

Abstract
Antigen-specific T cell receptor (TCR) gene transfer via patient-derived T cells is an attractive approach to cancer therapy, with the potential to circumvent immune regulatory networks. However, high-affinity tumour-specific TCR clonotypes are typically deleted from the available repertoire during thymic selection because the vast majority of targeted epitopes are derived from autologous proteins. This process places intrinsic constraints on the efficacy of T cell-based cancer vaccines and therapeutic strategies that employ naturally generated tumour-specific TCRs. In this study, we used altered peptide ligands and lentivirus-mediated transduction of affinity-enhanced TCRs selected by phage display to study the functional properties of CD8+ T cells specific for three different tumour-associated peptide antigens across a range of binding parameters. The key findings were: (i) TCR affinity controls T cell antigen sensitivity and polyfunctionality; (ii) supraphysiological affinity thresholds exist, above which T cell function cannot be improved; and (iii) T cells transduced with very high-affinity TCRs exhibit cross-reactivity with self-derived peptides presented by the restricting human leucocyte antigen. Optimal system-defined affinity windows above the range established for natural tumour-specific TCRs therefore allow the enhancement of T cell effector function without off-target effects. These findings have major implications for the rational design of novel TCR-based biologics underpinned by rigorous preclinical evaluation.
Keywords: adoptive therapy, cancer, T cell receptor, T cell, tumour antigen
Introduction
The vast majority of tumour-associated peptide antigens recognized by CD8+ T cells originate from autologous proteins and, as such, remain protected by immune tolerance mechanisms configured to prevent autoimmunity 1. Indeed, as a consequence of thymic selection, cognate T cell receptor (TCR) binding to these self-derived epitopes occurs at substantially lower affinities compared to pathogen-specific TCRs 2,3. This distinction probably explains why naturally generated T cell responses against cancer are largely non-protective 4. Moreover, the lack of high-affinity tumour-specific TCRs in the periphery may place intrinsic constraints on the efficacy of cancer vaccines designed to elicit T cell immunity.
Antigen-specific TCR gene transfer via patient-derived T cells is an attractive approach to cancer therapy, with the potential to break self-tolerance 5,6. Although only a limited number of trials have been conducted to date, evidence of clinical efficacy has generated substantial enthusiasm in the field 7–14. Importantly, these studies suggest that the use of engineered, affinity-enhanced TCRs may circumvent immune apathy towards tumour-derived antigens. Approximating affinity to mimic optimal pathogen-specific TCRs (KD = 0·1–10 μM) also makes sense from the biological perspective 2,15. Indeed, kinetic models of T cell activation propose that the potency of a peptide–major histocompatibility complex (pMHC) ligand is determined primarily by the duration of the TCR–pMHC interaction 16. However, it is difficult to disentangle the relative roles of affinity (KD) and TCR–pMHC dwell-time (t1/2) as determinants of ligand potency because, in most cases, these parameters are proportional 17. Thus, potent agonists generally bind with the highest affinities and longest half-lives 17–19, although many other parameters also affect T cell activation, antigen sensitivity and the response to TCR triggering 17,20.
In this study, we report a detailed analysis in experimental cancer systems probing the relationship between TCR–pMHC binding kinetics and T cell functionality. The data suggest that modest TCR affinity enhancements can substantially improve the functional profile of cognate T cells and indicate an optimal range for such modifications that varies across specificities.
Materials and methods
Peptides
All peptides were purchased in lyophilized form (Peptide Synthetics, Fareham, UK) and reconstituted in dimethyl sulphoxide (Sigma-Aldrich, Poole, UK) to a stock solution of 4 mg/ml. Working aliquots were prepared in RPMI medium supplemented with 100 U/ml penicillin, 100 μg/ml streptomycin and 2 mM L-glutamine (all from Life Technologies, Paisley, UK). Altered peptide ligands (APLs) were generated as described previously 18,21,22. Biophysical data are shown in Table 1.
Table 1.
Biophysical data for the ILA1 T cell receptor (TCR).
| Peptide | Primary sequence | Half-life (s) | KD eq (μM) | Kon (M−1 s−1) | Koff (s−1) | KD kin (μM) |
|---|---|---|---|---|---|---|
| 3G 18 | ILGKFLHWL | 14·7 | 3·7 | 1·6 × 104 | 4·7 × 10−2 | 2·9 |
| 3G8T 23 | ILGKFLHTL | 14·1 | 4 | 1·9 × 104 | 4·9 × 10−2 | 2·5 |
| 8T 18 | ILAKFLHTL | 7·3 | 27·6 | 4 × 103 | 9·5 × 10−2 | 23·75 |
| ILA 2,18 | ILAKFLHWL | 4·9a | 35·3a | 3·8 × 103a | 1·4 × 10−1a | 36a |
| 4L 24 | ILALFLHWL | 3·5 | 117 | 1·7 × 103 | 2 × 10−1 | 117 |
| 5Y 18,23 | ILAKYLHWL | 2·2 | 242 | 1·3 × 103 | 3·2 × 10−1 | 250 |
| 8E 18 | ILAKFLHEL | n.d. | >500 | n.a. | n.a. | n.a. |
KD = affinity; Kon = association rate; Koff = dissociation rate; n.d. = not determined; n.a. = too fast for reliable measurement. The index peptide is underlined. Variant amino acid residues are indicated in italics and bold type.
Average of combined studies.
T cell clone and target cell lines
The CD8+ clone ILA1, specific for the human telomerase reverse transcriptase (hTERT) peptide ILAKFLHWL (residues 540–548) restricted by human leucocyte antigen (HLA)-A*0201 (HLA-A2 from here onwards), was generated as described previously 21,22,25. Clonal cells were maintained in RPMI medium supplemented with 100 U/ml penicillin, 100 μg/ml streptomycin, 2 mM L-glutamine and 10% heat-inactivated fetal calf serum (FCS; Gibco/Invitrogen, Paisley, UK) (R10), together with 25 ng/ml interleukin (IL)-15 (PeproTech, Rocky Hill, NJ, USA), 200 IU/ml IL-2 (PeproTech) and 2·5% Cellkines (Helvetica Healthcare, Geneva, Switzerland). The following cell lines were cultured in R10: (i) HLA-A2+ C1R 26,27; (ii) Mel624·38 and Mel526 (melanoma; Thymed, Wendelsheim, Germany); (iii) HCT116 and HT144 [colorectal carcinoma; American Type Culture Company (ATCC), Manassas, VA, USA]; (iv) A375 and SKMel24 (melanoma; ATCC); (v) IM-9 [Epstein–Barr virus (EBV)-transformed B-LCL; ATCC]; (vi) SKMel37 (melanoma; Ludwig Institute for Cancer Research, Oxford, UK); and (vii) EJM [immunoglobulin (Ig)G lambda myeloma; DSMZ, Braunschweig, Germany]. NY-ESO-1+ HLA-A2+ tumour cell lines tested in this study were Mel624·38, Mel526, A375, SKMel37 and IM-9 28–31. MageA3+ HLA-A*0101+ (HLA-A1 from here onwards) tumour cell lines tested in this study were HCT116, HT144, A375, SKMel24 and EJM 28,30,32–34. The primary cell cultures HEP2 (HLA-A1+A2+ human hepatocyte; Sciencell, Carlsbad, CA, USA), SMC3 (HLA-A2+ human colonic smooth muscle; Sciencell), CIL-1 (HLA-A1+ human non-pigmented ciliary epithelium; Sciencell) and N9 (HLA-A2+ human epidermal melanocyte; ATCC) were maintained in their own proprietary media. HLA-A1+ 221 cells 35,36 were cultured in R10 supplemented with 800 μg/ml G418-sulphate (AG Scientific, San Diego, CA, USA).
Generation of CD8+ T cell cultures for lentivirus transduction
Blood bags were obtained from anonymous donors via the Welsh Blood Service (Pontyclun, UK) and confirmed seronegative for HIV-1. Lymphocytes were purified by standard density gradient centrifugation (Axis-Shield, Dundee, UK) and allotyped using αHLA-A1 and αHLA-A2 monoclonal antibodies (mAbs), as listed below. CD8+ T cells were positively selected using CD8 microbeads, purified through a magnetic-activated cell sorting (MACS) MS column (Miltenyi Biotec, Bisley, UK) and resuspended at 106 cells/well in R10 supplemented with IL-15, IL-2 and Cellkines, as above. Cells were activated overnight with αCD3/CD28 Dynabeads (Invitrogen) at a bead-to-cell ratio of 3 : 1 before lentivirus transduction.
Lentivirus generation and transduction of CD8+ T cells
Primary CD8+ T cells were transduced with lentivirus constructs expressing TCRs specific for the HLA-A2-restricted cancer-testis antigen NY-ESO-1157–165 (SLLMWITQC) or the HLA-A1-restricted melanoma antigen MageA3161–169 (EVDPIGHLY). Wild-type and high-affinity TCR mutants for NY-ESO-1157–165 were generated as described previously 37,38. High-affinity TCR mutants for MageA3161–169 were generated as part of this study. Biophysical data are shown in Table 2. The lentivirus transduction system was kindly provided by James Riley (University of Pennsylvania, PA, USA). Lentivirus particles were generated using a plasmid biosafe system as described previously 39. Briefly, lentivirus vector plasmids bearing each TCR construct were combined with the packaging plasmids pRSV.REV, pMDLg/pRRE and pVSG-V before transfection of 293T/17 cells (ATCC) with the Express-in reagent (Open Biosystems, Lafayette, CO, USA). Supernatant was collected after 24-h and 48-h incubations, and lentivirus particles were concentrated by ultracentrifugation. Activated primary CD8+ T cells (106 in 1 ml) were transduced with 1 ml of concentrated lentivirus. Three days later, transduction efficiency was determined by flow cytometry after staining with the relevant pMHCI tetramer and T cell antigen receptor variable (TCRV)β-specific mAb. Dynabeads were removed by magnet separation 5 days after transduction.
Table 2.
Biophysical data for the NY-ESO-1157–165 and MageA3161–169 T cell receptors (TCRs).
| Specificity (HLA restriction) | TCR (α/β) | Half-life (s) | KD (nM) | Kon (M−1 s−1) | Koff (s−1) |
|---|---|---|---|---|---|
| NY-ESO-1157–165 (HLA-A*0201) | wt/wt | 6·4 | 32000 | 4 × 104 | 1·28 × 10−1 |
| c259/wt | 19 | 730 | 4·9 × 104 | 3·6 × 10−2 | |
| c12/c2 | 240 | 450 | 9 × 103 | 4 × 10−3 | |
| wt/c51 | 720 | 25 | 5·4 × 104 | 1·4 × 10−3 | |
| c58/c61 | 68100 | 0·026 | 5·7 × 105 | 2·72 × 10−5 | |
| MageA3161–169 (HLA-A*0101) | wt/wt | n.a. | 300000 | n.a. | n.a. |
| a3c/wt | 0·7 | 55000 | 1·7 × 104 | 9·45 × 10−1 | |
| wt/b2a | 10·4 | 9430 | 1·2 × 104 | 7·7 × 10−2 | |
| a3a/wt | 6·1 | 6550 | 1·7 × 104 | 1·14 × 10−1 | |
| a3/wt | 66 | 190 | 5·5 × 104 | 1 × 10−2 | |
| wt/b2 | 120 | 169 | 3·3 × 104 | 6 × 10−3 |
KD = affinity; Koff = dissociation rate; Kon = association rate; HLA = human leucocyte antigen; n.a. = too fast for reliable measurement; TCR = T cell receptor; wt = wild-type.
Monoclonal antibodies, amine reactive dyes and tetrameric complexes
Directly conjugated mAbs were sourced as follows: (i) αCD3-PacificBlue, αCD8-allophycocyanin-H7 (APCH7), αCD19-AmCyan, αCD107a-fluorescein isothiocyanate (FITC), anti-macrophage inflammatory protein-1β-phycoerythrin (αMIP-1β-PE), anti-tumour necrosis factor-α-peridinin chlorophyll protein-cyanin 5·5 (αTNF-α-PerCPCy5.5), anti-interferon (αIFN)-γ-PECy7 and αIL-2-APC (BD Biosciences, San Jose, CA, USA); (ii) αTCRVβ5a-FITC (Thermo Scientific, Hemel Hempstead, UK); (iii) αTCRVβ5·1-FITC and αTCRVβ13·1-FITC (Beckman Coulter, High Wycombe, UK); (iv) αHLA-A2-FITC (Serotec, Kidlington, UK); and (v) αCD19-BV510 (BioLegend, London, UK). Dead cells were excluded from the analysis using live/dead fixable Aqua (Life Technologies). A purified mAb specific for HLA-A1/A36 was coupled to mouse αIgM-PE (Abcam, Cambridge, UK). Soluble pMHCI tetramers were generated as described previously 40,41.
Intracellular cytokine staining
T cells were resuspended at 106 cells/ml in RPMI medium supplemented with 100 U/ml penicillin, 100 μg/ml streptomycin, 2 mM L-glutamine and 2% FCS (R2) overnight. Target cells were pulsed with peptide as indicated for 1 h and washed before the addition of T cells at an effector-to-target ratio of 1 : 2. In experiments with CD8+ T cell clones, target cells were pulsed with the index peptide and each APL as listed in Table 1. In experiments with transduced CD8+ T cells, target cells were pulsed only with the index peptide (unless indicated otherwise). The irrelevant HLA-A2-restricted peptides HIV-1 Gag77–85 (SLYNTVATL) and influenza A matrix protein M158–66 (GILGFVFTL) were used as specificity controls 42,43. Experiments with tumour targets were conducted in the absence of exogenous peptide. In all assays, 1 μl/ml brefeldin A (Sigma-Aldrich), 0·35 μl/ml monensin (BD Biosciences) and 5 μl/ml αCD107a-FITC (where indicated) were added and the cultures were incubated for 6 h. After antigen stimulation, cells were washed and stained sequentially with live/dead fixable Aqua and mAbs directed against surface-expressed molecules. Cells were then fixed/permeabilized using a Cytofix/Cytoperm kit (BD Biosciences) and stained intracellularly for CD3, TCRVβ, MIP-1β, TNF-α, IFN-γ and IL-2, as indicated. Five effector functions were measured for experiments with the ILA1 clone. Four effector functions were measured for experiments with transduced T cells, as the relevant αTCRVβ mAb replaced αCD107a in the staining panel. Soluble αCD3 mAb (2 μg/ml) or phytohaemagglutinin (PHA) (1 μg/ml) were used as positive controls for activation. Stained cells were acquired using a FACS Canto II flow cytometer (BD Biosciences) with DIVA software (TreeStar Inc., Ashland, OR, USA). Fluorescence minus one (FMO) controls were used to check compensation and determine cell population gates. Events were gated as CD8+ (for the ILA clone) or relevant TCRVβ+ CD8+ (for transduced T cells) prior to effector function analysis after Boolean gating using FlowJo software version 7·6.4 (TreeStar Inc.). spice software version 5·33 was used for data presentation and analysis 44.
Measurement of soluble factors by ELISA
T cells were resuspended in R2 overnight. Target cells were pulsed with peptide as indicated for 1 h and washed before the addition of T cells at an effector-to-target ratio of 1 : 2. After overnight incubation, cells were pelleted and the culture supernatant was harvested for measurement of MIP-1β, TNF-α, IFN-γ and IL-2 by enzyme-linked immunosorbent assay (ELISA), according to the manufacturer's protocol (R&D Systems, Abingdon, UK).
Measurement of soluble factors by Luminex
Transduced T cells were defrosted, incubated in R2 for 2 h and added to target cells at an effector-to-target ratio of 1 : 2. After 24 h, the culture supernatant was harvested, centrifuged and stored at −20°C. Soluble factors were measured according to the manufacturer's instructions using a Human Cytokine 25-plex Panel (Invitrogen) with a Luminex® 200™ analyzer (Luminex, Austin, TX, USA).
Statistical analysis
The parametric Pearson's correlation was used to determine the strength of linear relationships between two variables. Correlations were determined using Prism version 6 software for Mac OSX (GraphPad Prism, Ashburton, UK). Statistically significant differences between concatenated polyfunctionality pies were determined using the permutation test in spice version 5·33 over 10 000 repetitions 44.
Results
CD8+ T cell polyfunctionality correlates with TCR–pMHC affinity
To study the relationship between T cell polyfunctionality and TCR–pMHC affinity, we made use of the HLA-A2-restricted hTERT-specific CD8+ T cell clone ILA1 18,45. The ILA1 TCR is known to bind a series of agonist APLs with affinity (KD) values ranging from 2·5 μM to >500 μM and half-life (t1/2) durations estimated from 1 s to a measured time of 14·7 s (Table 1). Importantly, this monoclonal system allows standardized comparisons as a function of TCR–pMHC binding. Moreover, all seven peptide ligands used in this study bind similarly to HLA-A2 18.
The ILA1 clone was stimulated with peptide-pulsed HLA-A2+ C1R cells across a range of concentrations for each ligand, and five different effector functions (CD107a, MIP-1β, TNF-α, IFN-γ and IL-2) were measured simultaneously by flow cytometry. At the highest peptide concentration, TCR–pMHC affinity correlated significantly with T cell polyfunctionality (KD versus five functions: Pearson's r = −0·99, P < 0·0001; KD versus four functions: Pearson's r = −0·93, P = 0·0027) (Fig. 1a). The lowest-affinity ligand (8E) elicited primarily single (MIP-1β) or dual (MIP-1β and TNF-α) function cells, although less easily triggered outputs were apparent at higher peptide concentrations. Higher-affinity ligands induced progressively greater numbers of polyfunctional cells at any given peptide concentration in accordance with a sensitivity hierarchy for each effector output (Fig. 1b). Moreover, the quantity of each soluble factor (MIP-1β, TNF-α, IFN-γ and IL-2) released into the supernatant mirrored the corresponding cellular percentages (Fig. 2a,b). Thus, under standardized conditions controlled for potentially confounding factors, TCR–pMHC affinity dictates the functional profile of cognate CD8+ T cells by regulating peptide sensitivity.
Fig 1.

Activation of the human leucocyte antigen (HLA)-A2-restricted ILA1 CD8+ T cell clone with biophysically characterized altered peptide ligands (APLs). ILA1 cells were stimulated with peptide-pulsed HLA-A2+ C1R target cells as indicated. Five functional readouts [CD107a, macrophage inflammatory protein (MIP)-1β, tumour necrosis factor (TNF)-α, interferon (IFN)-γ and interleukin (IL)-2] were measured by flow cytometry. (a) Overview of functional profiles. Pie chart segments represent the fraction of cells expressing the number of functions indicated in the key. Serial gating was performed as follows: (i) doublets were excluded in a forward scatter (FSC)-A versus FSC-W display; (ii) artefacts and fluorochrome aggregates were removed by Boolean analysis; (iii) viable CD3+ CD8+ cells were selected; and (iv) individual functions were identified. Combination gates were exported into spice software version 5·33 for further analysis. (b) Detailed analysis of functional profiles at peptide concentrations of 10−5 M. The pie charts are extended with arcs defining expressed functions as indicated in the key. The index peptide is denoted in red. Data shown are representative of three independent experiments. The corresponding biophysical parameters are listed in Table 1.
Fig 2.

Production of soluble factors in response to stimulation of the human leucocyte antigen (HLA)-A2-restricted ILA1 CD8+ T cell clone with biophysically characterized altered peptide ligands (APLs). (a) ILA1 cells were stimulated with peptide-pulsed HLA-A2+ C1R target cells as indicated. Supernatant was assayed for macrophage inflammatory protein (MIP)-1β, tumour necrosis factor (TNF)-α, interferon (IFN)-γ and interleukin (IL)-2 by enzyme-linked immunosorbent assay (ELISA). (b) Percentage frequencies of ILA1 cells expressing MIP-1β, TNF-α, IFN-γ and IL-2 determined by flow cytometry as described in Fig. 1.
TCR-transduced T cell polyfunctionality is dependent on donor and target cells
The above results reinforce the concept that TCR–pMHC affinity and/or half-life critically determine T cell antigen sensitivity and suggest that the functional profile of cancer-specific CD8+ T cells can be augmented via the manipulation of TCR binding parameters. To explore this possibility, we examined the impact of affinity enhancement on TCR-transduced functional outputs.
In initial experiments, CD8+ T cells from three different donors were transduced with a previously characterized HIV-1-specific TCR 15. The functional profiles observed across a range of cognate antigen concentrations differed substantially between donors (Supporting information, Fig. S1). Moreover, in accordance with a previous study 46, we found that antigen sensitivity varied across different target cells pulsed with identical concentrations of exogenous peptide (data not shown). Consequently, we compared only results obtained in parallel using identical target cells and the same parental CD8+ T cell donor (unless stated otherwise).
Affinity enhancement augments the functional profile of cancer-specific TCR-transduced CD8+ T cells
Next, we generated affinity-enhanced TCRs specific for two cancer-associated peptide epitopes: (i) NY-ESO-1157–165 (SLLMWITQC) restricted by HLA-A2; and (ii) MageA3161–169 (EVDPIGHLY) restricted by HLA-A1 (Table 2). CD8+ T cells from HLA-matched donors transduced with the highest affinity TCRs for NY-ESO-1 (c58/c61, KD = 26 pM) and MageA3 (a3/wt and wt/b2, KD = 190 nM and 169 nM, respectively) became non-viable in culture shortly after transduction. In contrast, HLA-A2− T cells transduced with the NY-ESO-1 c58/c61 TCR and HLA-A1− T cells transduced with the MageA3 a3/wt or wt/b2 TCRs were viable, but became activated in response to targets expressing the restricting HLA molecule without the addition of cognate peptide (data not shown). This self-reactivity probably explains the non-viability, via fratricide, of transduced T cells bearing the targeted HLA complex 47. Consequently, T cells engineered to express such super-enhanced TCRs are not likely to be useful in the therapeutic setting.
In subsequent experiments, we examined the specificity of HLA-A2+ CD8+ T cells transduced with more modestly enhanced NY-ESO-1-specific TCRs (Fig. 3). Four functional outputs (MIP-1β, TNF-α, IFN-γ and IL-2) were measured in response to cognate peptide exposure. Non-transduced CD8+ cells were used to control for non-specific activation via endogenous TCRs. Transduced CD8+ T cells were generated with the wild-type TCR and each of the following three affinity-enhanced variants: (i) c259/wt (KD = 730 nM, t1/2 = 19 s; fold increase: KD = 44, t1/2 = 3); (ii) c12/c2 (KD = 450 nM, t1/2 = 4 min; fold increase: KD = 71, t1/2 = 38); and (iii) wt/c51 (KD = 25 nM, t1/2 = 12 min; fold increase: KD = 1280, t1/2 = 113). All transduced cells expressed equivalent levels of TCR as determined by staining with the relevant αTCRVβ mAb and cognate pMHCI tetramer (data not shown).
Fig 3.
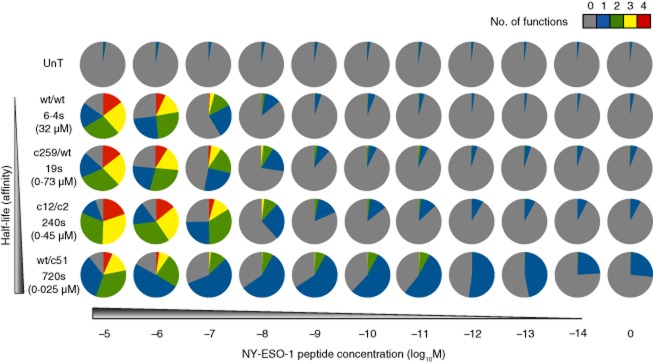
Activation of human leucocyte antigen (HLA)-A2-restricted NY-ESO-1-specific T cell receptor (TCR)-transduced CD8+ T cells by peptide-pulsed target cells. HLA-A2+ CD8+ T cells transduced with the indicated NY-ESO-1-specific TCRs were stimulated with HLA-A2+ C1R target cells pulsed with the cognate SLLMWITQC peptide across a range of concentrations. Four functional readouts [macrophage inflammatory protein (MIP)-1β, tumour necrosis factor (TNF)-α, interferon (IFN)-γ and interleukin (IL)-2] were measured by flow cytometry. Pie chart segments represent the fraction of cells expressing the number of functions indicated in the key. The gating strategy followed that described for Fig. 1a, with the exception that T cell antigen receptor variable (TCRV)β13·1 expression was used to identify transduced T cells where appropriate. Data shown are representative of two independent experiments. The corresponding biophysical parameters are listed in Table 2. UnT = non-transduced.
No specific responses to SLLMWITQC peptide-pulsed HLA-A2+ C1R target cells were observed in the absence of TCR transduction. In contrast, CD8+ T cells transduced with the wild-type, c259/wt or c12/c2 TCRs displayed detectable chemokine/cytokine production above background at cognate peptide concentrations as low as 10−11 M. Even greater antigen sensitivity was observed for the wt/c51 TCR, which elicited functional outputs at peptide concentrations down to 10−13 M. A positive association was observed between TCR affinity and the degree of polyfunctionality for CD8+ T cells transduced with the wild-type, c259/wt and c12/c2 TCRs. This was most evident at peptide concentrations in the region of 10−7 M. Furthermore, as in the ILA1 system, effector functions were elicited in a defined order (MIP-1β > TNF-α > IFN-γ > IL-2) with increasing TCR affinity (Supporting information, Fig. S2a). However, the proportion of wt/c51 TCR-transduced CD8+ T cells expressing three or four functions at equivalent peptide concentrations was substantially smaller compared with each of the other NY-ESO-1-specific TCRs, suggesting that the 1280-fold affinity enhancement lies beyond an optimum for the recognition of cognate antigen at the cell surface. In addition, T cells bearing the wt/c51 TCR exhibited background activation in the form of MIP-1β release. This contrasts with a previous study in which the wt/c51 TCR did not show any off-target activity 48. The most likely explanation for this discrepancy relates to the fact that MIP-1β release by CD8+ T cells is substantially more sensitive to antigen density than most other readouts of effector function 49. It is therefore important to examine a wide range of specific outputs when testing TCRs that may subsequently be used in vivo. Collectively, these data show that there is a TCR affinity window, exemplified by the c259/wt TCR, which allows improved recognition of a tumour-associated epitope in the absence of apparent cross-reactivity with other self-derived pMHC molecules.
To extend these findings, we performed similar experiments in the HLA-A1-restricted MageA3 system (Fig. 4). Transduced HLA-A1+ CD8+ T cells were generated with the wild-type TCR and each of the following three affinity-enhanced variants: (i) a3c/wt (KD = 55 μM, t1/2 = 0·7 s; fold increase: KD = 5·5); (ii) wt/b2a (KD = 9·43 μM, t1/2 = 10·4 s; fold increase: KD = 32); and (iii) a3a/wt (KD = 6·55 μM, t1/2 = 6·1 s; fold increase: KD = 46). Again, all transduced cells expressed equivalent levels of TCR (data not shown). As in the NY-ESO-1 system, there was a positive association between TCR affinity and peptide sensitivity. Greater proportions of polyfunctional CD8+ T cells, compared to wild-type, were observed with the a3c/wt and a3a/wt TCRs. In contrast, wt/b2a elicited functional profiles that were not substantially enhanced with respect to the wild-type TCR. It is notable that, despite a shorter half-life, the a3a/wt TCR elicited higher frequencies of cells producing two or more functions compared to the wt/b2a TCR. Marginally increased non-specific activation, reflected predominantly by MIP-1β release, was observed with both of these higher-affinity TCRs. Nonetheless, the hierarchy of effector functions (MIP-1β > TNF-α > IFN-γ/IL-2) elicited by increasing TCR affinity for the cognate pMHCI complex remained intact (Supporting information, Fig. S2b).
Fig 4.
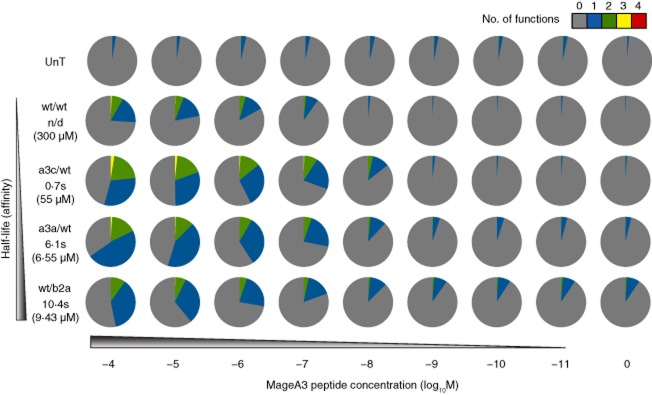
Activation of human leucocyte antigen (HLA)-A1-restricted MageA3-specific T cell receptor (TCR)-transduced CD8+ T cells by peptide-pulsed target cells. HLA-A1+ CD8+ T cells transduced with the indicated MageA3-specific TCRs were stimulated with HLA-A1+ 221 target cells pulsed with the cognate EVDPIGHLY peptide across a range of concentrations. Four functional readouts [macrophage inflammatory protein (MIP)-1β, tumour necrosis factor (TNF)-α, interferon (IFN)-γ and interleukin (IL)-2] were measured by flow cytometry. Pie chart segments represent the fraction of cells expressing the number of functions indicated in the key. The gating strategy followed that described for Fig. 1a, with the exception that T cell antigen receptor variable (TCRV)β5·1 expression was used to identify transduced T cells where appropriate. Data shown are representative of two independent experiments. The corresponding biophysical parameters are listed in Table 2. UnT = non-transduced.
Overall, these results demonstrate that TCR affinity enhancement can improve both T cell antigen sensitivity and the number of effector functions elicited at a given peptide concentration. However, such improvements in T cell function will only be relevant clinically if they extend to the recognition of tumour targets.
High-affinity cancer-specific TCRs mediate enhanced functional recognition of tumour cells
Tumour cells are known to express a range of epitope densities at the cell surface 1,31,50,51. We therefore examined the functional profile of NY-ESO-1-specific TCR-transduced CD8+ T cells in response to five different NY-ESO-1+ HLA-A2+ tumour cell lines: (i) Mel624·38, A375, SKMel37 and Mel526 (melanoma); and (ii) IM-9 (EBV-transformed B-LCL). Two NY-ESO-1− primary cell cultures were used as negative controls: (i) HEP2 (HLA-A1+A2+ hepatocyte); and (ii) SMC3 (HLA-A2+ colonic smooth muscle). Background MIP-1β production relative to non-transduced controls was observed with the c12/c2 and wt/c51 TCRs, most markedly in response to SMC3 primary cells (Fig. 5a). The Mel624·38 and A375 tumour cell lines were recognized robustly by all NY-ESO-1-specific TCR-transduced CD8+ T cells. In contrast, Mel526 was poorly recognized, most probably because it presents fewer than 10 copies of the cognate SLLMWITQC peptide per cell 31. Notably, the c259/wt and c12/c2 TCRs elicited comparable functional profiles despite a pronounced (∼12-fold) difference in binding half-life. Moreover, statistical analysis across all five tumour cell lines confirmed the functional enhancement conferred by these TCRs. For example, concatenated pies for c12/c2 were significantly different compared to wt/wt (P = 0·047; Wilcoxon's signed-rank test), and the fraction of cells that produced both MIP-1β and TNF-α was significantly higher for c12/c2 in the same comparison (P = 0·047; Wilcoxon's signed-rank test).
Fig 5.
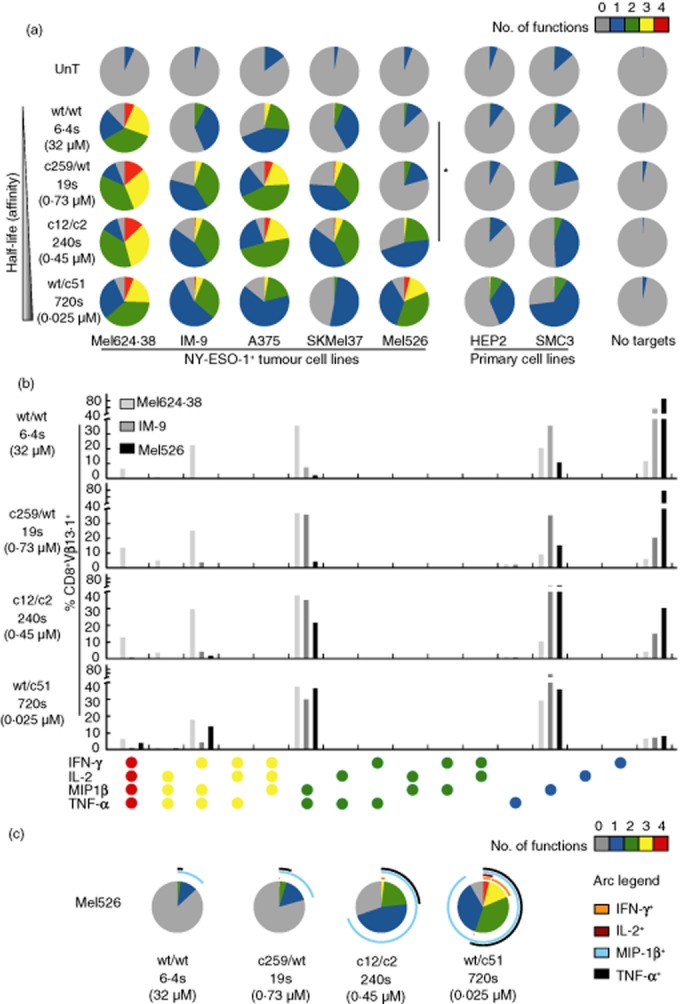
Stimulation of human leucocyte antigen (HLA)-A2-restricted NY-ESO-1-specific T cell receptor (TCR)-transduced CD8+ T cells with tumour cell lines. (a) Non-transduced (UnT) or NY-ESO-1-specific TCR-transduced CD8+ T cells were stimulated with NY-ESO-1+ HLA-A2+ tumour cell lines (Mel624·38, IM-9, A375, SKMel37 and Mel526) or NY-ESO-1− HLA-A2+ primary cells (HEP2 and SMC3). Four functional readouts [macrophage inflammatory protein (MIP)-1β, tumour necrosis factor (TNF)-α, interferon (IFN)-γ and interleukin (IL)-2] were measured by flow cytometry. Pie chart segments represent the fraction of cells expressing the number of functions indicated in the key. The gating strategy followed that described for Fig. 1a, with the exception that T cell antigen receptor variable (TCRV)β13·1 expression was used to identify transduced T cells where appropriate. *P < 0·05 across concatenated pies. (b) Combinations of functions elicited by stimulation of NY-ESO-1-specific TCR-transduced CD8+ T cells with the tumour cell lines Mel624·38, IM-9 and Mel526. (c) Detailed analysis of functional profiles elicited by stimulation of NY-ESO-1-specific TCR-transduced CD8+ T cells with Mel526 tumour cells. The pie charts are extended with arcs defining expressed functions as indicated in the key. Data shown are representative of two independent experiments. The corresponding biophysical parameters are listed in Table 2.
To extend these findings, we examined the functional profile of MageA3-specific TCR-transduced CD8+ T cells in response to five different MageA3+ HLA-A1+ tumour cell lines: (i) HCT116 and HT144 (colorectal carcinoma); (ii) A375 and SKMel24 (melanoma); and (iii) EJM (multiple myeloma). Two MageA3− primary cell cultures were used as negative controls: (i) HEP2 (HLA-A1+A2+ hepatocyte); and (ii) CIL-1 (HLA-A1+ non-pigmented ciliary epithelium). Affinity-enhanced TCR-transduced cells recognized all tumour targets robustly, eliciting higher frequency responses with increased polyfunctional profiles compared to wt/wt TCR-transduced cells (Fig. 6a). These differences were statistically significant. For example, concatenated pie comparisons across all five tumour cell lines showed that the affinity-enhanced TCRs elicited significantly different functional responses compared to wt/wt (a3c/wt, P = 0·009; a3a/wt, P = 0·006; wt/b2a, P = 0·001; Wilcoxon's signed-rank test). Clear differences were also apparent with respect to individual combinations of functions (Fig. 6b,c). For example, quadruple function cells were more common in the responses elicited by affinity-enhanced TCRs compared to wt/wt (a3c/wt, P = 0·047; a3a/wt, P = 0·047; wt/b2a, P = 0·016; Wilcoxon's signed-rank test). Similarly, triple function cells producing MIP-1β, TNF-α and IL-2 were more often observed with affinity-enhanced TCRs compared to wt/wt (P = 0·016 for all comparisons versus a3c/wt, a3a/wt and wt/b2a; Wilcoxon's signed-rank test). Background responses against the primary cells, predominantly in the form of MIP-1β production, were observed with wt/b2a and, to a lesser extent, a3a/wt TCR-transduced cells; no such off-target effects were apparent for either the wt/wt or the a3c/wt TCRs.
Fig 6.
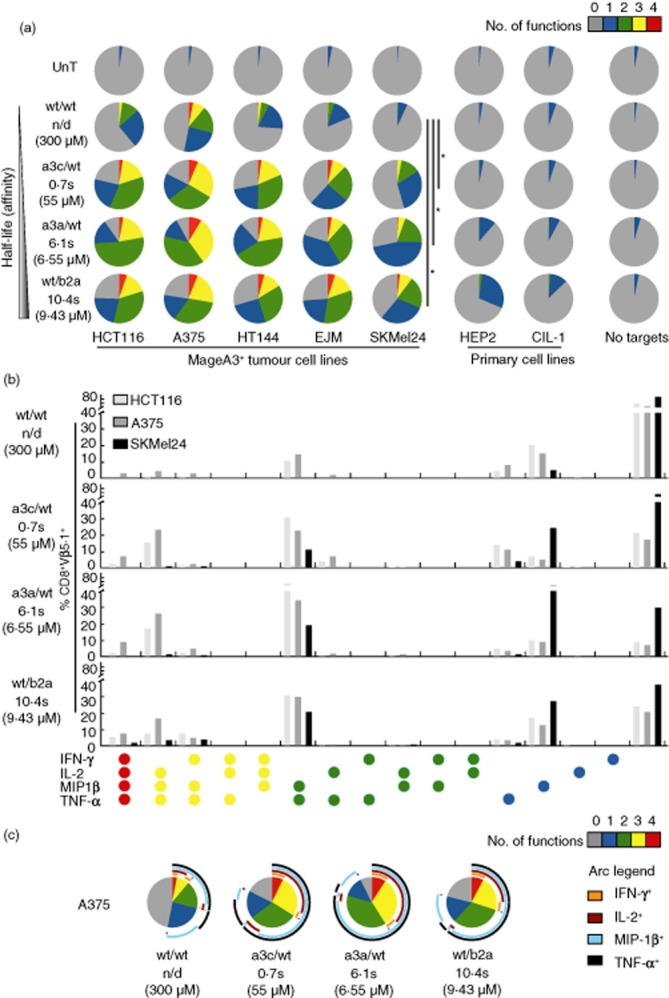
Stimulation of human leucocyte antigen (HLA)-A1-restricted MageA3-specific T cell receptor (TCR)-transduced CD8+ T cells with tumour cell lines. (a) Non-transduced (UnT) or MageA3-specific TCR-transduced CD8+ T cells were stimulated with MageA3+ HLA-A1+ tumour cell lines (HCT116, A375, HT144, EJM and SKMel24) or MageA3− HLA-A1+ primary cells (HEP2 and CIL-1). Four functional readouts [macrophage inflammatory protein (MIP)-1β, tumour necrosis factor (TNF)-α, interferon (IFN)-γ and interleukin (IL)-2] were measured by flow cytometry. Pie chart segments represent the fraction of cells expressing the number of functions indicated in the key. The gating strategy followed that described for Fig. 1a, with the exception that T cell antigen receptor variable (TCRV)β5·1 expression was used to identify transduced T cells where appropriate. *P < 0·05 across concatenated pies. (b) Combinations of functions elicited by stimulation of MageA3-specific TCR-transduced CD8+ T cells with the tumour cell lines HCT116, A375 and SKMel24. (c) Detailed analysis of functional profiles elicited by stimulation of MageA3-specific TCR-transduced CD8+ T cells with A375 tumour cells. The pie charts are extended with arcs defining expressed functions as indicated in the key. Data shown are representative of two independent experiments. The corresponding biophysical parameters are listed in Table 2.
Across both the NY-ESO-1 and MageA3 systems, the combinations of effector functions displayed by the various TCR-transduced CD8+ T cells in response to tumour targets reflected the hierarchy observed with peptide-pulsed targets (Supporting information, Fig. S2). In particular, dual function cells produced predominantly MIP-1β and TNF-α, while triple function cells expressed primarily IFN-γ in addition (Fig. 5b,c and 6b,c). Thus, polyfunctionality is augmented by TCR affinity enhancement in accordance with strict thresholds that are maintained in a cell-intrinsic manner.
Affinity-enhanced cancer-specific TCRs augment lymphokine production by transduced CD8+ T cells
The results presented above describe chemokine/cytokine production at the single-cell level. However, it is highly likely that the overall amount of each lymphokine will have an important effect on tumour-specific immunity. To address this issue, we used Luminex technology to measure the secretion of six soluble factors [MIP-1α, granulocyte-macrophage colony-stimulating factor (GM-CSF), MIP-1β, TNF-α, IFN-γ and IL-2] in both the NY-ESO-1 and MageA3 systems.
CD8+ T cells transduced with NY-ESO-1-specific TCRs were stimulated with the IM-9 (NY-ESO-1+ HLA-A2+ EBV-transformed B-LCL) tumour cell line or N9 (NY-ESO-1− HLA-A2+ epidermal melanocyte) primary cells. Affinity-enhanced TCR-transduced cells produced greater levels of each soluble factor in response to IM-9 stimulation compared to wt/wt TCR-transduced cells (Fig. 7a). In contrast, lymphokine production was negligible in response to N9 stimulation.
Fig 7.
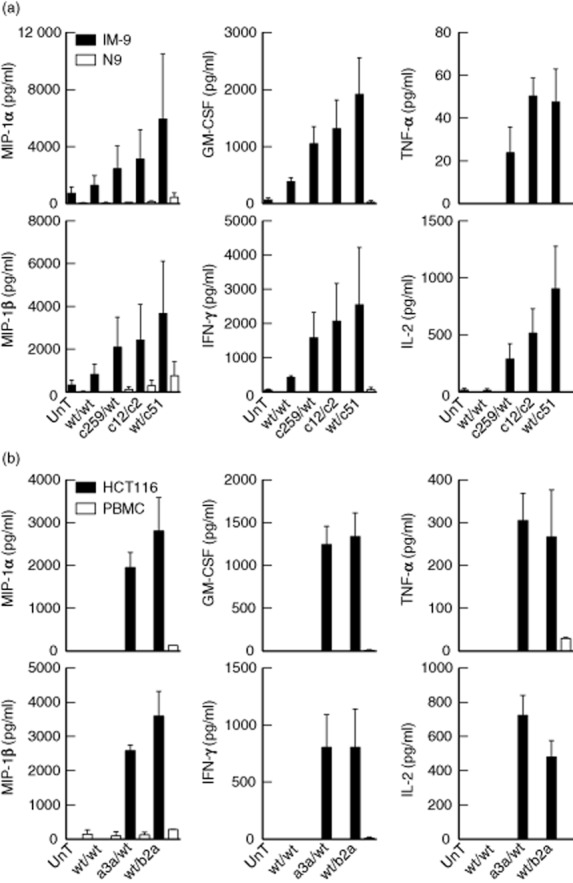
Production of soluble factors in response to stimulation of NY-ESO-1-specific or MageA3-specific T cell receptor (TCR)-transduced CD8+ T cells with tumour cell lines. (a) NY-ESO-1-specific TCR-transduced CD8+ T cells were stimulated with IM-9 tumour cells [NY-ESO-1+ human leucocyte antigen (HLA)-A2+] or N9 melanocytes (NY-ESO-1− HLA-A2+). (b) MageA3-specific TCR-transduced CD8+ T cells were stimulated with HCT116 tumour cells (MageA3+ HLA-A1+) or HLA-A1+ PBMCs. Supernatant was assayed for the indicated soluble factors by Luminex. Data shown are representative of two independent experiments. Error bars depict the standard deviation between triplicate samples. UnT = non-transduced.
CD8+ T cells transduced with MageA3-specific TCRs were stimulated with the HCT116 (MageA3+ HLA-A1+ colorectal carcinoma) tumour cell line or HLA-A1+ peripheral blood mononuclear cells (PBMCs). No responses were detected for non-transduced CD8+ T cells or wt/wt TCR-transduced cells (Fig. 7b). In contrast, the higher-affinity a3a/wt and wt/b2a TCR-transduced cells were activated by HCT116 stimulation. Minimal lymphokine production was observed in response to HLA-A1+ PBMCs.
Collectively, these data show that affinity-enhanced TCRs can improve CD8+ T cell functionality en masse, in line with the augmented profiles observed at the single-cell level.
Discussion
Thymic selection evolved to generate a T cell repertoire that enables protective immunity while avoiding potentially dangerous self-reactivity 4. As a consequence, it is difficult for the immune system to discriminate between self-derived and endogenous tumour-associated antigens, many of which are expressed to some extent by healthy cells. Indeed, naturally occurring T cells that recognize tumour-associated antigens typically display low levels of avidity and sensitivity, in contrast to their pathogen-specific counterparts 3. These intrinsic differences in the available T cell repertoire may explain, at least in part, the widespread failure of cancer vaccines.
Accumulating data indicate that the functional capabilities of antigen-specific T cell populations can determine the outcome of immune responses against infectious agents, such as HIV-1, Leishmania major and Mycobacterium tuberculosis 52–61. Moreover, in a cancer model, T cells with enhanced peptide sensitivity and tumour recognition were found to display a distinctive Th1 cytokine profile 62. The emerging consensus from such studies therefore postulates that the most effective T cell responses are highly antigen-sensitive and deploy multiple effector functions 63. These correlates present two distinct problems for the generation of effective immunity against cancer. First, tumour-associated peptide antigens are generally present at fewer than 50 copies per cell 31,51. Secondly, as described above, tumour-associated antigen-specific TCRs bind weakly to cognate pMHC molecules 2,3. This latter feature is amenable to manipulation, specifically in the setting of engineered TCR gene transfer therapy.
In the present study, we investigated the functional properties of engineered cancer-specific TCRs with affinities that approximate those measured for the best anti-viral TCRs 2,3,15. The key findings were: (i) TCR–pMHC monomeric affinity/half-life correlates with T cell antigen sensitivity and polyfunctionality; (ii) an affinity threshold exists, above which T cell function is not improved by further enhancement; and (iii) T cells transduced with super-enhanced high-affinity TCRs exhibit cross-reactivity with self-derived peptides presented by the restricting HLA molecule. Collectively, these data identify an optimal affinity window for cancer-specific TCRs that allows the enhancement of T cell effector function without non-specific activation.
It is notable that a TCR–pMHC binding affinity/half-life ceiling was observed in every system tested, beyond which no further functional enhancements were apparent. Activation of the ILA1 clone with the 3G peptide, which has the longest half-life of all APLs in this model, resulted in a weaker functional profile compared to the other high-affinity ligands (3G8T and 8T). Similarly, in the transduced T cell systems, the highest-affinity TCRs for both NY-ESO-1 and MageA3 elicited fewer polyfunctional responses compared to those with more modest affinity enhancements. These findings are consistent with data from other groups. For example, Irving et al. characterized a set of nine NY-ESO-1-specific TCRs with affinities ranging from 21 μM to 0·015 μM 64. Maximal T cell responses were observed at the lower end of this range and functional outputs reached a plateau at affinities between 1 μM and 5 μM. At the molecular level, this effect appeared to correlate with SHP-1 expression levels regulated as a function of TCR affinity 65. Concordantly, we identified c259/wt as the optimal NY-ESO-1-specific TCR (KD = 0·73 μM). However, our study also demonstrates that different affinity windows are optimal across different systems. In the case of MageA3, the optimal TCR affinity lies approximately 10-fold below that observed in the NY-ESO-1 system.
Previous reports have described affinity threshold phenomena in other systems. In one study, a single cancer-specific TCR was used with mimotopes to generate a range of affinities up to 4-fold higher than the wild-type interaction 66. Although the highest-affinity mimotopes elicited the most vigorous responses (IFN-γ production and proliferation) in vitro, optimal anti-tumour activity in vivo was observed after vaccination with the intermediate affinity mimotopes. Two studies in the NY-ESO-1 system, spanning a range of TCR affinities up to 1430-fold greater than wild-type, reached similar conclusions 38,48. In particular, target cell lysis was found to increase as a function of affinity up to a threshold of ∼7 μM, beyond which further increments diminished killing activity. These findings were confirmed recently using a series of seven gp100-specific TCRs 67. Excessive affinity enhancement has also been shown to impair T cell function in the 2C system 68,69. In addition, a recent study using APL vaccination in a diabetes/ovalbumin (OVA) mouse model showed that TCR affinity dictates not only the magnitude of T cell activation and target cell conjugation, but also the differentiation status of responding T cells 70. Similar observations have been reported for pathogen-specific TCRs 71,72. Thus, each system is constrained by an affinity ceiling, the parameters for which can be highly variable 73.
In the present study, we identified optimal TCR affinities in the NY-ESO-1 and MageA3 systems, defined across multiple functional readouts in the absence of non-specific reactivity. Earlier studies with the wild-type NY-ESO-1-specific TCR showed that transduced CD8+ T cells produced IFN-γ and lysed target cells in an antigen-specific manner 74. Genetically modified cancer-specific T cells have also been tested in the clinic 7. In this particular case, the investigators isolated a melanoma antigen recognized by T cells-1 (MART-1)-specific TCR from a tumour-infiltrating lymphocyte (TIL) clone derived from a patient who cleared metastatic melanoma after autologous TIL transfer. Reinfusion of CD8+ T cells transduced with this naturally occurring TCR showed promising efficacy in a cohort of 17 study subjects with melanoma. Two patients demonstrated near-complete tumour regression and were declared clinically disease-free 21 months after treatment. In vitro studies examining cancer-specific TCRs with enhanced binding parameters have followed 48,75,76, generating enthusiasm for various therapeutic applications. In a recent trial, objective clinical responses were observed using the NY-ESO-1-specific c259/wt TCR in 67% of patients (n = 6) with synovial cell sarcoma and 45% of patients (n = 11) with melanomas expressing the NY-ESO-1 antigen 10. Collectively, these studies underline the remarkable potential of optimally designed TCR gene transfer as an effective therapeutic option against cancer.
In summary, our data demonstrate that tumour-specific TCRs can be engineered in a system-specific manner to improve effector cell function without substantial off-target effects. These findings have major implications for the rational design of novel TCR-based biologicals underpinned by rigorous preclinical evaluation within each antigen specificity to mitigate the risk of cross-reactivity 77,78.
Acknowledgments
We thank Nathaniel Liddy, Peter Molloy and Yi Li (Immunocore Ltd, Abingdon, UK) for phage display selection of affinity-enhanced TCRs, and James Riley (University of Pennsylvania, PA, USA) for provision of lentivirus vectors. The NY-ESO-1-specific TCR sequence was kindly provided by Vincenzo Cerundolo (University of Oxford, Oxford, UK); the MageA3-specific TCR sequence was kindly provided by Pierre Coulie (University of Louvain, Louvain, Belgium). The SKMel37 melanoma cell line was a kind gift from the Ludwig Institute for Cancer Research; all experiments performed with SKMel37 were conducted at Adaptimmune Ltd (Abingdon, UK). M. P. T. was supported by a fellowship from the National Institute for Social Care and Health Research (UK). D. A. P. and A. K. S. are Wellcome Trust Senior Investigators. Additional funding was provided by Adaptimmune Ltd.
Disclosures
A. B. G., J. E. B., L. M., A. D. B., N. J. P. and B. K. J. are employees of Adaptimmune Ltd. The other authors have no financial conflicts of interest.
Author contributions
M. P. T., A. B. G., J. E. B., B. K. J., D. A. P., K. L. and A. K. S. conceived and designed the study; M. P. T., A. B. G., J. S. B. and K. L. performed experiments; A. D. B. and N. J. P. prepared affinity-enhanced TCRs; M. P. T., A. B. G., L. M. and K. L. analysed data; M P. T., A. B. G., J. E. B., B. K. J., D. A. P., K. L. and A. K. S. wrote the manuscript.
Supporting Information
Additional Supporting information may be found in the online version of this article at the publisher's web-site:
Figure S1. The impact of donor origin on the functional profile of T cell receptor (TCR)-transduced CD8+ T cells.
Figure S2. Combinations of functions elicited by stimulation of T cell receptor (TCR)-transduced CD8+ T cells with peptide-pulsed target cells.
References
- Boon T, Coulie PG, Van den Eynde BJ, van der Bruggen P. Human T cell responses against melanoma. Annu Rev Immunol. 2006;24:175–208. doi: 10.1146/annurev.immunol.24.021605.090733. [DOI] [PubMed] [Google Scholar]
- Cole DK, Pumphrey NJ, Boulter JM, et al. Human TCR-binding affinity is governed by MHC class restriction. J Immunol. 2007;178:5727–5734. doi: 10.4049/jimmunol.178.9.5727. [DOI] [PubMed] [Google Scholar]
- Aleksic M, Liddy N, Molloy PE, et al. Different affinity windows for virus and cancer-specific T-cell receptors: implications for therapeutic strategies. Eur J Immunol. 2012;42:3174–3179. doi: 10.1002/eji.201242606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savage PA, Davis MM. A kinetic window constricts the T cell receptor repertoire in the thymus. Immunity. 2001;14:243–252. doi: 10.1016/s1074-7613(01)00106-6. [DOI] [PubMed] [Google Scholar]
- Marr LA, Gilham DE, Campbell JD, Fraser AR. Immunology in the clinic review series; focus on cancer: double trouble for tumours: bi-functional and redirected T cells as effective cancer immunotherapies. Clin Exp Immunol. 2012;167:216–225. doi: 10.1111/j.1365-2249.2011.04517.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunert A, Straetemans T, Govers C, et al. TCR-engineered T cells meet new challenges to treat solid tumors: choice of antigen, T cell fitness, and sensitization of tumor milieu. Front Immunol. 2013;4:363. doi: 10.3389/fimmu.2013.00363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan RA, Dudley ME, Wunderlich JR, et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science. 2006;314:126–129. doi: 10.1126/science.1129003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson LA, Morgan RA, Dudley ME, et al. Gene therapy with human and mouse T-cell receptors mediates cancer regression and targets normal tissues expressing cognate antigen. Blood. 2009;114:535–546. doi: 10.1182/blood-2009-03-211714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt TM, Ragnarsson GB, Greenberg PD. T cell receptor gene therapy for cancer. Hum Gene Ther. 2009;20:1240–1248. doi: 10.1089/hum.2009.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins PF, Morgan RA, Feldman SA, et al. Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. J Clin Oncol. 2011;29:917–924. doi: 10.1200/JCO.2010.32.2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkhurst MR, Yang JC, Langan RC, et al. T cells targeting carcinoembryonic antigen can mediate regression of metastatic colorectal cancer but induce severe transient colitis. Mol Ther. 2011;19:620–626. doi: 10.1038/mt.2010.272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong JJ, Rosenberg SA, Dudley ME, et al. Successful treatment of melanoma brain metastases with adoptive cell therapy. Clin Cancer Res. 2010;16:4892–4898. doi: 10.1158/1078-0432.CCR-10-1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan RA, Chinnasamy N, Abate-Daga D, et al. Cancer regression and neurological toxicity following anti-MAGE-A3 TCR gene therapy. J Immunother. 2013;36:133–151. doi: 10.1097/CJI.0b013e3182829903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagisetty KH, Morgan RA. Cancer therapy with genetically-modified T cells for the treatment of melanoma. J Gene Med. 2012;14:400–404. doi: 10.1002/jgm.2636. [DOI] [PubMed] [Google Scholar]
- Varela-Rohena A, Molloy PE, Dunn SM, et al. Control of HIV-1 immune escape by CD8 T cells expressing enhanced T-cell receptor. Nat Med. 2008;14:1390–1395. doi: 10.1038/nm.1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKeithan TW. Kinetic proofreading in T-cell receptor signal transduction. Proc Natl Acad Sci USA. 1995;92:5042–5046. doi: 10.1073/pnas.92.11.5042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone JD, Chervin AS, Kranz DM. T-cell receptor binding affinities and kinetics: impact on T-cell activity and specificity. Immunology. 2009;126:165–176. doi: 10.1111/j.1365-2567.2008.03015.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laugel B, van den Berg HA, Gostick E, et al. Different T cell receptor affinity thresholds and CD8 coreceptor dependence govern cytotoxic T lymphocyte activation and tetramer binding properties. J Biol Chem. 2007;282:23799–23810. doi: 10.1074/jbc.M700976200. [DOI] [PubMed] [Google Scholar]
- Bridgeman JS, Sewell AK, Miles JJ, Price DA, Cole DK. Structural and biophysical determinants of alphabeta T-cell antigen recognition. Immunology. 2012;135:9–18. doi: 10.1111/j.1365-2567.2011.03515.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wooldridge L, Lissina A, Cole DK, van den Berg HA, Price DA, Sewell AK. Tricks with tetramers: how to get the most from multimeric peptide–MHC. Immunology. 2009;126:147–164. doi: 10.1111/j.1365-2567.2008.02848.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laugel B, Boulter JM, Lissin N, et al. Design of soluble recombinant T cell receptors for antigen targeting and T cell inhibition. J Biol Chem. 2005;280:1882–1892. doi: 10.1074/jbc.M409427200. [DOI] [PubMed] [Google Scholar]
- Whelan JA, Dunbar PR, Price DA, et al. Specificity of CTL interactions with peptide–MHC class I tetrameric complexes is temperature dependent. J Immunol. 1999;163:4342–4348. [PubMed] [Google Scholar]
- van den Berg HA, Ladell K, Miners K, et al. Cellular-level versus receptor-level response threshold hierarchies in T-cell activation. Front Immunol. 2013;4:250. doi: 10.3389/fimmu.2013.00250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melenhorst JJ, Scheinberg P, Chattopadhyay PK, et al. Detection of low avidity CD8+ T cell populations with coreceptor-enhanced peptide–major histocompatibility complex class I tetramers. J Immunol Methods. 2008;338:31–39. doi: 10.1016/j.jim.2008.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole DK, Yuan F, Rizkallah PJ, et al. Germ line-governed recognition of a cancer epitope by an immunodominant human T-cell receptor. J Biol Chem. 2009;284:27281–27289. doi: 10.1074/jbc.M109.022509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storkus WJ, Howell DN, Salter RD, Dawson JR, Cresswell P. NK susceptibility varies inversely with target cell class I HLA antigen expression. J Immunol. 1987;138:1657–1659. [PubMed] [Google Scholar]
- Purbhoo MA, Boulter JM, Price DA, et al. The human CD8 coreceptor effects cytotoxic T cell activation and antigen sensitivity primarily by mediating complete phosphorylation of the T cell receptor zeta chain. J Biol Chem. 2001;276:32786–32792. doi: 10.1074/jbc.M102498200. [DOI] [PubMed] [Google Scholar]
- Bownds S, Tong-On P, Rosenberg SA, Parkhurst M. Induction of tumor-reactive cytotoxic T-lymphocytes using a peptide from NY-ESO-1 modified at the carboxy-terminus to enhance HLA-A2.1 binding affinity and stability in solution. J Immunother. 2001;24:1–9. doi: 10.1097/00002371-200101000-00001. [DOI] [PubMed] [Google Scholar]
- Schultz-Thater E, Noppen C, Gudat F, et al. NY-ESO-1 tumour associated antigen is a cytoplasmic protein detectable by specific monoclonal antibodies in cell lines and clinical specimens. Br J Cancer. 2000;83:204–208. doi: 10.1054/bjoc.2000.1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atanackovic D, Arfsten J, Cao Y, et al. Cancer-testis antigens are commonly expressed in multiple myeloma and induce systemic immunity following allogeneic stem cell transplantation. Blood. 2007;109:1103–1112. doi: 10.1182/blood-2006-04-014480. [DOI] [PubMed] [Google Scholar]
- Purbhoo MA, Sutton DH, Brewer JE, et al. Quantifying and imaging NY-ESO-1/LAGE-1-derived epitopes on tumor cells using high affinity T cell receptors. J Immunol. 2006;176:7308–7316. doi: 10.4049/jimmunol.176.12.7308. [DOI] [PubMed] [Google Scholar]
- Kim KH, Choi JS, Kim IJ, Ku JL, Park JG. Promoter hypomethylation and reactivation of MAGE-A1 and MAGE-A3 genes in colorectal cancer cell lines and cancer tissues. World J Gastroenterol. 2006;12:5651–5657. doi: 10.3748/wjg.v12.i35.5651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchand M, van Baren N, Weynants P, et al. Tumor regressions observed in patients with metastatic melanoma treated with an antigenic peptide encoded by gene MAGE-3 and presented by HLA-A1. Int J Cancer. 1999;80:219–230. doi: 10.1002/(sici)1097-0215(19990118)80:2<219::aid-ijc10>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- Imro MA, Dellabona P, Manici S, et al. Human melanoma cells transfected with the B7-2 co-stimulatory molecule induce tumor-specific CD8+ cytotoxic T lymphocytes in vitro. Hum Gene Ther. 1998;9:1335–1344. doi: 10.1089/hum.1998.9.9-1335. [DOI] [PubMed] [Google Scholar]
- Shimizu Y, DeMars R. Production of human cells expressing individual transferred HLA-A,-B,-C genes using an HLA-A,-B,-C null human cell line. J Immunol. 1989;142:3320–3328. [PubMed] [Google Scholar]
- Celis E, Tsai V, Crimi C, et al. Induction of anti-tumor cytotoxic T lymphocytes in normal humans using primary cultures and synthetic peptide epitopes. Proc Natl Acad Sci USA. 1994;91:2105–2109. doi: 10.1073/pnas.91.6.2105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Bennett AD, Zheng Z, et al. High-affinity TCRs generated by phage display provide CD4+ T cells with the ability to recognize and kill tumor cell lines. J Immunol. 2007;179:5845–5854. doi: 10.4049/jimmunol.179.9.5845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins PF, Li YF, El-Gamil M, et al. Single and dual amino acid substitutions in TCR CDRs can enhance antigen-specific T cell functions. J Immunol. 2008;180:6116–6131. doi: 10.4049/jimmunol.180.9.6116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parry RV, Rumbley CA, Vandenberghe LH, June CH, Riley JL. CD28 and inducible costimulatory protein Src homology 2 binding domains show distinct regulation of phosphatidylinositol 3-kinase, Bcl-xL, and IL-2 expression in primary human CD4 T lymphocytes. J Immunol. 2003;171:166–174. doi: 10.4049/jimmunol.171.1.166. [DOI] [PubMed] [Google Scholar]
- Lissina A, Ladell K, Skowera A, et al. Protein kinase inhibitors substantially improve the physical detection of T-cells with peptide-MHC tetramers. J Immunol Methods. 2009;340:11–24. doi: 10.1016/j.jim.2008.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wooldridge L, van den Berg HA, Glick M, et al. Interaction between the CD8 coreceptor and major histocompatibility complex class I stabilizes T cell receptor-antigen complexes at the cell surface. J Biol Chem. 2005;280:27491–27501. doi: 10.1074/jbc.M500555200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sewell AK, Harcourt GC, Goulder PJ, Price DA, Phillips RE. Antagonism of cytotoxic T lymphocyte-mediated lysis by natural HIV-1 altered peptide ligands requires simultaneous presentation of agonist and antagonist peptides. Eur J Immunol. 1997;27:2323–2329. doi: 10.1002/eji.1830270929. [DOI] [PubMed] [Google Scholar]
- Gotch F, Rothbard J, Howland K, Townsend A, McMichael A. Cytotoxic T lymphocytes recognize a fragment of influenza virus matrix protein in association with HLA-A2. Nature. 1987;326:881–882. doi: 10.1038/326881a0. [DOI] [PubMed] [Google Scholar]
- Roederer M, Nozzi JL, Nason MC. SPICE: exploration and analysis of post-cytometric complex multivariate datasets. Cytometry A. 2011;79:167–174. doi: 10.1002/cyto.a.21015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purbhoo MA, Li Y, Sutton DH, et al. The HLA A*0201-restricted hTERT(540–548) peptide is not detected on tumor cells by a CTL clone or a high-affinity T-cell receptor. Mol Cancer Ther. 2007;6:2081–2091. doi: 10.1158/1535-7163.MCT-07-0092. [DOI] [PubMed] [Google Scholar]
- Gonzalez PA, Carreno LJ, Coombs D, et al. T cell receptor binding kinetics required for T cell activation depend on the density of cognate ligand on the antigen-presenting cell. Proc Natl Acad Sci USA. 2005;102:4824–4829. doi: 10.1073/pnas.0500922102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leisegang M, Wilde S, Spranger S, et al. MHC-restricted fratricide of human lymphocytes expressing survivin-specific transgenic T cell receptors. J Clin Invest. 2010;120:3869–3877. doi: 10.1172/JCI43437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid DA, Irving MB, Posevitz V, et al. Evidence for a TCR affinity threshold delimiting maximal CD8 T cell function. J Immunol. 2010;184:4936–4946. doi: 10.4049/jimmunol.1000173. [DOI] [PubMed] [Google Scholar]
- Price DA, Sewell AK, Dong T, et al. Antigen-specific release of beta-chemokines by anti-HIV-1 cytotoxic T lymphocytes. Curr Biol. 1998;8:355–358. doi: 10.1016/s0960-9822(98)70138-1. [DOI] [PubMed] [Google Scholar]
- Barrow C, Browning J, MacGregor D, et al. Tumor antigen expression in melanoma varies according to antigen and stage. Clin Cancer Res. 2006;12:764–771. doi: 10.1158/1078-0432.CCR-05-1544. [DOI] [PubMed] [Google Scholar]
- Liddy N, Bossi G, Adams KJ, et al. Monoclonal TCR-redirected tumor cell killing. Nat Med. 2012;18:980–987. doi: 10.1038/nm.2764. [DOI] [PubMed] [Google Scholar]
- Betts MR, Exley B, Price DA, et al. Characterization of functional and phenotypic changes in anti-Gag vaccine-induced T cell responses and their role in protection after HIV-1 infection. Proc Natl Acad Sci USA. 2005;102:4512–4517. doi: 10.1073/pnas.0408773102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makedonas G, Hutnick N, Haney D, et al. Perforin and IL-2 upregulation define qualitative differences among highly functional virus-specific human CD8 T cells. PLOS Pathog. 2010;6:e1000798. doi: 10.1371/journal.ppat.1000798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genesca M, Rourke T, Li J, et al. Live attenuated lentivirus infection elicits polyfunctional simian immunodeficiency virus Gag-specific CD8+ T cells with reduced apoptotic susceptibility in rhesus macaques that control virus replication after challenge with pathogenic SIVmac239. J Immunol. 2007;179:4732–4740. doi: 10.4049/jimmunol.179.7.4732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boaz MJ, Waters A, Murad S, Easterbrook PJ, Vyakarnam A. Presence of HIV-1 Gag-specific IFN-gamma+IL-2+ and CD28+IL-2+ CD4 T cell responses is associated with nonprogression in HIV-1 infection. J Immunol. 2002;169:6376–6385. doi: 10.4049/jimmunol.169.11.6376. [DOI] [PubMed] [Google Scholar]
- Kannanganat S, Ibegbu C, Chennareddi L, Robinson HL, Amara RR. Multiple-cytokine-producing antiviral CD4 T cells are functionally superior to single-cytokine-producing cells. J Virol. 2007;81:8468–8476. doi: 10.1128/JVI.00228-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almeida JR, Sauce D, Price DA, et al. Antigen sensitivity is a major determinant of CD8+ T-cell polyfunctionality and HIV-suppressive activity. Blood. 2009;113:6351–6360. doi: 10.1182/blood-2009-02-206557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darrah PA, Patel DT, De Luca PM, et al. Multifunctional TH1 cells define a correlate of vaccine-mediated protection against Leishmania major. Nat Med. 2007;13:843–850. doi: 10.1038/nm1592. [DOI] [PubMed] [Google Scholar]
- Derrick SC, Yabe IM, Yang A, Morris SL. Vaccine-induced anti-tuberculosis protective immunity in mice correlates with the magnitude and quality of multifunctional CD4 T cells. Vaccine. 2011;29:2902–2909. doi: 10.1016/j.vaccine.2011.02.010. [DOI] [PubMed] [Google Scholar]
- Betts MR, Nason MC, West SM, et al. HIV nonprogressors preferentially maintain highly functional HIV-specific CD8+ T cells. Blood. 2006;107:4781–4789. doi: 10.1182/blood-2005-12-4818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daucher M, Price DA, Brenchley JM, et al. Virological outcome after structured interruption of antiretroviral therapy for human immunodeficiency virus infection is associated with the functional profile of virus-specific CD8+ T cells. J Virol. 2008;82:4102–4114. doi: 10.1128/JVI.02212-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilde S, Sommermeyer D, Leisegang M, et al. Human antitumor CD8+ T cells producing Th1 polycytokines show superior antigen sensitivity and tumor recognition. J Immunol. 2012;189:598–605. doi: 10.4049/jimmunol.1102165. [DOI] [PubMed] [Google Scholar]
- Appay V, Douek DC, Price DA. CD8+ T cell efficacy in vaccination and disease. Nat Med. 2008;14:623–628. doi: 10.1038/nm.f.1774. [DOI] [PubMed] [Google Scholar]
- Irving M, Zoete V, Hebeisen M, et al. Interplay between T cell receptor binding kinetics and the level of cognate peptide presented by major histocompatibility complexes governs CD8+ T cell responsiveness. J Biol Chem. 2012;287:23068–23078. doi: 10.1074/jbc.M112.357673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebeisen M, Baitsch L, Presotto D, et al. SHP-1 phosphatase activity counteracts increased T cell receptor affinity. J Clin Invest. 2013;123:1044–1056. doi: 10.1172/JCI65325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMahan RH, McWilliams JA, Jordan KR, Dow SW, Wilson DB, Slansky JE. Relating TCR–peptide–MHC affinity to immunogenicity for the design of tumor vaccines. J Clin Invest. 2006;116:2543–2551. doi: 10.1172/JCI26936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong S, Malecek K, Johnson LA, et al. T-cell receptor affinity and avidity defines antitumor response and autoimmunity in T-cell immunotherapy. Proc Natl Acad Sci USA. 2013;110:6973–6978. doi: 10.1073/pnas.1221609110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chervin AS, Stone JD, Soto CM, et al. Design of T-cell receptor libraries with diverse binding properties to examine adoptive T-cell responses. Gene Ther. 2013;20:634–644. doi: 10.1038/gt.2012.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soto CM, Stone JD, Chervin AS, et al. MHC-class I-restricted CD4 T cells: a nanomolar affinity TCR has improved anti-tumor efficacy in vivo compared to the micromolar wild-type TCR. Cancer Immunol Immunother. 2013;62:359–369. doi: 10.1007/s00262-012-1336-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King CG, Koehli S, Hausmann B, Schmaler M, Zehn D, Palmer E. T cell affinity regulates asymmetric division, effector cell differentiation, and tissue pathology. Immunity. 2012;37:709–720. doi: 10.1016/j.immuni.2012.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueno T, Tomiyama H, Fujiwara M, Oka S, Takiguchi M. Functionally impaired HIV-specific CD8 T cells show high affinity TCR-ligand interactions. J Immunol. 2004;173:5451–5457. doi: 10.4049/jimmunol.173.9.5451. [DOI] [PubMed] [Google Scholar]
- Thomas S, Xue SA, Bangham CR, Jakobsen BK, Morris EC, Stauss HJ. Human T cells expressing affinity-matured TCR display accelerated responses but fail to recognize low density of MHC-peptide antigen. Blood. 2011;118:319–329. doi: 10.1182/blood-2010-12-326736. [DOI] [PubMed] [Google Scholar]
- Boulter JM, Schmitz N, Sewell AK, Godkin AJ, Bachmann MF, Gallimore AM. Potent T cell agonism mediated by a very rapid TCR/pMHC interaction. Eur J Immunol. 2007;37:798–806. doi: 10.1002/eji.200636743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Zheng Z, Robbins PF, Khong HT, Rosenberg SA, Morgan RA. Primary human lymphocytes transduced with NY-ESO-1 antigen-specific TCR genes recognize and kill diverse human tumor cell lines. J Immunol. 2005;174:4415–4423. doi: 10.4049/jimmunol.174.7.4415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JL, Morgan AJ, Stewart-Jones G, et al. Ca2+ release from the endoplasmic reticulum of NY-ESO-1-specific T cells is modulated by the affinity of TCR and by the use of the CD8 coreceptor. J Immunol. 2010;184:1829–1839. doi: 10.4049/jimmunol.0902103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kronig H, Hofer K, Conrad H, et al. Allorestricted T lymphocytes with a high avidity T-cell receptor towards NY-ESO-1 have potent anti-tumor activity. Int J Cancer. 2009;125:649–655. doi: 10.1002/ijc.24414. [DOI] [PubMed] [Google Scholar]
- Cameron BJ, Gerry AB, Dukes J, et al. Identification of a Titin-derived HLA-A1-presented peptide as a cross-reactive target for engineered MAGE A3-directed T cells. Sci Transl Med. 2013;5:197ra103. doi: 10.1126/scitranslmed.3006034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linette GP, Stadtmauer EA, Maus MV, et al. Cardiovascular toxicity and titin cross-reactivity of affinity-enhanced T cells in myeloma and melanoma. Blood. 2013;122:863–871. doi: 10.1182/blood-2013-03-490565. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. The impact of donor origin on the functional profile of T cell receptor (TCR)-transduced CD8+ T cells.
Figure S2. Combinations of functions elicited by stimulation of T cell receptor (TCR)-transduced CD8+ T cells with peptide-pulsed target cells.