

Abstract
Genome-wide association studies have greatly improved our understanding of the genetic basis of disease risk. The fact that they tend not to identify more than a fraction of the specific causal loci has led to divergence of opinion over whether most of the variance is hidden as numerous rare variants of large effect, or common variants of very small effect. Here I review 20 arguments for and against each of these models of the genetic basis of complex traits, and conclude that both classes of effect can be reconciled readily.
Soon after the ascension of genome-wide association studies (GWAS) as the pre-eminent tool for discovering polymorphic genes that influence disease susceptibility and quantitative traits, the field of genetics developed three major outlooks on the architecture of complex traits. When GWAS began the field was dominated by the simple common disease-common variant (CD-CV) hypothesis1–4 has been refuted in light of the so-called ‘missing heritability problem’, namely the observation that loci detected by GWAS almost without exception explain a small minority of the inferred genetic variance5,6: it is simply not the case that a few dozen loci of moderate effect and intermediate frequency each explain several percent of disease risk in a population, as is typically observed in crosses or pedigrees. Since then, the genetic component has been attributed instead to one of three causes: a large number of small-effect common variants across the entire allele frequency spectrum (the infinitesimal model)7,8, a large number of large-effect rare variants (the rare allele model)9, or some combination of genotypic, environmental and epigenetic interactions (the broad-sense heritability model)10,11. Figure 1 shows the expected distribution of genome-wide association profiles under each of these three models and the CD-CV model.
Figure 1. Different expected signatures from GWAS for four models of disease.

Each plot shows the approximate expected distribution of SNP effects for a modest study of 2,000 cases and controls. The Y-axis is the percent of the variance for a trait or disease liability in the population explained by each SNP (note that standard Manhattan plots typically show the significance instead, represented as the negative log10 of the p-value) and the X-axis is the location of tens of thousands of SNPs along the chromosome. In the common disease-common variant (CD-CV) model, a small number of moderate effect loci would produce very strong signals, each of which explains several percent of the genetic variance. Note the expanded scale of the y-axis here relative to the other plots. In the rare allele model, causal variant effects (yellow dots) may be large in a few individuals but are not common enough to explain much variance or result in genome-wide significance. The infinitesimal model by contrast does produce some significant peaks due to small effects of common variants, and in each case several SNPs within an LD-block associate with the trait. Finally, it can be argued that if associations are only seen in some environments (green and orange signals, bottom right), then in a mixed population the overall effect will be reduced at such loci (arrowheads), and fewer associations will be detected, explaining less of the variance.
GWAS are neither powered nor designed to detect variation under any of these models on a consistent basis, so there is as yet insufficient empirical data to resolve the debate. In all likelihood, each of the three genetic architectures contributes, possibly to different degrees, to different diseases or traits. However, since the heritability of risk is generally estimated as narrow sense variance, interaction effects should be considered secondary to the main effects of common and rare variants. The purpose of this article is therefore to review 20 arguments that support or refute the infinitesimal and rare allele models. This is not a comprehensive survey, but rather an overview of five arguments in favour and five arguments against each of these two models, drawn from theory and data from diverse categories of disease. Table 1 lists each of the arguments, roughly according to their relative strength.
Table 1.
The 20 Arguments
| For Rare Alleles |
| Evolutionary theory predicts that disease alleles should be rare. |
| Empirical population genetic data shows that deleterious variants are rare. |
| Rare copy number variants contribute to several complex psychological disorders. |
| Many rare familial disorders are due to rare alleles of large effect. |
| Synthetic associations may explain common variant effects. |
| Con Rare Alleles |
| Simulation of GWAS is not consistent with rare variant explanations. |
| GWAS associations are consistent across populations. |
| Sibling recurrence rates are greater than postulated rare variant effect sizes. |
| Epidemiological transitions cannot be attributed to rare variants. |
| Rare variants do not obviously have additive effects. |
| For Common Alleles |
| GWAS has successfully identified thousands of common variants. |
| Model organism research supports common variant contributions to complex phenotypes. |
| Variation in endophenotypes is almost certainly due to common variants. |
| The infinitesimal model is standard quantitative genetic theory. |
| Common variants collectively capture the majority of the genetic variance in GWAS. |
| Con Common Alleles |
| What accounts for the missing heritability? |
| Demographic phenomena suggest more than a simple common variant model. |
| The QTL paradox. |
| Absence of blending inheritance. |
| Very few common variants for disease have been functionally validated. |
The Models
Infinitesimal model: many variants of small effect
By infinitesimal model, I mean the proposition that common variants are among the major source of genetic variance for disease susceptibility and continuous traits, where hundreds or thousands of loci contribute in each case. The loci detected by GWAS are merely the largest effect sizes drawn from a Poisson or similar distribution. If half a dozen common variants explain 10% of risk in the population, the remainder is attributable to a myriad of variants that each explain considerably less than 1% and have genotype relative risks less than 1.112. Figure 2a shows that affected individuals will tend to carry a slight excess of risk variants, since the overall distribution of the number of risk alleles per affected individual is skewed relative to unaffecteds. If these follow the same distribution of allele frequencies as neutral variation, then they will include a large number of rare variants as well. Ultimately, every gene contributes to every trait, but with effect sizes so small that it would take samples greater than the population size of the species to detect them. In practice, as shown by the massive meta-analyses of GWAS for height and body mass index (BMI)13,14, each involving several hundred thousand people, it is unlikely that more than a few hundred loci will ever be confirmed for most diseases, and these will not necessarily explain even half of the genetic variance. The term infinitesimal borrows from the initial formulation of quantitative genetic theory by RA Fisher7. Here it simply signifies the idea that the heritability is not so much missing, as hidden beneath the significance thresholds used to define risk alleles with high confidence15.
Figure 2. Expected distribution of Risk Variants.
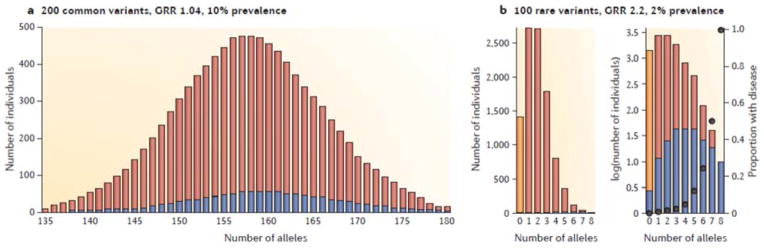
(a) The approximate frequency distribution of risk alleles in cases (blue) and controls (red) under the infinitesimal model for a disease with high heritability and 10% prevalence. For this particular parameterization I assumed 200 loci with risk allele frequencies from 0.1 to 0.9, but skewed toward lower frequencies. Each risk allele is assumed to increase the probability of disease additively by 1.04 relative to the overall risk of 10%. The frequency distribution in cases is skewed to the right, but note that the median number of risk alleles in affected individuals is only slightly greater than in unaffecteds. (b) An approximate frequency distribution of risk allele frequencies under a multiplicative rare allele model for a disease with high heritability and 1% prevalence. This parameterization assumes 100 loci, each with a risk allele frequency of 1%, such that each risk allele increases the probability of disease 2.5 fold over a background risk of 0.2%. The vast majority of unaffecteds carry at least one allele (the yellow bar show the expected number of individuals without any risk alleles). The inset shows the same figure on the log scale, emphasizing how relative risk increases with the number of variants carried. Note that the measured per allele GRR across the population in the presence of 100 other alleles is ~1.15, much smaller than the 2.5 fold multiplicative risk due to a single variant. For higher risks (say 5-fold) and 100 alleles, the frequencies must be very rare (~0.1%) for a disease prevalence of 1%, and affecteds will only carry one or two risk alleles.
Rare allele model: many rare alleles of large effect
The alternative view is that most of the variance for certain complex diseases is due to relatively highly penetrant rare variants, the allele frequency of which is typically less than 1%, most of which are recently derived alleles in the human population. Under this model, expressivity may be modified by other loci or the environment16,17, but the notion is that the rare susceptibility genotype is largely responsible for disease. The rare allele model generally refers to dominant effects, due either to haploinsufficient or gain of function alleles, where risk is elevated 2-fold or more over background. Under these conditions, penetrance need not be anywhere near 100%. In fact, as shown in Figure 2b, the vast majority of unaffected individuals are expected to carry one or more risk alleles. The notion is that a disease such as schizophrenia is actually a collection of hundreds, or possibly even thousands, of similar conditions attributable to rare variants at individual loci18. If each of these variants explains most of the risk in just a handful of people, their effects will not explain enough of the variance in a total population to be detected by standard GWAS procedures. The total number of loci that may contribute to any disease is a function of the baseline disease incidence, the number of rare variants per locus, and the genotypic relative risk (GRR, namely, effect size). For a disease with a high heritability of 80%, theory indicates that the number of contributing loci can range from over 500 with GRR of just 2 and one very rare variant per locus, to just a dozen loci with GRR of 5 and allele frequencies of 1% or if the disease is relatively common19.
Broad-sense heritability model: non-additive GxG and GxE interactions, and epigenetic effects
The broad-sense heritability model posits that additive contributions of common variants and large effects of rare variants are insufficient to explain the missing heritability. Proponents of this model point to a long history of detection of genotype-by-genotype interactions (epistasis) and genotype-by-environment interactions (G×E) in model organism quantitative genetic research20,21, and note the increasing number of studies documenting epigenetic effects22, notably parent-of-origin genetic contributions23,24 and inheritance of DNA methylation patterns25. The notion here is that since GWAS only measure the average effects of alleles across thousands of individuals, they are incapable of capturing heterogeneity of effect sizes at the family level that would be the hallmark of these broader components of the genetic architecture. Although broad-sense heritability is not considered further in this article (but see Box 1), I do not disregard its potential contribution. Rather, my purpose here is to contrast the two narrow sense models, as resolution of their contributions lays the foundation for consideration of other genetic mechanisms.
Box 1. Broad Sense Heritability Models.
Phenotypic variance is traditionally decomposed into genetic and environmental components, and heritability is defined as the ratio of the genetic to the total phenotypic variance, in a population8. The genetic component of phenotypic variance can be further decomposed into additive, dominance, and interaction effects. The additive component is the average effect of substituting one allele for the other, irrespective of whether dominance is present, namely whether the heterozygotes are closer in phenotype to one class of homozygote.
‘Broad sense heritability’ refers to the genetic effect including non-additive components, such as genotype-by-genotype interactions, also known as epistasis, genotype-by-environment interactions (G×E), and epigenetics. Epigenetics refers to the impact of chromatin modification on the effect of a genotype through DNA methylation, alteration of the histone code and, some would argue, microRNA expression.
Genome-wide association studies (GWAS) are not generally powered to detect epistasis or G×E121, and they are not designed to detect epigenetic influences122 (although recent sampling designs to detect parent-of-origin associations do provide evidence for them23). There are two major obstacles to detecting epistasis: very large samples are required to find sufficient individuals of each genotype combination to accurately measure small effects, and the number of comparisons scales exponentially with the number of interactions so the testing burden is enormous. Additionally, epistatic effects are generally thought to be likely to be small relative to main effects, and the GWAS literature provides few examples of departure from additivity. Similarly, there is as yet little evidence for environmental modification of genotype effects: a recent example relating to the protective effect of coffee consumption and Parkinson’s disease risk provides an exception108. One of the difficulties may be that the environment is difficult to define and measure, and it may be unique individual exposures that are more important than exposures shared by thousands of people.
A particular class of genetic interaction that has yet to attract full attention is deleterious intergenic compound heterozygosity, namely interactions between multiple rare variants that lead to disease. Whether and how often such interactions extend to heterozygous combinations of alleles has considerable implications for the potential of broad sense heritability to be a major component of disease susceptibility. If hundreds of mutations each at a frequency of less than 1% all affect a particular disease, then many more people will be doubly or triply heterozygous for various combinations, than are homozygous for any one variant. It is not known how often heterozygotes that individually do not associate with disease would be deleterious in combination. Synthetic genetic interaction screens in yeast123,124 have demonstrated that thousands of combinations of individually viable mutations can jointly be lethal, and similar examples have been documented in Drosophila melanogaster and Caenorhabditis elegans125,126.
Arguments in favour of the rare allele model
Evolutionary theory predicts that disease alleles should be rare
Perhaps the strongest argument for the rare allele model comes from evolutionary theory. Since disease is deleterious to fitness, variants that promote disease should be selected against, and hence disease-promoting variants should not be common3,26,27. The existence of disease-promoting variants reflects the balance between mutation creating new susceptibility variants, and selection preventing them from drifting into the population2,28. Mutation rates are sufficiently large that purifying selection cannot remove all deleterious variants, and those that have a modest effect on fitness (for example, if they influence late-onset diseases 29) may rise to allele frequencies of 1% or occasionally more, particularly if the effect is recessive. But selection is sufficiently efficient that even a fitness reduction of a fraction of a percent will keep allele frequencies from reaching common levels. The argument has been made that relaxed selection in modern humans may favor the accumulation of deleterious rare alleles, greatly increasing the prevalence of disease hundreds of generations into the future30.
Empirical population genetic data shows that deleterious variants are rare
It has been appreciated for some time that the distribution of minor allele frequencies (MAFs) is strongly skewed towards an excess of rare variants: over a third of all polymorphisms have frequencies below 5%31. Multiple factors contribute to this skewed distribution, but the finding from whole-exome sequence data that non-synonymous substitutions are even more significantly skewed towards low frequencies almost certainly reflects the operation of purifying selection32–34. As a class, amino acid substitutions appear to be deleterious. It need not follow that the reduction in fitness is due to promotion of chronic disease, and the observation does not equate to saying that all rare non-synonymous variants are deleterious. However, the finding is consistent with the theory that selection keeps fitness-reducing alleles at a large proportion of genes at low frequency35. It remains to be seen whether the same is true of regulatory polymorphisms36,37, since (despite the considerable technological achievements of the ENCODE project, http://www.genome.gov/10005107) we still lack efficient procedures for identifying enhancers and other regulatory regions that polymorphisms could disrupt38.
Many rare familial disorders are due to rare alleles of large effect
These are not restricted to conditions such as Cystic Fibrosis and Muscular Dystrophy that are solely due to rare, high penetrance Mendelian mutations. There are numerous chronic conditions that have familial analogs: well-known examples include rare variants promoting atherosclerosis through hypercholesterolemia39, the lesions responsible for Maturity Onset Diabetes of the Young (MODY)40, and the BRCA1 and BRCA2 breast cancer susceptibility mutations41. In fact, perusal of the Online Mendelian Inheritance in Man (OMIM) database provides examples of near-Mendelian cases of many relatively common disorders42. Probably the most comprehensive survey of this model is the demonstration that half of the cases of X-linked mental retardation can be ascribed to rare protein-coding mutations, which were discovered by comprehensive sequencing of X-chromosomal exons43. There is thus extensive precedent for rare variants contributing substantially to special cases of complex disease, including risk of infection44.
Rare copy number variants contribute to several complex psychological disorders
Copy number variants (CNV) are either hemizygous deletions or local duplications that result in 3 or even 4 copies of a locus45. Five percent of cases of schizophrenia and of autism have each been attributed to CNVs at fewer than a half dozen genomic locations46–48. These effects are less highly penetrant than Mendelian mutations, implying modification by the genetic background. There is no evidence to support the hypothesis that rare single nucleotide variants at the same loci are the major source of genetic variance9, but ongoing deep sequencing studies will quantify such effects. In the case of the ciliopathies, there is evidence from functional assays that rare variants can both promote and modify the severity of disease49. It is thus incontrovertible that rare variants contribute to disease risk, and that identification of such alleles is a powerful mode of genetic analysis. The question is whether they can account for more than a minor fraction of complex disease risk.
Synthetic associations may explain common variant effects
‘Synthetic association’ describes the situation where the association of a common variant with a disease is actually due to linkage disequilibrium between the common variant and several disease-promoting rare variants that happen to segregate on the same haplotype50. Thus, a common variant present in 20% of cases that mathematically explains one percent of disease susceptibility may actually simply report the activity of two or three rare variants that each substantially elevate risk in one or two percent of the cases. Until this year rare variants been excluded from the major whole genome genotyping platforms, so there has been no way to systematically document their contribution. Synthetic association is not expected to account for most of the missing heritability19, 51, but this type of effect must always be considered as an explanation for apparent common-variant effects52.
Arguments against the rare allele model
Analysis of GWAS data is not consistent with rare variant explanations
A major argument against rare variants as the predominant source of missing heritability comes from the analysis of allele frequency distributions of GWAS data19. Rare allele proponents point out that it is difficult to model the true distribution of rare variants, and that they are under-represented on genotyping arrays, which complicates the interpretation. Nevertheless, considerations of linkage disequilibrium using standard quantitative genetic theory19 place strong constraints on the number of rare variants and their range of effect sizes that would be compatible with them making a major contribution yet remaining undetected in GWAS, as explained in Figure 2. Furthermore, analyses of the distribution of risk allele frequencies across 8 traits argues that if anything minor allele frequencies are skewed to be greater than 0.2, providing strong empirical evidence that rare alleles are not alone responsible53. No-one doubts that some fraction of the total risk for any complex disease is due to rare alleles, but these studies argue that it is not the majority.
Sibling recurrence rates are greater than expected by the postulated effect sizes of rare variants
Heritability of disease is very often inferred from elevated sibling recurrence rates relative to incidence in unrelated individuals. Intuitively, if a disease has a prevalence of 1% in the general population, but 50% of siblings are affected, then the odds are elevated 50-fold in the family, whereas a single segregating rare variant with a 5-fold effect would only be expected to result in disease for 5% of carrier individuals. Sibling recurrence rates are generally much higher than can be attributed to rare variants with the postulated effect sizes17. If rare variants are contributing then they are doing so in the context of many other genetic variants in the pedigrees, as shown by a recent analysis of schizophrenia genetics54. Under multiplicative models, disease may cluster in families due to segregation of multiple rare variants that happen to be brought together in the pedigree. Nevertheless, the rare allele model must not only explain association data in populations, but also fit demographic distributions of disease within and among families, and there is little theory supporting the claim that they can do both55–57.
Rare variants do not obviously have additive effects
On the face of it, the widely documented additivity of genetic associations is inconsistent either with the dominance of rare variant effects, or the assumption that they interact multiplicatively within individuals to influence disease. However, it turns out that rare variants can easily induce apparently additive effects statistically, because the homozygotes for the tagging variant are twice as likely as the heterozygotes to carry the rare variant. Multiplicative interactions between rare variants cannot be measured in GWAS because of low power, but it will be important to establish mechanistically whether combinations of two or more such mutations increase risk in a linear or synergistic manner58,59. Compound heterozygosity for two different rare variants at one locus is well documented in diseases such as cystic fibrosis, hemochromatosis and sickle cell syndromes; extension of the concept to include intergenic multiple heterozygosity could represent a large source of genetic variance that is almost impossible to detect with current methods.
Epidemiological transitions cannot be attributed to rare variants
The fourth argument against rare variant effects is a demographic one, namely the changing prevalence of so many chronic diseases in a span of just two or three generations, and/or the known impact of environmental variables on risk. For example, diabetes and heart disease have greatly increased in incidence in India and China in the past 10 years60,61, an epidemiological transition that at best implies a change in penetrance of genetic effects attributable to any class of variant, rare or common, in the contemporary environment. Schizophrenia, a disease with very high heritability (inferred from twin studies) and for which very few replicated GWAS hits have been identified despite extensive scans, nevertheless shows such demographic influences as whether the parents live in rural or urban areas (disease rates are elevated in children born after migration to cities)62. Paternal age effects on psychological disease63 might be attributed to elevated mutation rates in sperm, but other hypotheses are equally compatible with the data, and maternal age effects operate in the opposite direction as younger mothers have higher likelihoods of having affected children64. In other words, rare variants alone cannot explain the demographic distribution of disease incidence.
GWAS associations are consistent across populations
An empirical argument against pervasive rare variant effects is that common variants are often consistent across populations — such as between Caucasians and Asians — despite differences in allele frequencies65,66. If rare variants are recently derived relative to the common variants, then they should be at different frequencies in Caucasians and Asians, and we would expect that they would only induce synthetic common variant associations in one of these populations, or at least would not tend to have the same magnitude of effect. This would be especially true of common variants that differ in frequency between the two populations. Under a mutation-selection balance model, it may be possible for different novel rare variants to have effects in each population, and fine mapping studies sometimes reveal subtle differences in the patterns of association [eg.67,68. Nevertheless, the simplest interpretation of the consistency of common variant effects is that they are actually due to the common variants themselves or to unobserved common variants in high LD across all populations.
Arguments in favour of the infinitesimal model
The infinitesimal model is standard quantitative genetic theory
Just as evolutionary theory provides a strong argument in favor of rare variants, standard quantitative genetic theory provides ample support for the infinitesimal model7,8. Whatever the causes of the maintenance of genetic variance may be, the consistent observation is that all diseases have significant heritability, and hence purifying selection has been unable to purge the population of disease-promoting variants2. At face value, the existence of dozens of susceptibility alleles for metabolic and immunological diseases with effect sizes that are just not detected for psychological diseases implies a difference in genetic architecture between the two categories of conditions. This may imply different intensities of purifying selection, although other models, including decanalization69, are also compatible with the data. Since the majority of the genetic variance remains unexplained, it is a priori just as likely to exist in the form of rare or common alleles, and the fact is that there is nothing about GWAS findings that is inconsistent with the infinitesimal model of many variants of very small effect across the full allele frequency spectrum. This model has served applied quantitative geneticists as well as evolutionary biologists for close to a century, and in a sense can be regarded as the null model that needs to be disproved before it is abandoned.
Common variants collectively capture the majority of the genetic variance in GWAS
Direct empirical support for the infinitesimal model comes from genomic variance analyses70, 71. Animal breeders have been using ‘genomic selection’ methods with great success for the past decade72, basing their selection of sires and dams on the overall predicted breeding value determined from the full set of genomic markers that capture variation distributed throughout the genome. Similarly in humans, by taking all nominally significant SNPs rather than just the GWAS significant ones, it is possible to capture much more of the genetic variance than is explained by the highly significant loci73,74 (see Box 2). A multivariate version of this approach, implemented by regression of phenotypic similarity on genetic relatedness, also implies that common variants capture the majority of the genetic variants71. Furthermore, partitioning of the genetic variance on a chromosome-by-chromosome basis, for a diverse set of traits, shows that the proportion of variance explained is consistently proportional to chromosome length75,76. Variance is distributed along all of the chromosomes, and is therefore attributable to hundreds of loci. Since common variants are used for the partitioning it is most parsimonious to conclude that they are responsible, and simulations of rare variants so distributed capture much less of the variance.
Box 2. Association Tests in Unrelated Individuals.
Genome-wide association studies (GWAS) generally start with comprehensive SNP-wise evaluation of the significance of the correlation between genotype and trait. For the most part, it is assumed that genotypic effects are ordered, namely that the heterozygotes will have intermediate risk or trait values.
As millions of tests are performed, the results must be adjusted to control for false positives, (usually using the conservative Bonferroni correction127), resulting in the standard GWAS threshold of 5×10−8. The significance of the test statistic is then plotted against SNP position along the chromosome (see Figure 1). For the most part, genotyped SNPs are thought to ‘tag’ the actual causal variant, so efforts are made to estimate (‘impute’) as many genotypes in the region of a GWAS hit as possible128, increasing the chances that the common causal variant is represented or that the effects of less common ones are better captured. Due to improved methods for imputation, most minor alleles down to 5% frequency can now be inferred with high accuracy, if the reference panel is appropriately matched for population structure129. Rare variants, particularly those at a frequency of less than 1%, need to be sequenced directly.
Although association tests are reasonably powered to detect common variants that have a genotypic relative risk of 1.2 or more in meta-analyses that include tens of thousands of individuals, detecting the effects of single rare variant remains problematic. Various score statistics are being developed that evaluate pools of rare variants in a part of a gene, or group of related genes, for over-representation in cases or controls119. These can be conditioned on prior estimation of the likelihood that a nucleotide substitution is deleterious. Functional tests are used increasingly to validate candidate causal polymorphisms.
Two additional strategies have been proposed to overcome the very high false negative rate in GWAS that results from adoption of the strict GWAS threshold. One is to generate a weighted sum of the contributions of all variants beyond nominal statistical thresholds observed in a discovery sample, and then ask whether it is predictive of risk or phenotype in a second sample53. In several instances where SNP-wise GWAS has uncovered few loci, this approach has provided clear evidence for polygenic risk, but it must be recognized that the high variance in estimation of effects results in noisy scores that, although highly significant in the replication sample, only capture over an order of magnitude less of the variance and are far from predictive. See74 for another related strategy. The second approach is to evaluate all SNPs simultaneously by multiple regression, or the elegant equivalent of regressing the similarity between individuals on their overall genetic similarity71, which gives rise to an estimate of the genetic variance explained by all SNPs. Partitioning of the genome into chromosome-sized units [75], or smaller, allows for estimation of the contribution of each region of the genome to the genotypic variance.
Variation in endophenotypes is almost certainly due to common variants
Threshold-dependent models77 postulate that disease is more likely to arise in individuals who have extreme values of underlying endophenotypes78,79. In many cases, causal common variants associated with a continuous endophenotype have been associated with disease [eg80–82], and in some cases these have been confirmed by in vitro biochemical assays for structural and regulatory effects [eg83–85]. eQTL analysis shows that gene expression and splicing are heavily influenced by common variants, possibly for the majority of transcripts86–88. To ignore these data is to deny that such transcriptional and metabolic variation is relevant to disease. It is of course possible that disease represents a discrete phase shift (in gene expression profiles, for example) that takes the organism outside the normal range of continuous variation. Thus, tumor samples have discrete transcriptome profile differences89,90, and it is not obvious that the normal variability is relevant to pathology. Similarly, at least some cortex samples from autistic brains converge on a common transcriptional profile91. By contrast, various blood disorders have been shown to correlate with extreme values for the major vectors of modules of gene expression92. More research on the relationship between endophenotypes and disease is needed, but most observers would consider it implausible that natural variation in physiology is irrelevant to variation in disease susceptibility, and would maintain that common variants are most likely responsible for continuously distributed physiological variation.
Model organism research supports common variant contributions to complex phenotypes
In model organism research, both pedigree analyses and genetic crosses in which linkage mapping is used to localize QTL almost always lead to the identification of multiple variants influencing the quantitative trait of interest20. This is as true of threshold-dependent characters and cryptic variation93 as it is of continuous variation. Furthermore, the phenomenon of transgressive segregation in mapping populations of mice and flies established from eight founder strains provides strong empirical support for the existence of common polygenes94–96. The overwhelming evidence from classical quantitative genetics is that traits are regulated by many loci with a wide range of effect sizes. A counterargument is that in any cross or pedigree, there is no information about the frequency of contributing QTL alleles in the population, so some fraction of the mapped factors are likely to be rare variants – and if the parents were selected from a base population, possibly most are unusual rare variants, including mutations that were unconsciously selected in the laboratory. In the past year, re-sequencing of evolved outbred populations of Drosophila melanogaster has provided strong support for selection on thousands of variants being responsible for changes in the highly complex traits of body size and fecundity97,98.
GWAS has successfully identified thousands of common variants99
While there has been a very public focus on the failure of GWAS to find the missing heritability5,6, the simplest explanation for this is that the expectations were based on unrealistic prior assumption of effect sizes. Once it is accepted that most alleles are associated with relative risks less than 1.2, it becomes clear that hundreds of thousands of individuals are required to identify more than a few dozen loci than explain more than 20% of the variance. In fact, sample sizes can be extrapolated from the range of variant effects in initial discovery samples12, and in the case of height, the GIANT consortium (http://www.broadinstitute.org/collaboration/giant/) confirmed the inferences from 30,000 individuals100 when they jumped to 180,000, finding an additional 180 or so loci14. Simply put, the data is generally consistent with the infinitesimal model to a first approximation, even where variants are yet to be identified. It is the common variant of moderate effect version of the CD-CV model that needs to be discarded.
Arguments against the infinitesimal model
The QTL paradox
Where are all of the QTL that are so consistently detected in pedigrees and in experimental crosses, when we transition to outbred populations? Why is it that 10 loci can each explain 5% of the genetic variance and most of the heritability in a cross between two strains, but in no case has GWAS yet found more than one locus with an effect size that large? Two explanations can be forwarded immediately: first, the QTL are actually rare variants that only contribute in that cross, and so are precisely the rare variants predicted by the rare allele theory; or second, each cross captures just a small fraction of the genetic variance in a population, so QTL with relatively large effects in one cross will be expected to be diluted in their contribution relative to other QTL when measured in the entire population. In many cases as well, the effect size estimated in the cross will be an overestimate due to the Beavis effect/winner’s curse101,102, while in an unknown proportion of cases, QTL will turn out to be due to multiple linked variants that coarse mapping fails to resolve into individual SNPs103. Nevertheless, there remains a general paradox that GWAS has found so few variants of moderate effect.
Absence of blending inheritance
A more pointed argument against the infinitesimal nature of effects is that it predicts less granularity in the distribution of risk and phenotypic trait variation than is often observed, though I am not aware of a quantitative assessment of this claim. In the infinitesimal model, disease risk ought to blend smoothly when unrelated people have children. The larger the number of alleles affecting a trait, the lower the among-individual variance should be under random mating since most individuals will share similar numbers of risk alleles. However, disease incidence and complex phenotypes generally cluster in families. A possible resolution of this conundrum is that the observed clustering of disease in families could be explained by stochastic variation in the number of susceptibility alleles: if two people happen to have more than the average number of small variants, so will their children. Furthermore, homophily, the tendency for couples to pair on the basis of shared attributes (including sub-clinical disease indicators), will tend to enrich for variants that promote those attributes104. Granularity of traits is difficult to document, but facial features provide a good example of a suite of traits that do not simply follow either blending or Mendelian inheritance105. Certain features such as the shape of the nose, location of the cheek bones, or curve of the lips, run strongly in families, appearing in distant relatives in patterns that are suggestive of large genetic effects. If such clustering is also true of endophenotypes then the infinitesimal model will be incomplete.
Demographic phenomena suggest more than a simple common variant model
As with the rare allele model, infinitesimal effects are also not consistent with a wide range of demographic effects that are indicative of GxE interactions and complex genetic interactions. Prime among these are (i) the pervasiveness of differences in disease risk between geographic areas that are not obviously explained by genetic differentiation, (ii) increasing burden of complex disease in the span of one or two generations (both of these phenomena are obvious upon browsing the CDC website at http://www/cdc/gov for incidence data), and (iii) conditioning of the risk for one disease on another disease in the same individual106,107. This does not counter the existence of thousands of small effect loci affecting each trait or disease risk profile, but it suggests that the narrow-sense genetic effects alone are unlikely to be sufficient explanation. It must be noted that there is very little evidence from GWAS for either GxE or GxG interactions108, but such effects could be mild at the level of individual associations and be below the power of detection.
Very few common variants for disease have been functionally validated
A technical argument against all common variant models is that association alone is insufficient evidence of function: correlation is not causation. Very few of the thousands of GWAS significant associations have been shown using molecular genetics, biochemistry or biophysics to be the actual risk variant38, 109. In this context, prudence suggests an open mind in each individual case as to whether the variant, another common variant in LD, or a series of less common variants of large effect synthetically associated may be responsible. The case of protection against anemia in chronic hepatitis C, where the initial GWAS discovery clearly captures two less common causal coding variants at the ITPA locus, is a good example110.
What accounts for the missing heritability?
Finally, there is the argument that there truly is missing heritability in GWAS studies. It is little appreciated that GWAS do not actually measure heritability (the ratio of genetic to total phenotypic variance in a population) but rather they measure just the genetic variance. Missing heritability is inferred with respect to heritability estimates that generally derive from family studies, where ironically direct estimates of the genetic contribution are lacking. Consequently, it is difficult to actually estimate how much of the variance GWAS-captured variants should explain in outbred populations. However, in the case of height, where heritability is as high as 80%, over 50% of the phenotype can be attributed to common variants using the mixed linear modeling approach71, and after adjustment for allele frequency skews and incomplete LD, essentially the entire genetic contribution has been ascribed to genotypic variation. Yet the same methods applied to BMI do not capture much more than half of the expected genotypic variance, and it is not clear that they are as efficient for schizophrenia, arthritis or intelligence75,76. In these cases, it can be argued that the infinitesimal model does not capture the full range of genetic variance, and that there is a true missing heritability problem that will need to be addressed with rare alleles and broad sense heritability components that were introduced in Box 1.
Conclusion
The true debate over the source of genetic variation for disease is not one of “is it rare or common variants”, or even of “how much does each class contribute”, but rather “how do they work together” 111. An interesting thought experiment is to ask whether a population of individuals who are only polymorphic for common variants differ in their common disease risk; or conversely whether rare variants are sufficient to explain the observed distribution of risk in the absence of common variants. The result of both experiments is likely to be negative, but the challenge is to devise strategies to test the hypotheses.
Empirically, there is ample support for both class of effect. As of June 2011, the NHGRI GWAS catalog lists 1,449 genome-wide significant associations with 237 traits and diseases, spread across all chromosome except the Y99. Some of these may be due to LD with rare variants, but parsimony suggests most are common variant effects. MutDB112(http://mutdb.org/) contains a much larger number of rare coding variants that are either associated with disease or predicted to be damaging. As more individuals are sequenced, many of these appear in healthy controls, but it must be recognized that even large effect alleles are subject to background modification and incomplete penetrance. Initial resequencing of genes identified by GWAS113 has produced mixed results: only 1 of 63 genes in an initial screen for inflammatory bowel disease found evidence for rare variant effects (and these were protective114, but see also115), whereas a significant excess of rare coding variants was found in 4 genes for hypertriglyceridemia116. Considerable resolution of the burden of deleterious rare variants will no doubt emerge in the next few years as whole exome and genome sequencing ramps up117,118. Interpretation will be complicated by the natural tendency to under-report negative results, by difficult statistical issues119, as well as the problem of how to define regulatory effects.
The typical resolution of the observation that disease is categorical (people are either cases or controls) yet genetic contributions are complex, is that disease is generally a threshold-dependent response superimposed over a continuous liability120. This interpretation actually provides a straightforward framework for integrating rare and common variant effects (Figure 4). Liability is quite likely to be established by the additive and infinitesimal contributions of hundreds of polymorphisms, each regulating a series of biochemical traits that impinge on the phenotype: metabolite abundance, gene expression, and hormone levels, for example. Disease arises either in individuals at the extremes of the liability scale who move beyond the threshold, or in individuals close to the threshold who are pushed into adversity through environmental and behavioral agents, or because they carry one or more rare variants. On this model, the rare variant can either increase or decrease function, and whether or not it associates with disease will be conditional on the background liability. The notion that even rare variants have variable penetrance thus blurs the distinction between infinitesimal and major-effect models of disease susceptibility.
Figure 4. Joint effects of rare and common variants.

A straight forward reconciliation of the effects of rare and common variants supposes that pervasive common variation influences the expression and activity of genes in pathways, establishing the background liability to disease that is then further modified by rare variants with larger effects. In this hypothetical example of central metabolism, standing variation results in some individuals having lower flux than others (left versus right; colored boxes imply enzyme activity differences from low, red, to high, green), but according to standard biochemical theory130, systems evolve such that most variation is accommodated within the healthy range. The impact of a rare variant that knocks out one copy of the enzyme indicated with the cross is conditional on this liability, pushing the individual on the left beyond the disease threshold, while the individual on the right can accommodate the mutation given higher activity elsewhere in glycolysis.
Figure 3. Inconsistency between GWAS results and rare variant expectations.
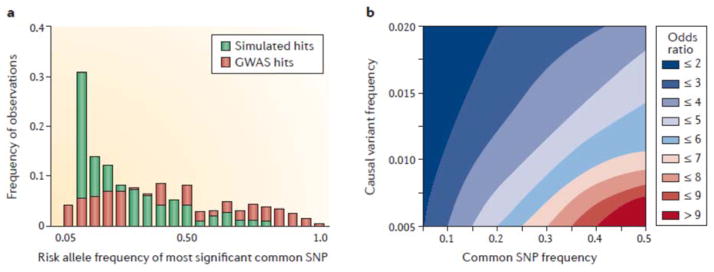
(A) The frequency distribution of risk allele frequencies (light red) for 414 common variant associations with 17 diseases is only slightly skewed toward lower frequency variants. By contrast, simulations, in this case assuming up to 9 rare causal variants inducing the common variant association with SNPs at the same frequency as observed on common genotyping platforms (light green bars) result in a marked left-skew with a peak for common variants whose frequency is less than 10%. (The skew is even stronger if only a single causal variant is responsible). The observed data is thus not immediately consistent with the rare variant model. (B) Part of the problem with synthetic associations is that they would explain too much heritability if they were pervasively responsible for common variant effects. This is due to the relationship between allele frequency, maximum possible LD, and the amount of variance explained [19]. The plot shows the expected odds ratio due to a rare variant of the indicated frequency (from 0.5% to 2%) if it increases the odds ratio at a common SNP (with which it is in maximum possible LD) 1.1-fold. Intermediate effect sizes (2<OR<5) require combined causal variant frequencies in excess of 1%. As the number of rare variants increases, the likelihood that they are in high LD with the common variant also drops, further reducing the probability that they can explain observed common variant association. Suppose a disease has a prevalence of 1%. Then 10 causal variants each at a frequency of 1% would result in 20% of people carrying a causal variant. If the penetrance is 5% then 1% of people would have the disease, and these 10 variants would completely explain the genetic risk. Similarly if 100 causal variants were each at 0.1% frequency, it would take ~10 such variants to induce each single common variant association with an observed OR of 1.1. If large GWAS detect dozens of such common loci, and they were actually due to LD with rare variants, then the heritability would be explained several times over. Alternatively, if hundreds of very rare causal variants are not in LD with common variants, we do not expect to see GWAS significant associations. Data from19.
Online Summary.
For the past couple of years, discussion of the so-called “missing heritability problem” has focused attention on the failure of genome-wide association studies (GWAS) to discover more than a minor fraction of the genetic variance for complex traits and disease susceptibility. However, it now appears that the problem is largely a result of the small contribution of most variants, either because the variants are too rare to contribute population-wide, or because the effect sizes of common variants are in general very small. This review presents five arguments for, and five against, each of these two models, and concludes that while the infinitesimal model is essentially correct, rare alleles of large effect almost certainly also make an essential contribution to risk of disease. Some of the more important arguments are:
Standard evolutionary and quantitative genetic theory both provide strong expectations for rare and common variant contributions
There is increasingly solid empirical evidence for both classes of contribution
Neither model can yet be said to provide compelling explanations for epidemiological transitions and other demographic phenomena including familial clustering and sibling resemblance
Mechanistic explanations for additive within-locus, and multiplicative between-locus, effects are ultimately desired to complement purely statistical description of effects
On either model, the majority of healthy individuals carry disease-associated alleles
While common variants may establish the background liability to many complex diseases, environmental and rare variant perturbations will often provide the extra impetus that pushe an individual over the disease threshold.
Acknowledgments
I particularly thank F. Vannberg, D. Goldstein, and P. Visscher for discussions and suggestions (clearly without attributing accountability), and the Georgia Institute of Technology and the US NIH for funding.
Glossary
- Common disease-common variant (CD-CV) hypothesis
The model that complex disease is largely attributable to a moderate number of common variants, each of which explain several percent of the risk in a population
- Genetic variance
The contribution of genotypic differences among individuals to phenotypic variation
- Heritability
The proportion of the phenotypic variance in a population that is due to genotypic differences among individuals
- Narrow sense variance
The additive component of the genetic variance, namely the average effect of substituting one allele for another at a locus
- Genotype relative risk
The ratio of the risk of disease between individuals with and without the genotype. A ratio of 1.1 equates to a 10% increase in risk
- Penetrance
The proportion of individuals with a mutation or risk variant who have the disease
- Expressivity
The severity of the disease in individuals who have the risk variant and disease; Genotype-by-genotype interactions (epistasis)¸ Refers to the situation in which the effect of one genotype is conditional on genotypes at one or more other unlinked loci
- Genotype-by-environment (G×E) interactions
Refers to the situation in which the effect of the genotype is conditional on the environment, which may include abiotic (temperature), biotic (viral exposure), and cultural/behavioral influences
- Parent-of-origin genetic contributions
Genetic effects that are only seen when the allele is transmitted either from the mother or from the father
- Purifying selection
Selection against genetic variants that reduce fitness, generally keeps deleterious alleles at a low frequency or removes them from the population
- ENCODE
Encyclopedia of DNA Elements
- Chronic disease
Medical conditions that develop slowly and persist, generally with a strong genetic component
- Ciliopathy
A class of diseases due to disruption of the cilium, a cellular organelle
- Linkage disequilibrium
Non-random association between genotypes, generally discussed in relation to loci that are closely located on a chromosome, for example within a gene
- Haplotype
A set of alleles that commonly segregate together, defined as regions of extended linkage disequilibrium, which in humans is often up to 100kb in length
- Mutation-selection balance
An evolutionary model that accounts for the maintenance of genetic variation as a balance between mutation generating variance, and purifying selection removing it
- Decanalization
The notion that genetic systems evolved to be buffered, but that large effect mutations or environmental change can overcome this buffering, increasing the genetic variance
- Genomic selection
The use of genetic markers spread throughout the genome to select individuals with desired predicted breeding values
- Predicted breeding value
The estimated phenotype of progeny of individuals that have a particular genotype
- Threshold-dependent model
A model that postulates that individuals who exceed some threshold value of a continuous physiological characteristic (‘liability’), have or are at high risk for disease
- Endophenotypes
Intermediate physiological or psychological traits, such as metabolite and transcript abundance, or a specific neuronal function
- eQTL analysis
Studies of the association between genotypes and gene expression (transcript abundance), leading to the detection of expression quantitative trait loci, or eQTL
- Cryptic variation
Genetic variation whose effects are only seen under perturbed conditions, such as in the presence a particular mutation or environmental exposure
- Transgressive segregation
The appearance of traits in the offspring that are more extreme than observed in either parent
- Beavis effect/winner’s curse
The observation that the effect sizes estimated in a discovery sample tend to be over-estimates of the true effect sizes since they typically receive the benefit of sampling variance in the same direction as the true effect, in order to exceed strict genome-wide significance levels
Biography
Autobiographical Statement
Greg Gibson recently moved to the Georgia Institute of Technology where he is using integrative genomics approaches to study the evolution of disease susceptibility and its implications for predictive health. He received his Ph.D. and postdoctoral training in Drosophila melanogaster developmental genetics, and developed his interest in evolutionary quantitative genetics for the most part while on the faculty at North Carolina State University. He has combined gene expression profiling and candidate gene association studies to elucidate the molecular basis of variation for complex traits, with a particular focus on cryptic variation and genotype-by-environment interactions.
Footnotes
Competing Interests Statement
The author declares no competing financial interests.
Greg Gibson’s homepage: http://www.gibsongroup.biology.gatech.edu/
References
- 1.Lander ES. The new genomics: global views of biology. Science. 1996;274:536–539. doi: 10.1126/science.274.5287.536. [DOI] [PubMed] [Google Scholar]
- 2.Reich DE, Lander ES. On the allelic spectrum of human disease. Trends Genet. 2001;17:502–510. doi: 10.1016/s0168-9525(01)02410-6. [DOI] [PubMed] [Google Scholar]
- 3.Pritchard JK, Cox NJ. The allelic architecture of human disease genes: common disease–common variant… or not? Hum Mol Genet. 2002;11:2417–2423. doi: 10.1093/hmg/11.20.2417. [DOI] [PubMed] [Google Scholar]
- 4.Botstein D, Risch N. Discovering genotypes underlying human phenotypes: past successes for Mendelian disease, future approaches for complex disease. Nature Genet. 2003;33:228–237. doi: 10.1038/ng1090. [DOI] [PubMed] [Google Scholar]
- 5.Maher B. Personal genomes: the case of the missing heritability. Nature. 2008;456:18–21. doi: 10.1038/456018a. [DOI] [PubMed] [Google Scholar]
- 6.Manolio TA, et al. Finding the missing heritability of complex diseases. Nature. 2009;461:747–753. doi: 10.1038/nature08494. A compendium of arguments relating to what may be the sources of missing heritability assembled by participants in an NIH workshop. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fisher RA. The Genetical Theory of Natural Selection. Oxford University Press; Oxford: 1930. [Google Scholar]
- 8.Visscher PM, Hill WG, Wray N. Heritability in the genomics era – errors and misconceptions. Nat Rev Genet. 2008;9:255–266. doi: 10.1038/nrg2322. An accessible modern introduction to the concept of heritability. [DOI] [PubMed] [Google Scholar]
- 9.Cirulli ET, Goldstein DB. Uncovering the roles of rare variants in common disease through whole-genome sequencing. Nat Rev Genet. 2010;11:415–425. doi: 10.1038/nrg2779. [DOI] [PubMed] [Google Scholar]
- 10.Feldman MW. The heritability hang-up. Science. 1975;190:1163–1168. doi: 10.1126/science.1198102. [DOI] [PubMed] [Google Scholar]
- 11.Eichler EE, et al. Missing heritability and strategies for finding the underlying causes of complex disease. Nat Rev Genet. 2010;11:446–450. doi: 10.1038/nrg2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Park JH, et al. Estimation of effect size distribution from genome-wide association studies and implications for future discoveries. Nat Genet. 2010;42:570–575. doi: 10.1038/ng.610. Discusses how the true number of associations and their effect sizes can be inferred from observed GWAS results. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Speliotes EK, et al. Association analyses of 249,796 individuals reveal 18 new loci associated with body mass index. Nat Genet. 2010;42:937–948. doi: 10.1038/ng.686. The largest GWAS meta-analysis to date shows how hundreds of complex variants influence continuous traits. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lango-Allen H, et al. Hundreds of variants clustered in genomic loci and biological pathways affect human height. Nature. 2010;467:832–838. doi: 10.1038/nature09410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gibson G. Hints of hidden heritability in GWAS. Nat Genet. 2010;42:558–560. doi: 10.1038/ng0710-558. [DOI] [PubMed] [Google Scholar]
- 16.Steinberg MH, Adewoye AH. Modifier genes and sickle cell anemia. Curr Opin Hematol. 2006;13:131–136. doi: 10.1097/01.moh.0000219656.50291.73. [DOI] [PubMed] [Google Scholar]
- 17.Bodmer W, Bonilla C. Common and rare variants in multifactorial susceptibility to common diseases. Nat Genet. 2008;40:695–701. doi: 10.1038/ng.f.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McClellan JM, Susser E, King M-C. Schizophrenia: a common disease caused by multiple rare alleles. Br J Psych. 2007;190:194–199. doi: 10.1192/bjp.bp.106.025585. [DOI] [PubMed] [Google Scholar]
- 19.Wray NR, Purcell SM, Visscher PM. Synthetic associations created by rare variants do not explain most GWAS results. PLoS Biology. 2011;9:e1000579. doi: 10.1371/journal.pbio.1000579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mackay TFC. The genetic architecture of quantitative traits. Annu Rev Genet. 2001;35:303–339. doi: 10.1146/annurev.genet.35.102401.090633. [DOI] [PubMed] [Google Scholar]
- 21.Mackay TFC, Stone EA. The genetics of quantitative traits: challenges and prospects. Nat Rev Genet. 2009;10:565–577. doi: 10.1038/nrg2612. [DOI] [PubMed] [Google Scholar]
- 22.Feinberg AP. Phenotypic plasticity and the epigenetics of human disease. Nature. 2007;447:433–440. doi: 10.1038/nature05919. [DOI] [PubMed] [Google Scholar]
- 23.Kong A, et al. Parental origin of sequence variants associated with complex diseases. Nature. 2009;462:868–874. doi: 10.1038/nature08625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Small KS, et al. Identification of an imprinted master trans regulator at the KLF14 locus related to multiple metabolic phenotypes. Nat Genet. 2011;43:561–564. doi: 10.1038/ng.833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jablonka E, Raz E. Transgenerational epigenetic inheritance: prevalence, mechanisms, and implications for the study of heredity and evolution. Quart Rev Biol. 2009;84:131–176. doi: 10.1086/598822. [DOI] [PubMed] [Google Scholar]
- 26.Bulmer MG. The effect of selection on genetic variability. Am Naturalist. 1971;105 [Google Scholar]
- 27.Barton NH, Turelli M. Evolutionary quantitative genetics: how little do we know? Annu Rev Genet. 1989;23:337–370. doi: 10.1146/annurev.ge.23.120189.002005. [DOI] [PubMed] [Google Scholar]
- 28.Bulmer MG. Maintenance of genetic variability by mutation-selection balance: a child’s guide through the jungle. Genome. 1989;31:761–767. [Google Scholar]
- 29.Charlesworth B. Fisher, Medawar, Hamilton and the evolution of aging. Genetics. 2000;156:927–931. doi: 10.1093/genetics/156.3.927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lynch M. Rate, molecular spectrum, and consequences of human mutation. Proc Natl Acad Sci USA. 2010;107:961–968. doi: 10.1073/pnas.0912629107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hartl DL, Clark AG. Principles of Population Genetics. 3. Sinauer Associates; Sunderland MA: 1998. [Google Scholar]
- 32.Cargill M, et al. Characterization of single-nucleotide polymorphisms in coding regions of human genes. Nat Genet. 1999;22:231–238. doi: 10.1038/10290. [DOI] [PubMed] [Google Scholar]
- 33.Kryukov GV, Pennacchio LA, Sunyaev SR. Most rare missense alleles are deleterious in humans: Implications for complex disease and association studies. Am JHum Genet. 2007;80:727–739. doi: 10.1086/513473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhu Q, et al. A genome-wide comparison of the functional properties of rare and common genetic variants in humans. Am J Hum Genet. 2011;88:458–468. doi: 10.1016/j.ajhg.2011.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lindblad-Toh K, et al. A high-resolution map of human evolutionary constraint using 29 mammals. Nature. 2011;478:476–482. doi: 10.1038/nature10530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wray GA. The evolutionary significance of cis-regulatory mutations. Nat Rev Genet. 8:206–216. doi: 10.1038/nrg2063. [DOI] [PubMed] [Google Scholar]
- 37.Montgomery SB, Lappalainen T, Gutierrez-Arcelus M, Dermitzakis ET. Rare and common regulatory variation in population-scale sequenced human genomes. PLoS Genet. 2011;7:e1002144. doi: 10.1371/journal.pgen.1002144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chorley BN, et al. Discovery and verification of functional single nucleotide polymorphisms in regulatory genomic regions: current and developing technologies. Mutat Res. 2008;659:147–157. doi: 10.1016/j.mrrev.2008.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Goldstein JL, Brown MS. The LDL Receptor locus and the genetics of familial hypercholesterolemia. Annu Rev Genet. 1979;13:259–289. doi: 10.1146/annurev.ge.13.120179.001355. [DOI] [PubMed] [Google Scholar]
- 40.Weedon MN, Frayling TM. Insights on pathogenesis of type 2 diabetes from MODY genetics. Curr Diabetes Rep. 2007;7:131–138. doi: 10.1007/s11892-007-0022-6. [DOI] [PubMed] [Google Scholar]
- 41.Easton DF, et al. A systematic genetic assessment of 1,433 sequence variants of unknown clinical significance in the BRCA1 and BRCA2 breast cancer-predisposition genes. Am J Hum Genet. 2007;81:873–883. doi: 10.1086/521032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hamosh A, et al. Online Mendelian Inheritance in Man (OMIM), a knowledgebase of human genes and genetic disorders. Nucl Acids Res. 2002;30:52–55. doi: 10.1093/nar/30.1.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tarpey PS, et al. A systematic, large-scale resequencing screen of X-chromosome coding exons in mental retardation. Nat Genet. 2009;41:535–543. doi: 10.1038/ng.367. One of the first whole exome sequencing studies designed to detect rare variants of large effect. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.George J, et al. Two human MYD88 variants, S34Y and R98C, interfere with MyD88-IRAK4-Myddosome assembly. J Biol Chem. 2011;286:1341–1353. doi: 10.1074/jbc.M110.159996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.McCarroll SA, Altshuler DA. Copy-number variation and association studies of human disease. Nat Genet. 2007;39:S37–S42. doi: 10.1038/ng2080. [DOI] [PubMed] [Google Scholar]
- 46.Stefansson H, et al. Large recurrent microdeletions associated with schizophrenia. Nature. 2008;455:232–236. doi: 10.1038/nature07229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sebat J, et al. Strong association of de novo copy number mutations with autism. Science. 2007;316:445–449. doi: 10.1126/science.1138659. The first demonstration that rare copy number variants associate with psychiatric disease. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cook EH, Jr, Scherer SW. Copy-number variations associated with neuropsychiatric conditions. Nature. 2008;455:919–923. doi: 10.1038/nature07458. [DOI] [PubMed] [Google Scholar]
- 49.Davis EE, et al. TTC21B contributes both causal and modifying alleles across the ciliopathy spectrum. Nat Genet. 2011;43:189–196. doi: 10.1038/ng.756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dickson SP, Wang K, Krantz I, Hakonarson H, Goldstein DB. Rare variants create synthetic genome-wide associations. PLoS Biol. 2010;8:e1000294. doi: 10.1371/journal.pbio.1000294. The argument that common variant associations may be due to linkage disequilibrium with rare variants. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Anderson CA, Soranzo N, Barrett JC, Zeggini E. Synthetic associations are unlikely to account for many common disease genome-wide association signals. PLoS Biology. 2011;9:e1000580. doi: 10.1371/journal.pbio.1000580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Goldstein DB. The importance of synthetic associations will only be resolved empirically. PLoS Biology. 2011;9:e1001008. doi: 10.1371/journal.pbio.1001008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Park J-H, et al. Distribution of allele frequencies and effect sizes and their inter-relationships for common genetic susceptibility variants. Proc Natl Acad Sci USA. 2011;108:18026–18031. doi: 10.1073/pnas.1114759108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ruderfer DM, et al. A family-based study of common polygenic variation and risk of schizophrenia. Mol Psych. 2011;16:887–888. doi: 10.1038/mp.2011.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Risch N. Linkage strategies for genetically complex traits: I. Multilocus models. Am J Hum Genet. 1990;46:222–228. [PMC free article] [PubMed] [Google Scholar]
- 56.Slatkin M. Genotype-specific risks as indicators of the genetic architecture of complex diseases. Am J Hum Genet. 2008;83:120–126. doi: 10.1016/j.ajhg.2008.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hemminki K, Bermejo JL. The ‘common disease-common variant’ hypothesis and familial risks. PLoS One. 2011;3:e2504. doi: 10.1371/journal.pone.0002504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Slatkin M. Exchangeable models of complex disease inheritance. Genetics. 2008;179:2253–2261. doi: 10.1534/genetics.107.077719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Moore JH, Williams SM. Epistasis and its implications for personal genetics. Am J Hum Genet. 2009;85:309–320. doi: 10.1016/j.ajhg.2009.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Amutha A, et al. Clinical profile of diabetes in the young seen between 1992 and 2009 at a specialist diabetes centre in south India. Prim Care Diabetes. 2011;5:223–229. doi: 10.1016/j.pcd.2011.04.003. [DOI] [PubMed] [Google Scholar]
- 61.Chan JC, et al. Diabetes in Asia: epidemiology, risk factors, and pathophysiology. JAMA. 2009;301:2129–2140. doi: 10.1001/jama.2009.726. [DOI] [PubMed] [Google Scholar]
- 62.Saha S, Chant D, Welham J, McGrath J. A systematic review of the prevalence of schizophrenia. PLoS Med. 2005;2:e141. doi: 10.1371/journal.pmed.0020141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Malaspina D, et al. Advancing paternal age and the risk of schizophrenia. Arch Gen Psych. 58:361–367. doi: 10.1001/archpsyc.58.4.361. [DOI] [PubMed] [Google Scholar]
- 64.Lopez-Castroman J, et al. Differences in maternal and paternal age between schizophrenia and other psychiatric disorders. Schizophr Res. 2010;116:184–190. doi: 10.1016/j.schres.2009.11.006. [DOI] [PubMed] [Google Scholar]
- 65.Waters KM, et al. Consistent association of type 2 diabetes risk variants found in Europeans in diverse racial and ethnic groups. PLoS Genet. 2010;6:e1001078. doi: 10.1371/journal.pgen.1001078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Shriner D, et al. Transferability and fine-mapping of genome-wide associated loci for adult height across human populations. PLoS One. 2009;4:e8398. doi: 10.1371/journal.pone.0008398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sim X, et al. Transferability of type 2 diabetes implicated loci in multi-ethnic cohorts from Southeast Asia. PLoS Genet. 2011;7:e1001363. doi: 10.1371/journal.pgen.1001363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Waters KM, et al. Generalizability of associations from prostate cancer genome-wide association studies in multiple populations. Cancer Epidemiol Biomarkers Rev. 2009;18:1285–1289. doi: 10.1158/1055-9965.EPI-08-1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gibson G. Decanalization and the origins of complex disease. Nat Rev Genet. 2009;10:134–140. doi: 10.1038/nrg2502. [DOI] [PubMed] [Google Scholar]
- 70.Schork NJ. Genome partitioning and whole genome analysis. Adv Genet. 2001;42:299–322. doi: 10.1016/s0065-2660(01)42030-x. [DOI] [PubMed] [Google Scholar]
- 71.Yang J, et al. Common SNPs explain a large proportion of the heritability for human height. Nat Genet. 42:565–569. doi: 10.1038/ng.608. Introduction of a multivariate approach to capturing the effects ofcommon variant associations genome-wide. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Goddard ME, Hayes BJ. Genomic selection. J Animal Breed Genet. 2007;124:323–330. doi: 10.1111/j.1439-0388.2007.00702.x. [DOI] [PubMed] [Google Scholar]
- 73.Purcell SM, et al. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature. 2009;460:748–752. doi: 10.1038/nature08185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.So H-C, Li M, Sham PC. Uncovering the total heritability explained by all true susceptibility variants in a genome-wide association study. Genet Epidemiol. 2011;35:447–456. doi: 10.1002/gepi.20593. [DOI] [PubMed] [Google Scholar]
- 75.Yang J, et al. Genome partitioning of genetic variation for complex traits using common SNPs. Nat Genet. 2011;43:519–525. doi: 10.1038/ng.823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Davies G, et al. Genome-wide association studies establish that human intelligence is highly heritable and polygenic. Mol Psych. 2011;16:996–1005. doi: 10.1038/mp.2011.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Falconer DS. Introduction to Quantitative Genetics. Chapter 18. Longman; New York: 1981. [Google Scholar]
- 78.Cannon TD, Keller MC. Endophenotypes in the genetic analysis of mental disorders. Annu Rev Clin Psychol. 2006;2:267–290. doi: 10.1146/annurev.clinpsy.2.022305.095232. [DOI] [PubMed] [Google Scholar]
- 79.Kendler KS, Meale MC. Endophenotype: a conceptual analysis. Mol Psych. 2010;15:789–797. doi: 10.1038/mp.2010.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kilpeläinen TO, et al. Genetic variation near IRS1 associates with reduced adiposity and an impaired metabolic profile. Nat Genet. 2011;43:753–760. doi: 10.1038/ng.866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bis JC, et al. Meta-analysis of genome-wide association studies from the CHARGE consortium identifies common variants associated with carotid intima media thickness and plaque. Nat Genet. 2011;43:940–947. doi: 10.1038/ng.920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Speliotes EK, et al. Genome-wide association analysis identifies variants associated with nonalcoholic fatty liver disease that have distinct effects on metabolic traits. PLoS Genet. 2011;7:e1001324. doi: 10.1371/journal.pgen.1001324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Musunuru K, et al. From noncoding variant to phenotype via SORT1 at the 1p13 cholesterol locus. Nature. 2010;466:714–719. doi: 10.1038/nature09266. Important case study of how to go from association study to molecular function of a specific variant. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wang Y, et al. Whole-genome association study identifies STK39 as a hypertension susceptibility gene. Proc Natl Acad Sci USA. 2009;106:226–231. doi: 10.1073/pnas.0808358106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Bertram L, Lill CM, Tanzi RE. The genetics of Alzheimer Disease: back to the future. Neuron. 2010;68:270–281. doi: 10.1016/j.neuron.2010.10.013. [DOI] [PubMed] [Google Scholar]
- 86.Cookson W, et al. Mapping complex disease traits with global gene expression. Nat Rev Genet. 2009;10:184–194. doi: 10.1038/nrg2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Majewski J, Pastinen T. The study of eQTL variations by RNA-Seq: from SNPs to phenotypes. Trends Genet. 2011;27:72–29. doi: 10.1016/j.tig.2010.10.006. [DOI] [PubMed] [Google Scholar]
- 88.Lalonde E, et al. RNA sequencing reveals the role of splicing polymorphisms in regulating human gene expression. Genome Res. 2011;21:545–554. doi: 10.1101/gr.111211.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Alizadeh AA, et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature. 2000;403:503–511. doi: 10.1038/35000501. [DOI] [PubMed] [Google Scholar]
- 90.Slavov N, Dawson KA. Correlation signature of the macroscopic states of the gene regulatory network in cancer. Proc Natl Acad Sci USA. 2009;106:4079–4084. doi: 10.1073/pnas.0810803106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Voineagu I, et al. Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature. 2011;474:380–384. doi: 10.1038/nature10110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Chaussabel D, et al. A modular analysis framework for blood genomics studies: application to systemic lupus erythematosus. Immunity. 2008;29:150–164. doi: 10.1016/j.immuni.2008.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Gibson G, Dworkin IM. Uncovering cryptic genetic variation. Nat Rev Genet. 5:681–690. doi: 10.1038/nrg1426. [DOI] [PubMed] [Google Scholar]
- 94.Aylor DL, et al. Genetic analysis of complex traits in the emerging Collaborative Cross. Genome Res. 2011;21:1213–12122. doi: 10.1101/gr.111310.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Philip VM, et al. Genetic analysis in the Collaborative Cross breeding population. Genome Res. 2011;21:1223–1238. doi: 10.1101/gr.113886.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Macdonald SJ, Long AD. Joint estimates of quantitative trait locus effect and frequency using synthetic recombinant populations of Drosophila melanogaster. Genetics. 2007;176:1261–1281. doi: 10.1534/genetics.106.069641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Burke MK, et al. Genome-wide analysis of a long-term evolution experiment with Drosophila. Nature. 2010;467:587–590. doi: 10.1038/nature09352. Evolve-and-resequence strategy to demonstrate the pervasive polygenic basis of complex traits. [DOI] [PubMed] [Google Scholar]
- 98.Turner TL, et al. Population-based resequencing of experimentally evolved populations reveals the genetic basis of body size variation in Drosophila melanogaster. PLoS Genet. 2011;7:e1001336. doi: 10.1371/journal.pgen.1001336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Manolio TA, Brooks LD, Collins FS. A HapMap harvest of insights into the genetics of common disease. J Clin Invest. 2008;118:1590–1605. doi: 10.1172/JCI34772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Weedon MN, et al. Genome-wide association analysis identifies 20 loci that influence adult height. Nat Genet. 2008;40:575–583. doi: 10.1038/ng.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Xu S. Theoretical basis of the Beavis effect. Genetics. 2003;165:2259–2268. doi: 10.1093/genetics/165.4.2259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Zhong R, Prentice RL. Correcting “winner’s curse” in odds ratios from genome-wide association findings for major complex human diseases. Genet Epidemiol. 2010;34:78–91. doi: 10.1002/gepi.20437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Pasyukova EG, Vieira C, Mackay TFC. Deficiency mapping of quantitative trait loci affecting longevity in Drosophila melanogaster. Genetics. 2000;156:1129–1146. doi: 10.1093/genetics/156.3.1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Fowler JH, Settle JE, Christakis NA. Correlated genotypes in friendship networks. Proc Natl Acad Sci USA. 2011;108:1993–1997. doi: 10.1073/pnas.1011687108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Jelenkovic A, Poveda A, Susanne C, Rebato E. Contribution of genetics and environment to craniofacial anthropometric phenotypes in Belgian nuclear families. Hum Biol. 2008;80:637–654. doi: 10.3378/1534-6617-80.6.637. [DOI] [PubMed] [Google Scholar]
- 106.Rzhetsky A, Wajngurt D, Park N, Zheng T. Probing genetic overlap among complex human phenotypes. Proc Natl Acad Sci USA. 2007;104:11694–11699. doi: 10.1073/pnas.0704820104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Ashley EA, et al. Clinical assessment incorporating a personal genome. Lancet. 2010;375:1525–1535. doi: 10.1016/S0140-6736(10)60452-7. Develops a strategy for integrating whole genome sequence and environmental exposure information to assess personal risk of disease. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Hamza TH, et al. Genome-wide gene-environment study identifies glutamate receptor gene GRIN2A as a Parkinson’s disease modifier gene via interaction with coffee. PLoS Genet. 2011;7:e1002237. doi: 10.1371/journal.pgen.1002237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Bauer RC, Stylianou IM, Rader DJ. Functional validation of new pathways in lipoprotein metabolism identified by human genetics. Curr Opin Lipidol. 2011;22:123–128. doi: 10.1097/MOL.0b013e32834469b3. [DOI] [PubMed] [Google Scholar]
- 110.Fellay J, et al. ITPA gene variants protect against anemia in patients treated for chronic hepatitis C. Nature. 2010;464:405–408. doi: 10.1038/nature08825. [DOI] [PubMed] [Google Scholar]
- 111.Schork N, Murray SS, Frazer K, Topol EJ. Common vs. rare allele hypotheses for complex disease. Curr Opin Genet Dev. 2009;19:212–219. doi: 10.1016/j.gde.2009.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Stenson PD, et al. The Human Gene Mutation Database: 2008 update. Genome Med. 2009;1:13. doi: 10.1186/gm13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Nejentsev S, Walker N, Riches D, Egholm M, Todd JA. Rare variants of IFIH1, a gene implicated in antiviral responses, protect against type 1 diabetes. Science. 2009;324:387–389. doi: 10.1126/science.1167728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Momozawa Y, et al. Resequencing of positional candidates identifies low frequency IL23R coding variants protecting against inflammatory bowel disease. Nat Genet. 2011;43:43–47. doi: 10.1038/ng.733. [DOI] [PubMed] [Google Scholar]
- 115.Rivas MA, et al. Deep resequencing of GWAS loci identifies independent rare variants associated with inflammatory bowel disease. Nat Genet. 2011;43:1066–1073. doi: 10.1038/ng.952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Johansen CT, Wang J, Lanktree MB, Cao H, McIntyre AD, et al. Excess of rare variants in genes identified by genome-wide association study of hypertriglyceridemia. Nat Genet. 2010;42:684–687. doi: 10.1038/ng.628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Ng SB, et al. Targeted capture and massively parallel sequencing of 12 human exomes. Nature. 2009;461:272–276. doi: 10.1038/nature08250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Lupski JR, Belmont JW, Boerwinkle E, Gibbs RA. Clan genomics and the complex architecture of human disease. Cell. 147:32–43. doi: 10.1016/j.cell.2011.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Bansal V, Libiger O, Torkamani A, Schork NJ. Statistical analysis strategies for association studies involving rare variants. Nat Rev Genet. 2010;11:773–785. doi: 10.1038/nrg2867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Rendel JM. Canalization and gene control. Academic Press; New York: 1967. [Google Scholar]
- 121.Bhattacharjee S, et al. Using principal components of genetic variation for robust and powerful detection of gene-gene interactions in case-control and case-only studies. Am J Hum Genet. 2010;86:331–342. doi: 10.1016/j.ajhg.2010.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Slatkin M. Epigenetic inheritance and the missing heritability problem. Genetics. 2009;182:845–850. doi: 10.1534/genetics.109.102798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Tong AH, et al. Systematic genetic analysis with ordered arrays of yeast deletion mutants. Science. 2001;294:2364–2368. doi: 10.1126/science.1065810. [DOI] [PubMed] [Google Scholar]
- 124.Costanzo M, et al. The genetic landscape of a cell. Science. 2010;327:425–431. doi: 10.1126/science.1180823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Lucchesi JC. Synthetic lethality and semi-lethality among functionally related mutants of Drosophila melanogaster. Genetics. 1968;59:37–44. doi: 10.1093/genetics/59.1.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Lehner B, et al. Systematic mapping of genetic interactions in Caenorhabditis elegans identifies common modifiers of diverse signaling program. Nat Genet. 2006;38:896–903. doi: 10.1038/ng1844. [DOI] [PubMed] [Google Scholar]
- 127.Duggal P, Gillanders PM, Holmes TN, Bailey-Wilson JE. Establishing an adjusted p-value threshold to control the family-wide type 1 error in genome-wide association studies. BMC Genomics. 2008;9:516. doi: 10.1186/1471-2164-9-516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Li Y, Willer C, Sanna S, Abecasis G. Genotype imputation. Annu Rev Genom Hum Genet. 2009;10:387–406. doi: 10.1146/annurev.genom.9.081307.164242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Shea J, et al. Comparing strategies to fine-map the association of common SNPs at chromosome 9p21 with type 2 diabetes and myocardial infarction. Nat Genet. 2011;43:801–805. doi: 10.1038/ng.871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Kacser H, Burns JA. The control of flux. Symp Soc Exp Biol. 1973;27:65–104. A theoretical argument for the recessivity of naturally occurring mutations affecting metabolism. [PubMed] [Google Scholar]