis (Adam17fl/flSox9-Cre mice; henceforth referred to as Adam17ΔSox9 mice) initially to study ADAM17 function in bones (Horiuchi et al., 2009) and hair follicles (Nagao et al., 2012). While Sox9 is expressed in a variety of tissues including lung, pancreas, brain, gut, and kidneys, its expression in skin is mostly limited to the epidermal component (Horiuchi et al., 2009).
Consistent with previous reports that utilized mice expressing Cre recombinase driven by the keratin 14 promoter to ablate Adam17 (Franzke et al., 2012; Murthy et al., 2012), Adam17ΔSox9 mice exhibited dry skin around 3 weeks after birth, which progressed to eczematous lesions with intense pruritus (Figure S1A, Supplemental Movie 1), recapitulating symptoms reported in patients with ADAM17 mutation (Blaydon et al., 2011). Lesional skin biopsies revealed mononuclear cell infiltrates including lymphocytes and mast cells (Figures 1A and S1B). Increased transepidermal water loss (TEWL) indicated barrier dysfunction (Figure 1B). Serum IgE and CCL17, the latter a T helper 2 (Th2) cell chemokine that reflects disease activity in human AD (Kakinuma et al., 2001), were elevated (Figures 1C and 1D).
Figure 1. Eczematous inflammation in ADAM17-deficient skin.

(A) Histopathology of skin biopsy from WT and Adam17fl/flSox9-Cre (Adam17ΔSox9) mice. Scale bars, 50 μm. (B–D) TEWL, serum IgE and serum CCL17/TARC values in WT and Adam17ΔSox9 mice (N=8). (E) Flow cytometry analysis of IFN-γ, IL-4 and IL-17A production in CD4+ cells from lymph nodes, and (F, G) IL-17A and IL-22 production by CD3+ cells in epidermis of WT and Adam17ΔSox9 mice (N=8). TCRγδhigh cells represent dendritic epidermal T cells, and TCRγδmid cells represent γδ T cells that have infiltrated epidermis from the dermis. Data in B–G are shown as mean ± SD. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001 as determined by Student’s t test. See also Figures S1.
Barrier disruption, eczema, and the mixture of humoral and cellular inflammation observed in Adam17ΔSox9 mice closely resembled features of human AD and genetic diseases that mimic AD. Owing to their unimpaired survival rate, these mice were advantageous over previously reported mice with Adam17 ablated in Keratin 14-expressing tissues (Franzke et al., 2012).
Consistent with the eczematous phenotype, cytokine expression analysis via flow cytometry in skin draining lymph nodes revealed increases in the numbers of Th1 and Th2 cells and prominent increases in Th17 cells (Figure 1E). Epidermal T cell composition was strikingly altered. Dendritic epidermal T cells, a resident Vγ5+ γδ T cell subset in epidermis, were replaced by the massive infiltration of Vγ5neg γδ TCRmid T cells and CD4+ T cells (Figure 1F). Approximately 20% of Vγ5neg γδ TCRmid T cells expressed Vγ4, but the majority did not (not shown). Cytokine analysis of infiltrating T cells revealed that epidermal CD4 T cells consisted of Th17 and Th22 cells, and the majority of Vγ5neg γδ TCRmid T cells produced interleukin-17 (IL-17) (γδT17), a fraction of which also produced IL-22 (Figure 1G).
In addition to Th1 and Th2 responses in AD, emerging reports demonstrate the contributions of Th17 and Th22 (Gittler et al., 2012; Koga et al., 2008; Nograles et al., 2009; Suarez-Farinas et al., 2013), suggesting that immune responses in AD are complex. Relative contributions of T helper subpopulations have not been explored in depth, but the prominent IL-17 responses in Adam17ΔSox9 mice seemed atypical in the context of eczematous dermatitis. To investigate the role of IL-17 and its associated cytokine IL-23, both of which are important in psoriatic inflammation, we crossed Adam17ΔSox9 mice to IL-17- and IL-23-deficient backgrounds. The lack of each cytokine had no effect on the onset of eczematous dermatitis (Figure S1C), demonstrating that the phenomenon observed herein was distinct from psoriasis and that IL-17 responses in Adam17ΔSox9 mice are likely to be secondary responses. Consistently, Adam17 ΔSox9 mice exhibited striking pruritus, dry skin and eczematous dermatitis, elevated IgE concentrations and a mixture of helper T cell responses, phenotypes that are remarkably similar to symptoms generally observed in AD and eczematous dermatitis in human genetic diseases.
Adam17ΔSox9 mice exhibit altered skin microbiota
Given the resemblance of the Adam17ΔSox9 mice phenotype to human eczematous dermatitis, we swabbed and cultured skin surfaces of 8 week-old Adam17ΔSox9 mice and WT littermates. Striking outgrowth of S. aureus was obtained from Adam17ΔSox9 mice cultures, but not from WT littermates (Figure 2A). Quantifying colony-forming units showed that S. aureus colonization began 3 to 4 weeks after birth and increased thereafter (Figure 2B). Scanning electron microscopy and immunofluorescence microscopy revealed S. aureus within stratum corneum and within follicular openings (Figures 2C and Figure S2A–S2C).
Figure 2. Staphylococcus aureus-associated dysbiosis in Adam17ΔSox9 mouse skin.

(A, B) Quantification of S. aureus cultured from facial skin swab samples of WT and Adam17ΔSox9 mice (N=8, mean ± SD). (C) Scanning electron microscopy on skin surface of WT and Adam17ΔSox9 mice. Scale bars, 10 μm. (D) Microbiota of WT and Adam17ΔSox9 mice analyzed at indicated time points after birth. Relative abundance of order-genera that represented > 1% of total 16S rRNA gene sequences with additional speciation of Corynebacterium and Staphylococcus genus. Results represent two independent experiments (N=3). See also Figures S2.
Recent advances in microbiome analyses have revealed that varieties of bacterial communities inhabit mammalian body surfaces (Grice et al., 2009) and shape gut and skin immunity (Ivanov et al., 2009; Naik et al., 2012). To characterize microbiota during the onset of eczematous inflammation, we performed microbiome analyses in WT or Adam17ΔSox9 mice over a period of 2 to 14 weeks after birth. We carried out 454 pyrosequencing of the V1–V3 region of bacterial 16S rRNA genes. All WT littermate mice exhibited identical microbiota, and underwent similar changes that began around week 8 after birth (Figure 2D), perhaps simulating bacterial community shifts observed in human physiologic development across Tanner stages (Oh et al., 2012). S. aureus was extremely rare among the WT skin microbiota.
Microbiota of Adam17ΔSox9 mice were indistinguishable from WT mice at week 2 after birth, but underwent dramatic changes at week 3 to 4 (Figure 2D). Emergence of Corynebacterium mastitidis was initially observed, followed by S. aureus at approximately week 6, which appeared to coincide with the aggravation of skin symptoms (Figure S2D). Of note, C. mastitidis disappeared and was completely replaced by Corynebacterium bovis, immediately after the emergence of S. aureus (Figure 2D). Therefore, the skin microbiota of Adam17ΔSox9 mice undergo a sequential change where dysbiosis starts with the emergence of C. mastitidis and with S. aureus and C. bovis predominating later. The finding that dysbiosis includes S. aureus and Corynebacterium species recapitulates that which is seen in both human eczematous dermatitis and AD.
Dysbiosis drives eczematous inflammation
The above findings in Adam17ΔSox9 mice provided a unique opportunity to address the role of dysbiosis during eczematous inflammation. Based on antibiotic sensitivity profiles of S. aureus (strain SAAS9) and C. bovis (strain CBAS9) isolates from Adam17ΔSox9 mice, we elected to treat Adam17ΔSox9 mice with cefazolin and enrofloxacin because of their anti-bacterial activity against the isolated strains (Table S1), established use in human (Schneider et al., 2013) or veterinarian clinical practice (Paradis et al., 1990), and solubility, which enabled continuous feeding of mice with these antibiotics through drinking water. After weaning, each group of Adam17ΔSox9 mice was separated into cages that were given drinking water with or without antibiotics.
In striking contrast to untreated Adam17ΔSox9 mice, antibiotic-treated mice were almost completely protected from development of eczematous lesions (Figure S3A). Clinical scores, TEWL and serum IgE concentrations also remained lower than their untreated controls (Figures 3A–3C). FACS analysis revealed a reduction in the numbers of Th2 and Th17 cells in skin draining lymph nodes (Figure 3D). Consistent with this data, serum concentrations of IL-1β, IL-6, IL-13, IL-17A and IL-17F were decreased after antibiotic treatment, although CCL17/TARC remained high (Figure S3B). Antibiotics also normalized epidermal γδ T cell constituents (Figure S3C). CD4+ T cell numbers in epidermis of antibiotic-treated Adam17ΔSox9 mice remained high, but these remaining CD4+ T cells produced less IL-4 and did not produce IL-17A or IL-22 (Figure 3E and Figure S3D). A potential explanation is that the CD4+ T cells were attracted to epidermis via CCL17 that remained highly expressed, but were not primed to produce cytokines owing to the decrease in epidermal bacterial load.
Figure 3. Antibiotics prevent eczematous inflammation and conserves bacterial diversity.

Adam17ΔSox9 mice were either treated or untreated with antibiotics cocktail (Abx) since weaning. See also Figure S3. (A–C) Clinical score, TEWL and serum IgE concentrations (N=7). A C shown as mean ± SD. *P<0.05, **P<0.01, ***P<0.001, ****P <0.0001 as determined by Student’s t test. (D) Flow cytometry analysis of IFN-γ, IL-4 and IL-17A production in CD4+ cells from lymph nodes, and (E) IL-17A and IL-22 production by CD3+ cells in epidermis of WT and Adam17ΔSox9 mice (N=7). (F) Microbiota of Abx-treated and untreated Adam17ΔSox9 and WT mice at indicated time points after birth. Relative abundance plots of order-genera that represented >1% of total 16S rRNA gene sequences with additional speciation of Corynebacterium and Staphylococcus genus of 4 representative mice for each time point. Results are representative of two experiments. (G) Shannon diversity index of Abx-treated and untreated Adam17ΔSox9 and WT mice from week 2 to 8 after birth. Each time point represents mean ± SEM. *P<0.05, **P<0.01, ***P<0.001 determined by Student’s t test and adjusted for multiple comparison by Benjamini and Hochberg correction. (H) Principal coordinates analysis (PCoA) of the microbial community structure (Theta similarity index) of Abx-treated and untreated Adam17ΔSox9 and WT mice. A shorter distance between points indicates higher similarity between the microbiome of samples represented by these points. Biplot arrows indicate the two most significant unique consensus taxonomies contributing to variation along axes 1 and 2 as determined by Spearman correlation. Length of biplot arrows reflects the contribution of that taxa to the top two axes: a, S. aureus (r=−0.73, P<1e-15); b, C. bovis (r=−0.77, P<1e-15). Spearman correlations and P-values refer to axis 1.
Microbiome analysis revealed higher bacterial diversity in antibiotic-treated Adam17ΔSox9 mice than in untreated mice, and dramatic reductions in the proportions of S. aureus were observed (Figures 3F and G and Figure S3E). C. bovis proportions were also reduced, but to a lesser extent when compared to the changes observed for S. aureus (Figures 3F and 3G and Figure S3E). The bacterial community structure in untreated Adam17ΔSox9 mice and controls diverged over time but was more similar after antibiotic treatment (Figures 3G and 3H).
We further investigated the association of dysbiosis with eczematous inflammation by performing protocol crossover studies. A group of Adam17ΔSox9 mice was left untreated up to week 10 after birth, and then subsequently received antibiotic therapy. Conversely, another group of Adam17ΔSox9 mice was maintained on antibiotics from week 3 to 10, which were discontinued thereafter (Figure S4A). Strikingly, initiation of antibiotics in previously untreated Adam17ΔSox9 mice extinguished pre-established eczematous dermatitis and inflammation (Figures 4A–C and Figure S4A) and reversed dysbiosis (Figure 4D and Figure S4B), indicating that targeting dysbiosis had a therapeutic effect. Furthermore, although mice receiving continuous antibiotics were prevented from developing eczematous lesions, withdrawal of antibiotics led to marked skin inflammation with rapid emergence of C. bovis and S. aureus (Figures 4A–D and Figures S4A and S4B). These data collectively establish dysbiosis as a pivotal event during the onset of eczematous dermatitis in Adam17ΔSox9 mice.
Figure 4. Antibiotics have therapeutic effect in Adam17ΔSox9 mice.

Adam17ΔSox9 mice were untreated from 3~9 weeks after birth, and were put on antibiotics from 10~13 weeks, or vice versa. See also Figure S4. (A, B) Clinical score and TEWL in Adam17ΔSox9 mice during the crossover protocol (N=7). (C) IL-17A and IL-22 production by CD3+ cells in epidermis of Adam17ΔSox9 mice during the crossover protocol (N=7). (D) Relative abundance of order-genera that represented >1% of total 16S rRNA sequences with additional speciation of Corynebacterium and Staphylococcus genus before and after crossover. The data in A–C are shown as the mean ± SD. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001 determined by Student’s t test.
S. aureus and Corynebacterium species contribute to eczematous dermatitis formation
We next explored which of the three bacterial species that constituted dysbiotic flora in Adam17ΔSox9 mice contributed to the pathogenesis of eczematous inflammation. Meta-analysis of clinical score, TEWL, and IgE in mice described in Figures 1 and 3 was performed. As anticipated from the association of S. aureus in AD, and concordant with our observation that S. aureus emerged immediately prior to the onset of eczematous dermatitis (Figure S2D), formation of eczematous dermatitis in Adam17ΔSox9 mice correlated with the proportion of S. aureus and C. bovis (Figures S5A and S5B). IgE concentrations correlated with only C. bovis frequency (Figure S5A). No positive correlation of C. mastitidis with any of the factors studied herein was found.
To assess the contribution of each bacterial species in vivo, we utilized Adam17ΔSox9 mice that were pre-treated with antibiotics. Adam17ΔSox9 mice treated with the antibiotic cocktail showed minimal signs of eczema (Figures S3A and S4A). After termination of treatment, there exists an almost 2-week period before overt appearance of eczematous dermatitis. Taking advantage of this window, we inoculated SAAS9, CBAS9, or a C. mastitidis strain (CMAS9, isolated from Adam17ΔSox9 mice) onto Adam17ΔSox9 mice immediately after termination of antibiotics (Figure S5C). Strikingly, and consistent with the meta-analysis, inoculation of mice with SAAS9 induced the most severe eczematous dermatitis (Figures 5A and S5D). Inoculation of CBAS9 also induced enhanced eczematous dermatitis, albeit to a lesser extent than SAAS9 (Figures 5A and S5D). Clinical scores of mice that were inoculated with CMAS9 were variable and did not reach statistical significance when compared to un-inoculated control Adam17ΔSox9 mice (Figures 5A and S5D).
Figure 5. Contribution of S. aureus, C. bovis and C. mastitidis to eczematous inflammation.
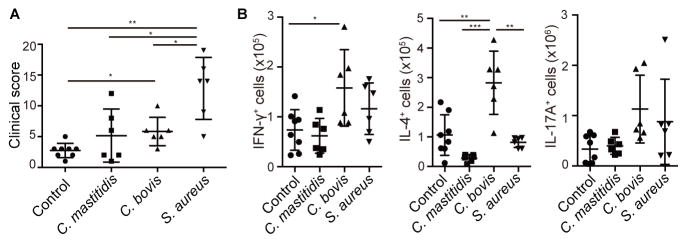
After withdrawal of antibiotic treatment form Adam17ΔSox9 mice that were pretreated with antibiotics, S. aureus, C. bovis and C. mastitidis strains were inoculated onto the mice. See also Figure S5. (A) Clinical scores of Adam17ΔSox9 mice after bacterial inoculation (N=6–8). (B) Flow cytometry analysis of IFN-γ, IL-4 and IL-17A production in CD4+ cells from lymph nodes of bacteria-inoculated Adam17ΔSox9 mice (N=6–8). The data are shown as the mean ± SD. *P<0.05, **P<0.01, ***P<0.001 determined by Student’s t test.
T helper responses in skin-draining lymph nodes were analyzed. Surprisingly, SAAS9-inoculation did not have an impact on the numbers of Th1 or Th2 cells (Figure 5B). In contrast, increased Th1 and Th2 responses, in particular the latter, were observed in skin-draining lymph nodes of mice inoculated with CBAS9 (Figure 5B). Although both SAAS9 and CBAS9 showed trends for enhancing Th17 responses, they did not reach not statistical significance. CMAS9 had no effect on T helper responses that were analyzed.
These results identified S. aureus as potent inducer of eczematous dermatitis via an immunological pathway that has yet to be determined. C. bovis prominently enhanced Th2 responses, providing explanation for elevated serum IgE concentrations in Adam17ΔSox9 mice. Taken together, S. aureus and C. bovis contribute to different aspects of eczematous inflammation in this model.
Ablation of EGFR-signaling allows dysbiosis
Important roles of EGFR signaling in regulating skin immune homeostasis have begun to be characterized, either in the context of AD (Saaf et al., 2012; Zhang et al., 2014) or EGFR-inhibitors (Lacouture, 2006). EGFR ligands are major substrates of ADAM17. ADAM17 cleaves the membrane-bound pro-forms of EGFR ligands including heparin-binding EGF-like growth factor and TGFα, and releases soluble forms to activate EGFR signaling pathways. It was recently shown that conditional ablation of EGFR from epidermis led to a skin inflammatory phenotype that was associated with barrier defects (Lichtenberger et al., 2013; Mascia et al., 2013). We therefore explored whether impaired EGFR signaling downstream of ADAM17 contributed to dysbiosis and eczematous dermatitis formation.
We generated Egfrfl/flSox9-Cre (EgfrΔSox9) mice that did not express Egfr in Sox9-expressing tissues. Consistent with other reports, EgfrΔSox9 mice exhibited eczematous phenotypes that were similar to, but slightly less severe than, those in Adam17ΔSox9 mice (Figures 6A and S6). TEWL and serum IgE concentrations were elevated in comparison to WT mice (Figures 6B and 6C). Inflammatory profiles were also similar to Adam17ΔSox9 mice, where EgfrΔSox9 mice exhibited mixed Th1, 2 and 17 accumulations in lymph nodes and Th17, Th22 and Tγδ17 inflammation in skin (Figures 6D and 6E) The numbers of IL-22-producing epidermal T cells were not as prominent in EgfrΔSox9 mice as in Adam17ΔSox9 mice (Figure 6E).
Figure 6. Ablation of EGFR-signaling induces dysbiosis and eczematous inflammation.

(A) Clinical scores in Adam17ΔSox9 and EgfrΔSox9 mice (N=7). See also Figure S6. (B, C) TEWL and serum IgE values in WT and EgfrΔSox9 mice (N=7). (D) Flow cytometry analysis of IFN-γ, IL-4 and IL-17A production in CD4+ cells from lymph nodes, and (E) IL-17A and IL-22 production by CD3+ cells in epidermis of WT and EgfrΔSox9 mice (N=7). (F, G) Quantification of S. aureus cultured from facial skin swab samples of WT and EgfrΔSox9 mice (N=7). (H) Microbiota of 8-week-old WT and Egfr17ΔSox9 mice.
Skin swab cultures from 8-week-old EgfrΔSox9 mice isolated S. aureus (Figure 6F and 6G), and microbiome analysis revealed the emergence of Corynebacterium spp and S. aureus (Figure 6H). Dysbiotic changes were less prominent than those of Adam17ΔSox9 mice, but nevertheless document impairment of EGFR signaling as an important pathway downstream of ADAM17 that allows the overgrowth of S. aureus and Corynebacterium species.
Taken together, ADAM17 regulates homeostasis of cutaneous microbial communities in an EGFR-dependent manner. Further elucidation of mechanisms downstream of EGFR signaling should provide information on molecules and cells that could be considered to be potential targets to normalize dysbiosis.
Langerhans cells mediate innate responses to S. aureus
In contrast to a colitis model in which gut dysbiosis and disease can be transferred to co-housed WT mice (Elinav et al., 2011), co-housing WT mice with Adam17ΔSox9 mice only led to temporal colonization of the former with S. aureus and did not transfer skin disease (data not shown). This suggested that S. aureus alone was insufficient to cause eczematous inflammation, and that it was incapable of persistently colonizing normal skin. Therefore, to determine whether S. aureus could induce immune responses through an impaired stratum corneum barrier, we utilized Flg−/− mice that have a defined barrier defect (Kawasaki et al., 2012). Filaggrin is a major structural protein in the stratum corneum, and FLG mutations are associated with human AD (Palmer et al., 2006). However, Flg−/− mice do not develop spontaneous dermatitis under normal SPF conditions, but exhibit enhanced percutaneous sensitization (Kawasaki et al., 2012). Epidermal tight junctions are intact in these mice (unpublished data).
S. aureus (SAAS9) isolated from Adam17ΔSox9 mice were topically inoculated onto the shaved back skin of Flg−/− and WT mice. Although topical inoculation did not induce overt dermatitis, it induced robust skin infiltration of TH17 and γδT17 cells into epidermis in Flg−/−, but not WT mice (Figure 7A), demonstrating that barrier disruption in the stratum corneum was sufficient for enhanced immune responses to topically inoculated S. aureus.
Figure 7. Langerhans cells mediate IL-17 response upon S. aureus inoculation.

(A) Flow cytometry analysis of epidermis-infiltrating CD4 T cells and γδ T cells in topical inoculation of S. aureus on WT, Flg−/− and Flg−/− Langerin-DTA mice (N=6). (B, C) Area of inflamed lesions and epidermis-infiltrating γδ T cells of WT, Flg−/− and Flg−/− Langerin-DTA mice inoculated with S. aureus via occlusive dressing technique. The data are shown as the mean ± SD. *P<0.05, **P<0.01 determined by Student’s t test. See also Figure S7.
Langerhans cells (LC) are unique dendritic cells in the epidermis (Merad et al., 2008). Through a series of studies, we have demonstrated that LC take up microbial antigens that have breached the stratum corneum, but exist outside of tight junctions, by extending their dendrites through intact tight junction barrier and induce antigen-specific humoral (IgG1) responses (Nagao et al., 2009; Kubo et al., 2009; Ouchi et al., 2011). When the epidermal barriers are breached during Candida infection, LC are capable of inducing Th17 responses (Igyarto et al., 2011).
During dysbiosis that occur in stratum corneum (Figures S2A–C), LC are advantageously positioned to acquire those bacteria through epidermal tight junctions. To determine if LC contributed to immunity against S. aureus, we generated Flg−/− mice that lacked LC, by crossing them to mice that express diphtheria toxin fragment A driven by the human langerin promoter (Langerin-DTA mice) (Kaplan et al., 2005). Of note, Th17 and γδT17 responses induced by topical SAAS9-inoculation were abrogated in Flg−/− Langerin-DTA mice (Figure 7A), demonstrating a role for LC in mediating IL-17 responses against S. aureus.
Because topical SAAS9 exposure did not generate dermatitis, we inoculated SAAS9 via an occlusive dressing technique that led to the formation of visible dermatitis in WT mice (Figure S7) that lasted for approximately 3 days. Consistent with the immune responses in the topical exposure protocol, dermatitis was aggravated in Flg−/− mice. However, dermatitis was attenuated in Flg−/− Langerin-DTA mice, and IL-17A responses were nearly abrogated (Figures 7B,7C and S7). IL-22 responses were not observed in any setting (Data not shown). The transient nature of dermatitis in this assay indicates that S. aureus and barrier disruption are not sufficient to cause chronic disease. Nevertheless, these data demonstrated that LC facilitated innate immune responses against S. aureus, presumably by acquiring components of S. aureus through the tight junction barrier. This process likely represents an early immunological event in the initial phases of dysbiosis and skin inflammation.
Discussion
Eczematous inflammation in Adam17ΔSox9 mice occurred in conjunction with a striking shift in microbiota, in which S. aureus and Corybacterium spp were major constituents. This dysbiotic pattern closely recapitulated those seen in human AD (Kong et al., 2012) and primary immunodeficiencies with eczematous dermatitis (Oh et al., 2013). In particular, S. aureus colonization is universal in AD patients, and it has been long debated whether emergence of S. aureus is a secondary by-product of chronic inflammation or whether it is a primary component of pathogenesis that actively contributes to eczema development. In Adam17ΔSox9 mice, dysbiosis was a pivotal event during eczematous dermatitis formation, and antibiotics-mediated normalization of dysbiosis both prevented onset and suppressed active inflammation. Furthermore, bacterial inoculation experiments in Adam17ΔSox9 mice revealed that eczematous inflammation could be accelerated by both S. aureus and C. bovis (S. aureus > C. bovis). C. bovis was the only agent that induced prominent Th2 responses, suggesting differential contribution of S. aureus and C. bovis to the complex eczematous phenotype. Generation of gnotobiotic mice would be required to determine whether each agent is sufficient to induce full-blown disease in the absence of each other, and to determine immunological pathways induced by S. aureus inoculation that are critical for eczematous dermatitis formation. Although a role for C. mastitidis was not revealed in the current study, it is attractive to hypothesize that this organism might enhance the growth of S. aureus and C. bovis.
The importance of IL-17 responses is well established in psoriasis in both mice and humans (Lowes et al., 2014). Recent studies report the involvement of IL-17 responses also in human AD (Koga et al., 2008; Suarez-Farinas et al., 2013), but its relative contribution to AD pathogenesis is not yet determined. Although Adam17ΔSox9 mice did not exhibit macroscopic features that were reminiscent of psoriasis, the robust IL-17 responses prompted us to exclude psoriatic inflammation. The lack of improvement in Adam17ΔSox9 mice in both IL-17 and IL-23 backgrounds indicate that the pathophysiology of the skin inflammation is distinct from that of psoriasis and that these cytokines are not essential during an eczematous response. It is possible that the IL-17 responses observed here arise secondarily to dysbiosis, and perhaps, might be a consequence of utilizing mouse models.
While mutations in the gene encoding the structural stratum corneum protein Filaggrin are considered major predisposing factors in “classic” AD (Irvine et al., 2011), a variety of other genes are involved, and it is possible that further elucidation of involved genes might subdivide this heterogeneous disease. Whether or not eczematous dermatitis seen in Adam17ΔSox9 mice can be extrapolated to human diseases remains to be determined. Importantly, the mouse model studied herein is an actual counterpart of a rare human disease that manifests eczematous dermatitis (Blaydon et al., 2011), and therefore represents a prototypic eczematous dermatitis model. Dry skin, pruritus, elevated serum IgE, and naturally occurring dysbiosis in Adam17ΔSox9 mice recapitulate the most important aspects of human eczematous dermatitis and AD.
A systematic review of human studies failed to provide strong evidence for recommending interventions to reduce S. aureus in AD (Bath-Hextall et al., 2010). However, outcomes of antibiotic therapy in humans may be variable due to unconfirmed presence of dysbiosis at the start of the studies and as of yet suboptimal combinations and/or dose of antibiotics. From this viewpoint, future studies might require pre-treatment microbiome analyses. Furthermore, intensified antibiotic cocktails might be necessary to target skin-colonizing S. aureus, as is done for Helicobacter pylori eradication (Chey and Wong, 2007; Malfertheiner et al., 2007). Thus, antibiotic data currently available for humans is inconclusive and requires further evaluation.
We demonstrated that targeting dysbiosis via antibiotic cocktails had dramatic effects on eczematous lesions of Adam17ΔSox9 mice. However, administering an antibiotic cocktail for a prolonged period of time, as we have done experimentally, is not likely to be a practical approach in patients. Long-term oral antibiotic therapy would have undesirable effects on microbiota in the gut and other distal sites (Cho et al., 2012; Willing et al., 2011) and could also select antibiotic-resistant bacteria (Sommer and Dantas, 2011; Wright, 2007). Meanwhile, bleach baths, a common clinical practice that is receiving increasing scientific interest, is recommended for patients with moderate to severe AD to reduce disease severity (Eichenfield et al., 2014; Huang et al., 2009; Leung et al., 2013), and could potentially be targeting dysbiosis. In combination with the findings in the current work, mechanisms of this classic therapy might deserve further exploration. The finding that dysbiosis occurs in part via impaired EGFR signaling might provide foundation for novel strategies to modify the skin microbiota.
In conclusion, the findings in this study document dysbiosis as a pathological factor that can drive eczematous inflammation in mice. Utilizing this unique model, it will be possible to further explore molecular mechanisms in epidermis that result in dysbiosis and inflammation, and to design strategies that normalize them long-term. Accomplishment of these in a skin-specific manner will lead to safe and effective therapies and should have profound impact on the treatment of AD, as well as other forms of eczematous dermatitis and perhaps skin rash associated with EGFR inhibitor use.
Experimental Procedures
Details of experimental procedures on microbiome analysis are included in Supplemental Information.
Mice
Adam17ΔSox9 mice and EgfrΔSox9 mice were generated by crossing Adam17flox/flox mice or Egfrflox/flox mice (kind gift from Dr. David Threadgill, University of North Carolina) with Sox9-Cre mice as previously described (Horiuchi et al., 2009; Nagao et al., 2012). Mice were backcrossed into the C57BL/6 background for at least 6 generations. IL-17−/− and IL-23p19−/− mice were generously provided by Dr. A. Yoshimura (Keio University School of Medicine, Tokyo, Japan). The generation of Flg−/− mice and Langerin-DTA mice has been described (Kaplan et al., 2005; Kawasaki et al., 2012). All mice were bred and housed in a specific pathogen free condition. All animal experimental procedures and study protocols were approved by the Ethical Committee for Animal Experiments of the Keio University School of Medicine.
Preparation of epidermal cell suspensions
Epidermal cell suspensions were prepared from mouse trunk skin. Shaved trunk skin was incubated with 0.15% trypsin and 0.27 mM EDTA at 37°C for 45 minutes. Epidermal cells were scraped off in 5% FBS PBS, washed and then filtered through cell strainers (BD).
Flow cytometry analysis and antibodies
For cytokine analysis, cell suspensions were incubated for 5 hours in complete RPMI with 50 ng/ml PMA and 500 ng/ml Ionomycin in the presence of 5 μg/ml Brefeldin A. Cells were stained with LIVE/DEAD Fixable Dead Cell Stain Kits (Invitrogen) and surface markers, followed by fixation and permeabilization with BD Cytofix/Cytoperm (BD) and intracellular cytokine staining. The following antibodies from Biolegend or eBioscience were used for analysis: anti-CD45 (30-F11), anti-CD3 (145-2C11), anti-CD4 (GK1.5), anti-TCRγδ (GL3), anti-IL-17A (TC11-18H10.1), anti-IL-4 (11B11), anti-IFN-γ (XMG1.2) and anti-IL-22 (1H8PWSR).
Antibiotic treatment
Mice were treated with 0.5 mg/ml cefazolin (Nichi-Iko Pharmaceutical Co., Ltd.) and 0.5mg/ml enrofloxacin (Bayer HealthCare) in drinking water.
Histological analysis and immunofluorescence microscopy
Skin from the facial area of sacrificed mice were taken for hematoxylin and eosin and toluidine blue staining Immunofluorescence microscopy was performed to detect S. aureus with biotinylated rabbit polyclonal anti-S. aureus antibody (abcam) which was detected with AlexaFluor 488-conjugated anti-biotin secondary antibody (Invitrogen).
Electron microscopy
For scanning electron microscopy, skin taken from the facial area was fixed in 2% glutaraldehyde, post-fixed with osmium tetroxide, replaced with isoamyl acetate and were observed by SV6600 (Hitachi). For transmission electron microscopy, skin samples were pre-fixed with 2% glutaraldehyde, post-fixed in osmium tetroxide and were embedded in epoxy resin. Ultrathin sections were prepared and were observed by JEM-1230 (JEOL).
IgE and Cytokine analysis
Serum total IgE were measured by Mouse IgE ELISA quantitative set (Bethyl Laboratories). Cytokine concentrations were measured by Milliplex Map Kit (Millipore), and serum CCL17/TARC were detected by Mouse CCL17/TARC Quantikine (R&D Systems).
Clinical score
Clinical severity of skin lesions was scored with a method adapted from a scoring index of canine atopic dermatitis (Plant et al., 2012). In brief, the clinical score was measured by assessing the severity and extent of lesions (erythema, excoriation, erosion, alopecia, lichenification and hyperpigmentation) at head and pinnae, forefeet, hind feet, ventral thorax and axillae, ventral abdomen and inguinal, and graded as 0 (none), 1 (mild), 2,3 (moderate), or 4,5 (severe and extensive lesions). The total index ranges from 0 to 50.
Trans epidermal water loss (TEWL)
TEWL was measured with a VAPOSCAN AS-VT100RS machine (Asahi Biomed).
Bacterial culture
Cultures were obtained with swabs from a cheek of mice and plated on Mannitol salt agar with egg yolk (BD) or Trypticase soy agar with 5% sheep blood (BD). To measure CFUs of S. aureus, a standard plate count method was performed. Briefly, skin swabs from a cheek of mice (1 cm2) were placed into 1 ml of PBS, and serial dilutions were prepared. 100 μl of each dilution was transferred on Mannitol salt agar with egg yolk plates (BD), and colonies were counted after 2 day-incubation. CFUs were calculated by dividing the number of colonies per plate by the dilution factor.
Bacterial isolation and inoculation onto Adam17 Sox9 mice
C. mastitidis and C. bovis were isolated from Adam17 Sox9 mice on Trypticase soy agar with 5% sheep blood plates (BD), and S. aureus was isolated by culturing on Mannitol salt agar with egg yolk plates (BD), for consistency. Isolated strains were designated as SAAS9 (S. aureus), CBAS9 (C. bovis), and CMAS9 (C. mastitidis). All analyzed S. aureus isolates had sequences that were identical to that of ST88 clone via multilocus sequence typing (MLST) (http://saureus.mlst.net), therefore likely representing a single clone. 10 colonies of each bacteria species were picked and dissolved in 0.4 ml sterile PBS. Bacteria containing PBS or control PBS were inoculated with cotton swabs on facial area of mice. The mice were pre-treated with antibiotics from 3 weeks after birth to 8–10 weeks, and inoculation of bacteria was started after withdrawal of antibiotic treatment. The inoculation was performed from day 0 to day 6, and the mice were analyzed on day 8.
SAAS9 inoculation onto Flg−/− mice
SAAS9 was placed into tryptic soy broth (BD) and grown overnight at 37°C in a shaking incubator. Exponential-phase bacteria were obtained after a one-hour subculture of a 1:50 dilution of the overnight culture. Bacterial concentrations were estimated by measuring the absorbance of at 600 nm. Bacterial cells were pelleted and washed 3 times in PBS. 4×107 colony forming units of SAAS9 in 100μl of PBS were topically inoculated on shaved back skin of mice. SAAS9 inoculation was performed at day 1, 3, 5, 7 and 9, and the mice were examined at day 10. For inoculation via occlusive dressing, 4×107 colony forming units of SAAS9 were applied, and the back skin of mice was covered with Tegaderm™ Roll (Sumitomo 3M Limited). The dressing was removed at day 4, and the mice were examined at day 7. Area of skin lesions were photographed and measured by Photoshop CS5.1 (Adobe).
Supplementary Material
Acknowledgments
This work was supported by Research for Prevention and Treatment of Immune/Allergic Diseases from the Ministry of Health, Labor and Welfare of Japan, Grant-in-Aid for JSPS Fellows and the US National Institutes of Health (NIH) NCI Intramural Research Programs. We thank Julie A. Segre and Mark C. Udey for helpful discussions; Cynthia Ng, Morgan Park, and Sean Conlan for underlying efforts.
Footnotes
Assession Numbers
The sequencing data have been submitted to NCBI BioProject (http://www.ncbi.nlm.nih.gov/bioproject) under accession number 30121.
Author Contributions
T.K. and K.N. designed the study, performed experiments and wrote the paper. M.G. and H.H.K performed experiments and discussed the data. K.H., H.K. provided mice and discussed the data. M.A. discussed data and provided administrative support. H.A. and D.H.K provided mice.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bath-Hextall FJ, Birnie AJ, Ravenscroft JC, Williams HC. Interventions to reduce Staphylococcus aureus in the management of atopic eczema: an updated Cochrane review. Br J Dermatol. 2010;163:12–26. doi: 10.1111/j.1365-2133.2010.09743.x. [DOI] [PubMed] [Google Scholar]
- Blaydon DC, Biancheri P, Di WL, Plagnol V, Cabral RM, Brooke MA, van Heel DA, Ruschendorf F, Toynbee M, Walne A, et al. Inflammatory skin and bowel disease linked to ADAM17 deletion. N Engl J Med. 2011;365:1502–1508. doi: 10.1056/NEJMoa1100721. [DOI] [PubMed] [Google Scholar]
- Blobel CP. ADAMs: key components in EGFR signalling and development. Nat Rev Mol Cell Biol. 2005;6:32–43. doi: 10.1038/nrm1548. [DOI] [PubMed] [Google Scholar]
- Chavanas S, Bodemer C, Rochat A, Hamel-Teillac D, Ali M, Irvine AD, Bonafe JL, Wilkinson J, Taieb A, Barrandon Y, et al. Mutations in SPINK5, encoding a serine protease inhibitor, cause Netherton syndrome. Nat Genet. 2000;25:141–142. doi: 10.1038/75977. [DOI] [PubMed] [Google Scholar]
- Chey WD, Wong BC. American College of Gastroenterology guideline on the management of Helicobacter pylori infection. Am J Gastroenterol. 2007;102:1808–1825. doi: 10.1111/j.1572-0241.2007.01393.x. [DOI] [PubMed] [Google Scholar]
- Cho I, Yamanishi S, Cox L, Methe BA, Zavadil J, Li K, Gao Z, Mahana D, Raju K, Teitler I, et al. Antibiotics in early life alter the murine colonic microbiome and adiposity. Nature. 2012;488:621–626. doi: 10.1038/nature11400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eichenfield LF, Tom WL, Berger TG, Krol A, Paller AS, Schwarzenberger K, Bergman JN, Chamlin SL, Cohen DE, Cooper KD, et al. Guidelines of care for the management of atopic dermatitis: section 2. Management and treatment of atopic dermatitis with topical therapies. J Am Acad Dermatol. 2014;71:116–132. doi: 10.1016/j.jaad.2014.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eilers RE, Jr, Gandhi M, Patel JD, Mulcahy MF, Agulnik M, Hensing T, Lacouture ME. Dermatologic infections in cancer patients treated with epidermal growth factor receptor inhibitor therapy. J Natl Cancer Inst. 2010;102:47–53. doi: 10.1093/jnci/djp439. [DOI] [PubMed] [Google Scholar]
- Elinav E, Strowig T, Kau AL, Henao-Mejia J, Thaiss CA, Booth CJ, Peaper DR, Bertin J, Eisenbarth SC, Gordon JI, et al. NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell. 2011;145:745–757. doi: 10.1016/j.cell.2011.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franzke CW, Cobzaru C, Triantafyllopoulou A, Loffek S, Horiuchi K, Threadgill DW, Kurz T, van Rooijen N, Bruckner-Tuderman L, Blobel CP. Epidermal ADAM17 maintains the skin barrier by regulating EGFR ligand-dependent terminal keratinocyte differentiation. J Exp Med. 2012;209:1105–1119. doi: 10.1084/jem.20112258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gittler JK, Shemer A, Suarez-Farinas M, Fuentes-Duculan J, Gulewicz KJ, Wang CQ, Mitsui H, Cardinale I, de Guzman Strong C, Krueger JG, et al. Progressive activation of T(H)2/T(H)22 cytokines and selective epidermal proteins characterizes acute and chronic atopic dermatitis. J Allergy Clin Immunol. 2012;130:1344–1354. doi: 10.1016/j.jaci.2012.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grice EA, Kong HH, Conlan S, Deming CB, Davis J, Young AC, Bouffard GG, Blakesley RW, Murray PR, Green ED, et al. Topographical and temporal diversity of the human skin microbiome. Science. 2009;324:1190–1192. doi: 10.1126/science.1171700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horiuchi K, Kimura T, Miyamoto T, Miyamoto K, Akiyama H, Takaishi H, Morioka H, Nakamura T, Okada Y, Blobel CP, et al. Conditional inactivation of TACE by a Sox9 promoter leads to osteoporosis and increased granulopoiesis via dysregulation of IL-17 and G-CSF. J Immunol. 2009;182:2093–2101. doi: 10.4049/jimmunol.0802491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang JT, Abrams M, Tlougan B, Rademaker A, Paller AS. Treatment of Staphylococcus aureus colonization in atopic dermatitis decreases disease severity. Pediatrics. 2009;123:e808–814. doi: 10.1542/peds.2008-2217. [DOI] [PubMed] [Google Scholar]
- Igyarto BZ, Haley K, Ortner D, Bobr A, Gerami-Nejad M, Edelson BT, Zurawski SM, Malissen B, Zurawski G, Berman J, et al. Skin-resident murine dendritic cell subsets promote distinct and opposing antigen-specific T helper cell responses. Immunity. 2011;35:260–272. doi: 10.1016/j.immuni.2011.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irvine AD, McLean WHI, Leung DYM. Filaggrin Mutations Associated with Skin and Allergic Diseases. N Engl J Med. 2011;365:1315–1327. doi: 10.1056/NEJMra1011040. [DOI] [PubMed] [Google Scholar]
- Ivanov II, Atarashi K, Manel N, Brodie EL, Shima T, Karaoz U, Wei D, Goldfarb KC, Santee CA, Lynch SV, et al. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell. 2009;139:485–498. doi: 10.1016/j.cell.2009.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakinuma T, Nakamura K, Wakugawa M, Mitsui H, Tada Y, Saeki H, Torii H, Asahina A, Onai N, Matsushima K, et al. Thymus and activation-regulated chemokine in atopic dermatitis: Serum thymus and activation-regulated chemokine level is closely related with disease activity. J Allergy Clin Immunol. 2001;107:535–541. doi: 10.1067/mai.2001.113237. [DOI] [PubMed] [Google Scholar]
- Kaplan DH, Jenison MC, Saeland S, Shlomchik WD, Shlomchik MJ. Epidermal Langerhans Cell-Deficient Mice Develop Enhanced Contact Hypersensitivity. Immunity. 2005;23:611–620. doi: 10.1016/j.immuni.2005.10.008. [DOI] [PubMed] [Google Scholar]
- Kawasaki H, Nagao K, Kubo A, Hata T, Shimizu A, Mizuno H, Yamada T, Amagai M. Altered stratum corneum barrier and enhanced percutaneous immune responses in filaggrin-null mice. J Allergy Clin Immunol. 2012;129:1538–1546. doi: 10.1016/j.jaci.2012.01.068. [DOI] [PubMed] [Google Scholar]
- Koga C, Kabashima K, Shiraishi N, Kobayashi M, Tokura Y. Possible pathogenic role of Th17 cells for atopic dermatitis. J Invest Dermatol. 2008;128:2625–2630. doi: 10.1038/jid.2008.111. [DOI] [PubMed] [Google Scholar]
- Kong HH, Oh J, Deming C, Conlan S, Grice EA, Beatson MA, Nomicos E, Polley EC, Komarow HD, Murray PR, et al. Temporal shifts in the skin microbiome associated with disease flares and treatment in children with atopic dermatitis. Genome Res. 2012;22:850–859. doi: 10.1101/gr.131029.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubo A, Nagao K, Amagai M. Epidermal barrier dysfunction and cutaneous sensitization in atopic diseases. J Clin Invest. 2012;122:440–447. doi: 10.1172/JCI57416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubo A, Nagao K, Yokouchi M, Sasaki H, Amagai M. External antigen uptake by Langerhans cells with reorganization of epidermal tight junction barriers. J Exp Med. 2009;206:2937–2946. doi: 10.1084/jem.20091527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacouture ME. Mechanisms of cutaneous toxicities to EGFR inhibitors. Nat Rev Cancer. 2006;6:803–812. doi: 10.1038/nrc1970. [DOI] [PubMed] [Google Scholar]
- Leung TH, Zhang LF, Wang J, Ning S, Knox SJ, Kim SK. Topical hypochlorite ameliorates NF-kappaB-mediated skin diseases in mice. J Clin Invest. 2013;123:5361–5370. doi: 10.1172/JCI70895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leyden JJ, Marples RR, Kligman AM. Staphylococcus aureus in the lesions of atopic dermatitis. Br J Dermatol. 1974;90:525–525. doi: 10.1111/j.1365-2133.1974.tb06447.x. [DOI] [PubMed] [Google Scholar]
- Lichtenberger BM, Gerber PA, Holcmann M, Buhren BA, Amberg N, Smolle V, Schrumpf H, Boelke E, Ansari P, Mackenzie C, et al. Epidermal EGFR controls cutaneous host defense and prevents inflammation. Sci Transl Med. 2013;5:199ra111. doi: 10.1126/scitranslmed.3005886. [DOI] [PubMed] [Google Scholar]
- Lowes MA, Suarez-Farinas M, Krueger JG. Immunology of psoriasis. Annu Rev Immunol. 2014;32:227–255. doi: 10.1146/annurev-immunol-032713-120225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malfertheiner P, Megraud F, O’Morain C, Bazzoli F, El-Omar E, Graham D, Hunt R, Rokkas T, Vakil N, Kuipers EJ. Current concepts in the management of Helicobacter pylori infection: the Maastricht III Consensus Report. Gut. 2007;56:772–781. doi: 10.1136/gut.2006.101634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mascia F, Lam G, Keith C, Garber C, Steinberg SM, Kohn E, Yuspa SH. Genetic ablation of epidermal EGFR reveals the dynamic origin of adverse effects of anti-EGFR therapy. Sci Transl Med. 2013;5:199ra110. doi: 10.1126/scitranslmed.3005773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merad M, Ginhoux F, Collin M. Origin, homeostasis and function of Langerhans cells and other langerin-expressing dendritic cells. Nat Rev Immunol. 2008;8:935–947. doi: 10.1038/nri2455. [DOI] [PubMed] [Google Scholar]
- Minegishi Y, Saito M, Tsuchiya S, Tsuge I, Takada H, Hara T, Kawamura N, Ariga T, Pasic S, Stojkovic O, et al. Dominant-negative mutations in the DNA-binding domain of STAT3 cause hyper-IgE syndrome. Nature. 2007;448:1058–1062. doi: 10.1038/nature06096. [DOI] [PubMed] [Google Scholar]
- Murthy A, Shao YW, Narala SR, Molyneux SD, Zuniga-Pflucker JC, Khokha R. Notch activation by the metalloproteinase ADAM17 regulates myeloproliferation and atopic barrier immunity by suppressing epithelial cytokine synthesis. Immunity. 2012;36:105–119. doi: 10.1016/j.immuni.2012.01.005. [DOI] [PubMed] [Google Scholar]
- Nagao K, Ginhoux F, Leitner WW, Motegi S, Bennett CL, Clausen BE, Merad M, Udey MC. Murine epidermal Langerhans cells and langerin-expressing dermal dendritic cells are unrelated and exhibit distinct functions. Proc Natl Acad Sci U S A. 2009;106:3312–3317. doi: 10.1073/pnas.0807126106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagao K, Kobayashi T, Ohyama M, Akiyama H, Horiuchi K, Amagai M. Brief report: requirement of TACE/ADAM17 for hair follicle bulge niche establishment. Stem Cells. 2012;30:1781–1785. doi: 10.1002/stem.1153. [DOI] [PubMed] [Google Scholar]
- Naik S, Bouladoux N, Wilhelm C, Molloy MJ, Salcedo R, Kastenmuller W, Deming C, Quinones M, Koo L, Conlan S, et al. Compartmentalized control of skin immunity by resident commensals. Science. 2012;337:1115–1119. doi: 10.1126/science.1225152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nograles KE, Zaba LC, Shemer A, Fuentes-Duculan J, Cardinale I, Kikuchi T, Ramon M, Bergman R, Krueger JG, Guttman-Yassky E. IL-22-producing “T22” T cells account for upregulated IL-22 in atopic dermatitis despite reduced IL-17-producing TH17 T cells. J Allergy Clin Immunol. 2009;123:1244–1252. e1242. doi: 10.1016/j.jaci.2009.03.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh J, Conlan S, Polley EC, Segre JA, Kong HH. Shifts in human skin and nares microbiota of healthy children and adults. Genome Med. 2012;4:77. doi: 10.1186/gm378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh J, Freeman AF, Program NCS, Park M, Sokolic R, Candotti F, Holland SM, Segre JA, Kong HH. The altered landscape of the human skin microbiome in patients with primary immunodeficiencies. Genome Res. 2013 doi: 10.1101/gr.159467.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouchi T, Kubo A, Yokouchi M, Adachi T, Kobayashi T, Kitashima DY, Fujii H, Clausen BE, Koyasu S, Amagai M, et al. Langerhans cell antigen capture through tight junctions confers preemptive immunity in experimental staphylococcal scalded skin syndrome. J Exp Med. 2011;208:2607–2613. doi: 10.1084/jem.20111718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer CN, Irvine AD, Terron-Kwiatkowski A, Zhao Y, Liao H, Lee SP, Goudie DR, Sandilands A, Campbell LE, Smith FJ, et al. Common loss-of-function variants of the epidermal barrier protein filaggrin are a major predisposing factor for atopic dermatitis. Nat Genet. 2006;38:441–446. doi: 10.1038/ng1767. [DOI] [PubMed] [Google Scholar]
- Paradis M, Lemay S, Scott DW, Miller WH, Wellington J, Panich R. Efficacy of Enrofloxacin in the Treatment of Canine Bacterial Pyoderma. Vet Dermatol. 1990;1:123–127. doi: 10.1111/j.1365-3164.1990.tb00090.x. [DOI] [PubMed] [Google Scholar]
- Peschon JJ, Slack JL, Reddy P, Stocking KL, Sunnarborg SW, Lee DC, Russell WE, Castner BJ, Johnson RS, Fitzner JN, et al. An essential role for ectodomain shedding in mammalian development. Science. 1998;282:1281–1284. doi: 10.1126/science.282.5392.1281. [DOI] [PubMed] [Google Scholar]
- Plant JD, Gortel K, Kovalik M, Polissar NL, Neradilek MB. Development and validation of the Canine Atopic Dermatitis Lesion Index, a scale for the rapid scoring of lesion severity in canine atopic dermatitis. Vet Dermatol. 2012;23:515–e103. doi: 10.1111/j.1365-3164.2012.01113.x. [DOI] [PubMed] [Google Scholar]
- Saaf A, Pivarcsi A, Winge MC, Wahlgren CF, Homey B, Nordenskjold M, Tengvall-Linder M, Bradley M. Characterization of EGFR and ErbB2 expression in atopic dermatitis patients. Arch Dermatol Res. 2012;304:773–780. doi: 10.1007/s00403-012-1242-4. [DOI] [PubMed] [Google Scholar]
- Schneider L, Tilles S, Lio P, Boguniewicz M, Beck L, LeBovidge J, Novak N, Bernstein D, Blessing-Moore J, Khan D, et al. Atopic dermatitis: a practice parameter update 2012. J Allergy Clin Immunol. 2013;131:295–299. e291–227. doi: 10.1016/j.jaci.2012.12.672. [DOI] [PubMed] [Google Scholar]
- Sommer MOA, Dantas G. Antibiotics and the resistant microbiome. Curr Opin Microbiol. 2011;14:556–563. doi: 10.1016/j.mib.2011.07.005. [DOI] [PubMed] [Google Scholar]
- Spergel JM, Paller AS. Atopic dermatitis and the atopic march. J Allergy Clin Immunol. 2003;112:S118–S127. doi: 10.1016/j.jaci.2003.09.033. [DOI] [PubMed] [Google Scholar]
- Suarez-Farinas M, Dhingra N, Gittler J, Shemer A, Cardinale I, de Guzman Strong C, Krueger JG, Guttman-Yassky E. Intrinsic atopic dermatitis shows similar TH2 and higher TH17 immune activation compared with extrinsic atopic dermatitis. J Allergy Clin Immunol. 2013;132:361–370. doi: 10.1016/j.jaci.2013.04.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willing BP, Russell SL, Finlay BB. Shifting the balance: antibiotic effects on host-microbiota mutualism. Nat Rev Microbiol. 2011;9:233–243. doi: 10.1038/nrmicro2536. [DOI] [PubMed] [Google Scholar]
- Wright GD. The antibiotic resistome: the nexus of chemical and genetic diversity. Nat Rev Microbiol. 2007;5:175–186. doi: 10.1038/nrmicro1614. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Xiao C, Gibson AM, Bass SA, Khurana Hershey GK. EGFR signaling blunts allergen-induced IL-6 production and Th17 responses in the skin and attenuates development and relapse of atopic dermatitis. J Immunol. 2014;192:859–866. doi: 10.4049/jimmunol.1301062. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.