

Abstract
G protein–coupled receptor (GPCR) signaling in osteoblasts (OBs) is an important regulator of bone formation. We previously described a mouse model expressing Rs1, an engineered constitutively active Gs-coupled GPCR, under the control of the 2.3-kb Col I promoter. These mice showed a dramatic age-dependent increase in trabecular bone of femurs. Here, we further evaluated the effects of enhanced Gs signaling in OBs on intramembranous bone formation by examining calvariae of 1- and 9-week-old Col1(2.3)/Rs1 mice and characterized the in vivo gene expression specifically occurring in osteoblasts with activated Gs G protein–coupled receptor signaling, at the cellular level rather than in a whole bone. Rs1 calvariae displayed a dramatic increase in bone volume with partial loss of cortical structure. By immunohistochemistry, Osterix was detected in cells throughout the inter-trabecular space while Osteocalcin was expressed predominantly in cells along bone surfaces, suggesting the role of paracrine mediators secreted from OBs driven by 2.3-kb Col I promoter could influence early OB commitment, differentiation, and/or proliferation. Gene expression analysis of calvarial OBs revealed that genes affected by Rs1 signaling include those encoding proteins important for cell differentiation, cytokines and growth factors, angiogenesis, coagulation, and energy metabolism. The set of Gs-GPCRs and other GPCRs that may contribute to the observed skeletal phenotype and candidate paracrine mediators of the effect of Gs signaling in OBs were also determined. Our results identify novel detailed in vivo cellular changes of the anabolic response of the skeleton to Gs signaling in mature OBs.
Keywords: G protein coupled receptor (GPCR), Osteoblasts (OBs), Mouse model, Gene expression analysis, Paracrine mediators, Intramembranous bone
Introduction
Osteoblasts (OBs) play a key role in control of skeletal homeostasis by influencing the initiation and extent of bone resorption and bone formation through complex mechanisms that are only partially understood. A number of factors contribute to this complexity. Fortunately, considerable progress has been made over the past few decades in defining the molecular events associated with the transition of committed osteoprogenitors to fully differentiated, post-proliferative OBs and osteocytes [1,2]. In particular, the roles of transcription factors, such as Runx2 and Osterix, in the progression of OB differentiation are well documented [3,4]. Much less clear are the roles of extrinsic factors, such as hormones, cytokines, and other elements of the skeletal microenvironment, in regulating OB commitment, differentiation, and function. Moreover, very little is known about how such extrinsic factors regulate OBs in different anatomic components.
One of the most important mechanisms for cellular response to extrinsic factors is through G protein signaling. G protein-coupled receptors (GPCRs) are highly relevant to OB differentiation and function, as evidenced by the anabolic skeletal response to parathyroid hormone (PTH) and prostaglandin E [5,6], two agents that act on GPCRs presented on OB lineage cells. Genetic studies in human and mice further support the key role played by G protein signaling [7–9]. We described a number of transgenic mouse models in which G protein coupled signaling has been manipulated in vivo in OBs that express the 2.3 kb-Col I promoter. These models include OB-specific expression of an engineered constitutively active Gs-coupled receptor, Rs1, and OB-specific expression of pertussis toxin (PTX) to block Gi signaling; both of which demonstrate an anabolic bone phenotype [10,11]. Mice expressing Rs1, showed an increase in bone accrual within the skull and in femur size, assessed by the whole body microCT analysis, with a dramatic age-dependent increase in trabecular bone with features resembling fibrous dysplasia. At 9 weeks of age, the male and female mutant mice showed dramatic increases (380%) in whole-body areal bone mineral density, as determined by dual-energy x-ray absorptiometry scanning. Histological assessment of femoral bones indicated that there was an increase in OB lineage cells, especially immature OBs, indicated by an expansion of cells expressing early OB markers, Runx2 and Osterix. Increased osteoclast activity was suggested by the large number of tartrate-resistant acid phosphatase (TRAP)-positive regions adjacent to the trabeculi within the lesions. Bone tissue quality; mineralization, composition, and maturity, of calvariae and femurs in Rs1 mice were also assessed by the complementary techniques of fourier transform infrared (FTIR) spectroscopy and synchrotron radiation micro-computed tomography (SRμCT) [12]. We demonstrated that mineral-to-matrix ratio and cross-link ratio were significantly lower in 6- and 15-week mutant bones. No differences in FTIR spectroscopic parameters were detected between the two anatomic sites despite the different bone-formation processes (endochondral vs. intramembranous). Tissue mineral density was also significantly lower in 3- and 9-week transgenic femoral diaphyses. The results indicate that continuous Gs activation in mature OBs lead to deposition of immature bone tissue with reduced mineralization.
The ability of Rs1 signaling in mature OBs to drive expansion of immature OBs could be mediated by paracrine factors, secreted from mature OBs that influence early OB commitment, differentiation, and/or proliferation. However, little is known about the in vivo cellular basis of the skeletal changes in response to enhanced Gs signaling in mature OBs.
In this study, we determined how Gs signaling in mature OBs affects bone formation by examining the Rs1 calvarial bone phenotype at 1 and 9 weeks of age and investigated the effect of enhanced OB Gs signaling on the OB transcriptome by examining alterations in gene expression in vivo in OBs from calvariae of 1-week-old Rs1 mice, compared to controls. The functionally related, differentially expressed genes, the Gs- and Gi-GPCRs, and candidate paracrine mediators that may contribute to the observed skeletal phenotype of the effect of Gs signaling in OBs were determined.
Methods
Transgenic mice
To examine the influence of Gs signaling in OBs, we used a mutated version of the Gs-coupled 5HT-4 serotonin receptor (Rs1) that has constitutive Gs signaling activity [13], together with the tetracycline-regulated system (Tet-off) to regulate the expression of Rs1 in vivo. The Col1(2.3)-tTA/TetO-Rs1 double transgenic mice (abbreviated Col1(2.3)/Rs1) were generated by crossing mice carrying the heterozygous TetO-Rs1 transgene with mice carrying the homozygous Col1(2.3)-tTA transgene. Endochondral bones of these mutant mice were well characterized as described [10,14,15].
In this study, we utilized green fluorescent protein (GFP)-based reporters to identify Rs1 expressing cells for investigating OB transcriptome analysis. We co-expressed a histone-GFP marker in OBs in vivo alone (abbreviated Col1(2.3)/GFP) or with Rs1 in triple transgenic mice (abbreviated Col1(2.3)/GFP/Rs1). This was accomplished by generating mice harboring a TetO-histone-GFP gene (mouse line Tg (TetO-HIST1H2BJ/GFP) 47Efu/J, Jackson Laboratory) with a TetO-Rs1 gene. Double transgenic mice were then crossed with mice homozygous for the 2.3 Col1-tTA genes to examine the skeletal effects of Gs signaling in mature OBs. Both transgenes driven by TetO were expressed in OBs only when the 2.3 Col1-tTA transgene was present. In this approach, 25% of the progeny were controls (express only histone-GFP) and 25% were experimental (express both histone-GFP and Rs1). All animals were maintained in the FVB/N background. Mice were on regular chow (LabDiet 5053; PMI Nutrition, St. Louise, MO) without doxycycline administration to allow transgene expression in experimental mice since conception. All protocols were approved by the Institutional Animal Care and Use Committee of the San Francisco Veterans Affairs Medical Center.
Calvarial bone histology
Calvariae collected for hematoxylin/eosin (H&E) staining and immunohistochemistry were fixed in 10% neutral buffered formalin (NBF; Fisher Scientific, Pittsburgh, PA) for 24 hours and stored at 4°C in 70% ethanol. Bones were decalcified in 10% EDTA at 4°C for 2–3 days before paraffin embedding, sectioning, and H&E staining. Assessment of calvarial bone was performed on 10-μm-thick sections at the midpoint of the parietal part.
Bone immunohistochemistry
Osteocalcin and Osterix were detected on formalin-fixed, paraffin-embedded sections (10 μm thickness) with primary antibodies from Takara Bio (Cat#M173) and AbCam (Cat#ab22552) as described [10]. Since Rs1 bears an N-terminal FLAG tag, the detection of Rs1 expressing OBs was done on alcohol-fixed, paraffin-embedded bone sections with a M1 FLAG primary antibody (F3040; Sigma-Aldrich) in M.O.M. diluents (dilute protein concentrate stock solution 1:13.5 in Tris-buffered saline). Detection of CD31, the endothelial cell marker, was performed using an Anti-Ig HRP detection kit (BD Pharmingen) following the manufacturer’s protocol.
Static and dynamic histomorphometry
Calvariae from 1- and 9-week-old mice were isolated at the time of euthanasia and fixed in 10% NBF for 1–2 days before embedding in methyl methacrylate and sectioning with Jung 2065 and 2165 microtomes (Leica, Bannockburn, IL). Sectioned bones were processed for Von Kossa staining as described [16]. For dynamic histomorphometry, 9-week-old mice were injected with 20 mg/kg of calcein (Sigma-Aldrich) 21 and 7 days before euthanasia and with 15 mg/kg of demeclocycline (Sigma-Aldrich) 2 days before euthanasia. Mice were euthanized at 9 weeks, and calvariae were isolated, fixed in 10% NBF, and stored in 70% ethanol. The undecalcified bone samples were embedded in methyl methacrylate. Histological assessment of calvarial bone was performed on 10-μm-thick sections at the midpoint of the parietal part (Fig. 1A). Mosaic-tiled images were acquired at ×20 magnification with a Zeiss Axioplan Imager M1 microscope (Carl Zeiss MicroImaging, Thornwood, NY) fitted with a motorized stage. The tiled images were stitched and converted to a single image with the Axiovision software (Carl Zeiss MicroImaging) before histomorphometry analyses.
Figure 1.

(A) Gross calvarial specimens from 1- and 9-week-old mice. White box represents the region of interest for histological assessment of calvariae. (B) H&E and VK images of 1-and 9-week-old Col1(2.3)/GFP/Rs1 mice compared to controls, Col1(2.3)/GFP mice. Mutant calvariae had a dramatic increase in trabecular bone volume with a distorted cortical structure. (C) Fluorescence imaging of calvariae showed a disorganized mineralization in the 9-week-old mutant calvariae. Red arrow represents the first (calcein; green) and second labels (demeclocycline; orange). (Scale bar, 50μm) Ca, central area; Pa, peripheral area; tb, trabeculi; bm, bone marrow
Separation of cell populations
Calvariae with OB-specific expression of histone-GFP either alone or together with Rs1 under control of the 2.3kb-Col I promoter were isolated from 1-week-old transgenic mice, pooled and divided into three groups according to their genotype (Col1(2.3) and Col1(2.3)/Rs1; Col1(2.3)/GFP; or Col1(2.3)/GFP/Rs1). After removal of the calvarial sutures, calvarial tissues were washed once with PBS and then subjected to four sequential, 30-minute digestions in an enzymatic mixture (1.5 U/ml collagenase P (Roche), 0.05% trypsin and 0.25 mM EDTA in PBS) at 37°C on a rocking platform (0.5–1 ml of mixture per calvaria). Cell fractions 2–4 were collected, pooled and resuspended in Dulbecco’s modified Eagle’s medium (DMEM; Life Technology) containing 10% fetal calf serum (FCS) and centrifuged. Then, cells were resuspended in 0.5–1 ml of 2% FCS in PBS and filtered through a 70-μm cell strainer. Individual cell populations were sorted based on GFP expression and fluorescent activated cell sorting (FACS) (BD FACSAria; BD Biosciences) with a 100-μm nozzle at the Cell Sorting Core at the San Francisco VA Medical Center. Cells from non-GFP-expressing mice (Col1(2.3) plus Col1(2.3)/Rs1) were used as controls to preset the sorting gate. Sorted cells were collected into 30% FCS in DMEM. A fraction of the sorted cell population (5×103) was used for FACS reanalysis by the same gating strategy in the sorted procedure to assure the maximum purity. Cell suspensions were kept cold during the entire sorting process to minimize changes in gene expression.
RNA isolation and microarray analysis
Total RNA from FACS-sorted cell populations (GFP-expressing cells from Col1(2.3)/GFP and Col1(2.3)/GFP/Rs1 group) were isolated immediately after sorting and purified using the Arcturus PicoPure RNA isolation kit (Applied Biosystems, Carlsbad, CA), followed by DNase treatment with the RNase-Free DNase Set (Qiagen, Valencia, CA), according to manufacturer’s instructions. The quantity and quality of total RNA were assessed by NanoDrop ND-1000 Spectrophotometer and Agilent 2100 Bioanalyzer (Agilent Technologies). The 28S/18S ratios of the RNA were in the range of 1.8–2.1, and the RNA Integrity Numbers were in the range of 8.8–10. Reverse transcription and amplification of isolated RNA into cDNA were performed using the NuGEN FFPE WTA kit (NuGEN, San Carlos, CA). The integrity of resultant cDNA was assessed using the Agilent 2100 Bioanalyzer and individual samples were further processed and hybridized to Affymetrix Mouse Gene 1.0 ST arrays (Affymetrix, Cleveland, OH) before scanning, according to the protocol in WT Sense Target Labeling Assay Manual from Affymetrix (Version 4; FS450_0007) at the UCSF Gladstone Genomics Core Facility. Expression analysis was performed on three separate OB preparations of RNA from each genotype. Data were normalized using Guanine Cytosine Robust Multi-Array Analysis (GC-RMA). Bayesian statistical analysis was carried out using Linear Models of Microarrays (LIMMA) [17] to identify statistically significant differentially expressed genes between Col1(2.3)/GFP and Col1(2.3)/GFP/Rs1. Moderated t-statistics and the associated p-values were calculated and p-values less than 0.01 were considered to be statistically significant. Comparison groups were annotated with statistically significant Gene Ontology (GO) term overrepresentation using the GO-Elite software packages [18]. Microarray data have been submitted to the Gene Expression Omnibus (NCBI, NIH) database (series accession number GSE57104; http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?token=idwzccomrjellqz&acc=GSE57104).
Quantitative real-time PCR
For gene expression analysis by quantitative real-time PCR (qPCR), we compared selected genes that were differentially expressed in Col1(2.3)/GFP/Rs1 to Col1(2.3)/GFP by microarray analysis. Gene expression was quantified by SYBR Green I-based qPCR utilizing SYBR Green PCR Master Mix (part#4309155, Applied Biosystems). Primers were synthesized by Elim Biopharmaceuticals, Inc (Supplemental Table 1). qPCR was carried out in ABI Prism 7300 real-time thermocycler (Applied Biosystems). The results were analyzed using the Sequence Detection System software (SDS) supplied with the thermocycler. All reactions were performed in triplicates from the different experiments, and the expression of target genes was displayed normalized to GAPDH. Expressed GPCRs were identified using the Taqman® real time Mouse GPCR Array (part#4378703, Applied Biosystems), according to the manufacturer’s instructions. Array plates were run on the ABI Prism 7900HT system, and the data were analyzed using the SDS 2.3 and RQ Manager 1.2 software provided by Applied Biosystems. Quantitative analysis of GPCR expression, including housekeeping gene (18S), was expressed as cycle threshold (Ct) values. Average Ct values <30 on a TaqMan Array considered positive reactions reflecting detectable cDNA target copies in the sample. So we selected a cutoff of Ct= 30. Data shown were from the average of three independent samples.
Bone marrow stromal cells (BMSCs) cultures
Primary mouse BMSCs were isolated from the femurs and tibiae of 10–12-week old WT mice. The cells were collected in primary culture medium (PCM) consisting of ∞-Modification of Eagle’s medium, 10% fetal bovine serum, 100U/ml penicillin, 100μg/ml streptomycin, and 0.25μg/ml Fungizone (Gibco Cell Culture), and plated onto 10-cm cell culture dishes at a density of 7×106 cells/dish. Cells were incubated at 37°C with 5% CO2 and maintained undisturbed for five days to allow for cell attachment. After that, PCM was removed along with all non-adherent cells and replaced with secondary osteogenic differentiation medium (SDM) (PCM containing 50μg/ml ascorbic acid and 3mM β-glycerolphosphate) to initiate OB differentiation. Thereafter, SDM was replaced every two or three days. Human PTH [1–34] (pPTH; #H-4835, Bachem) and forskolin (#F6886, Sigma) were prepared by dissolving in acetic acid (4.07μM final concentration) and ethanol (0.82% final concentration), respectively. Cells were exposed to 10−7M hPTH or forskolin at final concentration of 0.05 mM at day 28 for 24 hours. Total RNA from BMSCs cultures was isolated using PureLink Micro-to-Midi total RNA purification system (Cat#12183-018, Ambion) and further purified using RNeasy Mini Kit (Cat#74104, Qiagen). Reverse transcription was carried out using TaqMan Reverse Transcription Reagents (Part#N808-0234, Applied Biosystems). Fibroblast growth factor 9 (Fgf9) gene expression was quantified as described in qPCR section. The results of the experiments were confirmed by repeating the experiment three times.
Mineralization
BMSCs cultures were performed in the absence or presence of murine Fgf9 (SRP4057, Sigma-Aldrich). BMSCs were isolated from WT mice, collected in PCM and plated onto 10-cm cell culture dishes at a density of 7×106 cells/dish. After incubating BMSCs in PCM for five days, the medium was removed along with all non-adherent cells and SCM was replaced and changed every two or three days. 5 ng/ml murine Fgf9 was added to the culture system from day 0 to day 20. To assess mineralization, two percent silver nitrate solution (Sigma-Aldrich) was added to cell culture dishes on day 20 for Von Kossa (VK) staining and UV-crosslinked for 10 minutes. Stained cultures were scanned and quantified using Improvision Openlab software version 5.0.2. The results of the experiment were confirmed by repeating the experiment three times.
Statistical analysis
All qPCR data were evaluated using a two-tailed Student’s t tests assuming equal variance. Data were presented as the mean ± SD. Statistical significance was taken as p<0.05.
Results
Bone histomorphometry
Mice expressing Rs1 displayed notable asymmetric thickening of the calvariae starting at one week of age, and the observed phenotypes became more severe with age (Fig. 1A). By histological assessment, Rs1 calvariae had a dramatic increase in trabecular bone volume with a distorted cortical structure. High-magnification images of 9-week-old Rs1 calvariae revealed a large number of cells with stromal-type morphology between trabeculae with many appearing stacked on and near the trabecular surfaces. Bone marrow elements appeared to be scattered in small areas between the trabecular bones (Fig. 1B). Bone formation rates and patterns in 9-week-old samples were examined by dynamic histomorphometry. However, we could not quantify bone formation rate in the mutant mice due to the disorganized nature of bone formation, which displayed a diffuse labeling pattern consistent with the rapid bone formation (Fig. 1C).
Bone immunohistochemistry
Osterix expression was readily detected by immunohistochemistry in cells throughout the intertrabecular space in Rs1 calvariae, but not in controls (Fig. 2A). OBs that attached to cortical bone surfaces in control calvariae might be fully mature and express only low levels of Osterix. Osteocalcin was expressed predominantly in cells along bone surfaces in mutant and control mice (Fig. 2B). Immunohistochemistry with an antibody against the FLAG tag on Rs1 identified a significant number of cells within the Rs1 calvariae located along bone surfaces at the same location as Osteocalcin-expressing cells (Fig. 2C). They represented maturing OBs, based on the known expression patterns of the 2.3-kb Col I promoter fragment [19] and on the abundant Osteocalcin in the bone lesions. Endothelial cells, identified by CD31 expression, were broadly distributed within the expanded calvarial tissue of Col1(2.3)/Rs1 mice (Fig. 2D). The immunohistochemistry results indicated that many of the uniform cells in the mutant bone lesions are in the OB lineage with an increase in normal-looking blood vessel structures.
Figure 2.
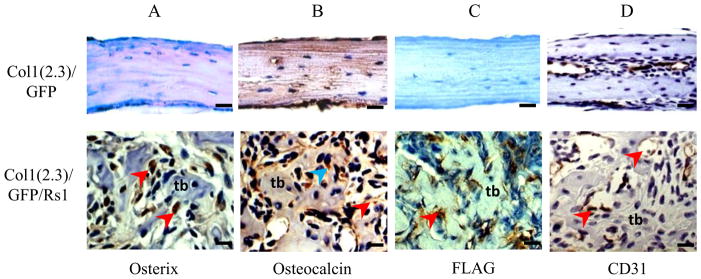
High-magnification immunohistochemistry of decalcified Col1(2.3)/GFP/Rs1 (central area) and control (whole area) calvariae from 9-week-old mice. (A) Osterix immunohistochemistry on formalin-fixed samples, demonstrating significant nuclear staining (brown; red arrowheads) by cells throughout the Col1(2.3)/GFP/Rs1 bony lesion mostly between the trabeculi. (B) Osteocalcin immunohistochemistry on formalin-fixed samples, demonstrating that cytoplasmic (brown; blue arrowhead) and extracellular (brown; red arrowhead) osteocalcin are present in the bone lesions, confirming the presence of mature osteoblasts. (C) FLAG immunohistochemistry on ethanol-fixed samples showed cytoplasmic staining of Rs1 expression only in Col1(2.3)/GFP/Rs1 calvarial sections (brown; red arrowhead). (D) CD31 immunohistochemistry on formalin-fixed samples, demonstrating diffuse positivity of blood vessels and sinusoids in the mutant clavaria (brown; red arrowheads). (Scale bar 100μm) tb, tabeculi
Isolation of GFP-expressing cell population
Cell populations obtained by enzymatic digestion of calvariae from 1-week-old double and triple transgenic GFP reporter (Col1(2.3)/GFP/Rs1 mice vs. Col1(2.3)/GFP mice) were FACS sorted for GFP expression to separate two cell populations (GFP-positive and GFP-negative cells) in each genotype. GFP-negative cells, representing various non-OB cell lineages mixed with osteoprogenitor and immature OBs, were clearly separated from the GFP-positive cell populations. Approximately 10% of the isolated cells are typically GFP positive. Sorted populations were 97–99% pure by FACS reanalysis (Fig. 3A).
Figure 3.

(A) FACS analysis of GFP expression of cells obtained by enzymatic digestion of neonatal calvariae isolated from transgenic mice. Cell populations were reanalyzed for purity. (B) qPCR analysis of osteoblast and osteoclast marker genes with RNA isolated from GFP negative population and GFP positive population from 3 Rs1 expression OBs and 3 controls after FACS sorting. Data were normalized to GAPDH RNA expression in the same sample. *P<0.01 (C) qPCR analysis with RNA isolated from GFP positive population from 3 Rs1 expression OBs and 3 controls after FACS sorting. Data were normalized to GAPDH RNA expression in the same sample. Rs1 expression was detected only in Rs1 group.
Identifying GPCRs expressed on OBs and genes differentially expressed in Rs1 mice
We compared the gene expression profiles in mature OBs from transgenic mice and controls from three biological replicates of each genotype. The identifiable cell populations were processed through RNA extraction, in vitro transcription, cDNA labeling and array hybridization. The Affymetrix Mouse Gene 1.0 ST Arrays were used to determine differences in gene expression. Each array contains a total of 26,166 coding transcripts. Significantly regulated genes were clustered using the GO Tree Machine, and functional analysis was done with GO-Elite software. We identified 358 differentially expressed transcripts if p ≤ 0.01 was set (moderate t-statistics). Small magnitudes of differential expression were detected, ranging from 0.26-fold to 4.63-fold changes in expression. Applying a threshold of a greater than 1.5-fold change in expression level revealed a filtered list of 133 genes, 114 of which were associated with functional annotations in the GO-Elite software (52 upregulated genes and 62 downregulated genes, Table 1).
Table 1.
Genes differentially expressed in Rs1 expressing OBs compared to controls
| Gene symbol | Gene name | Mean ratio (Rs1/control) | p-value |
|---|---|---|---|
| Growth factor | |||
| Figf | c-fos induced growth factor | 2.08 | 1.6×10−4 |
| Fgf9 | fibroblast growth factor 9 | 0.63 | 4.0×10−5 |
| Transcription factor and regulator | |||
| Dennd4a | DENN/MADD domain containing 4A | 1.99 | 1.1×10−3 |
| Ldb2 | LIM domain binding 2 | 1.79 | 6.6×10−3 |
| Hmgb3 | high mobility group box 3 | 1.72 | 8.3×10−3 |
| Mlx | MAX-like protein X | 1.61 | 9.0 ×10−5 |
| Asxl2 | additional sex combs like 2 (Drosophila) | 0.65 | 4.1×10−3 |
| Supt7l | suppressor of Ty 7 (S. cerevisiae)-like | 0.63 | 1.6×10−3 |
| Tceal5 | transcription elongation factor A (SII)-like 5 | 0.62 | 9.2×10−3 |
| Ddx1 | DEAD (Asp-Glu-Ala-Asp) box polypeptide 1 | 0.58 | 4.0×10−5 |
| Nfxl1 | nuclear transcription factor, X-box binding-like 1 | 0.56 | 9.6×10−4 |
| Zbtb20 | zinc finger and BTB domain containing 20 | 0.53 | 4.1×10−3 |
| Samd9l | sterile alpha motif domain containing 9-like | 0.44 | 5.1×10−3 |
| Bex1 | brain expressed gene 1 | 0.37 | 8.5×10−4 |
| Cell cycle regulator | |||
| Fbxo5 | F-box protein 5 | 1.97 | 8.4×10−3 |
| Tubb3 | tubulin, beta 3 | 1.93 | 1.3×10−3 |
| Cdc2a | cell division cycle 2 homolog A (S. pombe) | 1.84 | 1.0×10−3 |
| Lsm10 | U7 snRNP-specific Sm-like protein LSM10 | 1.69 | 5.0×10−3 |
| Bub1b | budding uninhibited by benzimidazoles 1 homolog, beta (S. cerevisiae) | 1.65 | 6.8×10−3 |
| Casc5 | cancer susceptibility candidate 5 | 1.57 | 7.4×10−3 |
| Prc1 | protein regulator of cytokinesis 1 | 1.56 | 3.2×10−3 |
| Anln | anillin, actin binding protein | 1.55 | 5.1×10−3 |
| Elovl1 | elongation of very long chain fatty acids (FEN1/Elo2, SUR4/Elo3, yeast)-like 1 | 1.54 | 3.5×10−3 |
| Ndc80 | NDC80 homolog, kinetochore complex component (S. cerevisiae) | 1.53 | 7.5×10−3 |
| Cdkn2b | cyclin-dependent kinase inhibitor 2B (p15, inhibits CDK4) | 1.53 | 3.5×10−3 |
| Cdca2 | cell division cycle associated 2 | 1.51 | 1.3×10−3 |
| Cspp1 | centrosome and spindle pole associated protein 1 | 0.26 | 3.0×10−3 |
| Regulation of apoptosis | |||
| Bard1 | BRCA1 associated RING domain 1 | 1.78 | 5.3×10−3 |
| Trp53inp1 | transformation related protein 53 inducible nuclear protein 1 | 0.61 | |
| Casp4 | caspase 4, apoptosis-related cysteine peptidase | 0.48 | 6.8×10−3 |
| Trim13 | tripartite motif-containing 13 | 0.44 | 9.9×10−3 |
| Clec2d | C-type lectin domain family 2, member | 0.43 | 7.8×10−3 |
| Casp1 | caspase 1 | 0.26 | 1.6×10−4 |
| Enzyme activity | |||
| Car8 | carbonic anhydrase 8 | 1.85 | 8.2×10−3 |
| Slpi | Secretory leukocyte peptidase inhibitor | 1.81 | 4.4×10−3 |
| Adat2 | adenosine deaminase, tRNA-specific 2, TAD2 homolog (S. cerevisiae) | 1.64 | 1.4×10−3 |
| Xdh | xanthine dehydrogenase | 1.62 | 9.3×10−3 |
| Sae1 | SUMO1 activating enzyme subunit 1 | 1.55 | 1.1×10−3 |
| Got1 | glutamate oxaloacetate transaminase 1, soluble | 1.52 | 5.0×10−5 |
| Ctdspl2 | CTD (carboxy-terminal domain, RNA polymerase II, polypeptide A) small phosphatase like 2 | 0.66 | 3.8×10−3 |
| Dub2a | deubiquitinating enzyme 2a | 0.64 | 4.4×10−3 |
| Car2 | carbonic anhydrase 2 | 0.62 | 8.7×10−3 |
| Gstm1 | glutathione S-transferase, mu 1 | 0.60 | 8.0×10−3 |
| Ppp1r12b | protein phosphatase 1, regulatory (inhibitor) subunit 12B | 0.48 | 5.9×10−3 |
| Lipid and carbohydrate metabolic process | |||
| Lipi | Lipase, member I | 4.63 | 9.0×10−5 |
| Idi1 | isopentenyl-diphosphate delta isomerase | 2.21 | 1.6×10−3 |
| Fabp3 | fatty acid binding protein 3, muscle and heart | 1.69 | 2.2×10−4 |
| Me1 | malic enzyme 1, NADP(+)-dependent, cytosolic | 1.60 | 7.7×10−3 |
| Gpr116 | G protein-coupled receptor 116 | 0.49 | 8.3×10−3 |
| Transporter | |||
| Slc35a1 | solute carrier family 35 (CMP-sialic acid transporter), member 1 | 1.83 | 1.8×10−3 |
| Efha1 | EF hand domain family A1 | 1.67 | 1.5×10−3 |
| Arfgef2 | ADP-ribosylation factor guanine nucleotide | 0.66 | 1.4×10−3 |
| Trappc2l | trafficking protein particle complex 2-like | 0.63 | 2.3×10−3 |
| Mfsd7b | major facilitator superfamily domain containing 7B | 0.53 | 2.2×10−3 |
| Atp6v1b2 | ATPase, H+ transporting, lysosomal V1 subunit B2 | 0.53 | 1.0×10−3 |
| Atp6v0d2 | ATPase, H+ transporting, lysosomal V0 subunit D2 | 0.26 | 6.7×10−3 |
| Kinase activity | |||
| Prkrip1 | Prkr interacting protein 1 (IL11 inducible) | 1.66 | 3.6×10−3 |
| Lmnb1 | lamin B1 | 1.54 | 1.8×10−4 |
| Fgd4 | FYVE, RhoGEF and PH domain containing 4 | 0.62 | 4.2×10−3 |
| Adrbk2 | adrenergic receptor kinase, beta 2 | 0.61 | 3.5×10−3 |
| Pip4k2a | phosphatidylinositol-5-phosphate 4-kinase, type II, alpha | 0.61 | 2.4×10−3 |
| Ptpn3 | protein tyrosine phosphatase, non-receptor type 3 | 0.44 | 4.2×10−3 |
| Protein binding | |||
| Mrpl17 | mitochondrial ribosomal protein L17 | 2.27 | 5.5×10−3 |
| Iqcb1 | IQ calmodulin-binding motif containing1 | 0.62 | 2.9×10−3 |
| Ppfia2 | protein tyrosine phosphatase, receptor type, f polypeptide (PTPRF), interacting protein (liprin), alpha 2 | 0.59 | 3.2×10−3 |
| Cytoskeleton | |||
| Dync2li1 | dynein cytoplasmic 2 light intermediate chain 1 | 1.71 | 1.0×10−3 |
| Plekhh2 | pleckstrin homology domain containing, family H (with MyTH4 domain) member2 | 0.64 | 3.0×10−3 |
| Syne2 | synaptic nuclear envelope 2 | 0.30 | 5.0×10−5 |
| Solute carrier | |||
| Slc38a4 | solute carrier family 38, member 4 | 1.91 | 3.3×10−3 |
| Slc25a19 | solute carrier family 25 (mitochondrial thiamine pyrophosphate carrier), member 19 | 0.58 | 5.7×10−3 |
| Slc5a3 | solute carrier family 5 (inositol transporters), member 3 | 0.50 | 2.4×10−4 |
| Immune system | |||
| Cd1d1 | CD1d1 antigen | 1.89 | 4.2×10−3 |
| Srgn | Serglycin | 0.48 | 3.9×10−3 |
| Organogenesis | |||
| Rbp4 | Retinol binding protein 4, plasma | 1.69 | 4.7×10−4 |
| Lmbr1 | limb region 1 | 0.53 | 7.9×10−3 |
| Aard | alanine and arginine rich domain containing protein | 0.51 | 4.0×10−5 |
| Nucleoside/nucleotide metabolic process | |||
| Mtap | methylthioadenosine phosphorylase | 1.53 | 3.0×10−3 |
| Gpatch8 | G patch domain containing 8 | 0.62 | 2.7×10−3 |
| Rbm47 | RNA binding motif protein 47 | 0.60 | 6.9×10−3 |
| Gimap6 | GTPase, IMAP family member 6 | 0.57 | 8.4×10−3 |
| Rab30 | RAB30, member RAS oncogene family | 0.57 | 3.2×10−3 |
| Gtpbp10 | GTP-binding protein 10 (putative) | 0.45 | 3.1×10−3 |
| Membrane protein | |||
| Tmem43 | transmembrane protein 43 | 1.62 | 6.8×10−3 |
| Tm4sf1 | transmembrane 4 superfamily member 1 | 1.61 | 2.0×10−5 |
| Neto2 | neuropilin (NRP) and tolloid (TLL)-like 2 | 0.66 | 9.5 ×10−4 |
| Tm4sf19 | transmembrane 4 L six family member19 | 0.64 | 6.1×10−3 |
| Tspan13 | tetraspanin 13 | 0.63 | 9.4×10−3 |
| Fam171b | family with sequence similarity 171, member B | 0.59 | 2.8×10−3 |
| Tspan7 | tetraspanin 7 | 0.53 | 7.6×10−3 |
| Coagulation cascades | |||
| Thbd | Thrombomodulin | 0.57 | 1.7×10−4 |
| Erythrocyte homeostasis | |||
| Fech | Ferrochelatase | 0.61 | 6.1×10−3 |
| Ossification | |||
| Ostn | Osteocrin | 0.31 | 3.7×10−3 |
| Signal transduction | |||
| Rras2 | related RAS viral (r-ras) oncogene homolog 2 | 2.05 | 8.1×10−4 |
| Srgap3 | SLIT-ROBO Rho GTPase activating protein 3 | 0.65 | 4.8×10−4 |
| Angiogenesis | |||
| Mfge8 | Milk fat globule-EGF factor 8 protein | 1.50 | 4.2×10−3 |
| Cell adhesion molecule | |||
| Pcdhb17 | protocadherin beta 17 | 1.68 | 3.8×10−3 |
| Ion channel | |||
| Kcnj15 | potassium inwardly-rectifying channel, subfamily J, member 15 | 1.98 | 7.7×10−4 |
| Kcnk1 | potassium channel, subfamily K, member 1 | 1.53 | 2.2×10−4 |
| Wdfy2 | WD repeat and FYVE domain containing2 | 0.65 | 5.2×10−3 |
| Zfp788 | zinc finger protein 788 | 0.63 | 5.2×10−3 |
| Phf20l1 | PHD finger protein 20-like 1 | 0.63 | 1.9×10−3 |
| Zfp763 | zinc finger protein 763 | 0.55 | 1.5×10−3 |
| Matrix protein | |||
| Ecm1 | extracellular matrix protein 1 | 1.61 | 1.9×10−3 |
| Chad | Chondroadherin | 0.65 | 9.0×10−3 |
| Cytokine secretion | |||
| Irf1 | interferon regulatory factor 1 | 0.60 | 3.2×10−3 |
| Lcp2 | lymphocyte cytosolic protein 2 | 0.47 | 8.1×10−3 |
| Other | |||
| Lgals2 | lectin, galactose-binding, soluble 2 | 3.31 | 9.0 ×10−5 |
| Fv1 | Friend virus susceptibility 1 | 2.11 | 1.0 ×10−4 |
| Fam84a | family with sequence similarity 84, member A | 1.80 | 1.2×10−3 |
| Mpv17 | Mpv17 transgene, kidney disease mutant | 1.72 | 7.7×10−3 |
| Thada | thyroid adenoma associated | 1.63 | 2.4×10−4 |
| Myg1 | melanocyte proliferating gene 1 | 1.55 | 3.5×10−3 |
| Prosc | proline synthetase co-transcribed | 0.62 | 8.1×10−3 |
| Hmcn1 | hemicentin 1 | 0.36 | 5.0×10−3 |
Values are mean ratio of the Rs1 vs. control among the 6 samples, P<0.01 in microarray data
The largest group of functionally related, differentially expressed genes was those involved in cell cycle and transcriptional regulation. Others included genes associated with apoptosis, enzyme activity, transporter and membrane protein. We observe no changes in genes associated with skeletal signaling pathways (i.e., Wnt/β catenin signaling, BMP, or IGF signaling pathways). Because our transgenic mouse model supported the concept of non-cell-autonomous effects of OB Gs signaling, we sought to identify candidate paracrine mediators by determining the effect of Rs1 on the expression level of genes encoding secreted proteins. We identified 13 regulated genes, from those in Table 1, that encode secreted proteins.
Furthermore, we identified a set of GPCRs expressed on mature OBs. Gs signaling activity in the unperturbed mature OB is determined by the aggregate activity of this complement of receptors, which also determine the sensory inputs to which Gs signaling activity can respond. No differences in GPCRs expression were observed in the two groups. These results are summarized in Table 2.
Table 2.
Gs- and Gi-coupled GPCRs expressed in FACS-sorted osteoblasts ranked by abundance (validated by Taqman real-time GPCR Array at Ct number below 30)
| Gene symbol | Gene name | Ct number (18S=11) | Class | Endogenous ligand(s) | Coupling |
|---|---|---|---|---|---|
| S1pr1 | sphingosine-1-phosphate receptor 1 | 17 | A | sphingosine-1-phosphate, sphingosylphosphorylcholine | Gi/Go |
| S1pr2 | sphingosine-1-phosphate receptor 2 | 20 | A | sphingosine-1-phosphate, sphingosylphosphorylcholine | Gs, Gq/G11, G12/G13 |
| Lpar1 | lysophosphatidic acid receptor 1 | 20 | A | lysophosphatidic acid | Gi/Go, Gq/G11, G12/G13 |
| Lpar6 | lysophosphatidic acid receptor 6 | 20 | A | lysophosphatidic acid | Gs, Gi/Go, G12/G13 |
| Ccr1 | chemokine (C-C motif) receptor 1 | 20 | A | CCL15, CCL23 | Gi/Go |
| Ptger4 | prostaglandin E receptor 4 (subtype EP4) | 20 | A | PGE2, PGF2α, PGI2, PGD2 | Gs/Gi/Go |
| Gpr30 | G-protein coupled receptor 30 | 21 | A | 17β-estradiol | Gi/Go, Gs |
| Agtr1a | angiotensin II receptor, type 1a | 21 | A | angiotensin I, angiotensin II, angiotensin III | Gi/Go, Gq/G11 |
| Pth1r | parathyroid hormone 1 receptor | 22 | B | Parathyroid hormone, parathyroid hormone related peptide | Gs, Gq/G11 |
| F2rl2 | coagulation factor II (thrombin) receptor-like 2 | 22 | A | thrombin | Gi/Go, Gq/G11, G12/G13 |
| Gpr1 33 | G protein-coupled receptor 133 | 22 | Adhesion | orphan | Gs |
| Aplnr | Apelin receptor | 23 | A | Apelin-13, apelin-17, apelin-36, [Pyr1]apelin-13 | Gi/Go |
| Lpar4 | lysophosphatidic acid receptor 4 | 24 | A | lysophosphatidic acid | Gs, Gi/Go, Gq/G11, G12/G13 |
| Gabbr1 | gamma-aminobutyric acid (GABA) B receptor, 1 | 25 | C | GABA | Gi/Go |
| Lpar3 | lysophosphatidic acid receptor 3 | 26 | A | lysophosphatidic acid | Gi/Go, Gq/G11 |
| Gpr34 | G protein-coupled receptor 34 | 26 | A | lysophosphatidylserine | Gi/Go |
| Gpr68 | G protein-coupled receptor 68 | 26 | A | protons | Gi/Go, Gq/G11 |
| Cxcr4 | chemokine (C-X-C motif) receptor 4 | 27 | A | stroma cell-derived factor 1 | Gi/Go |
| Ptger2 | prostaglandin E receptor 2 (subtype EP2) | 27 | A | PGE2, PGF2α, PGI2, PGD2 | Gs |
| Cmklr1 | chemokine-like receptor 1 | 27 | A | resolving E1, chemerin | Gi/Go |
| Ccr8 | chemokine (C-C motif) receptor 8 | 27 | A | CCL1, CCL8 | Gi/Go |
| Gpbar1 | G protein-coupled bile acid receptor 1 | 28 | A | lithocholic acid, deoxycholic acid, chenodeoxycholic acid, cholic acid | Gs |
| S1pr3 | sphingosine-1-phosphate receptor 3 | 29 | A | sphingosine-1-phosphate, sphingosylphosphorylcholine | Gi/Go, Gq/G11, G12/G13 |
| Sstr2 | somatostatin receptor 2 | 29 | A | SRIF-14, SRIF-28, CST-17 | Gi/Go, Gq/G11 |
| Adra2a | adrenergic receptor, alpha 2a | 29 | A | adrenaline, noradrenaline | Gi/Go, Gs |
| Gpr84 | G protein-coupled receptor 84 | 29 | A | medium-chain-length fatty acid | Gi/Go |
| Cxcr6 | chemokine (C-X-C motif) receptor 6 | 29 | A | CXCL16 | Gi/Go |
Validation of microarray results by real-time qPCR
qPCR was performed to verify the effectiveness of the sorting process and validated the microarray results. We analyzed the mRNA levels of the transgene and OB markers in GFP positive cells compared to GFP-negative cells from control mice. As expected, early OB marker genes, Runx2 and Osterix, were highly expressed in GFP-negative population, whereas mature OB marker genes, Col1a1 and Osteocalcin, were abundant in GFP-positive cells (Fig. 3B). Rs1 mRNA expression was detected only in OBs from Rs1mice, and its expression was absent in GFP-positive population from control mice (Fig. 3C). These findings suggest that the sorted GFP positive population represents mainly differentiating OBs that express the tetO-responsive GFP and Rs1 genes. We were able to validate by qPCR that 10 of 13 regulated genes that encode secreted proteins were differentially expressed, and the direction of the changes was consistent with the microarray results (Table 3). The difference in the fold change between the two techniques likely reflects difference in sensitivity of the methods.
Table 3.
Validating expression of gene-coded secreted proteins changes in microarrays and qPCR
| Gene symbol | Gene name | Fold change | |
|---|---|---|---|
| Microarray# | qPCR | ||
| Lipi | Lipase, member I | 4.6 | 8.3* |
| Figf | c-fos induced growth factor | 2.1 | 15.7* |
| Slpi | Secretory leukocyte peptidase inhibitor | 1.8 | 7.0* |
| Rbp4 | Retinol binding protein 4, plasma | 1.7 | 1.7* |
| Ecm1 | Extracellular matrix protein 1 | 1.6 | 2.0 |
| Mfge8 | Milk fat globule-EGF factor 8 protein | 1.5 | 8.1* |
| Chad | Chondroadherin | 0.7 | 0.3* |
| Comp | Cartilage oligomeric matrix protein | 0.7 | 0.6 |
| Fgf9 | Fibroblast growth factor 9 | 0.6 | 0.8* |
| Thbd | Thrombomodulin | 0.6 | 0.06* |
| Car2 | Carbonic anhydrase 2 | 0.6 | 0.3* |
| Srgn | Serglycin | 0.5 | 0.5 |
| Ostn | Osteocrin | 0.3 | 0.05* |
The values in microarray and qPCR analysis are presented as fold changes between three Rs1 and three control mice.
P<0.01 in microarray and
P≤0.05 in qPCR
Effects of PTH and forskolin treatment on Fgf9 expression in BMSC cultures
We used primary mouse OBs culture to assess whether Fgf9 is a direct target of Gs signaling. We found that exposure of BMSCs with PTH or with forskolin, to raise cyclic AMP levels, markedly down-regulated the expression of Fgf9 compared to controls (−64% and −72% respectively, p<0.05) (Fig. 4A), suggesting that Fgf9 expression maybe directly regulated by Gs signaling in OBs.
Figure 4.
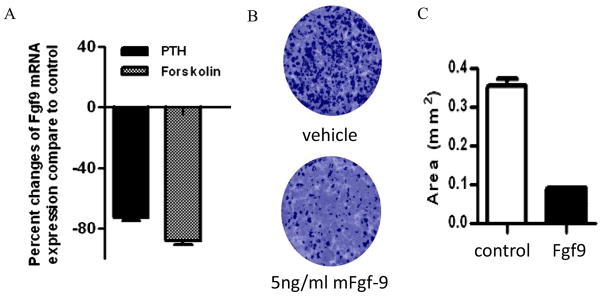
(A) Effects of PTH and forskolin treatment on Fgf9 expression in primary BMSC cultures. Data were normalized to GAPDH RNA expression in the same sample and expressed as percent changes compared to controls. (B) Effect of continuous treatment of BMSCs with murine Fgf9 assessed by VK staining. VK-positive mineralized nodules were stained black. (C) Exposure of Fgf9 from Day 0 to day 20 increased VK staining area. Total colony area positive for VK staining was quantified as area (mm2). The results of these assays were confirmed by repeating the experiment three times. All data are shown as mean ± SD. *: p<0.05.
Effect of treatment of BMSCs with mouse FGF9
The effect of Fgf9 on osteogenesis was assessed in cell culture system. When added Fgf9 directly to mouse BMSCs grown under osteogenic conditions, Fgf9 dramatically suppressed (by>80%) the formation of mineralized nodules, assessed by VK staining, compared to controls (Fig. 4B).
Discussion
We previously reported that strong basal Gs signaling by the Rs1 RASSL expressed in OBs driven by the 2.3-kb Col1 promoter fragment resulted in a dramatic age-dependent increase in endochondral bone formation [10,14,15]. In this study, we investigated in detail the effects of increased Gs signaling in mature OBs on intramembranous bone formation in neonatal and growing Rs1 transgenic mice. The phenotype of Rs1 calvariae resembled that seen in Rs1 femoral bones, indicating that Gs signaling in mature OBs is sufficient to initiate dramatic and similar skeletal responses in both endochrondral and intramembranous bones. The histological analyses demonstrated that Rs1 expression produces an expansion of osteoprogenitor cells that do not express Rs1, supporting the notion that the effect of Rs1 signaling in mature OBs alters bone formation by regulating the expression of factors that influence the early commitment, proliferation, and/or differentiation of osteoprogenitors.
To determine the cellular response in vivo by which mature OBs regulate bone formation in response to Gs signaling, we utilized a microarray approach to examine Rs1-induced alterations in the OB transcriptome. The approach was used to identify an approximate snapshot of the OB transcriptome at the time of sacrifice by isolating the OB population that expresses Rs1 by GFP labeling, without the use of cell culture. We compared gene expression between control OBs and OBs-expressing Rs1. Successful isolation of mature OBs population was confirmed by abundant expression of differentiating OB marker genes, such as Osteocalcin and alkaline phosphatase, in GFP-positive cells. In addition, we compared our data to the findings of Paic et al. [20] who utilized dual GFP receptor mice in which OBs were identified by expression of GFP (cyan) driven by 2.3 kb of the Col1a promoter (Col2.3 cyan). We found a good correlation of OB marker genes and genes associated with OB differentiation from our control OBs and their Col2.3cyan positive OBs (r = 0.74) (Supplemental Fig. 1). These results further validate the efficacy of our procedure for isolating mature OBs.
Interestingly, we found that the magnitude of changes seen in Rs1-expressing OBs was relatively small despite the striking skeletal phenotype. Since the mice were about 1 week old and still in a phase of rapid skeletal growth, the anabolic program in OBs was presumably highly active in wild-type animals, and that might obscured the effects of additional anabolic signaling in response to Gs activation. It is also possible that compensatory signals from the other cells in the bone environment might have attenuated the response of mature OBs to chronic Gs signaling. However, microarray analysis revealed that genes involved in cell cycle and transcriptional regulation were the most changed in mature OBs in response to enhanced Gs signaling.
We then focused on identifying candidate paracrine mediators that were differentially expressed in mature OBs expressing Rs1. Of 13 such regulated genes, 10 were validated by qPCR although the magnitude of changes did not always quantitatively correspond to the microarray results. Interestingly, seven genes (Lipi, Rbp4, Chad, Mfge8, Fgf9, Car2 and Thbd) had at least half site cyclic AMP responsive elements (CREs) within 5 kb of the transcriptional start site, and two of them (Fgf9 and Thbd) had predicted functional CREs in their promoters, as reported by Zhang et al. [21] This implies that these validated genes are potential downstream transcriptional targets of Gs signaling.
Products of some of the validated genes have been reported to have roles in bone cell differentiation and function. For instance, carbonic anhydrase 2 (Car2) is essential for bone resorption and osteoclast differentiation [22,23]. Osteocrine (Ostn) and chrondroadherin (Chad) are reported to be important for bone development. Ostn is expressed in OBs, muscle and fat cells, and OB-specific Ostn-overexpressing mice display elongated long bones from enhanced proliferation and differentiation of growth plate chrondrocytes [24]. Chad is highly expressed in cartilaginous tissues with lower levels in bone and tendon [25,26]. Chad null mice displayed widening of the epiphyseal growth plate with possible impaired of hypertrophic differentiation of chondrocytes [27]. Ostn and Chad expression were significantly downregulated in our model of increase Gs signaling in OB. Nonetheless, we did not observe any appreciable chrondrocyte phenotypes of long bones in vivo. The contribution of Ostn and Chad to the bone changes in Rs1 model has to be further investigated.
Chronic inflammatory processes are often associated with bone loss [28,29]. We found upregulation of secretory leukocyte peptidase inhibitor (Slpi) expression in our transgenic OBs. Slpi downregulates the synthesis of tumor necrosis factors alpha suggesting that it may act as a proinflammation mediator [30]. Recently, Sharma et al. [31] reported downregulation of this gene in axial spondyloarthropathy, an effect that might be important role in the pathogenesis of this disease that often responds markedly to tumor necrosis factors inhibition. Slpi might participate in paracrine mediators effecting osteoclasts. The coagulation system influences bone mineralization [32]. We identified thrombomodulin (Thbd), a gene involved in blood coagulation, to be significantly downregulated by Gs signaling in OBs. Delvaeye et al. [33] showed that Thbd binds to C3b and factor H and negatively regulates complement. It also accelerates the inactivation of anaphylatoxins C3a and C5a and provides protection against complement activation. It remains to establish the role of coagulation system in bone environment in our mouse model. The microarray analysis has also highlighted previously unknown significant effects of Gs signaling in OBs on molecular effectors related to glucose and lipid metabolism, Lipi and Rbp4. Interplay between bone metabolism and glucose or fat metabolism have been extensively addressed [34–36]. Condition that alters genes related to energy metabolism may influence skeletal homeostasis.
At least three validated genes involved in cell growth and angiogenesis were significantly altered in Rs1 expressing OBs: c-fos induced growth factor (Figf; also known as VEGF-D) and milk fat globule-EGF factor 8 protein (Mfge8) were upregulated, and fibroblast growth factor 9 (Fgf9) was downregulated. Angiogenesis and bone formation are coupled during skeletal development and fracture healing [37–39]. Besides playing a role in vascular and lymphatic endothelial cells homeostasis, VEGF-D has been implicated in OB differentiation [40]. During mouse development VEGF-D expression was detected in the OBs of the growth plate, periosteal layer of the developing vertebral column and in the dental mesenchyme close to the enamel epithelium where dentin and enamel matrices are deposited, suggesting that VEGF-D might play a role in intramembranous and long-bone formation [40,41]. VEGF-D/FGFR-3 signaling is a strong candidate to participate in the downstream effects of OB Gs signaling on bone.
Among secreted factors differentially expressed in Rs1 mutants, none has previously been reported to be related to OB-specific Gs signaling. Fgf9 is particularly enticing because it has been identified as having a role during skeletal development and fracture healing [42]. In addition, Fgf9 expression was also significantly altered in OB-specific blockade of Gi-coupled signaling (PTX model) (Data not shown). We demonstrated that Fgf9 expression was markedly down-regulated in the BMSCs treated with forskolin or PTH to raise cyclic AMP levels. Moreover, Continuous treatment of BMSCs with Fgf9 diminished OB mineralization. Taken together, these results support the possibility that alteration in Fgf9 expression induced by G protein signaling in OBs contribute to the changes of bone mass in the Rs1 model. Further studies assessing the bone phenotypes in OB-specific Fgf9 KO mice might needed to clarify the issue.
The balance of Gs- and Gi-GPCR signaling is an important regulator of bone formation. The role of Gi signaling in OBs may be, at least in part, due to antagonized increase in cyclic AMP levels in response to Gs coupled signaling. The microarray results allowed us to assess the relative expression of GPCRs in mature OBs. We identified 27 Gs-and/or Gi-coupled GPCRs whose expressions on OBs were validated by Taqman real-time GPCR Array at Ct number below 30. Many of these receptors have known or suspected roles in OB and osteocyte differentiation and function. Not surprisingly, parathyroid hormone 1 receptor (Pth1r) was highly expressed in this OB population. For many of the identified OB GPCRs, their role in regulating normal bone development and skeletal function is not known. Definitive assessment of the role of specific GPCRs in mature OBs will require evaluation of the effects of targeted deletion.
Several groups investigated gene expression profiles by microarray analysis in OB cell lines [43] and in whole-bone samples [44,45] in response to exogenous parathyroid hormone. This is the first study to date examining alterations in gene expression in vivo in enhanced OB Gs signaling. We found only a small number of differentially expressed genes that overlap with their results (i.e., Car2, Fech, Ecm1, Mfge8, Slpi, Tm4sf1, Car8), consistent with the notion that there are different OB transcriptome effects, depending on the timing and source of Gs activation.
In conclusion, our study provides a useful tool to evaluate the in vivo gene expression specifically occurring in OBs with activated Gs-GPCR signaling, at the cellular level rather than in a whole bone. The study revealed that cell cycle and transcriptional regulation were the most highly enriched pathways. Complete set of GPCRs expressed in OBs and new candidate paracrine mediators in response to chronic Gs activation in OBs were also indentified. All these regulated genes could be used for further functional studies. Biological validation of the role of these mediators may lead to new therapeutic targets for enhancing bone formation in osteoporosis and for reversing bone disease associated with excess Gs signaling (fibrous dysplasia, hyperparathyroidism).
Supplementary Material
Highlights.
OB expression of an engineered Gs-coupled receptor dramatically increases bone mass.
We investigated the alterations in gene expression in vivo in enhanced OB Gs signaling.
Genes in cell cycle and transcription were increased in enhanced OB Gs signaling.
GPCRs and paracrine mediators of the effect of Gs signaling in OBs were determined.
Acknowledgments
Funding support: This work was supported by grants to RAN from NIH (DK072071) and the Department of Veterans Affairs (Merit Review grant 1I01BX001496), to ECH from the NIH (AR056299) and the National Osteoporosis Foundation (2008–19).
We thank Richard Kao, Zhiqiang cheng and Yongmei Wang for valuable technical assistance and discussion. We also thank the FACS Core Facility at the San Francisco VA Medical Center for FACS analysis and the Gladstone Genomics Core for microarray expression analysis. This work was supported by grants to RAN from NIH (DK072071) and the Department of Veterans Affairs (Merit Review grant 1I01BX001496), to ECH from the NIH (AR056299) and the National Osteoporosis Foundation (2008–19).
Abbreviations
- GPCR
G protein–coupled receptor
- OBs
osteoblasts
- TRAP
tartrate-resistant acid phosphatase
- FTIR
fourier transform infrared
- SRμCT
synchrotron radiation micro-computed tomography
- GFP
green fluorescent protein
- qPCR
quantitative real-time polymerase chain reaction
- Ct
cycle threshold
- FACS
fluorescent activated cell sorting
- PCM
primary culture medium
- SDM
secondary osteogenic differentiation medium
- BMSCs
bone marrow stromal cells
- VK
Von Kossa
- CRE
cyclic AMP responsive elements
Footnotes
Disclosures: The authors have nothing to disclose.
Authors’ contributions: LW, SMM, and RAN contributed to study design; ECH and BRC provided mice; LW, LPW, WDL, and DO performed experiments; LW collected, analyzed and interpreted all data; LW prepared the manuscript. LPW, ECH, and RAN supervised analyses and edited the manuscript. All authors gave conceptual advice, read and approved final version of manuscript. LW takes responsibility for the integrity of the data analysis.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Lalita Wattanachanya, Email: lalita_md@yahoo.com.
Liping Wang, Email: lipingwang05@yahoo.com.
Susan M. Millard, Email: susan.millard@mater.uq.edu.au.
Wei-Dar Lu, Email: weidar_lu@yahoo.com.
Dylan O’Carroll, Email: dylancocarroll@gmail.com.
Edward C. Hsiao, Email: Edward.Hsiao@ucsf.edu.
Bruce R. Conklin, Email: bconklin@gladstone.ucsf.edu.
Robert A. Nissenson, Email: Robert.Nissenson@ucsf.edu.
References
- 1.Soltanoff CS, Yang S, Chen W, Li YP. Signaling networks that control the lineage commitment and differentiation of bone cells. [accessed January 25, 2015];Crit Rev Eukaryot Gene Expr. 2009 19(1):1–46. doi: 10.1615/CritRevEukarGeneExpr.v19.i1.10. http://www.ncbi.nlm.nih.gov/pubmed/19191755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Heino TJ, Hentunen TA. Differentiation of osteoblasts and osteocytes from mesenchymal stem cells. [accessed January 25, 2015];Curr Stem Cell Res Ther. 2008 3(2):131–45. doi: 10.2174/157488808784223032. http://www.eurekaselect.com/82545/article. [DOI] [PubMed] [Google Scholar]
- 3.Franceschi RT, Ge C, Xiao G, Roca H, Jiang D. Transcriptional regulation of osteoblasts. [accessed January 25, 2015];Cells Tissues Organs. 2009 189(1–4):144–52. doi: 10.1159/000151747. http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3512205/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lian JB, Stein GS, Javed A, et al. Networks and hubs for the transcriptional control of osteoblastogenesis. [accessed January 25, 2015];Rev Endocr Metab Disord. 2006 7(1–2):1–16. doi: 10.1007/s11154-006-9001-5. http://link.springer.com/article/10.1007%2Fs11154-006-9001-5. [DOI] [PubMed] [Google Scholar]
- 5.Dobnig H, Turner RT. The effects of programmed administration of human parathyroid hormone fragment (1–34) on bone histomorphometry and serum chemistry in rats. [accessed January 25, 2015];Endocrinology. 1997 138(11):4607–12. doi: 10.1210/endo.138.11.5505. http://press.endocrine.org/doi/pdf/10.1210/endo.138.11.5505. [DOI] [PubMed] [Google Scholar]
- 6.Genetos DC, Yellowley CE, Loots GG. Prostaglandin E2 signals through PTGER2 to regulate sclerostin expression. [accessed January 25, 2015];PLoS One. 2011 6(3):e17772. doi: 10.1371/journal.pone.0017772. http://dx.plos.org/10.1371/journal.pone.0017772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sakamoto A, Chen M, Nakamura T, Xie T, Karsenty G, Weinstein LS. Deficiency of the G-protein alpha-subunit G(s)alpha in osteoblasts leads to differential effects on trabecular and cortical bone. [accessed January 25, 2015];J Biol Chem. 2005 280(22):21369–75. doi: 10.1074/jbc.M500346200. http://www.jbc.org/cgi/pmidlookup?view=long&pmid=15797856. [DOI] [PubMed] [Google Scholar]
- 8.Calvi LM, Sims NA, Hunzelman JL, et al. Activated parathyroid hormone/parathyroid hormone-related protein receptor in osteoblastic cells differentially affects cortical and trabecular bone. [accessed January 25, 2015];J Clin Invest. 2001 107(3):277–86. doi: 10.1172/JCI11296. http://www.jci.org/articles/view/11296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.O’Brien CA, Plotkin LI, Galli C, et al. Control of bone mass and remodeling by PTH receptor signaling in osteocytes. [accessed January 25, 2015];PLoS One. 2008 3(8):e2942. doi: 10.1371/journal.pone.0002942. http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2491588/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hsiao EC, Boudignon BM, Chang WC, et al. Osteoblast expression of an engineered Gs-coupled receptor dramatically increases bone mass. [accessed January 25, 2015];Proc Natl Acad Sci. 2008 105(4):1209–14. doi: 10.1073/pnas.0707457105. http://www.pnas.org/cgi/pmidlookup?view=long&pmid=18212126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Peng J, Bencsik M, Louie A, et al. Conditional expression of a Gi-coupled receptor in osteoblasts results in trabecular osteopenia. [accessed January 25, 2015];Endocrinology. 2008 149(3):1329–37. doi: 10.1210/en.2007-0235. http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2275363/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kazakia GJ, Speer D, Shanbhag S, et al. Mineral composition is altered by osteoblast expression of an engineered Gs-coupled receptor. [accessed January 25, 2015];Ca;cif Tissue Int. 2011 89:10–20. doi: 10.1007/s00223-011-9487-z. http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3110278/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Claeysen S, Joubert L, Sebben M, Bockaert J, Dumuis A. A single mutation in the 5-HT4 receptor (5-HT4-R D100(3.32)A) generates a Gs-coupled receptor activated exclusively by synthetic ligands (RASSL) [accessed January 25, 2015];J Biol Chem. 2003 278(2):699–702. doi: 10.1074/jbc.C200588200. http://www.jbc.org/content/278/2/699.full. [DOI] [PubMed] [Google Scholar]
- 14.Hsiao EC, Millard SM, Louie A, Huang Y, Conklin BR, Nissenson RA. Ligand-mediated activation of an engineered gs g protein-coupled receptor in osteoblasts increases trabecular bone formation. [accessed January 25, 2015];Mol Endocrinol. 2010 24(3):621–31. doi: 10.1210/me.2009-0424. http://press.endocrine.org/doi/abs/10.1210/me.2009-0424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hsiao EC, Boudignon BM, Halloran BP, Nissenson RA, Conklin BR. Gs G protein-coupled receptor signaling in osteoblasts elicits age-dependent effects on bone formation. [accessed January 25, 2015];J Bone Miner Res. 2010 25(3):584–93. doi: 10.1002/jbmr.3. http://onlinelibrary.wiley.com/doi/10.1002/jbmr.3/pdf. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Iwaniec UT, Yuan D, Power RA, Wronski TJ. Strain-dependent variations in the response of cancellous bone to ovariectomy in mice. [accessed January 25, 2015];J Bone Miner Res. 2006 21(7):1068–74. doi: 10.1359/JBMR.060402. http://onlinelibrary.wiley.com/doi/10.1359/jbmr.060402/pdf. [DOI] [PubMed] [Google Scholar]
- 17.Smyth GK. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. [accessed January 25, 2015];Stat Appl Genet Mol Biol. 2004 3:Article3. doi: 10.2202/1544-6115.1027. http://www.degruyter.com/doi/10.2202/1544-6115.1027. [DOI] [PubMed] [Google Scholar]
- 18.Zambon AC, Gaj S, Ho I, et al. GO-Elite: a flexible solution for pathway and ontology over-representation. [accessed January 25, 2015];Bioinformatics. 2012 28(16):2209–10. doi: 10.1093/bioinformatics/bts366. http://bioinformatics.oxfordjournals.org/content/28/16/2209.long. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dacic S, Kalajzic I, Visnjic D, Lichtler AC, Rowe DW. Col1a1-driven transgenic markers of osteoblast lineage progression. [accessed January 25, 2015];J Bone Miner Res. 2001 16(7):1228–36. doi: 10.1359/jbmr.2001.16.7.1228. http://onlinelibrary.wiley.com/doi/10.1359/jbmr.2001.16.7.1228/abstract. [DOI] [PubMed] [Google Scholar]
- 20.Paic F, Igwe JC, Nori R, et al. Identification of differentially expressed genes between osteoblasts and osteocytes. [accessed January 25, 2015];Bone. 2009 45(4):682–92. doi: 10.1016/j.bone.2009.06.010. http://www.thebonejournal.com/article/S8756-3282(09)01634-2/abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang X, Odom DT, Koo SH, et al. Genome-wide analysis of cAMP-response element binding protein occupancy, phosphorylation, and target gene activation in human tissues. [accessed January 25, 2015];Proc Natl Acad Sci USA. 2005 102(12):4459–64. doi: 10.1073/pnas.0501076102. http://www.pnas.org/content/102/12/4459.long. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lehenkari P, Hentunen TA, Laitala-Leinonen T, Tuukkanen J, Vaananen HK. Carbonic anhydrase II plays a major role in osteoclast differentiation and bone resorption by effecting the steady state intracellular pH and Ca2+ [accessed January 25, 2015];Exp Cell Res. 1998 242(1):128–37. doi: 10.1006/excr.1998.4071. http://www.sciencedirect.com/science/article/pii/S001448279894071X. [DOI] [PubMed] [Google Scholar]
- 23.Margolis DS, Szivek JA, Lai LW, Lien YH. Phenotypic characteristics of bone in carbonic anhydrase II-deficient mice. [accessed January 25, 2015];Calcif Tissue Int. 2008 82(1):66–76. doi: 10.1007/s00223-007-9098-x. http://link.springer.com/article/10.1007%2Fs00223-007-9098-x. [DOI] [PubMed] [Google Scholar]
- 24.Thomas G, Moffatt P, Salois P, et al. Osteocrin, a novel bone-specific secreted protein that modulates the osteoblast phenotype. [accessed January 25, 2015];J Biol Chem. 2003 278(50):50563–71. doi: 10.1074/jbc.M307310200. http://www.jbc.org/content/278/50/50563.long. [DOI] [PubMed] [Google Scholar]
- 25.Shen Z, Gantcheva S, Mansson B, Heinegard D, Sommarin Y. Chondroadherin expression changes in skeletal development. [accessed January 25, 2015];Biochem J. 1998 330 ( Pt 1):549–57. doi: 10.1042/bj3300549. http://www.biochemj.org/bj/330/0549/bj3300549.htm. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mizuno M, Fujisawa R, Kuboki Y. Bone chondroadherin promotes attachment of osteoblastic cells to solid-state substrates and shows affinity to collagen. [accessed January 25, 2015];Calcif Tissue Int. 1996 59(3):163–7. doi: 10.1007/s002239900103. http://www.ncbi.nlm.nih.gov/pubmed/8694892. [DOI] [PubMed] [Google Scholar]
- 27.Hessle L, Stordalen GA, Wenglen C, et al. The skeletal phenotype of chondroadherin deficient mice. [accessed January 25, 2015];PLoS One. 2013 8(6):e63080. doi: 10.1371/journal.pone.0063080. http://journals.plos.org/plosone/article?id=10.1371/journal.pone.0063080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hardy R, Cooper MS. Bone loss in inflammatory disorders. [accessed January 25, 2015];J Endocrinol. 2009 201(3):309–20. doi: 10.1677/JOE-08-0568. http://joe.endocrinology-journals.org/content/201/3/309.long. [DOI] [PubMed] [Google Scholar]
- 29.Schett G, Kiechl S, Weger S, et al. High-sensitivity C-reactive protein and risk of nontraumatic fractures in the Bruneck study. [accessed January 25, 2015];Arch Intern Med. 2006 166(22):2495–501. doi: 10.1001/archinte.166.22.2495. http://archinte.jamanetwork.com/article.aspx?articleid=769532. [DOI] [PubMed] [Google Scholar]
- 30.Wang N, Thuraisingam T, Fallavollita L, Ding A, Radzioch D, Brodt P. The secretory leukocyte protease inhibitor is a type 1 insulin-like growth factor receptor-regulated protein that protects against liver metastasis by attenuating the host proinflammatory response. [accessed January 25, 2015];Cancer Res. 2006 66(6):3062–70. doi: 10.1158/0008-5472.CAN-05-2638. http://cancerres.aacrjournals.org/content/66/6/3062.long. [DOI] [PubMed] [Google Scholar]
- 31.Sharma SM, Choi D, Planck SR, et al. Insights in to the pathogenesis of axial spondyloarthropathy based on gene expression profiles. [accessed January 25, 2015];Arthritis Res Ther. 2009 11(6):R168. doi: 10.1186/ar2855. http://arthritis-research.com/content/11/6/R168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Krueger T, Westenfeld R, Schurgers L, Brandenburg V. Coagulation meets calcification: the vitamin K system. [accessed January 25, 2015];Int J Artif Organs. 2009 32(2):67–74. doi: 10.1177/039139880903200202. http://www.ncbi.nlm.nih.gov/pubmed/19363777. [DOI] [PubMed] [Google Scholar]
- 33.Delvaeye M, Conway EM. Coagulation and innate immune responses: can we view them separately? [accessed January 25, 2015];Blood. 2009 114(12):2367–74. doi: 10.1182/blood-2009-05-199208. http://www.bloodjournal.org/content/114/12/2367.long?sso-checked=true. [DOI] [PubMed] [Google Scholar]
- 34.Clemens TL, Karsenty G. The osteoblast: an insulin target cell controlling glucose homeostasis. [accessed January 25, 2015];J Bone Miner Res. 2011 26(4):677–80. doi: 10.1002/jbmr.321. http://onlinelibrary.wiley.com/doi/10.1002/jbmr.321/abstract. [DOI] [PubMed] [Google Scholar]
- 35.Ferron M, Wei J, Yoshizawa T, Del, et al. Insulin signaling in osteoblasts integrates bone remodeling and energy metabolism. [accessed January 25, 2015];Cell. 2010 142(2):296–308. doi: 10.1016/j.cell.2010.06.003. http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2910411/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Reid IR, Evans MC, Cooper GJ, Ames RW, Stapleton J. Circulating insulin levels are related to bone density in normal postmenopausal women. [accessed January 25, 2015];Am J Physiol. 1993 265(4 Pt 1):E655–9. doi: 10.1152/ajpendo.1993.265.4.E655. http://ajpendo.physiology.org/cgi/pmidlookup?view=reprint&pmid=8238341. [DOI] [PubMed] [Google Scholar]
- 37.Brandi ML, Collin-Osdoby P. Vascular biology and the skeleton. [accessed January 25, 2015];J Bone Miner Res. 2006 21(2):183–92. doi: 10.1359/JBMR.050917. http://onlinelibrary.wiley.com/doi/10.1359/JBMR.050917/pdf. [DOI] [PubMed] [Google Scholar]
- 38.Kanczler JM, Oreffo RO. Osteogenesis and angiogenesis: the potential for engineering bone. [accessed January 25, 2015];Eur Cell Mater. 2008 15:100–14. doi: 10.22203/ecm.v015a08. http://www.ecmjournal.org/journal/papers/vol015/pdf/v015a08.pdf. [DOI] [PubMed] [Google Scholar]
- 39.Ytrehus B, Carlson CS, Lundeheim N, et al. Vascularisation and osteochondrosis of the epiphyseal growth cartilage of the distal femur in pigs--development with age, growth rate, weight and joint shape. [accessed January 25, 2015];Bone. 2004 34(3):454–65. doi: 10.1016/j.bone.2003.07.011. http://www.thebonejournal.com/article/S8756-3282(03)00335-1/pdf. [DOI] [PubMed] [Google Scholar]
- 40.Orlandini M, Spreafico A, Bardelli M, et al. Vascular endothelial growth factor-D activates VEGFR-3 expressed in osteoblasts inducing their differentiation. [accessed January 25, 2015];J Biol Chem. 2006 281(26):17961–7. doi: 10.1074/jbc.M600413200. http://www.jbc.org/content/281/26/17961.long. [DOI] [PubMed] [Google Scholar]
- 41.Avantaggiato V, Orlandini M, Acampora D, Oliviero S, Simeone A. Embryonic expression pattern of the murine figf gene, a growth factor belonging to platelet-derived growth factor/vascular endothelial growth factor family. [accessed January 25, 2015];Mech Dev. 1998 73(2):221–4. doi: 10.1016/s0925-4773(98)00049-5. http://www.ncbi.nlm.nih.gov/pubmed/9622638. [DOI] [PubMed] [Google Scholar]
- 42.Björn Behra B, Leuchta P, Longakera MT, et al. Fgf-9 is required for angiogenesis and osteogenesis in long bone repair. [accessed January 25, 2015];PNAS. 2010 107(26):11853–8. doi: 10.1073/pnas.1003317107. http://www.pnas.org/content/107/26/11853.long. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Qin L, Qiu P, Wang L, et al. Gene expression profiles and transcription factors involved in parathyroid hormone signaling in osteoblasts revealed by microarray and bioinformatics. [accessed January 25, 2015];J Biol Chem. 2003 278(22):19723–31. doi: 10.1074/jbc.M212226200. http://www.jbc.org/content/278/22/19723.long. [DOI] [PubMed] [Google Scholar]
- 44.Li X, Liu H, Qin L, et al. Determination of dual effects of parathyroid hormone on skeletal gene expression in vivo by microarray and network analysis. [accessed January 25, 2015];J Biol Chem. 2007 282(45):33086–97. doi: 10.1074/jbc.M705194200. http://www.jbc.org/cgi/pmidlookup?view=long&pmid=17690103. [DOI] [PubMed] [Google Scholar]
- 45.Onyia JE, Helvering LM, Gelbert L, et al. Molecular profile of catabolic versus anabolic treatment regimens of parathyroid hormone (PTH) in rat bone: an analysis by DNA microarray. [accessed January 25, 2015];J Cell Biochem. 2005 95(2):403–18. doi: 10.1002/jcb.20438. http://onlinelibrary.wiley.com/doi/10.1002/jcb.20438/abstract. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.