

Abstract
Diabetes impedes vascular repair and causes vasoregression in the brain after stroke, but mechanisms underlying this response are still unclear. We hypothesized that excess peroxynitrite formation in diabetic ischemia/reperfusion (I/R) injury inactivates the p85 subunit of phosphoinositide 3-kinase (PI3K) by nitration and diverts the PI3K–Akt survival signal to the p38–mitogen-activated protein kinase apoptosis pathway. Nitrotyrosine (NY), Akt and p38 activity, p85 nitration, and caspase-3 cleavage were measured in brains from control, diabetic (GK), or metformin-treated GK rats subjected to sham or stroke surgery and in brain microvascular endothelial cells (BMVECs) from Wistar and GK rats subjected to hypoxia/reoxygenation injury. GK rat brains showed increased NY, caspase-3 cleavage, and p38 activation and decreased Akt activation. Metformin attenuated stroke-induced nitrative signaling in GK rats. GK rat BMVECs showed increased basal nitrative stress compared with controls. A second hit by hypoxia/reoxygenation injury dramatically increased the nitration of p85 and activation of p38 but decreased Akt. These effects were associated with impairment of angiogenic response and were restored by treatment with the peroxynitrite scavenger 5,10,15,20-tetrakis(4-sulfonatophenyl)porphyrinato iron III chloride or the nitration inhibitor epicatechin. Our results provide evidence that I/R-induced peroxynitrite inhibits survival, induces apoptosis, and promotes peroxynitrite as a novel therapeutic target for the improvement of reparative angiogenesis after stroke in diabetes.
Introduction
Diabetes increases the risk and amplifies the severity of cerebral complications such as stroke and cognitive impairment (1,2). As in other complications of diabetes, such as retinopathy, nephropathy, or coronary artery disease, micro- and macrovascular disease is believed to play an important role in the pathogenesis and progression of these cerebral complications. Acute ischemic stroke is the fourth leading cause of death but the number one cause of adult disability in the United States (3,4). Therefore, therapeutic strategies to improve functional outcome by stimulating the brain’s recovery mechanisms are highly important. Since brain function is heavily dependent on constant cerebral blood flow, enhancement of angiogenesis by proangiogenic agents and/or stem cells is being evaluated as a therapeutic modality in experimental models of stroke. Our understanding of how diabetes affects vascular plasticity and integrity of the brain, especially after stroke, is limited.
In a series of studies we showed that diabetes causes increased, yet dysfunctional, neovascularization in the cerebrovascular bed in type 2 diabetic Goto-Kakizaki (GK) rats and ob/ob mice (5–7). These changes were mediated, in part, by an increased vascular endothelial growth factor (VEGF) angiogenic signal and diabetes-induced oxidative stress (5–7). However, this augmented angiogenesis was associated with poor vessel wall maturity, as indicated by reduced pericytes along with increased numbers of nonperfused vessels and permeability. When ischemia/reperfusion (I/R) was superimposed on this pathology, animals developed greater bleeding into the brain and exhibited worse functional outcome compared with controls (8–10). A follow-up study showed that diabetes not only impairs reparative neovascularization after stroke but also impedes the recovery because of vascular regression (6). Moreover, glycemic control with metformin after stroke improved neurovascular repair and improved functional outcome (6). The molecular mechanisms underlying vascular regression after stroke in diabetes remain unclear.
Reactive oxygen species (ROS)/reactive nitrogen species (RNS) act as signaling molecules of growth factors to promote angiogenesis, but the emerging redox window concept postulates that there is a delicate balance, and the level of ROS/RNS is critical for the angiogenic response (11,12). In the current study we tested the hypothesis that I/R injury in a diabetic setting causes the formation of excess peroxynitrite, which diverts phosphoinositide 3-kinase (PI3K)–Akt survival signals to p38–mitogen-activated protein kinase (MAPK) apoptosis via nitration and inactivation of the regulatory p85 subunit of PI3K. We further postulated that metformin attenuates this response.
Research Design and Methods
Animals
All animal procedures were carried out in accordance with the National Institutes of Health guidelines under protocols approved by Georgia Regents University and Charlie Norwood VA Medical Center. Wistar rats were purchased from Harlan (Indianapolis, IN), and GK rats were purchased from Taconic Biosciences (Hudson, NY). Blood glucose was measured during the light cycle every day. An additional group of GK rats were treated with metformin (300 mg/kg/day in drinking water) after stroke until death. Since animals do not eat and blood glucose levels decrease to normal levels immediately after stroke, treatment was started when blood glucose reached >140 mg/dL (on average 36 h after stroke) so hypoglycemia was not caused in the acute stroke period, which can affect outcome. Animals were housed individually, and water consumption was measured to optimize the dosage of metformin. Blood glucose and body weight data are summarized in Table 1.
Table 1.
Blood glucose and body weight of animals
| Body weight† (g) | Initial blood glucose (mg/dL) | Blood glucose at death† (mg/dL) | |
|---|---|---|---|
| Wistar rats | 318 ± 6 | <105 | 94 ± 7.5 |
| GK rats | 271 ± 7 | >140 | 197 ± 8.5* |
| GK rats + metformin treatment | 299 ± 12 | >140 | 91 ± 5.9 |
*P < 0.05.
†Data are mean ± SEM.
Method of Ischemic Stroke
Control or diabetic male rats (250–300 g, 10–12 weeks old) were randomized to sham or stroke surgery achieved by occlusion of the middle cerebral artery (MCAO), as we reported previously (6,13). Ischemia was induced for a period of 90 min, and reperfusion was achieved by removal of the filament. Animals were returned to their home cages after MCAO and given easy access to food and water. On day 14, animals were injected with 500 μL of 50 mg/mL fluorescein isothiocyanate–dextran (molecular weight 2,000,000; Sigma-Aldrich, St. Louis, MO) through the jugular vein under pentobarbital anesthesia to visualize blood vessels. Brains were processed in 4% paraformaldehyde (24 h) and 30% sucrose in PBS for immunohistochemical studies.
Immunolocalization Studies
Images were acquired from regions of interest (ROIs) B (anterior to the middle cerebral artery [MCA] comprising the frontal sensory cortex, bregma 3 to 1), C (medial where the MCA branches out to supply the frontal motor cortex, bregma 1 to −1), and D (posterior to the MCA comprising the parietal cortex, bregma −1 to −3) (Fig. 1A, indicated by yellow squares). An overall representation of these regions was obtained by imaging ROIs from three different sections obtained from one slice. Optical density values were obtained from each ROI, and a mean value of three images from each section was calculated. Each measurement from one animal comprised an average of nine images from the cortical region. Brain sections were exposed overnight to monoclonal anti-nitrotyrosine antibody (Millipore, Billerica, MA); polyclonal anti-cleaved caspase-3 (Calbiochem; EMD Millipore, Darmstadt, Germany); monoclonal anti-phospho-Akt (Cell Signaling Technology, Beverly, MA); or monoclonal anti-phospho-p38 (Cell Signaling Technology). All the primary antibodies were specific for rats and used in 1:200 dilutions of 1% BSA in 0.3% Triton X-100 in PBS. The sections were blocked using 0.1% horse serum dissolved in 1% BSA in 0.3% Triton X-100 in PBS. Then the sections were incubated with 1:500 dilutions of Texas-red conjugated goat anti-mouse or goat anti-rabbit antibodies (Invitrogen, Carlsbad, CA) for 2 h. Images were acquired using an Axiovert 200 microscope (Carl Zeiss MicroImaging, Thornwood, NY). Images were analyzed blindly for optical density using ImageJ software (http://imagej.nih.gov/ij/index.html).
Figure 1.

Diabetes increases basal and poststroke nitrotyrosine (NY) levels in the cerebrovasculature and brain parenchyma. Wistar (Wis) and GK rats were subjected to 90 min of MCAO. On day 14, brains were assessed for NY levels via slot-blot and immunohistochemistry. A: Depiction of coronal brain section C and ROIs (yellow boxes) that were used for immunoblotting and immunohistochemistry studies. B: Three representative images (top) and cumulative data (bottom) for nitrotyrosine levels in brain homogenates, as measured by slot-blot. C: Representative immunohistochemistry images for NY signal and localization to cerebrovasculature. Animals were injected with fluorescein isothiocyanate (FITC)–dextran to visualize blood vessels and reacted against NY antibody. Sham GK rats showed higher cerebrovascular NY levels, as detected by both methods, compared with control Wis rats. I/R injury amplified NY formation in both groups. Treatment of diabetic GK rats with metformin (GK Stroke Met) significantly reduced NY levels compared with GK animals with stroke. Results are shown as mean ± SEM. ΩP < 0.0001, GK vs. Wis rats; *P < 0.0001, stroke vs. sham; #P < 0.003 GK rats treated with metformin vs. GK rats (n = 6–8).
Cell Apoptosis
Fixed brain sections were immobilized on electrostatic adherence glass slides (Travigen, Gaithersburg, MD) and treated for in situ detection of apoptosis according to the manufacturer’s protocol (TACS 2 TDT-DAB In situ Apoptosis Detection Kit; Travigen). Positive cells were blindly counted using an Axiovert 200 microscope (Carl Zeiss). Results are presented as the mean number of positive cells (n = 6–8 per group).
Isolation of Protein from Formalin-Fixed Brain Tissue
Protein was isolated from fixed brains using the Qproteome FFPE Tissue Kit (Qiagen, Valencia, CA), according to the manufacturer’s protocol. The Qproteome FFPE Tissue Kit reverses the cross-linking process and delivers full-length proteins ready for analysis by Western blotting or protein array, with subsequent immunodetection.
Isolation of Primary Brain Microvascular Endothelial Cells from Wistar or GK Rats
Brain microvascular endothelial cells (BMVECs) were isolated as described previously (5). Experiments were performed using cells between passages 4 and 6. Cells were switched to serum-free medium 6 h before cell migration assay. The peroxynitrite decomposition catalyst 5,10,15,20-tetrakis(4-sulfonatophenyl)porphyrinato iron III chloride (FeTPPs; EMD Millipore) was used at a concentration of 5 µmol/L, whereas the nitration inhibitor epicatechin was used at a concentration of 200 μmol/L. Metformin was used at a 5 mmol/L concentration.
Hypoxia Treatment
BMVECs were grown to 80% confluence and then switched to serum-free medium and placed in a hypoxic chamber (0.2% O2, 5% CO2) for 10 h, followed by 18 h of normoxia (21% O2, 5% CO2) to mimic ischemia. The 0.2% O2 concentration was chosen for hypoxia (0.2% oxygen) exposure based on previous research showing that 0.2% O2 generates significant peroxynitrite without inducing toxicity in BMVECs. The hypoxia (0.2% oxygen) exposure was performed in a humidified incubator modified by installing a PROOX model 110 oxygen regulator (Biospherix, Redfield, NY). The oxygen concentration was monitored continuously using the PROOX oxygen sensor.
Nitrotyrosine Slot-Blot Detection
Relative amounts of proteins nitrated on tyrosine residues were measured using the slot-blot technique, as described previously (14).
Immunoprecipitation and Western Blot Analysis
Cells were harvested after various treatments and lysed in modified radioimmunoprecipitation assay buffer (Millipore). Total protein (50 µg) was boiled in 6× Laemmli sample buffer, separated on a 10–12% SDS-PAGE by electrophoresis, transferred to a nitrocellulose membrane, and stained with a specific antibody. All primary antibodies were rat specific: monoclonal anti-nitrotyrosine antibody (Millipore), polyclonal anti-cleaved caspase-3 (EMD Millipore), monoclonal anti-phospho-Akt, polyclonal anti-Akt, anti-cleaved poly(ADP-ribose) polymerase (PARP), polyclonal anti-p38, or monoclonal anti-phospho-p38 (Cell Signaling Technology). For immunoprecipitation, cell lysate (200 µg) was incubated with anti-p85 antibody (5 µg) and A/G agarose beads (Santa Cruz Biotechnology, Santa Cruz, CA) overnight. The precipitated proteins were analyzed by SDS-PAGE and blotted with antibodies against anti-nitrotyrosine or anti-p85 for equal loading. Primary antibodies were detected using a horseradish peroxidase–conjugated antibody and enhanced chemiluminescence (GE Healthcare, Piscataway, NJ). Relative optical densities of immunoreactivity were determined by densitometry software (Alpha Innotech, ProteinSimple, San Jose, CA).
Angiogenic Assays
The wound-healing and tube formation assays were performed as described previously (15).
Data Analysis
Data are presented as mean ± SEM. Data were assessed for normality, and the assumptions of ANOVA and a log transformation were used when needed. The effect of I/R on diabetic animals was assessed using a 2-disease (Wistar vs. GK) by 2-stroke (sham vs. I/R) ANOVA; a significant interaction would indicate a differential effect on outcomes of stroke dependent on disease status. Comparison of the effect of metformin treatment on diabetic animals was determined using a one-way ANOVA based on data from animals that experienced strokes (Wistar, GK, and GK + metformin). Data from the in vitro cell studies were analyzed using a 2-disease (Wistar vs. GK) by 2-oxygenation (normoxia vs. hypoxia) ANOVA to determine the effect of oxygen deprivation; a significant interaction would indicate a differential effect dependent on disease. Data from cells subjected to oxygen deprivation were then analyzed using a series of 2-disease (Wistar vs. GK) by 2-treatment (none vs. FeTPPs or epicatechin) ANOVAs to determine the effect of reoxygenation; a significant interaction would indicate a differential effect due to disease status. SAS software version 9.3 (SAS Inc., Cary, NC) was used for all analyses. Statistical significance was determined at α < 0.05, and a Tukey post hoc test was used to compare means from significant ANOVAs.
Results
Diabetes Increases Basal and Poststroke Nitrotyrosine Levels in the Cerebrovasculature and Brain Parenchyma
GK rats expressed higher basal levels of nitrotyrosine in both cerebrovasculature and brain parenchyma compared with control Wistar rats (Fig. 1B and C). I/R injury amplified nitrotyrosine formation in both Wistar and GK rats. Treatment of GK rats with metformin significantly reduced nitrotyrosine levels compared with untreated GK rats (Fig. 1B and C).
Stroke Increases Apoptosis in Diabetic Rat Brains
Immunoblotting studies showed that GK rats showed a higher basal level of caspase-3 activation compared with sham Wistar rats (Fig. 2A). I/R injury significantly increased caspase-3 cleavage in both Wistar and GK rats compared with sham (Fig. 2A). Treatment of diabetic GK rats with metformin significantly reduced caspase-3 activation (Fig. 2A). To further confirm I/R-induced cell death after stroke, we measured apoptosis using TUNEL assay. Our results illustrate a significant interaction between diabetes and I/R injury such that the effect of stroke is amplified in diabetes (Fig. 2B). Treatment of diabetic GK rats with metformin significantly reduced TUNEL-positive cells compared with the GK vehicle group (Fig. 2B). Immunohistochemical studies for cleaved caspase-3 showed a significant interaction between diabetes and I/R injury. While I/R injury caused a twofold increase in caspase-3 cleavage in Wistar rats, the impact was greater in GK rats (Fig. 2C).
Figure 2.

Stroke increases apoptosis in diabetic rat brains. A: Western blot (left) and cumulative data (right) for cleaved caspase-3 in brain homogenates from Wistar (Wis) and GK rats. Sham GK rats showed a higher basal level of caspase-3 activation (ΩP < 0.05 vs. Wis Sham). I/R injury increased caspase-3 cleavage in both Wis and GK rats (*P < 0.05 vs. control). Treatment of diabetic GK rats with metformin (GK Stroke Met) significantly reduced caspase-3 activation compared with GK animals with stroke (#P < 0.001 vs. GK rats with stroke). B: Apoptosis was further confirmed using TUNEL assay on brain sections. At baseline there was no difference between the control and diabetes groups, but I/R injury significantly increased the positive cell count (white arrows) in GK rats but not in controls (**P = 0.0019, disease and intervention interaction). Treatment of diabetic GK rats with metformin significantly reduced TUNEL-positive cells compared with GK animals with stroke (#P < 0.001 vs. GK rats with stroke). Results are shown as mean ± SEM (n = 6–8). C: Representative images of (top and middle) and cumulative data (bottom) from coronal sections used in Fig. 1A that were stained for cleaved caspase-3. Sham GK rats showed a higher basal level of caspase-3 activation (ΩP < 0.05 vs. Wis Sham). I/R injury increased caspase-3 cleavage in both groups, but there was a disease and intervention interaction such that caspase-3 activation was greater in diabetic GK rats (**P = 0.0001). Treatment with metformin significantly reduced caspase-3 activation compared with the untreated GK stroke group (#P < 0.0001 vs. GK rats with stroke). FITC, fluorescein isothiocyanate.
Stroke Activates Proapoptotic Pathways in Diabetic Rats via Increased Tyrosine Nitration
Akt activation, detected by immunohistochemistry, was decreased (Fig. 3A) but the pP38 signal was increased (Fig. 3B) in brain sections from diabetic animals. Metformin treatment prevented these changes. These results were confirmed by immunoblotting, which showed decreased pAkt and increased pP38 levels in diabetes (Fig. 3C and D). Treatment with metformin significantly restored Akt activation compared with the untreated GK group (Fig. 3C and D). Immunoprecipitation of brain lysate with antibody against the p85 subunit of PI3K followed by immunoblotting against nitrotyrosine antibody showed a significant increase in the nitration of p85 in sham GK rats, as well as in control and diabetic rats with stroke (Fig. 3E).
Figure 3.


Stroke activates proapoptotic pathways in diabetic rats via increased tyrosine nitration of the p85 subunit. Representative immunohistochemistry images (top and middle) and cumulative data (bottom) for Akt (A) and p38 (B) localization and staining intensity show decreased survival and increased proapoptotic markers in the cerebrovasculature in diabetes at baseline and after stroke (*P < 0.001 vs. Wistar [Wis]). Treatment with metformin restored Akt activation compared with untreated GK rats after stroke (#P < 0.05). **P < 0.05 vs. sham. C: Representative immunoblots (left) and cumulative data (right) of Akt activation in Wis and GK rat brain homogenates at baseline and after stroke. Our results showed that GK rats showed significantly reduced levels of activated Akt compared with Wis rats (*P < 0.05). Treatment with metformin significantly elevated Akt activation compared with GK rats after stroke (#P < 0.05). D: Representative immunoblots (left) and cumulative data (right) of p38 activation in Wis and GK rat brain homogenates at baseline and after stroke. Our results showed that GK rats showed significantly elevated levels of activated P38 compared with Wis (*P < 0.05). Treatment with metformin significantly prevented p38 activation compared with GK rats after stroke (#P < 0.05). E: Representative immunoprecipitation experiments showing the nitration of p85 subunit. Brain homogenates were immunoprecipitated with p85 antibody followed by immunoblotting against nitrotyrosine (NY). Nitrated p85 levels were higher in GK rats (*P < 0.05 vs. Wis Sham), and stroke increased them even more (**P < 0.05 vs. sham). Metformin treatment prevented this increase in p85 nitration (#P < 0.05). Results are shown as mean ± SEM (n = 4–8). FITC, fluorescein isothiocyanate; I.B, immunoblotting; I.P, immunoprecipitation.
Hypoxia/Reoxygenation Increases Endothelial Cell Nitrative Stress in Diabetes
To mimic the in vivo I/R injury induced by stroke, we optimized an in vitro cell culture technique. BMVECs isolated from both Wistar and GK rats were subjected to oxygen deprivation for 10 h, followed by 18 h of normoxia. Cells were treated at reoxygenation with either FeTPPs to scavenge peroxynitrite or epicatechin to inhibit nitration. As shown in Fig. 4A and B, cells from diabetic GK rats showed greater nitrotyrosine levels under normoxic conditions. There was a disease and intervention effect such that hypoxia/reoxygenation augmented nitrotyrosine formation in the GK BMVECs more than in the control cells. Treatment with FeTPPs prevented hypoxia/reoxygenation-induced nitrative stress in cells from GK rats (Fig. 4A and B). Moreover, treatment with the nitration inhibitor caused a similar reduction of nitrotyrosine formation in BMVECs from diabetic animals (Fig. 4A and B).
Figure 4.
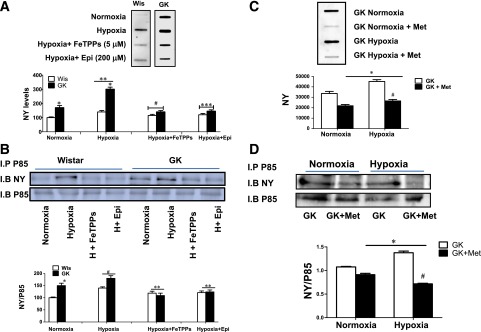
Hypoxia/reoxygenation increases endothelial cell nitrative stress and nitration of the p85 subunit of PI3K in diabetes. BMVECs isolated from Wistar (Wis) and GK rats were subjected to oxygen deprivation (oxygen <0.2%) for 10 h, followed by 18 h of normoxia (oxygen 21%). Cells were treated with either the peroxynitrite decomposition catalyst FeTPPs (5 μmol/L) or the nitration inhibitor epicatechin (Epi; 200 μmol/L) at reoxygenation. A: Representative images of total nitrotyrosine (NY) levels as detected by slot-blot analysis in BMVECs from control and GK rats. Cells from diabetic GK rats showed greater NY levels under basal normoxic conditions compared with controls (*P < 0.05 vs. Wis). A 2 (Wis and GK) × 2 (normoxia and hypoxia) ANOVA showed that hypoxia/reoxygenation increased NY formation in GK but not Wis endothelial cells (**P = 0.0008). Treatment with FeTPPs prevented hypoxia/reoxygenation-induced nitrative stress in cells from GK rats but had no effect on cells from Wis rats (#P < 0.0001), indicating a disease and treatment interaction. Treatment with Epi caused a similar reduction in NY formation in BMVECs from GK rats (***P = 0.0008). B: Representative images of homogenates immunoprecipitated (I.P) with p85 antibody and then immunoblotted (I.B) against NY antibody to assess p85 tyrosine nitration and p85 antibody for loading. GK rat cells had a higher p85 nitration at baseline and after hypoxia (H) compared with controls (*P = 0.006 vs. Wis). Hypoxia/reoxygenation caused a significant increase in both Wis and GK rat cells (#P = 0.0006). A 2 (Wis and GK) × 2 (vehicle vs. treatment) ANOVA showed that treatment with FeTPPs or Epi prevented hypoxia/reoxygenation-induced p85 nitration in both Wis and GK rat cells (**P < 0.0001). Results are shown as mean ± SEM (n = 6–8). C: GK rat BMVECs treated with 5 mmol/L metformin significantly reduced NY formation. Treatment with metformin prevented hypoxia/reoxygenation-induced nitrative stress in GK rats (*P < 0.05 vs. GK rats; #P < 0.01 vs. GK rat hypoxia). D: Metformin decreased p85 nitration in GK rat BMVECs under normoxic conditions and after hypoxia compared with controls. *P < 0.05 vs. GK rats; #P < 0.01 vs. GK rat hypoxia). Results are shown as mean ± SEM (n = 4–6).
Hypoxia/Reoxygenation Increases Nitration of the p85 Subunit of PI3K
Cells from GK rats had a higher basal level of p85 nitration, and exposing BMVECs to hypoxia/reoxygenation caused a significant increase in cell types from both Wistar and GK rats (Fig. 4B). Treatment with FeTPPs or epicatechin prevented p85 nitration (Fig. 4B). Treatment of GK endothelial cells with metformin (5 mmol/L) significantly decreased nitrotyrosine formation (Fig. 4C) and improved Akt activation (Supplementary Fig. 1) under normoxic and hypoxic conditions. Immunoprecipitation studies showed that metformin significantly decreased p85 nitration at normoxic as well as hypoxic conditions (Fig. 4D).
Hypoxia/Reoxygenation Inhibits Akt Survival and Activates p38 MAPK Apoptosis Pathways
Cells from GK rats had relatively less phosphorylated Akt at baseline. Exposing BMVECs to hypoxia/reoxygenation significantly decreased Akt phosphorylation in cells from both Wistar and GK rats (Fig. 5A and B). Treatment with FeTPPs or the nitration inhibitor had a greater effect on Akt activation in cells from GK rats compared with cells from Wistar rats (Fig. 5A and B). As shown in Fig. 5C and D, BMVECs from GK rats showed 2.6-fold higher basal p38 activation. Hypoxia/reoxygenation further increased p38 activation in BMVECs from both Wistar and GK rats (Fig. 5C and D). Treatment with FeTPPs or epicatechin decreased p38 activation, and this was more pronounced in cells from GK rats (Fig. 5C and D).
Figure 5.

Hypoxia/reoxygenation inhibits Akt survival and activates p38 MAPK. Akt and p38 activation were measured by immunoblot analysis in BMVECs isolated from both Wistar (Wis) and GK rats subjected to hypoxia (H) (10 h) and reoxygenation (18 h). Cells were treated with either the peroxynitrite decomposition catalyst FeTPPs (5 μmol/L) or the nitration inhibitor epicatechin (Epi; 200 μmol/L) at reoxygenation. A and B: Cells from diabetic GK rats showed lower Akt activation under basal normoxic as well as hypoxic conditions compared with controls (*P < 0.05). Hypoxia/reoxygenation further decreased Akt phosphorylation in both groups (#P < 0.0001). A 2 (Wis and GK rats) × 2 (vehicle vs. treatment) ANOVA indicated a disease and treatment interaction such that treatment with FeTPPs or Epi prevented this decrease in Akt phosphorylation to a greater extent in GK rat cells (**P < 0.0001). C and D: Cells from diabetic GK rats showed higher phosphorylation of p38 under basal normoxic and hypoxic conditions compared with controls (*P < 0.05). Hypoxia increased p38 activation in both groups (#P < 0.0001). Treatment with FeTPPs or Epi indicated an interaction showing that either treatment is more effective in GK rat cells (**P = 0.0089 and ***P = 0.0029). Results are shown as mean ± SEM (n = 6–8).
Hypoxia/Reoxygenation Aggravates Endothelial Cell Apoptosis in Diabetes
Baseline levels of cleaved caspase-3 were higher in cells from GK rats, and hypoxia caused a significant increase in caspase cleavage in cells from both control and GK rats (Fig. 6A and B). Treatment with FeTPPs or epicatechin decreased caspase cleavage, and this was more pronounced in cells from GK rats. In parallel, BMVECs from GK rats showed higher basal levels of PARP cleavage, which was further augmented by I/R injury (Fig. 6A and C). Treatment with FeTPPs or epicatechin was more effective in cells from GK rats (Fig. 6A and C).
Figure 6.

Hypoxia/reoxygenation aggravates endothelial cell apoptosis in diabetes. Cleaved caspase-3 and PARP were measured by immunoblot as indices of apoptosis in BMVECs isolated from both Wistar (Wis) and GK rats subjected to hypoxia (H) and reoxygenation. Cells were treated with either the peroxynitrite decomposition catalyst FeTPPs (5 μmol/L) or the nitration inhibitor epicatechin (Epi; 200 μmol/L) at reoxygenation. A and B: Cleaved caspase-3 levels are higher in GK rat cells at baseline and hypoxia compared with controls (*P < 0.05). Hypoxia/reoxygenation caused a significant increase in caspase cleavage in both control and GK rat cells (#P < 0.0001). Treatment with FeTPPs or Epi decreased caspase cleavage compared with the hypoxia groups, and this was more pronounced in GK rat cells (**P < 0.0001). C: In parallel, GK rat BMVECs showed higher PARP cleavage at baseline and hypoxia compared with controls (*P < 0.05). Hypoxia/reoxygenation caused a significant increase in cleaved PARP in both control and GK rat cells (#P < 0.0001). Treatment with FeTPPs or Epi decreased caspase cleavage compared with the hypoxia groups, and this was more pronounced in GK rat cells (**P < 0.0001). Results are shown as mean ± SEM (n = 6–8).
Hypoxia/Reoxygenation Impairs Angiogenic Response
Under normoxic conditions, cells from GK rats display greater angiogenic potential, indicated by increased tube length (Fig. 7A and B) and cell migration (Fig. 7C), confirming our previous studies (5). A hypoxic insult significantly reduced the angiogenic properties of BMVECs, and the reduction was larger in cells of GK rats (Fig. 7A–C). Treatments that scavenge peroxynitrite or prevent nitration recovered angiogenic properties in GK rat cells (Fig. 7A–C).
Figure 7.

Hypoxia/reoxygenation impairs angiogenic response. The angiogenic signal was assessed using tube formation and cell migration assays. For tube formation assay, BMVECs isolated from both Wistar (Wis) and GK rats were added to a reduced growth factor matrigel and subjected to hypoxia (H) and reoxygenation. Cells were assessed for mean tube length. Representative images in panel A and cumulative data in panel B show that, under normoxic conditions, GK rat cells display greater angiogenic potential, indicated by increased tube length (*P < 0.05). Hypoxic insult significantly reduces the angiogenic properties of BMVECs in both Wis and GK rat cells, and this effect was more pronounced in cells from GK rats (#P = 0.001). Treatment with FeTPPs or epicatechin (Epi) recovered angiogenic properties in GK rat cells (**P < 0.0001). C: A similar pattern was observed in cell migration. Results are shown as mean ± SEM (n = 6–8). EC, endothelial cell.
Discussion
Diabetes worsens the outcome of acute ischemic stroke, which is the leading cause of adult disability in the U.S. Given that the brain is a highly vascularized organ, receiving 20% of the cardiac output, the cerebrovasculature is likely to play an important role in stroke injury and recovery. We have shown that there is dramatic vascular loss in the brain of diabetic animals, but not control animals, after stroke. Building on this foundation, this study addressed the potential mechanisms of cerebral vasoregression in diabetes. We provide evidence that 1) I/R injury of the brain in the diabetic setting increases nitrative stress; 2) increased nitrative stress causes the nitration of the regulatory subunit of PI3K, p85, and the nitration of the p85 subunit diverts the downstream Akt survival signal to the apoptotic p38 MAPK pathway; 3) increased nitrative stress also impairs the angiogenic properties critical for repair; 4) a peroxynitrite decomposition catalyst or a nitration inhibitor prevents nitrative stress and resulting endothelial apoptosis in diabetes; and, finally, 5) metformin treatment in the period after stroke prevents nitrative stress–mediated endothelial apoptosis in vivo and in vitro.
Diabetes augments acute neurovascular injury and worsens stroke outcome, but the impact of diabetes on neurovascular restoration and recovery after stroke is far less clear (11,16). Accumulating evidence suggests that vascular and neuronal repair in the brain are coupled. Creation of an angiogenic microenvironment precedes and promotes axonal remodeling, as well as neuroblast migration along new blood vessels, emphasizing the importance of the cerebrovasculature in repair and recovery (17,18). Enhancement of angiogenesis is considered a therapeutic modality to improve functional outcome after stroke. The limited nature of the evidence of cerebral neovascularization after stroke, however, calls for caution. Ye et al. (19) reported that increased angiopoietin (Ang)-2 and decreased Ang-1 levels promote immature vessel formation in the infarcted hemisphere in type 1 diabetic rats, leading to poor functional outcome. Another study showed that angiogenesis is impaired in type 2 diabetic GK rats after stroke, and this is associated with decreased VEGF and increased angiostatin signaling (20). We also reported impaired angiogenesis as well as vascular regression in GK rats 14 days after stroke (6). This study also showed that metformin treatment in the recovery phase after stroke prevented vasoregression and improved functional recovery. Collectively, these studies suggest that impaired angiogenesis means not only failure to stimulate new vessel formation but also failure to stimulate stable and functional new vessels in both type 1 and type 2 models of diabetes. Thus optimization of the angiogenic response to generate fully functional vessels or signals to promote neurovascular restoration is likely to be the key to successful recovery after diabetic stroke. Building on this strong foundation, the current study used type 2 diabetic GK rats that are amenable to treatment by oral hypoglycemic agents. Our results unravel oxidative stress–dependent mechanisms of endothelial cell death in the brain in diabetes if an ischemic injury is superimposed, and it identifies peroxynitrite as a potential target to improve neurovascular restoration and functional recovery.
Angiogenesis is a complex process. While numerous anti- and proangiogenic growth factors are involved, it is widely accepted that VEGF-A plays a central role in the regulation of angiogenesis. Activation of VEGF receptor-2 is followed by a cascade of downstream signaling to promote endothelial cell survival, proliferation, and migration and prevent endothelial apoptosis. VEGF activation of VEGF receptor-2 transduces the antiapoptotic signal via the PI3K/Akt signaling pathway (21,22), and ROS/RNS act as signaling molecules, mediating this effect (14,15). We previously reported that VEGF mediates an angiogenic response in BMVECs from diabetic GK rats in a peroxynitrite-dependent manner (5). In the current study, however, we found peroxynitrite-mediated apoptosis in the same cells after hypoxia/reoxygenation. The redox state of the microenvironment is important for the angiogenic response. Therapeutic approaches to enhance angiogenesis failed in ischemic cardiovascular disease (12,23). As recently reviewed, one potential reason was identified as patients with coronary artery disease being vastly different from the young and healthy animals used for preclinical studies (24). These studies led to the “redox window” concept, which postulates that mild but not severe increases in ROS and RNS are needed to induce angiogenesis. The current study provides supportive evidence for this concept and also suggests that therapeutic angiogenesis may not be achievable or desirable in the diabetes setting. Indeed, Chen et al. (25) showed that cell therapy with bone marrow stromal cells improved repair and functional recovery in control but not diabetic animals, where the approach worsened blood-brain barrier integrity.
Tyrosine nitration is one of the stable posttranslational modifications produced via the nitration of tyrosine residues by peroxynitrite (14,26). The formation of excess nitrotyrosine has been previously reported in ischemic models of transient MCAO/reperfusion, as well as in other brain injury models (13,27–35). Our results are in agreement with other studies that showed that diabetes increases peroxynitrite and tyrosine nitration (7,36–39). In the current study we examined the nitration of the p85 subunit of PI3K as a target that determines the fate of endothelial cells in the diabetic stroke setting, since previous studies suggested that nitration of this regulatory subunit acts as a switch in modulating cell survival (14,40,41). Our in vivo and in vitro data suggest that preventing nitration of the p85 subunit using peroxynitrite prevents endothelial apoptosis, and that FeTPPs or epicatechin can be used to improve endothelial survival and recovery after ischemic brain injury in diabetes.
We reported that glycemic control with metformin prevents cerebrovascular remodeling, as well as dysfunctional neovascularization, in diabetes (7,10). In a recent study metformin treatment started after vascular disease was established was able to reverse diabetes-mediated structural changes in the cerebrovasculature (42,43). Although clinical trials focusing on the impact of glycemic control on cardiovascular outcomes had mixed results, tight glycemic control has been an effective treatment for the prevention of microvascular complications such as diabetic nephropathy and retinopathy (44–49). Metformin is a first-line drug commonly used to treat patients with type 2 diabetes. We demonstrated that glycemic control induced by metformin in rats that had not achieved prior glycemic control attenuated vasoregression after stroke and improved functional outcomes, including cognitive deficits (6). In the current study we provide evidence that metformin treatment reduces nitrative stress in vivo, suggesting that hyperglycemia contributes to stroke-induced oxidative stress. Interestingly, however, metformin reduces nitrotyrosine formation and nitration and improves Akt phosphorylation in endothelial cells in vitro, suggesting that metformin has direct antioxidant effects in addition to its glucose-lowering actions, both of which may contribute to improved endothelial cell survival and angiogenic response. Further studies with other hypoglycemic agents are warranted to clarify the contribution of glycemic control to the improvement of vascular repair after stroke.
Our studies have the following limitations. First, with the exception of VEGF, we did not measure other angiogenic/antiangiogenic factors in our model. Our data (Supplementary Fig. 2) show that VEGF levels are higher in cells from GK rats at baseline, and while hypoxia/reoxygenation increases VEGF in control cells, there is no further increase in GK rat cells. Given that VEGF works in concert with Ang-2 and Ang-1, the angiogenic/antiangiogenic profile needs to be investigated further in future studies. Along the same lines, improvement of endothelial cell survival by metformin may not be solely due to enhancement of VEGF and/or other angiogenic signaling. For example, augmented insulin receptor substrate-1 signaling by metformin may contribute to Akt-mediated survival. Second, this study investigated the effect of peroxynitrite scavenging and inhibition of nitration on angiogenic response only in the in vitro model and did not use FeTPPs or epicatechin as a therapeutic strategy in animals in the recovery period after stroke. Third, this study did not measure vascularization indices and functional outcome parameters after stroke because our previous study reported these measurements. Nevertheless, our results are important because they identify potential molecular targets to prevent vasoregression that occurs after I/R in diabetes and suggest that excess nitrative stress in the diabetic stroke setting acts as a switch, diverting protective survival signaling to an apoptotic pathway (Fig. 8). Our results show that metformin reduces nitrative stress and improves endothelial survival in the period after stroke, possibly via direct antioxidant and glucose-lowering effects. Based on this solid foundation, studies investigating the impact of peroxynitrite scavenging or inhibition of nitration alone or in combination with glycemic control on vascular repair and functional recovery after stroke will be of high translational value.
Figure 8.

A schematic representation of the proposed mechanism by which I/R injury in the diabetic setting increases peroxynitrite and nitrative stress, resulting in tyrosine nitration of p85, the regulatory subunit of PI3K. I/R diverts the downstream Akt survival signal to the apoptotic p38 MAPK pathway, which increases endothelial apoptosis and decreases cerebral angiogenesis. EC, endothelial cell; NY, nitrotyrosine.
Supplementary Material
Article Information
Funding. This work was supported in part by an American Heart Association Postdoctoral Fellowship (14POST19580004 to M.A.); American Heart Association Predoctoral Fellowships (12PRE11300001 to M.C. and 13PRE17090026 to S.H.); Veterans Affairs Merit Awards (BX000347, BX00891) and National Institutes of Health award (NS-063965) to S.C.F.; and a Veterans Affairs Research Career Scientists award and National Institutes of Health awards (NS-070239, NS-083559) to A.E. A.E. is a Research Career Scientist at the Charlie Norwood Veterans Administration Medical Center in Augusta, GA.
Duality of Interest. No potential conflicts of interest relevant to this article were reported.
Author Contributions. M.A. collected and analyzed the data and wrote the manuscript. R.P. and W.L. collected the data. M.C. and S.H. collected and analyzed the data. M.H.J. performed statistical analyses and reviewed the manuscript. S.C.F. reviewed the manuscript. A.E. wrote and reviewed the manuscript. A.E. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Footnotes
This article contains Supplementary Data online at http://diabetes.diabetesjournals.org/lookup/suppl/doi:10.2337/db14-1423/-/DC1.
References
- 1.Ergul A, Alhusban A, Fagan SC. Angiogenesis: a harmonized target for recovery after stroke. Stroke 2012;43:2270–2274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kalaria RN. Vascular basis for brain degeneration: faltering controls and risk factors for dementia. Nutr Rev 2010;68(Suppl. 2):S74–S87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bansal S, Sangha KS, Khatri P. Drug treatment of acute ischemic stroke. Am J Cardiovasc Drugs 2013;13:57–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ergul A, Li W, Elgebaly MM, Bruno A, Fagan SC. Hyperglycemia, diabetes and stroke: focus on the cerebrovasculature. Vascul Pharmacol 2009;51:44–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Prakash R, Somanath PR, El-Remessy AB, et al. Enhanced cerebral but not peripheral angiogenesis in the Goto-Kakizaki model of type 2 diabetes involves VEGF and peroxynitrite signaling. Diabetes 2012;61:1533–1542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Prakash R, Li W, Qu Z, Johnson MA, Fagan SC, Ergul A. Vascularization pattern after ischemic stroke is different in control versus diabetic rats: relevance to stroke recovery. Stroke 2013;44:2875–2882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Prakash R, Johnson M, Fagan SC, Ergul A. Cerebral neovascularization and remodeling patterns in two different models of type 2 diabetes. PLoS One 2013;8:e56264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ergul A, Elgebaly MM, Middlemore ML, et al. Increased hemorrhagic transformation and altered infarct size and localization after experimental stroke in a rat model type 2 diabetes. BMC Neurol 2007;7:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kozak A, Ergul A, El-Remessy AB, et al. Candesartan augments ischemia-induced proangiogenic state and results in sustained improvement after stroke. Stroke 2009;40:1870–1876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Elgebaly MM, Prakash R, Li W, et al. Vascular protection in diabetic stroke: role of matrix metalloprotease-dependent vascular remodeling. J Cereb Blood Flow Metab 2010;30:1928–1938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ergul A, Abdelsaid M, Fouda AY, Fagan SC. Cerebral neovascularization in diabetes: implications for stroke recovery and beyond. J Cereb Blood Flow Metab 2014;34:553–563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yun J, Rocic P, Pung YF, et al. Redox-dependent mechanisms in coronary collateral growth: the “redox window” hypothesis. Antioxid Redox Signal 2009;11:1961–1974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kelly-Cobbs AI, Prakash R, Li W, et al. Targets of vascular protection in acute ischemic stroke differ in type 2 diabetes. Am J Physiol Heart Circ Physiol 2013;304:H806–H815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Abdelsaid MA, Pillai BA, Matragoon S, Prakash R, Al-Shabrawey M, El-Remessy AB. Early intervention of tyrosine nitration prevents vaso-obliteration and neovascularization in ischemic retinopathy. J Pharmacol Exp Ther 2010;332:125–134 [DOI] [PubMed] [Google Scholar]
- 15.Abdelsaid MA, El-Remessy AB. S-glutathionylation of LMW-PTP regulates VEGF-mediated FAK activation and endothelial cell migration. J Cell Sci 2012;125:4751–4760 [DOI] [PubMed] [Google Scholar]
- 16.Ergul A, Kelly-Cobbs A, Abdalla M, Fagan SC. Cerebrovascular complications of diabetes: focus on stroke. Endocr Metab Immune Disord Drug Targets 2012;12:148–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ding Q, Ying Z, Gómez-Pinilla F. Exercise influences hippocampal plasticity by modulating brain-derived neurotrophic factor processing. Neuroscience 2011;192:773–780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Thored P, Wood J, Arvidsson A, Cammenga J, Kokaia Z, Lindvall O. Long-term neuroblast migration along blood vessels in an area with transient angiogenesis and increased vascularization after stroke. Stroke 2007;38:3032–3039 [DOI] [PubMed] [Google Scholar]
- 19.Ye Y, Wang G, Wang H, Wang X. Brain-derived neurotrophic factor (BDNF) infusion restored astrocytic plasticity in the hippocampus of a rat model of depression. Neurosci Lett 2011;503:15–19 [DOI] [PubMed] [Google Scholar]
- 20.Zhu M, Bi X, Jia Q, Shangguan S. The possible mechanism for impaired angiogenesis after transient focal ischemia in type 2 diabetic GK rats: different expressions of angiostatin and vascular endothelial growth factor. Biomed Pharmacother 2010;64:208–213 [DOI] [PubMed] [Google Scholar]
- 21.Fujio Y, Walsh K. Akt mediates cytoprotection of endothelial cells by vascular endothelial growth factor in an anchorage-dependent manner. J Biol Chem 1999;274:16349–16354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gerber HP, McMurtrey A, Kowalski J, et al. Vascular endothelial growth factor regulates endothelial cell survival through the phosphatidylinositol 3′-kinase/Akt signal transduction pathway. Requirement for Flk-1/KDR activation. J Biol Chem 1998;273:30336–30343 [DOI] [PubMed] [Google Scholar]
- 23.Costa PZ, Soares R. Neovascularization in diabetes and its complications. Unraveling the angiogenic paradox. Life Sci 2013;92:1037–1045 [DOI] [PubMed] [Google Scholar]
- 24.Boodhwani M, Sellke FW. Therapeutic angiogenesis in diabetes and hypercholesterolemia: influence of oxidative stress. Antioxid Redox Signal 2009;11:1945–1959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen J, Ye X, Yan T, et al. Adverse effects of bone marrow stromal cell treatment of stroke in diabetic rats. Stroke 2011;42:3551–3558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Radi R. Nitric oxide, oxidants, and protein tyrosine nitration. Proc Natl Acad Sci U S A 2004;101:4003–4008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gürsoy-Ozdemir Y, Can A, Dalkara T. Reperfusion-induced oxidative/nitrative injury to neurovascular unit after focal cerebral ischemia. Stroke 2004;35:1449–1453 [DOI] [PubMed] [Google Scholar]
- 28.Tan S, Bose R, Derrick M. Hypoxia-ischemia in fetal rabbit brain increases reactive nitrogen species production: quantitative estimation of nitrotyrosine. Free Radic Biol Med 2001;30:1045–1051 [DOI] [PubMed] [Google Scholar]
- 29.Gürsoy-Ozdemir Y, Bolay H, Saribaş O, Dalkara T. Role of endothelial nitric oxide generation and peroxynitrite formation in reperfusion injury after focal cerebral ischemia. Stroke 2000;31:1974–1980; discussion 1981 [DOI] [PubMed] [Google Scholar]
- 30.Bailey DM, Taudorf S, Berg RM, et al. Increased cerebral output of free radicals during hypoxia: implications for acute mountain sickness? Am J Physiol Regul Integr Comp Physiol 2009;297:R1283–R1292 [DOI] [PubMed] [Google Scholar]
- 31.Sirinyan M, Sennlaub F, Dorfman A, et al. Hyperoxic exposure leads to nitrative stress and ensuing microvascular degeneration and diminished brain mass and function in the immature subject. Stroke 2006;37:2807–2815 [DOI] [PubMed] [Google Scholar]
- 32.Sun L, Yang L, Fu Y, et al. Capacity of HSYA to inhibit nitrotyrosine formation induced by focal ischemic brain injury. Nitric Oxide 2013;35:144–151 [DOI] [PubMed] [Google Scholar]
- 33.Coucha M, Li W, Johnson MH, Fagan SC, Ergul A. Protein nitration impairs the myogenic tone of rat middle cerebral arteries in both ischemic and nonischemic hemispheres after ischemic stroke. Am J Physiol Heart Circ Physiol 2013;305:H1726–H1735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hall ED. Antioxidant therapies for acute spinal cord injury. Neurotherapeutics 2011;8:152–167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hall ED, Wang JA, Miller DM. Relationship of nitric oxide synthase induction to peroxynitrite-mediated oxidative damage during the first week after experimental traumatic brain injury. Exp Neurol 2012;238:176–182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang XL, Rainwater DL, Leone A, Mahaney MC. Effects of diabetes on plasma nitrotyrosine levels. Diabet Med 2004;21:577–580 [DOI] [PubMed] [Google Scholar]
- 37.El-Remessy AB, Al-Shabrawey M, Khalifa Y, Tsai NT, Caldwell RB, Liou GI. Neuroprotective and blood-retinal barrier-preserving effects of cannabidiol in experimental diabetes. Am J Pathol 2006;168:235–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.El-Remessy AB, Behzadian MA, Abou-Mohamed G, Franklin T, Caldwell RW, Caldwell RB. Experimental diabetes causes breakdown of the blood-retina barrier by a mechanism involving tyrosine nitration and increases in expression of vascular endothelial growth factor and urokinase plasminogen activator receptor. Am J Pathol 2003;162:1995–2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chao XD, Ma YH, Luo P, et al. Up-regulation of heme oxygenase-1 attenuates brain damage after cerebral ischemia via simultaneous inhibition of superoxide production and preservation of NO bioavailability. Exp Neurol 2013;239:163–169 [DOI] [PubMed] [Google Scholar]
- 40.el-Remessy AB, Bartoli M, Platt DH, Fulton D, Caldwell RB. Oxidative stress inactivates VEGF survival signaling in retinal endothelial cells via PI 3-kinase tyrosine nitration. J Cell Sci 2005;118:243–252 [DOI] [PubMed] [Google Scholar]
- 41.Hellberg CB, Boggs SE, Lapetina EG. Phosphatidylinositol 3-kinase is a target for protein tyrosine nitration. Biochem Biophys Res Commun 1998;252:313–317 [DOI] [PubMed] [Google Scholar]
- 42.Abdelsaid M, Kaczmarek J, Coucha M, Ergul A. Dual endothelin receptor antagonism with bosentan reverses established vascular remodeling and dysfunctional angiogenesis in diabetic rats: relevance to glycemic control. Life Sci 18 January 2014 [Epub ahead of print] [DOI] [PMC free article] [PubMed]
- 43.Abdelsaid M, Ma H, Coucha M, Ergul A. Late dual endothelin receptor blockade with bosentan restores impaired cerebrovascular function in diabetes. Life Sci 2014;118:263–267 [DOI] [PMC free article] [PubMed]
- 44.Duckworth WC. Hyperglycemia and cardiovascular disease. Curr Atheroscler Rep 2001;3:383–391 [DOI] [PubMed] [Google Scholar]
- 45.Lund SS, Tarnow L, Stehouwer CD, et al. Impact of metformin versus repaglinide on non-glycaemic cardiovascular risk markers related to inflammation and endothelial dysfunction in non-obese patients with type 2 diabetes. Eur J Endocrinol 2008;158:631–641 [DOI] [PubMed] [Google Scholar]
- 46.Adler AI, Stratton IM, Neil HA, et al. Association of systolic blood pressure with macrovascular and microvascular complications of type 2 diabetes (UKPDS 36): prospective observational study. BMJ 2000;321:412–419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.UK Prospective Diabetes Study (UKPDS) Group. Effect of intensive blood-glucose control with metformin on complications in overweight patients with type 2 diabetes (UKPDS 34). Lancet 1998;352:854–865 [PubMed] [Google Scholar]
- 48.Schernthaner G. Diabetes and Cardiovascular Disease: Is intensive glucose control beneficial or deadly? Lessons from ACCORD, ADVANCE, VADT, UKPDS, PROactive, and NICE-SUGAR. Wien Med Wochenschr 2010;160:8–19 [DOI] [PubMed] [Google Scholar]
- 49.Laakso M. Hyperglycemia and cardiovascular disease in type 2 diabetes. Diabetes 1999;48:937–942 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.