

Abstract
Background
Stent thrombosis (ST), a postinterventional complication with a mortality rate of 50%, has an incidence that rises precipitously in patients at risk. Chronic renal failure and end-stage renal disease have emerged as particularly strong ST risk factors, yet the mechanism remains elusive. Tissue factor (TF) is a crucial mediator of injury-related thrombosis and has been implicated for ST. We posit that uremia modulates TF in the local vessel wall to induce postinterventional thrombosis in patients with end-stage renal disease.
Methods and Results
As a model of the de-endothelialized, postinterventional state, we exposed primary human vascular smooth muscle cells (vSMCs) pretreated with uremic serum (obtained from ESRD patients on hemodialysis) to coronary-like blood flow. vSMC TF expression, activity, stability, and posttranslational modification were examined after vSMCs were treated with uremic serum or solutes. We found significantly greater clot formation after uremic serum exposure, which was substantially reduced with the prior treatment with anti-TF neutralizing antibody. Uremic sera induced 2- to 3-fold higher TF expression and activity in vSMCs independent of diabetes mellitus. Relevant concentrations of isolated uremic solutes such as indole-3-acetic acid (3.5 μg/mL), indoxyl sulfate (25 μg/mL), and uric acid (80 μg/mL) recapitulated these effects in cell culture and the flow loop model. We show further that TF undergoes ubiquitination at baseline and that uremic serum, indole-3-acetic acid, and indoxyl sulfate significantly prolong TF half-life by inhibiting its ubiquitination.
Conclusions
The uremic milieu is profoundly thrombogenic and upregulates vSMC TF levels by increasing TF stability and decreasing its ubiquitination. Together, these data demonstrate for the first time that the posttranslational regulation of TF in uremia may have a causative role in the increased ST risk observed in uremic patients. These data suggest that interventions that reduce vSMC TF may help to prevent ST and that uremic solutes should be considered as novel risk factors for ST in patients with chronic renal failure.
Keywords: angioplasty, coagulation, coronary stent, kidney, risk factors, thrombus, tissue
Tissue factor (TF) is critical to a range of pathological vascular processes including atherosclerosis, neointimal hyperplasia, and coagulation.1–5 After coronary intervention, be it balloon angioplasty or stent implantation, TF exposed on the denuded vascular smooth muscle cell (vSMC) surface serves as a key precipitant of injury-related local clot formation and stent thrombosis (ST),4,6 a potentially lethal event that kills up to half of patients.7,8 ST occurs with bare-metal stents or drug-eluting stents8,9 and may be influenced by a variety of stent design–, procedure-, patient-, and lesion-related factors. Chronic renal failure (CRF) and end-stage renal disease (ESRD) in particular are associated with poor outcomes.9–13
In 1 study, 1-year cardiac mortality after coronary intervention increased from 1.9% to 15.2% in patients with CRF (serum creatinine >3.0 mg/dL) and 10.0% for ESRD dialysis patients.14 The risk of ST rises 6.5-fold in patients with renal failure, and uremia predisposes to early (within 30 days), late (between 31 days and 1 year), and very late (≥1 year) ST.11,12
Multiple studies in CRF patients have shown increased circulating levels of regulators of thrombosis such as soluble TF, activated factor VIII, von Willebrand factor, TF pathway inhibitor, and fibrinogen.15–18 However, the effects of uremia on local TF activity in various vascular cell types remain scant.4,6 Although uremic serum is known to increase TF expression in endothelial cells (ECs),17 its effect on exposed vSMCs, the key driver of ST, remains unknown.
We examined the effect of the uremic milieu on vSMC TF protein kinetics, regulation, and function, hypothesizing that the uremic milieu inflicts a hypercoagulable state by modulating TF in vSMCs. Because uremic manifestations are thought to arise from the direct effects of retained uremic solutes on cells,19 we examined TF expression and activity in vSMCs treated with uremic serum and different uremic solutes at concentrations observed in CRF/ESRD patients19,20 (Table 1). An integrated assessment of uremia-enhanced thrombogenicity was performed with the use of an established flow model designed to emulate the postinterventional state by exposing vSMCs to blood in a relevant coronary-like hemodynamic environment.21 In this model, direct contact of blood with exposed vSMCs emulates the postinterventional reactive vascular status.21,22 Because de-endothelialized vSMCs persist over a prolonged time irrespective of stent type, our model is valid for late/very late ST due to both bare-metal and drug-eluting stents.
Table 1.
Uremic Solute Concentrations20
| Water soluble | |
| Urea | 1200 |
| Creatinine | 60 |
| Oxalic acid | 5 |
| Water insoluble | |
| Uric acid | 80 |
| Protein bound | |
| Indole-3-acetic acid | 3.5 |
| Indoxyl sulfate | 25 |
| Homocysteine | 2.7 |
| P-cresol | 10 |
Data are expressed as micrograms per milliliter.
Methods
Detailed methods regarding cell lysis, immunoblotting, immunoprecipitation, immunofluorescence, TF stability assay, ubiquitination, activity assay, enzyme-linked immunosorbent assay, reverse transcription polymerase chain reaction, uremic solutes, and vSMC injury model are described in the online-only Data Supplement.
Human Subjects and Serum Collection
Uremic sera from ESRD patients and control subjects were pooled as described previously.23 Briefly, the patients with ESRD on hemodialysis were recruited randomly from a pool of 150 patients at the DeVita Hemodialysis Center (Boston, MA). The protocol was approved by institutional review boards of both Boston University Medical Center and Massachusetts Institute of Technology. Patients aged 20 to 75 years were recruited over a 6-month period and were excluded if their hemoglobin concentration was <8 g/dL. Informed consent was obtained, and 10 mL of blood was collected before the next subsequent hemodialysis. Control sera were from Research Blood Component Inc (Boston, MA) for sex- and ethnicity-matched subjects. Blood was collected in Vacutainer tubes, and serum was separated by centrifuging clotted blood at 1100g for 10 minutes at room temperature to obtain serum. Blood urea nitrogen, creatinine, and glucose were assayed in all patients, and control sera were excluded if creatinine was >1.0 mg/dL.
Cell Lines
vSMCs (ATCC) were grown in Dulbecco’s modified Eagle’s medium–low glucose with 5% fetal calf serum, 1% penicillin, and streptomycin. Human coronary artery endothelial cells (Promocell) were grown in endothelial growth medium-2 (Promocell, Germany), as described previously.22 THP-1 cells (ATCC) were grown in Roswell Park Memorial Institute medium with 5% fetal bovine serum and 0.05 mmol/L 2-mercaptoethanol.
Flow Loop Tubes
A flow loop consists of Silastic tubes coated with human vSMCs subjected to a coronary flow condition that emulates the endovascular intervention ex vivo and serves as a screening tool to examine thrombosis in various vascular beds.21 Before cell seeding, 4-cm-long, 1/8-inch internal diameter Tygon tubes (Saint-Gobain, France) were prepared as described previously22 and injected with 1·106 cells per milliliter vSMCs into fibronectin-coated tubes and cultured for 36 hours under axial rotation at 10 rph, 37°C, 5% CO2.22
vSMC-coated tubes were exposed to control or uremic serum or uremic solutes for 24 hours. The segments were explanted, gently flushed to remove the media, and positioned in the reactive site flow loop model as described.21 Fresh whole blood was collected from healthy volunteers in a 10% acid/citrate dextrose solution. Immediately before testing, a 100-mmol/L CaCl2/75-mmol/L MgCl2 solution was added to the blood (70-μL solution per 1 mL blood) and loaded into the flow loops. After 10-minute runs, the loops were emptied and flushed with 60 mL Tyrode buffer (0.01 mol/L HEPES, 0.75 mmol/L MgCl2) to remove nonadherent material. The clot was lysed with 1% Triton-X solution for 20 minutes. Lactate dehydrogenase and hemoglobin were measured with the use of Quantichrome heme assay and Cyto-Tox 96 nonradioactive cytotoxicity assay (Promega) as measures of erythrocyte content and total clot mass, respectively.
TF neutralizing assay was performed as described previously.24 The vSMC-coated tubes exposed to uremic serum or uremic solutes for 24 hours were treated with isotype control or anti-TF neutralizing antibody (aTF) (50 μg/mL) (American Diagnostica) for 1 hour before the flow loop experiment.
Statistical Analysis
In all figures, data are expressed as mean±SEM. A nonparametric Kruskal-Wallis test, followed by a Scheffé post hoc analysis of the original measured values normalized to their corresponding controls, was conducted to determine statistical differences between values. Values of P<0.05 were considered significant.
Results
Patient Characteristics
All but 1 of the 26 ESRD patients approached consented to participate in the study, and none were excluded for anemia. One blood sample was excluded because it failed to clot as a result of an error in blood collection. Two samples from the 26 volunteers were excluded because their serum creatinine levels were 1.5 and 6.8 mg/dL. Baseline demographic characteristics of patients with normal renal function and those with ESRD (Table 2) were generally identical, although ESRD patients were aged 47±10 (range, 27–73) years, predominantly black men, and hypertensive. Half of the subjects with ESRD were diabetics. As expected, blood urea nitrogen, creatinine, and glucose levels were significantly higher and hemoglobin levels were significantly lower in uremic patients.
Table 2.
Patient Characteristics
| Control Group (n=24) | Uremic Group (n=24) | P | |
|---|---|---|---|
| Age, y* | 24–59 (42) | 29–73 (47) | 0.01 |
| Sex | |||
| Male | 19 | 19 | |
| Female | 5 | 5 | |
| Ethnic background | |||
| Black | 20 | 20 | |
| Hispanic | 3 | 3 | |
| Asian Indian | 1 | 1 | |
| White | 0 | 0 | |
| Cause of renal failure | |||
| Diabetes mellitus | 0 | 12 | |
| Hypertension | 0 | 5 | |
| Lupus nephritis | 0 | 1 | |
| Unknown | 0 | 6 | |
| Blood pressure, mm Hg† | |||
| Systolic | 113.4±8.5 | 152.65±11.32 | 0.006 |
| Diastolic | 69.45±4.9 | 84.95±6.9 | 0.004 |
| Hemoglobin, g/dL† | 13.9±1.04 | 10.69±1.39 | 0.007 |
| Blood urea nitrogen, mg/dL† |
15.2±2.73 | 70.45±12.32 | 0.0002 |
| Serum creatinine, mg/dL† |
0.81±0.11 | 6.96±1.43 | 0.0003 |
| Serum glucose, mg/dL† |
77.65±8.99 | 100±27.28 | 0.0001 |
Data are expressed as range (mean).
Data are expressed as mean±SD.
vSMCs Have the Highest Amount of Endogenous TF
Because coronary intervention affects many cell types, we examined relative levels of TF in vSMCs, human coronary artery ECs, and THP-1 cells, an acute myelomonocytic leukemia cell line considered to be a model for monocytes (Figure IA and IB in the online-only Data Supplement). TF abundance was examined by immunoblotting and corroborated with immunofluorescence (Figure IA and IB in in the online-only Data Supplement). vSMCs had at least 2- to 3-fold higher TF abundance compared with human coronary artery ECs and THP-1 cells. TF localization was found mainly at the plasma membrane and cytosol. With identical light exposure, virtually no TF was detected in human coronary artery ECs (Figure IB in the online-only Data Supplement). Increased baseline vSMC TF is consistent with the highest amount of clot formation observed with the vSMC-coated compared with the fibronectin- or EC-coated flow loop tubes (Figure IC in the online-only Data Supplement). The high level of vSMC TF expression and its recognized role in ST validated its further use in our study.
Uremic Serum Induces Higher Clotting in Ex Vivo Flow Loop Model
Visible clot was higher in uremic serum–treated tubes (Figure 1A). Because vSMC TF is a crucial component in the initiation of the coagulation cascade after injury,25 we examined the specific role of TF. The uremia-induced clotting was substantially reduced with aTF pretreatment (Figure 1A). Inhibition of TF significantly reduced hemoglobin and lactate dehydrogenase as markers of red blood cell number and thrombus mass (Figure 1A). Because aTF exclusively inhibits the activity of surface-exposed TF,24 these data indicate that uremic serum enhances the expression and activity of surface TF and that TF is the major pathogenic mediator of uremia-induced clot formation.
Figure 1.

Uremia is highly thrombogenic milieu increasing tissue factor (TF) expression and activity in vascular smooth muscle cells (vSMCs). A, Ex vivo thrombogenicity of uremic serum that is significantly abrogated by TF neutralization. Top, vSMCs grown on fibronectin-coated tubes were exposed to pooled 5% control or uremic serum for 24 hours. The tubes were then subjected to flow loop system with blood flowing on surface. A representative visual examination of clot after stopping the system is shown. Bottom, Hemoglobin (Hgb) and lactate dehydrogenase (LDH) values of the thrombus from the tubes treated with uremic serum and control serum are shown. Mean of 9 tubes is shown. *P<0.01 for hemoglobin and P<0.02 for LDH of uremic compared with control serum. Similarly, hemoglobin and LDH values of thrombus from the uremic serum–exposed tubes pretreated with control (control aB) or anti-TF neutralizing (aTF) antibody are shown. Mean of 6 tubes is shown. #P<0.01 for hemoglobin and P<0.02 for LDH of aTF compared with control antibody. Error bars=SEM. B, Uremic serum induces expression of TF. vSMC 1·105 cells seeded in 6-well plates and exposed to 5% and 10% of control (Con) and uremic (Ur) serum. Cell lysates prepared after 24 hours were immunoblotted for TF and pancadherin antibodies. Representative blot from 5 experiments is shown. Right, Densitometry analysis was performed on TF bands with ImageJ with the use of pancadherin to normalize the signal. Average of 5 experiments is shown. P<0.01 for 5% and P<0.005 for 10% uremic serum compared with control. Error bars=SEM. C, Immunofluorescence of TF in response to control and uremic serum. vSMCs treated for 24 hours with 5% control and uremic serum were fixed and stained for TF. Nuclei were stained with DAPI. D, Nondiabetic serum also increases TF expression. Cell lysates of vSMCs treated with 5% pooled control or nondiabetic or diabetic uremic serum were probed for TF and pancadherin. Representative blot of 3 experiments is shown. The TF band normalized with pancadherin. Average of 3 experiments is shown. P<0.02 for non–diabetes mellitus (DM) and P<0.01 for DM uremic serum compared with control. Error bars=SEM. E, TF activity increased with uremic serum. vSMCs 1·105 seeded in 6-well plates and exposed to 5% and 10% of control and uremic serum are shown. Cell lysates prepared after 24 hours were subjected to TF activity measurements. TF activity was normalized to number of vSMCs. Mean of 3 experiments performed in duplicate is shown. Error bars=SEM. WB indicates westernblot.
TF Abundance and Biological Activity Are Higher in vSMCs Treated With Uremic Serum
We validated the aforementioned finding in vSMCs treated with uremic serum. Exposure of uremic serum for 24 hours increased TF expression 1.8- to 3.3-fold with an increasing amount of uremic serum (Figure 1B). Increased TF expression was detected by immunofluorescence in the membrane and in the cytosol (Figure 1C). Half of our patient population had diabetes mellitus (DM). Because DM independently affects clotting,26 which can confound the effect of uremia, we separately examined the diabetic and nondiabetic uremic sera. Immunoblotting revealed an increase in TF after exposure to diabetic or nondiabetic uremic serum alike (Figure 1D). Increase in TF expression was correlated with an increase in biological activity. TF activity also increased significantly in vSMCs exposed to 5% uremic serum (control, 59.8±11.8 versus uremic, 125.8±12.5 pmol/L per 105 cells; P<0.05) or 10% serum (control, 72.3±10.7 versus uremic, 140.7±3.7 pmol/L per 105 cells; P<0.05) (Figure 1E). Intriguingly, increasing the serum concentration from 5% to 10% control or uremic sera resulted in only 19% and 11% increase in TF activity, respectively; thus, doubling the serum concentration did not result in a proportionate increase in TF activity.
Specific Uremic Solutes Increase TF Expression and Activity, Which is Further Augmented by Injury
Retained solutes are assumed to exert the effects of the uremic milieu. Therefore, we examined whether individual uremic solutes might be responsible for the increased TF levels and activity observed in vSMCs exposed to uremic serum. Among water-soluble uremic solutes, urea, creatinine, and oxalic acid had no effect on TF expression (Figure 2A). Water-insoluble uremic solutes doubled TF expression (Figure 2A and 2B) and activity (Figure 2E; TF activity rose from 12.2±0.5 to 22.4±0.9 pmol/L per 105 cells; P<0.05). Protein-bound uremic solutes such as indole-3-acetic acid (IA) and indoxyl sulfate (IS) increased TF expression (Figure 2C and 2D) and activity by 2-fold (Figure 2E; TF activity: control, 12.2±0.5; IA, 23.2±5.9 pmol/L per 105 cells; P<0.05; and IS, 28.7±3.0 pmol/L per 105 cells; P<0.01). The IA, IS, and uric acid (UA) increased TF predominantly in the cytosol and the membrane (Figure 2B and 2D). Thus, IA-, IS-, and UA-mediated increased TF activity tracked consistently with TF protein expression. vSMC scratch injury assay is an in vitro counterpart of the endovascular interventional injury model. TF expression almost doubled with scratch injury, consistent with previously published in vivo observations.27 Pretreatment of uremic solutes further augmented injury-induced TF expression, ranging from 40% with UA to almost 100% with IS (Figure 3E).
Figure 2.

Complexity of uremic solutes to increase tissue factor (TF) levels and activity in vascular smooth muscle cells (vSMCs). A, Uric acid increases TF expression. vSMCs were treated with water-soluble and -insoluble uremic solutes at concentrations observed in patients with chronic renal failure for 24 hours. Cell lysates were probed for TF and pancadherin. Cells treated with water or NaOH served as control. Representative blot from 3 experiments is shown. Densitometry analysis was performed on TF bands with the use of pancadherin to normalize the signal. Average of 3 experiments is shown. P<0.001 for uric acid compared with NaOH. Error bars=SEM. Crea indicates creatinine; and Oxal, oxalic acid. B, Immunofluorescence of TF. vSMCs treated with uremic solutes for 24 hours were fixed and stained with TF. DAPI was used to stain nuclei. C, Indole-3-acetic acid (IA) and indoxyl sulfate (IS) increase TF expression in vSMCs. Cell lysates of vSMCs treated with protein-bound uremic solutes were probed for TF and pancadherin antibodies. Cells treated with media containing human serum albumin served as control. Representative blot from 3 experiments is shown. Densitometry analysis was performed as above. Average of 3 experiments is shown. P<0.02 for IA and P<0.01 for IS compared with human serum albumin (HSA) (control). Error bars=SEM. D, Immunofluorescence of TF. vSMCs treated with protein-bound uremic solutes were fixed and stained with TF. DAPI was used to stain nuclei. E, Increase in TF activity in vSMCs treated with IA, IS, and uric acid. vSMCs 1·105 seeded in 6-well plates and exposed to exposed to uremic solutes serum are shown. Cell lysates prepared after 24 hours were subjected to TF activity measurements. TF activity was normalized to number of vSMCs. Mean of 3 experiments performed in duplicates is shown. Error bars=SEM. DMSO indicates dimethyl sulfoxide; and Hom, homocysteine.
Figure 3.

Temporal, dose-dependent, and injury-induced increase in vascular smooth muscle cell (vSMC) tissue factor (TF) with uremic solutes. A, Uric acid (UA)–induced upregulation of TF occurs within 4 hours of treatment. Lysates from vSMCs treated with NaOH and UA were harvested at different time periods and immunoblotted with TF and pancadherin antibodies. Representative blot from 3 experiments is shown. Densitometry analysis was performed. Average of 3 experiments is shown. P<0.01 for 4- and 24-hour UA and P<0.02 for 48-hour UA compared with NaOH. Error bars=SEM. B, Doubling of TF with concentrations of UA observed in patients with chronic renal failure. vSMCs treated with UA with increasing concentrations for 24 hours are shown. Representative blot from 3 experiments is shown. Densitometry analysis was performed. Average of 3 experiments is shown. P<0.02 for 80 μg/mL and P<0.001 for 100 μg/mL UA compared with NaOH. Error bars=SEM. C, Indole-3-acetic acid (IA) and indoxyl sulfate (IS) induce TF expression within 4 hours of treatment. vSMCs treated with IA and IS were harvested at different time intervals, and lysates were immunoblotted. Cells treated with human serum albumin served as control. Representative blot from 3 experiments is shown. Densitometry analysis was performed. Average of 3 experiments is shown. P<0.01 for 4, P<0.001 for 24, and P<0.01 for 48 hours for IA compared with human serum albumin; and P<0.01 for 4, 24, and 48 hours for IS compared with human serum albumin. Error bars=SEM. D, Increase in TF expression with increasing dose of IA and IS. vSMCs treated with IA and IS with increasing concentrations for 24 hours were harvested for immunoblotting. Cells treated with human serum albumin served as control. Representative blot from 3 experiments is shown. Densitometry analysis was performed. Average of 3 experiments is shown. P<0.05 for 1.75 μg/mL and P<0.01 for 3.25 μg/mL IA compared with human serum albumin; P<0.01 for 12.5 and 25 μg/mL IS compared with human serum albumin. Error bars=SEM. E, Injury further augments upregulation of TF. vSMCs 1·106 treated with uremic solutes for 24 hours were injured with a cell scraper. The cells were washed and incubated with fresh medium without uremic solutes and harvested after 2 hours. Lysates were probed with the use of TF and pancadherin antibodies. Representative blot from 4 experiments is shown. Densitometry analysis was performed. Average of 4 experiments is shown. All P values refer to TF increase without vs with scratch injury. P<0.02 for control, P<0.01 for IA and IS, and P<0.05 for uric acid. Error bars=SEM. WB indicates westernblot.
Increase in Thrombogenicity With IA-, IS-, and UA-Treated vSMCs in the Flow Loop Model
Because UA, IA, and IS increased TF protein abundance and activity (Figure 2A through 2E), we examined whether these solutes also induce thrombogenicity in our ex vivo flow loop model. Compared with control, UA treatment resulted in significantly higher clot formation as assessed through qualitative visualization as well as quantitative hemoglobin and lactate dehydrogenase measurements (Figure 4A). Inhibiting the surface TF activity with the use of aTF significantly abrogated clot formation in flow loop models (Figure 4A). Similarly, IA- and IS-treated flow loops exhibited significantly higher thrombogenicity, which was substantially reduced by aTF (Figure 4B and 4C). Overall, because the individual uremic solutes regulate TF expression, activity, and thrombogenicity, these observations suggest that uremia directly and independently increases thrombogenicity and that specific uremic solutes are accountable for the thrombogenicity in the uremic milieu.
Figure 4.

Uric acid, indole-3-acetic acid (IA), and indoxyl sulfate (IS) increase thrombogenicity and recapitulate the effect of uremic serum. A, Ex vivo thrombogenicity of uric acid on vascular smooth muscle cell (vSMC)–coated flow loop model. Top, vSMCs exposed to uric acid for 24 hours. The tubes were then subjected to the flow loop system. A representative visual examination of clot is shown. Bottom, Hemoglobin and lactate dehydrogenase (LDH) values of the thrombus from the tubes treated with uric acid and NaOH are shown. Mean of 9 tubes is shown. *Hemoglobin P<0.001 and LDH P<0.001 for uric acid compared with NaOH. Hemoglobin and LDH values of thrombus from the uric acid–exposed tubes pretreated with control (control aB) or anti-TF neutralizing (aTF) antibody are also shown. Mean of 6 tubes is shown. #P<0.01 for hemoglobin and P<0.02 for LDH of aTF compared with control antibody. Error bars=SEM. B, Ex vivo thrombogenicity of IA on vSMC-coated flow loop model. Top, vSMCs treated with IA and for 24 hours were subjected to flow loop. A representative visual examination of clot is shown. Bottom, Hemoglobin and LDH values of the thrombus from the tubes treated with IA and human serum albumin (control) are shown. Mean of 9 tubes is shown. *Hemoglobin P<0.05 and LDH P<0.05 for IA compared with human serum albumin. Hemoglobin and LDH values of thrombus from the IA-exposed tubes pretreated with control antibody or aTF antibody are also shown. Mean of 6 tubes. #P<0.05 for hemoglobin and P<0.05 for LDH for aTF compared with control antibody. Error bars=SEM. C, IS-induced thrombus was significantly inhibited by surface TF neutralization. Top, vSMCs treated with IA for 24 hours were subjected to flow loop. A representative visual examination of clot is shown. Bottom, Hemoglobin and LDH values of the thrombus from the tubes treated with IS and human serum albumin (control) are shown. Mean of 9 tubes is shown. *Hemoglobin P<0.05 and LDH P<0.05 for IA compared with human serum albumin. Hemoglobin and LDH values of thrombus from the IS-exposed tubes pretreated with control antibody or aTF antibody are also shown. Mean of 6 tubes is shown. #P<0.05 for hemoglobin and P<0.05 for LDH for aTF compared with control antibody. Error bars=SEM.
Uremic Serum or Solutes Regulate TF Posttranslationally
It is conceivable that TF can be regulated at multiple levels including transcriptional, translational, or posttranslational levels. Because uremic serum upregulates EC TF mRNA,17 we examined whether the same effect was observed in vSMCs (Figure IIA in the online-only Data Supplement). Treatment of vSMCs with 5% control, nondiabetic, or diabetic uremic serum had no effect on TF mRNA at different time points. Because uremic serum does not regulate TF mRNA, we examined whether TF may be regulated posttranslationally at a level of degradation similar to that of other integral membrane proteins.28 TF degradation and stability were measured by TF half-life after blocking protein translation.29 Uremic serum significantly prolonged TF half-life (Figure 5A) independent of the diabetic state. We then examined whether individual uremic solutes recapitulate this effect of uremic serum on TF stability. Although UA increased basal TF levels, there was no increase in TF half-life (Figure 5B). On the other hand, TF half-life was significantly prolonged to 3.5 hours with IA and IS (Figure 5C).
Figure 5.

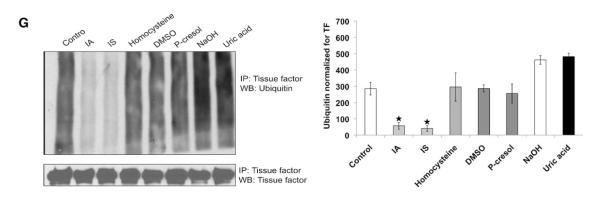
Uremic serum stabilizes tissue factor (TF) by inhibiting its ubiquitination, and indole-3-acetic acid (IA) and indoxyl sulfate (IS) recapitulate its effect on TF. A, Both nondiabetic and diabetic uremic sera increase TF half-life. Vascular smooth muscle cells (vSMCs) treated with pooled 5% control or nondiabetic or diabetic uremic sera for 24 hours are shown. Emetine 20 μmol/L was used for indicated time. The cells were harvested, and TF expression was analyzed by immunoblotting. Representative blot from 3 experiments is shown. Densitometry was performed on half-life study with the use of Image J, and pancadherin was used to normalize the TF signal. Average of 3 experiments is shown. *P value is 0.03 for both the DM and non-DM sera treated vSMCs compared to the control. Error bars=SEM. DM indicates diabetes mellitus. B, Uric acid does not affect half-life of TF. vSMCs treated with NaOH and uric acid for 24 hours were treated with emetine as above. Representative blot from 3 experiments is shown. Densitometry was performed on half-life study with Image J, and pancadherin was used to normalize the TF signal. *P value is 0.02 for both the IA and IS treated vSMCs compared to the control. Average of 3 experiments is shown. Error bars=SEM. C, IA and IS prolong half-life of TF. vSMCs exposed to protein-bound solutes and control (treated with human serum albumin [HSA]) for 24 hours were treated with emetine for indicated time. The cells were harvested, and TF expression was analyzed by immunoblotting. Representative blot from 3 experiments is shown. Densitometry was performed on half-life study shown in Figure 4F with the use of Image J, and pancadherin was used to normalize the TF signal. *P value is 0.02 for both the IA and IS treated vSMCs compared to the control. Average of 3 experiments is shown. Error bars=SEM. D, Ubiquitination of endogenous TF. vSMCs 1·106 were treated with MG132. TF was immunoprecipitated (IP) with the use of TF antibody and immunoblotted with ubiquitin antibodies. Immunoprecipitated TF is shown as input. Representative immunoblot from 3 experiments is shown. Densitometry analysis was performed on ubiquitin smear normalized with immunoprecipitated TF with the use of Image J. Average of 3 experiments is shown. P<0.05 for ubiquitin without vs with MG132. Error bars=SEM. E, Uremic serum inhibits TF ubiquitination. vSMCs treated with 5% pooled control or nondiabetic or diabetic uremic sera for 24 hours were lysed after treatment with MG132. TF ubiquitination was examined as described above. Representative immunoblot from 3 experiments is shown. Densitometry analysis was performed on ubiquitin smear as described above. Average of 3 experiments is shown. P<0.05 for non-DM and DM uremic compared with control serum. Error bars=SEM. F, Water-soluble uremic solutes have no effect on TF ubiquitination. vSMCs were treated with uremic solutes for 24 hours and 10 μM MG132 for the last 6 hours. Water-treated sample served as control. Representative immunoblot from 2 experiments is shown. Densitometry analysis was performed as described above. Average of 2 experiments is shown. Error bars=SEM. G, IA and IS inhibit TF ubiquitination. vSMCs were treated with protein-bound uremic solutes or uric acid solutes for 24 hours and MG132. TF was immunoprecipitated as above. Human albumin or NaOH served as control. Representative immunoblot from 3 experiments is shown. Densitometry analysis was performed as described above. Average of 3 experiments is shown. P<0.05 for IA and IS compared with human serum albumin (control). Error bars=SEM. DMSO indicates dimethyl sulfoxide; and WB, westernblot.
IA and IS Suppress Ubiquitination of Endogenous TF
Ubiquitination followed by proteasomal degradation is a fundamental process regulating protein stability and degradation.28 We first examined whether TF, like other membrane proteins, undergoes ubiquitination.28 Proteasome inhibitor treatment of vSMCs uncovered baseline ubiquitination of TF, which appears as a smear (Figure 5D). Consistent with TF half-life, uremic sera (non-DM and DM), IA, and IS inhibited TF ubiquitination (Figure 5E). On the other hand, those uremic solutes, such as UA, that had no effect of TF stability did not alter its ubiquitination (Figure 5F and 5G). These data indicate that IA and IS stabilize TF by inhibiting its ubiquitination.
Discussion
Patients with uremia are at significantly higher risk of atherothrombotic events, particularly after vascular intervention.13,30–33 In the present report, we show that uremic serum induces a profoundly thrombogenic state mediated in large part by regulation of vSMC TF expression and activity. Uremic serum has been documented to raise TF expression in ECs,17 which does not account for the substantial increase in thrombosis in the injured state, in which ECs are often lost. We now demonstrate a predominant effect on vSMCs. We further define the role of specific uremic solutes that may account for the effect of uremic serum on TF, distinguish the effects of protein-bound and water-soluble and -insoluble uremic solutes, link the thrombotic impact of specific uremic solutes to ubiquitination, examine the TF biology in the presence of these solutes compounded by injury, and describe a different and novel mechanism of vSMC TF regulation by uremic solutes.
Uremia Regulates vSMC TF Independent of DM
DM is a major cause of CRF and ESRD and increases thrombogenicity by elevating soluble TF in serum and TF expression in ECs.26 Thus, DM could pose a confounding effect in evaluating the independence of uremia on thrombogenicity. However, uremic sera increased TF expression, stabilized TF half-life, and reduced its ubiquitination in patient cohorts both with and without DM and to a similar extent. These findings indicate that the effect of uremia on TF is independent of DM. This conclusion is further supported by the fact that uremic solutes such as IA, IS, and UA (those solutes upregulating TF expression and activity) are elevated irrespective of the cause of renal failure or associated disease states. Our observations are consistent with prior studies such as the Registry of Stent Thrombosis for Review and Reevaluation (RESTART) trial, in which CRF (estimated glomerular filtration rate <30 mL/min) emerged as an independent risk factor in multivariate analysis for ST even in cases in which 45% of patients had DM.11
Complexity of Effects of Uremic Solutes on TF
Uremic solutes are thought to mediate the protean manifestations of renal failure. In general, they are the products of intermediate metabolism or retained proteins that failed to be fully excreted.34 There are numerous candidate uremic toxins, including β2-microglobulin, leptin, advanced glycosylation end products, p-cresol, IA, IS, and homocysteine.19 We examined the effects of some of the uremic solutes previously linked to vascular dysfunction.32 Although some solutes, in particular those that are water soluble, had no significant effect on TF levels, others led to dramatic increases in TF expression and activity. IA, IS, and UA in particular account for the effect of intact serum and act synergistically with vSMC injury in upregulating TF, albeit by different mechanisms. Protein-bound uremic solutes such as IA and IS increased the stability of TF by interfering with its ubiquitination process and increasing TF expression. Such protein binding may facilitate solute-TF binding and regulation of TF. For example, IA and IS share an indole ring that may interfere in TF binding with its putative E3 ubiquitin ligase, a possibility that remains to be explored. It also remains to be determined whether IA- and IS-mediated inhibition of ubiquitination is specific for TF or affects ubiquitination of other substrates.
The mechanism of UA-mediated TF regulation remains unclear. UA is a unique uremic solute, being neither protein bound nor water soluble at high physiological concentrations.35 Although UA increases TF levels and activity, we found no effect on TF mRNA or protein stability. Because protein production under steady state conditions is a combination of transcription, translation, and degradation, it is possible that UA regulates TF translation. It is also plausible that UA binds to TF and modifies its interactions with other proteins, such as factor VII, thereby augmenting its activity. In addition, UA may affect TF indirectly through regulating inflammatory pathways.2,35
These observations support a ST model in renal failure in which the uremic milieu increases basal TF levels and activity in vSMCs largely through regulating TF stability and ubiquitination. On vascular intervention, injury-induced increase in vSMC TF is further augmented in the presence of uremic solutes. This enhancement of local TF levels and activity in exposed vSMCs in combination with increased circulating levels of procoagulants15–17 such as activated factor VII18 in uremic patients exacerbates local coagulation, culminating in ST. All of these possibilities warrant further investigation and are active areas of investigation in our laboratory. Collectively, the present study illustrates the complexity of the effects of uremic solutes on TF regulation.
Levels of TF Regulation
TF mRNA is transcribed and translated with a leader peptide, which undergoes cleavage at the membrane, thus targeting TF to the plasma membrane.6 TF may be increased by regulation of any of these processes. Tumor necrosis factor-α, thrombin, and endotoxin induce TF expression in vSMCs6 by stimulating the TF promoter to induce transcription of TF mRNA via the mitogen-activated protein kinase and phosphoinositide-3 kinase pathways.6 Interestingly, we found no effect of uremic serum or uremic solutes on TF mRNA. This is in contrast to the findings of Serradell et al,17 who demonstrated increased TF mRNA in ECs exposed to uremic serum. Our observation of vSMC TF regulation on uremic solute exposure at an early 4-hour time point (Figure 3A and 3C) suggests a posttranscriptional mechanism and further distinguishes these findings from previous work with ECs.17
Protein degradation/stability, an important determinant of its function, is a highly regulated process. Many membrane proteins, including ligand-bound receptors, undergo lysosomal- or proteasome-mediated degradation. Proteins are usually tagged for proteasomal destruction by covalent attachment of ubiquitin, a small, compact protein, which is a 3-step process.28 This study confirms that tightly regulated protein such as TF undergoes ubiquitination under basal conditions. Those solutes such as IA and IS that regulate TF stability inhibited TF ubiquitination, whereas others that did not regulate TF stability had no effect on TF ubiquitination (Figure 5B and 5F). These data support the functional paradigm that the mechanisms of TF regulation are complex and are cell type and mediator specific.6
Other Clinical Correlates
Interestingly, treatment of vSMCs with increasing concentrations of uremic serum from 5% to 10% did not increase TF activity proportionate to TF expression (Figure 1E). A range of possibilities arises, including the potential contributions of other local thromboregulatory factors or the existence of multiple TF states. Numerous studies have shown increased circulating levels of various antithrombotic factors such as TF pathway inhibitor in uremic plasma.15,16 Although we did not specifically examine the effect of uremia on potential thrombotic inhibitors in the local milieu, it is plausible that increasing the uremic serum concentration may simultaneously induce expression of thrombotic inhibitors in vSMCs. It is conceivable that uremic serum may also modulate TF activity states. TF is thought to exist in 2 states: a low-activity state (also called encrypted) and a high-activity state (also called de-encrypted).1 It is plausible that uremic serum may alter the TF encryption-de-encrypted balance to prevent proportionate augmentation of TF activity. Because TF neutralizing antibody inhibits the activity of de-encrypted/active TF on the surface of vSMCs, our data suggest that the uremic milieu may increase the amount of decrypted TF.
TF level almost doubled with the solute concentrations found in patients with mild to moderate CRF20,36 and then reached a plateau with no further increase at higher levels of uremic solutes (Figure 3B and 3D). These data may suggest that even mild CRF can have a potent effect on thrombogenesis and that increasing uremic solute concentrations with the deterioration of renal function may not result in a further increase in vSMC TF expression or ST risk. This inference is consistent with the observations that worsening renal function beyond mild to moderate CRF (creatinine >1.6–2.0 mg/dL) did not further increase complications such as ST or worsen the outcomes after coronary intervention.14,37
Limitations
This investigation has several limitations, particularly in regard to the natural inability to precisely match control and uremic populations, the use of pooled serum, focus on a limited set of uremic solutes, and consideration of single pools of TF. Our dialysis population mirrored the expected demographics and therefore included older patients with significantly higher blood pressure than the healthy control patient group. Such a difference is unavoidable. The predominance of black subjects in our study reflects the presence of these subjects in Boston Medical Center in the inner city area of Boston. Data may therefore be biased toward the black population, but this is, if anything, an underrepresented population in most clinical trials. In this investigation, we used the pooled serum because we wanted to first examine the class effect of uremia on postinterventional thrombosis. Ideally, future studies will examine far larger study groups and the individual patient serum compared with age-, sex-, and ethnic background–matched controls. Several drugs can exert an effect on clotting. The fact that our control group consisted of unidentified subjects for whom the medication record was unavailable represents a potential limitation of this study. We examined only a subset of uremic solutes that are known to have vascular effects,34 and those solutes too were examined separately because our objective was to understand the independent effect of each solute. We consider that uremic serum is a mixture of a host of uremic solutes and hence should represent the combined effect of all of the solutes. It is likely that other factors in uremia may also play a role in vSMC TF. Nonetheless, the description of the effects and mechanism of action of uremic solutes on TF remains unique. Finally, we measured TF expression, activity, stability, and ubiquitination in whole cell lysate. Future studies may be able to distinguish the 3 distinct cellular pools in vSMCs, including membrane-bound: encrypted and de-encrypted, and intracellular TF.24
Implications of Uremia Regulation of TF
This study has several important clinical diagnostic and therapeutic implications. The finding that IA, IS, and UA link TF to ST in uremia offers the potential for novel biomarkers for assessing ST risk in CRF patients, and the finding that TF stability is linked to ubiquitination offers novel putative drug targets. These findings have broad implications that extend well beyond the phenomenon of ST as well. TF plays a crucial role in the development of atherosclerosis and its thrombotic complications,2,3 and accelerated atherosclerosis is a feature of uremic vascular disease.32 TF may now link uremia and uremic solutes to the progressive disease observed in CRF patients. TF overexpression in vSMCs has also been shown to augment neointimal hyperplasia in models of coronary intervention through both coagulant- and noncoagulant-mediated pathways.5 In addition to implications in the coronary vascular bed, such augmentation of TF levels with uremia may significantly affect other vascular beds. For example, neointimal hyperplasia and clotting are among the most common causes of hemodialysis access failure or suboptimal fistula performance, and therefore therapies directed toward IA, IS, and UA may well improve access success and survival.38,39 UA has long been implicated in hypertension, atherosclerosis, preeclampsia, and renal disease.35 The regulation by UA of TF in vSMCs may add novel insights linking such pathogenic mechanisms. Further exploration of the rich interactions between uremic solutes and vSMCs could well lead to novel mechanistic, diagnostic, and therapeutic insights into the vascular biology of patients with renal failure, a subgroup that is consistently plagued with poor cardiovascular outcomes.
Supplementary Material
Data Supplement Figure 1. TF expression is highest in vSMCs among different vascular cell-types.
Data Supplement Figure 2. Uremic serum does not later TF message
CLINICAL PERSPECTIVE.
Advances in cardiovascular therapeutics and techniques have expanded the pool of patients amenable for percutaneous intervention, and, increasingly, higher-risk patients with comorbidities such as concomitant chronic renal failure undergo complex interventions. However, these subsets are often at higher risk for treatment failures, and chronic renal failure and end-stage renal disease have been implicated as leading risk factors in endovascular stent thrombosis, a condition associated with a nearly 50% mortality rate. Although uremia has long been recognized as thrombogenic, the mechanisms and markers of postinterventional thrombosis remain poorly elucidated, thus limiting our ability to risk stratify and treat these patients effectively. In this study, we show that uremia, independent of diabetes mellitus (the leading cause of chronic renal failure), mediates postinterventional thrombosis by upregulating local tissue factor levels in vascular smooth muscle cells, which are the principal cells exposed to blood after endovascular manipulation. We identify specific and novel role of uremic solutes in regulating tissue factor through ubiquitination and subsequent degradation. Our data indicate that vascular smooth muscle cell tissue factor is the critical mediator of enhanced thrombogenicity in the postinterventional environment when exposed to uremia, offering a potential therapeutic target in these patients. We further identify specific uremic solutes in the postinterventional milieu that can be explored as biomarkers to stratify thrombotic risk in patients with chronic renal failure before intervention.
Acknowledgments
The authors thank Dr Laura Dember for help with the institutional review board application at Boston University Medical Center and Drs Laura Dember, Adam Segal, Jasvinder Bhatia, and Aylit Schultz (Boston University School of Medicine) for identifying potential participants. The authors thank Drs Richard Cohen, Markus Bachschmid, and Jean Francis (Boston University School of Medicine) for their insightful comments. We thank Pablo Santomá for help in flow loop models and Chrystelle Kiang and Carine J. Moezinia (Massachusetts Institute of Technology) for cell culture work.
Sources of Funding This work was funded in part by grant R01 GM-49039 (Dr Edelman), National Institutes of Health/National Institute of Diabetes and Digestive and Kidney Diseases grant K08 DK080946 (Dr Chitalia), Young Investigator Award of the National Kidney Foundation USA (Dr Chitalia), grant BFU2009-09804 (Dr Balcells) from Spain’s Ministerio de Ciencia e Innovación, Barcelona Chamber of Commerce (Dr Balcells), Fundació Empreses IQS and POSIMAT (Dr Balcells), grant FI-DGR 2011 (Dr Martorell) from Generalitat de Catalunya, Spain, and grant NIH, National Center For Research Center grant 5P51RR000168-48, and FDA Presidential Fellowship Award (Dr Bosch) and CIMIT Y11-177 (Dr Kolandaivelu).
Footnotes
Disclosures None.
This study was presented in part at the annual meeting of the American Society of Nephrology, November 8-13, 2011, Philadelphia, PA. Guest Editor for this article was Daniel I. Simon, MD.
The online-only Data Supplement is available with this article at http://circ.ahajournals.org/lookup/suppl/doi:10.1161/CIRCULATIONAHA.112.118174/-/DC1.
References
- 1.Butenas S, Orfeo T, Mann KG. Tissue factor in coagulation: which? where? when? Arterioscler Thromb Vasc Biol. 2009;29:1989–1996. doi: 10.1161/ATVBAHA.108.177402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cimmino G, D’Amico C, Vaccaro V, D’Anna M, Golino P. The missing link between atherosclerosis, inflammation and thrombosis: is it tissue factor? Expert Rev Cardiovasc Ther. 2011;9:517–523. doi: 10.1586/erc.11.40. [DOI] [PubMed] [Google Scholar]
- 3.Westrick RJ, Bodary PF, Xu Z, Shen YC, Broze GJ, Eitzman DT. Deficiency of tissue factor pathway inhibitor promotes atherosclerosis and thrombosis in mice. Circulation. 2001;103:3044–3046. doi: 10.1161/hc2501.092492. [DOI] [PubMed] [Google Scholar]
- 4.Gomez K, McVey JH. Tissue factor initiated blood coagulation. Front Biosci. 2006;11:1349–1359. doi: 10.2741/1888. [DOI] [PubMed] [Google Scholar]
- 5.Hasenstab D, Lea H, Hart CE, Lok S, Clowes AW. Tissue factor overexpression in rat arterial neointima models thrombosis and progression of advanced atherosclerosis. Circulation. 2000;101:2651–2657. doi: 10.1161/01.cir.101.22.2651. [DOI] [PubMed] [Google Scholar]
- 6.Steffel J, Lüscher TF, Tanner FC. Tissue factor in cardiovascular diseases: molecular mechanisms and clinical implications. Circulation. 2006;113:722–731. doi: 10.1161/CIRCULATIONAHA.105.567297. [DOI] [PubMed] [Google Scholar]
- 7.Motovska Z, Knot J, Widimsky P. Stent thrombosis: risk assessment and prevention. Cardiovasc Ther. 2010;28:e92–100. doi: 10.1111/j.1755-5922.2010.00186.x. [DOI] [PubMed] [Google Scholar]
- 8.Stähli BE, Camici GG, Tanner FC. Drug-eluting stent thrombosis. Ther Adv Cardiovasc Dis. 2009;3:45–52. doi: 10.1177/1753944708096280. [DOI] [PubMed] [Google Scholar]
- 9.Park DW, Park SW, Park KH, Lee BK, Kim YH, Lee CW, Hong MK, Kim JJ, Park SJ. Frequency of and risk factors for stent thrombosis after drug-eluting stent implantation during long-term follow-up. Am J Cardiol. 2006;98:352–356. doi: 10.1016/j.amjcard.2006.02.039. [DOI] [PubMed] [Google Scholar]
- 10.Iakovou I, Schmidt T, Bonizzoni E, Ge L, Sangiorgi GM, Stankovic G, Airoldi F, Chieffo A, Montorfano M, Carlino M, Michev I, Corvaja N, Briguori C, Gerckens U, Grube E, Colombo A. Incidence, predictors, and outcome of thrombosis after successful implantation of drug-eluting stents. JAMA. 2005;293:2126–2130. doi: 10.1001/jama.293.17.2126. [DOI] [PubMed] [Google Scholar]
- 11.Kimura T, Morimoto T, Kozuma K, Honda Y, Kume T, Aizawa T, Mitsudo K, Miyazaki S, Yamaguchi T, Hiyoshi E, Nishimura E, Isshiki T. RESTART Investigators. Comparisons of baseline demographics, clinical presentation, and long-term outcome among patients with early, late, and very late stent thrombosis of sirolimus-eluting stents: observations from the Registry of Stent Thrombosis for Review and Reevaluation (RESTART) Circulation. 2010;122:52–61. doi: 10.1161/CIRCULATIONAHA.109.903955. [DOI] [PubMed] [Google Scholar]
- 12.Lasala JM, Cox DA, Dobies D, Baran K, Bachinsky WB, Rogers EW, Breall JA, Lewis DH, Song A, Starzyk RM, Mascioli SR, Dawkins KD, Baim DS. ARRIVE 1 and ARRIVE 2 Participating Physicians. Drug-eluting stent thrombosis in routine clinical practice: two-year outcomes and predictors from the TAXUS ARRIVE registries. Circ Cardiovasc Interv. 2009;2:285–293. doi: 10.1161/CIRCINTERVENTIONS.109.852178.109.852178. [DOI] [PubMed] [Google Scholar]
- 13.Lambert ND, Sacrinty MT, Ketch TR, Turner SJ, Santos RM, Daniel KR, Applegate RJ, Kutcher MA, Sane DC. Chronic kidney disease and dipstick proteinuria are risk factors for stent thrombosis in patients with myocardial infarction. Am Heart J. 2009;157:688–694. doi: 10.1016/j.ahj.2009.01.009. [DOI] [PubMed] [Google Scholar]
- 14.Ting HH, Tahirkheli NK, Berger PB, McCarthy JT, Timimi FK, Mathew V, Rihal CS, Hasdai D, Holmes DR., Jr Evaluation of long-term survival after successful percutaneous coronary intervention among patients with chronic renal failure. Am J Cardiol. 2001;87:630–633. A9. doi: 10.1016/s0002-9149(00)01442-9. [DOI] [PubMed] [Google Scholar]
- 15.Mercier E, Branger B, Vecina F, Al-Sabadani B, Berlan J, Dauzat M, Fourcade J, Gris JC. Tissue factor coagulation pathway and blood cells activation state in renal insufficiency. Hematol J. 2001;2:18–25. doi: 10.1038/sj.thj.6200072. [DOI] [PubMed] [Google Scholar]
- 16.Dubin R, Cushman M, Folsom AR, Fried LF, Palmas W, Peralta CA, Wassel C, Shlipak MG. Kidney function and multiple hemostatic markers: cross sectional associations in the multi-ethnic study of atherosclerosis. BMC Nephrol. 2011;12:3. doi: 10.1186/1471-2369-12-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Serradell M, Díaz-Ricart M, Cases A, Zurbano MJ, Aznar-Salatti J, López-Pedret J, Ordinas A, Escolar G. Uremic medium disturbs the hemostatic balance of cultured human endothelial cells. Thromb Haemost. 2001;86:1099–1105. [PubMed] [Google Scholar]
- 18.Kario K, Matsuo T, Matsuo M, Koide M, Yamada T, Nakamura S, Sakata T, Kato H, Miyata T. Marked increase of activated factor VII in uremic patients. Thromb Haemost. 1995;73:763–767. [PubMed] [Google Scholar]
- 19.Vanholder R, Argilés A, Baurmeister U, Brunet P, Clark W, Cohen G, DeDeyn PP, Deppisch R, Descamps-Latscha B, Henle T, Jorres A, Massy ZA, Rodriguez M, Stegmayr B, Stenvinkel P, Wratten ML. Uremic toxicity: present state of the art. Int J Artif Organs. 2001;24:695–725. [PubMed] [Google Scholar]
- 20.Dou L, Bertrand E, Cerini C, Faure V, Sampol J, Vanholder R, Berland Y, Brunet P. The uremic solutes p-cresol and indoxyl sulfate inhibit endothelial proliferation and wound repair. Kidney Int. 2004;65:442–451. doi: 10.1111/j.1523-1755.2004.00399.x. [DOI] [PubMed] [Google Scholar]
- 21.Kolandaivelu K, Swaminathan R, Gibson WJ, Kolachalama VB, Nguyen-Ehrenreich KL, Giddings VL, Coleman L, Wong GK, Edelman ER. Stent thrombogenicity early in high-risk interventional settings is driven by stent design and deployment and protected by polymer-drug coatings. Circulation. 2011;123:1400–1409. doi: 10.1161/CIRCULATIONAHA.110.003210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Balcells M, Martorell J, Olivé C, Santacana M, Chitalia V, Cardoso AA, Edelman ER. Smooth muscle cells orchestrate the endothelial cell response to flow and injury. Circulation. 2010;121:2192–2199. doi: 10.1161/CIRCULATIONAHA.109.877282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chitalia VC, Murikipudi S, Indolfi L, Rabadi L, Valdez R, Franses JW, Edelman ER. Matrix-embedded endothelial cells are protected from the uremic milieu. Nephrol Dial Transplant. 2011;26:3858–3865. doi: 10.1093/ndt/gfr337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schecter AD, Giesen PL, Taby O, Rosenfield CL, Rossikhina M, Fyfe BS, Kohtz DS, Fallon JT, Nemerson Y, Taubman MB. Tissue factor expression in human arterial smooth muscle cells: TF is present in three cellular pools after growth factor stimulation. J Clin Invest. 1997;100:2276–2285. doi: 10.1172/JCI119765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Butenas S, Orfeo T, Mann KG. Tissue factor activity and function in blood coagulation. Thromb Res. 2008;122(suppl 1):S42–S46. doi: 10.1016/S0049-3848(08)70018-5. [DOI] [PubMed] [Google Scholar]
- 26.Bogdanov VY, Osterud B. Cardiovascular complications of diabetes mellitus: the tissue factor perspective. Thromb Res. 2010;125:112–118. doi: 10.1016/j.thromres.2009.06.033. [DOI] [PubMed] [Google Scholar]
- 27.Marmur JD, Rossikhina M, Guha A, Fyfe B, Friedrich V, Mendlowitz M, Nemerson Y, Taubman MB. Tissue factor is rapidly induced in arterial smooth muscle after balloon injury. J Clin Invest. 1993;91:2253–2259. doi: 10.1172/JCI116452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Staub O, Rotin D. Role of ubiquitylation in cellular membrane transport. Physiol Rev. 2006;86:669–707. doi: 10.1152/physrev.00020.2005. [DOI] [PubMed] [Google Scholar]
- 29.Chitalia VC, Foy RL, Bachschmid MM, Zeng L, Panchenko MV, Zhou MI, Bharti A, Seldin DC, Lecker SH, Dominguez I, Cohen HT. Jade-1 inhibits Wnt signalling by ubiquitylating beta-catenin and mediates Wnt pathway inhibition by pVHL. Nat Cell Biol. 2008;10:1208–1216. doi: 10.1038/ncb1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ocak G, Verduijn M, Vossen CY, Lijfering WM, Dekker FW, Rosend-aal FR, Gansevoort RT, Mahmoodi BK. Chronic kidney disease stages 1-3 increase the risk of venous thrombosis. J Thromb Haemost. 2010;8:2428–2435. doi: 10.1111/j.1538-7836.2010.04048.x. [DOI] [PubMed] [Google Scholar]
- 31.London GM. Vascular disease and atherosclerosis in uremia. Nefrologia. 2005;25(suppl 2):91–95. [PubMed] [Google Scholar]
- 32.Brunet P, Gondouin B, Duval-Sabatier A, Dou L, Cerini C, Dignat-George F, Jourde-Chiche N, Argiles A, Burtey S. Does uremia cause vascular dys-function? Kidney Blood Press Res. 2011;34:284–290. doi: 10.1159/000327131. [DOI] [PubMed] [Google Scholar]
- 33.Fort J. Chronic renal failure: a cardiovascular risk factor. Kidney Int Suppl. 2005;99:S25–S29. doi: 10.1111/j.1523-1755.2005.09906.x. [DOI] [PubMed] [Google Scholar]
- 34.Vanholder R, VanLaecke S, Glorieux G. The middle-molecule hypothesis 30 years after: lost and rediscovered in the universe of uremic toxicity? J Nephrol. 2008;21:146–160. [PubMed] [Google Scholar]
- 35.Feig DI, Kang DH, Johnson RJ. Uric acid and cardiovascular risk. N Engl J Med. 2008;359:1811–1821. doi: 10.1056/NEJMra0800885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Neri L, RoccaRey LA, Lentine KL, Hinyard LJ, Pinsky B, Xiao H, Dukes J, Schnitzler MA. Joint association of hyperuricemia and reduced GFR on cardiovascular morbidity: a historical cohort study based on laboratory and claims data from a national insurance provider. Am J Kidney Dis. 2011;58:398–408. doi: 10.1053/j.ajkd.2011.04.025. [DOI] [PubMed] [Google Scholar]
- 37.Rubenstein MH, Sheynberg BV, Harrell LC, Schunkert H, Bazari H, Palacios IF. Effectiveness of and adverse events after percutaneous coronary intervention in patients with mild versus severe renal failure. Am J Cardiol. 2001;87:856–860. doi: 10.1016/s0002-9149(00)01526-5. [DOI] [PubMed] [Google Scholar]
- 38.Hakim RM, Himmelfarb J. Hemodialysis access failure: a call to action: revisited. Kidney Int. 2009;76:1040–1048. doi: 10.1038/ki.2009.318. [DOI] [PubMed] [Google Scholar]
- 39.Lee T, Roy-Chaudhury P. Advances and new frontiers in the pathophysiology of venous neointimal hyperplasia and dialysis access stenosis. Adv Chronic Kidney Dis. 2009;16:329–338. doi: 10.1053/j.ackd.2009.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Supplement Figure 1. TF expression is highest in vSMCs among different vascular cell-types.
Data Supplement Figure 2. Uremic serum does not later TF message