

Abstract
Embryonic development depends on spatial and temporal control of gene function, and deciphering the molecular mechanisms that underlie pattern formation requires methods for perturbing gene expression with similar precision. Emerging chemical technologies can enable such perturbations, as exemplified by the use of caged morpholino (cMO) oligonucleotides to photo-inactivate genes in zebrafish embryos with spatiotemporal control. This chapter describes general principles for cMO design and methods for cMO assembly in three steps from commercially available reagents. Experimental techniques for the microinjection and photoactivation of these reagents are described in detail, as well as the preparation and application of caged fluorescein dextran (cFD) for labeling irradiated cells. Using these protocols, cMOs can be effective tools for functional genomic studies in zebrafish and other model organisms.
I. Introduction
During embryonic patterning, genetic programs are precisely coordinated to create complex tissues and organs. Genome sequencing and forward genetic screens have revealed an extensive list of patterning genes, many of which are expressed in a tissue-specific manner at discrete time points in embryo development. One of the remaining challenges in developmental biology is to understand how these genes act in space and time to coordinate embryogenesis by stereotypically modulating cellular functions. Toward that goal, several chemical, optical, and/or genetic approaches for conditional gene regulation have been developed, and such technologies have provided key insights into the molecular mechanisms that underlie tissue patterning and function (Gradinaru et al.; Ouyang and Chen; Shestopalov and Chen, 2008).
As evident in this issue of Methods in Cell Biology, the zebrafish is ideally suited for visualizing vertebrate ontogeny, since its embryos and larvae are optically transparent and develop rapidly ex utero. However, methods for regulating endogenous gene function in zebrafish are underdeveloped relative to other model organisms; inducible RNA interference technologies have exhibited limited efficacy thus far (Blidner et al., 2008; Zhao et al., 2008), and targeted genetic knockouts are limited to zinc-finger nucleases (Remy et al., 2010). In lieu of these approaches, synthetic oligonucleotides such as morpholinos (MOs) have been employed as antisense reagents in zebrafish embryos (Shestopalov and Chen, 2010). MO oligomers, typically 25 nucleotides in length, display DNA bases from a morpholine ring system and are connected by a phosphorodiamidate backbone (Summerton, 1999). Due to this non-natural structure, MOs are resistant to nucleases and persist in zebrafish embryos for up to four days (Bill et al., 2009; Nasevicius and Ekker, 2000). When MOs are injected into zebrafish prior to the eight-cell stage, they become uniformly distributed throughout the embryo and constitutively block either RNA splicing or translation, depending on the targeted RNA sequence.
While MOs are widely used to interrogate gene function in zebrafish, the utility of these reagents is limited by their constitutive activity, as gene expression is inhibited immediately after microinjection. Conventional MOs therefore are less effective for studying genes that are required for embryonic survival and/or have pleiotropic functions during embryonic patterning. To overcome this limitation, we recently developed cMOs that can be activated by 360-nm light, taking advantage of the transparency of zebrafish embryos (Shestopalov et al., 2007). This was achieved by tethering a complementary MO-derived inhibitor to the 25-base targeting sequence through a dimethoxynitrobenzyl (DMNB) group-containing photocleavable linker (Fig. 1). The resulting intramolecular hairpin suppresses hybridization of the targeting sequence to its complementary RNA, and cMOs are significantly less active in vivo than their 25-base counterparts. Linker cleavage with 360-nm light converts the cMO hairpin into an intermolecular MO duplex that is energetically favored to dissociate, allowing the targeting MO to alter RNA splicing or translation. Similar caging strategies have been previously described for regulating RNA function in vivo (Tang et al., 2007; Tang et al., 2008), and this general approach differs from other oligonucleotide-caging technologies that target individual nucleoside bases (Young et al., 2008), the phosphate backbone (Ando et al., 2001), the oligonucleotide termini (Nguyen et al., 2006), or use an excess of complementary caged RNA oligomer (Tomasini et al., 2009). The cMO technology is advantageous to other methods for light-controlled antisense as it uses the well-established MO antisense scaffold, a single photo-cleavable group, and a stoichiometric amount of MO-regulating oligonucleotide (Shestopalov and Chen, 2010). Cells containing photoactivated cMOs can be traced with a variety of light-inducible fluorescent proteins (Lippincott-Schwartz and Patterson, 2008) or dextran-conjugated caged fluorophores, such as cFD (Kozlowski and Weinberg, 2000) or caged rhodamine dextran (Gee et al., 2001).
Figure 1. Schematic representation of the cMO hairpin technology.
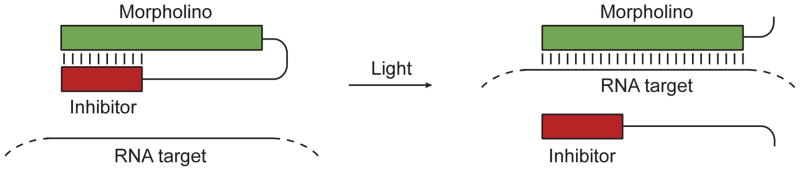
MO/RNA hybridization can be abrogated by tethering a complementary inhibitor MO via a photocleavable linker. Irradiation with 360-nm light leads to inhibitor dissociation, allowing the targeting MO to base pair with RNA containing a complementary sequence. Adapted with permission (Ouyang et al., 2009; Copyright 2009, American Chemical Society).
While our initial published results established the general principle of using caged oligonucleotides to conditionally regulate in vivo gene expression (Shestopalov et al., 2007), there were limitations to generalizable cMO implementation. First, our initial studies required preparation of the inhibitory MO oligomer and its appendant photocleavable linker through solid-phase chemistry, since conventional MOs amenable to functionalization at the 5′ end were not commercially available at that time. These procedures are laborious and time-consuming, encumbering the synthesis and evaluation of other cMOs. Second, generalizable guidelines for the design of hairpin cMOs were not evident, as only one inhibitory sequence and structural configuration was tested. To resolve these challenges we synthesized a DMNB-based bifunctional linker that can be used to conjugate the targeting MO and its complementary inhibitor in only three steps, starting with appropriately functionalized, commercially available MO oligomers (Ouyang et al., 2009). We have utilized this optimized synthetic route to systematically analyze the in vitro thermodynamics and in vivo efficacy of various cMO hairpin structures and established design criteria for optimizing cMO activity profiles. These advances have enabled us to prepare photoactivatable reagents targeting the zebrafish genes no tail a (ntla) (Halpern et al., 1993), heart of glass (heg) (Mably et al., 2003), floating head (flh) (Talbot et al., 1995), ets variant gene 2 (etv2) (Sumanas and Lin, 2006), SRY-box containing gene 32 (sox32) (Alexander et al., 1999), and α–catenin (ctnna) (Cerda et al., 1999). We further demonstrated the versatility of our caging approach by replacing the DMNB chromophore with a bromohydroxyquinoline (BHQ) group, which has a significantly greater cross section for two-photon excitation (Fedoryak and Dore, 2002; Ouyang et al., 2009).
In this chapter, we describe detailed methods for the design, synthesis, and utilization of cMO hairpins based on our most recent publication (Ouyang et al., 2009). We also describe synthesis of cFD from commercially available precursors for co-injection with cMOs to enable tracking of the irradiated cells.
II. Design and Synthesis of cMOs
A. Design of cMOs
As described above, cMOs are composed of a 25-base, RNA-targeting MO tethered to a complementary MO oligonucleotide, thereby generating a stem-loop structure through intramolecular base pairing that abrogates its binding to RNA targets (Fig. 1). Linker photolysis converts the cMO hairpin into a MO duplex with significantly weaker, intermolecular base-pairing interaction that permits duplex dissociation and MO/RNA hybridization. For optimum performance, the cMO hairpin should therefore exhibit certain biophysical properties. There must be enough intramolecular binding energy within the hairpin to minimize strand exchange and intermolecular binding to RNA. However, cMO hairpins with too much intramolecular binding energy, once photolyzed, will form MO duplexes with significant intermolecular binding energy and strand-exchange with RNA will be inhibited. This delicate balance of binding energy also underscores the requirement for an inhibitory oligomer with a MO backbone, as cMOs containing DNA or RNA inhibitors can exhibit high basal gene-silencing activities, presumably due to hydrolysis of the phosphate backbone in vivo (S. Sinha and J. K. Chen, unpublished observations).
To establish general principles for cMO design, we systematically investigated the thermodynamic properties of MO/MO and MO/RNA base pairing, both in vitro and in vivo. The intra- and intermolecular interactions of MO duplexes were compared, and as a result of these studies, Eq. 1 was developed as a general formula for calculating the melting temperature (Tm) for intermolecular MO duplexes (Ouyang et al., 2009).
| Eq. 1 |
We find that this simple formula holds true for MO duplexes containing up to 16 base pairs, and longer duplexes may need further analysis of nearest-neighbor effects for accurate Tm estimation. We also determined that cMO hairpins exhibit optimum efficacy in vivo when they are based on MO duplexes with a Tm of 41–44 °C and adopt blunt-ended rather than staggered stem-loops. For example, we found that an optimum cMO hairpin against ntla should be made from the following MOs:
For other MO sequences, bases can be added or removed from the 5′ end of the inhibitor MO to achieve a MO duplex Tm of 41–44 °C, along with removal of the 3′-most base in the inhibitor sequence, if necessary. Typical inhibitor MOs are 10–13 bases in length.
B. Synthesis of a Propargyl-Functionalized, Photocleavable Crosslinker
The bifunctional, photocleavable crosslinker can be synthesized from commercially available 6-nitrovertaldehyde in ten steps (Scheme 1A). Detailed procedures for steps 1–5 (Shestopalov et al., 2007) and 6–10 (Ouyang et al., 2009) have been published, and the synthetic yield for step 5 can be further improved by performing the reaction in a pressure tube at 80 °C. Although the cis and trans forms of the crosslinker carbamate (compounds after step 7) are separable by silica gel chromatography, it is not necessary to isolate the individual stereoisomers them since they will slowly interconvert. The final product was column-purified to remove all trace impurities and stored as a lyophilized solid in single-reaction aliquots (1.1 mg for reactions on 100 nmol scale) at −20 °C to prevent NHS ester hydrolysis during freeze-thaw cycles. All reactions and synthetic manipulations were done under ambient light without noticeable decomposition, but the photolabile carbamates were shielded from light and kept at −20 °C during long-term storage.
Scheme 1. Crosslinker syntheses.

Synthetic procedures for preparing the propargyl-functionalized, photocleavable crosslinker (A) and azide-functionalized crosslinker (B). TMS = trimethylsilane, Ts = tosyl, DIPEA = diisopropyl ethyl amine, CDI = carbonyl diimidizole, DSC = disuccinimidyl carbonate. Adapted with permission (Ouyang et al., 2009; Copyright 2009, American Chemical Society).
C. Synthesis and Purification of cMOs
1. Synthesis of an Azide-Functionalized Crosslinker
Preparation of an azide- and NHS ester-containing crosslinker can be accomplished in two steps from commercially available 3-iodopropionic acid (Scheme 1B), and detailed synthetic procedures have been published (Shestopalov et al., 2007). The final product was stored dry in single-use aliquots (0.5 mg for 100-nmol-scale reactions) at −20 °C to minimize hydrolysis of the activated ester.
2. NHS Ester Coupling with Amine-Modified MO Oligomers
To construct cMO hairpins, MO oligomers with a 3′ amine modification and inhibitory MO oligomers with a 5′ amine modification were obtained from Gene-Tools, LLC in 300-nmol quantities and acylated with the appropriate NHS-containing linkers (Scheme 2). Each MO was dissolved in 300 μL of water. In general, we recommend using a water bath sonicator to help dissolve MOs. Actual oligomer concentration was determined according to the manufacturer’s instructions by diluting 2 μL of oligomer solution 40-fold in 0.1 M HCl, heating to 100 °C for 1 min to dissociate aggregates, cooling to ambient temperature, and measuring absorbance at 265 nm using a NanoDrop™ UV-VIS spectrometer. MO solutions prepared in this manner are typically in the 0.7–0.9 mM concentration range.
Scheme 2. cMO synthesis.

(i) Azide-functionalized crosslinker, 0.1 M sodium borate (pH 8.5), DMSO, 80–90% after purification. (ii) Propargyl-functionalized, photocleavable crosslinker, 0.1 M sodium borate (pH 8.5), DMSO, 70–90% after purification. (iii) sodium ascorbate, TBTA, CuI, 0.1 M potassium phosphate (pH 8.0), DMSO, 10–25% after HPLC purification. Adapted with permission (Ouyang et al., 2009; Copyright 2009, American Chemical Society).
All MO conjugation reactions and manipulations were carried out in standard 1.5-mL polypropylene microcentrifuge tubes. Prior to acylation, the amine-functionalized MOs were lyophilized to dryness, dissolved in 0.1 M Na2B4O7 (pH 8.5), heated to 100 °C for 1 min to dissociate aggregates, and allowed to cool to ambient temperature. To prepare the 3′ azide 25-base targeting MO, one 0.5-mg aliquot of NHS ester-functionalized azide was dissolved in 20 μL of DMSO and added to the aqueous solution containing 100 nmol of amine-modified MO in 0.1 M Na2B4O7 buffer. The 5′ amine-modified inhibitor MO was functionalized with the DMNB-containing crosslinker in a similar manner; one 1.1-mg aliquot of the propargyl-functionalized NHS ester was dissolved in 15 μL of DMSO and added to the buffered solution containing 100 nmol of the inhibitor MO. In this latter case, a cloudy suspension initially forms and later disappears as the reaction proceeds. Coupling reactions were vortexed overnight in foil-wrapped microcentrifuge tubes, resulting in quantitative conversion to the acylated MOs.
3. Purification of Functionalized MO Oligomers
The 3′ azide-functionalized targeting MO was purified from unreacted NHS ester and its hydrolyzed byproduct by passing the reaction mixture through a disposable size-exclusion column. One illustra NAP™-10 column (GE Life Sciences 17–0854) was typically used for each 100 nmol-scale reaction mixture, according to the manufacturer’s instructions. Columns were equilibrated with water and the reaction mixture was diluted to 1 mL with water prior to loading onto the column. The MO-containing eluent was lyophilized to dryness and dissolved in 100 μL water, and the final MO concentration was determined as described in Section C2. This purification method typically yielded at least 50 nmol of azide-functionalized MO.
The inhibitor MO-crosslinker conjugate was more difficult to purify as the relatively large DMNB-containing linker is not completely removed by size-exclusion column chromatography. Therefore, following product elution from the NAP™-10 column and lyophilization (as described above for the 25-base targeting MO), the inhibitor MO was dissolved in 400 μL of water and acidified with 4 μL of glacial acetic acid to protonate any hydrolyzed linker. The aqueous solution was washed three times with chloroform (400 μL) and twice with ethyl acetate (400 μL) to remove any unreacted or hydrolyzed crosslinker. These washes were performed in standard microcentrifuge tubes by vortexing the aqueous/organic mixture for 30 sec followed by centrifugation (16,000 g, 1 min) and disposal of the organic layer. The aqueous/organic interface, often containing cloudy precipitate, was carried through to increase purification yield. Care should be taken not to dispose of the MO-containing aqueous layer during the ethyl acetate washes, as unlike chloroform, ethyl acetate is less dense then water. Following the organic washes, the aqueous layer acidity was quenched by adding 20 μL of 10% ammonium hydroxide, and MO solution alkalinity was confirmed by spotting 0.5 μL of the aqueous solution on pH paper. Failure to basify the MO solution results in oligonucleotide degradation during lyophilization. The mixture of MO, ammonium acetate, and water was lyophilized to dryness, and residual ammonium acetate was removed by two cycles of MO solubilization in 200 μL of water and lyophilization to dryness. The purified inhibitory MO was then dissolved in 400 μL of 0.1 M KH2PO4 (pH 8), and oligomer concentration was spectroscopically quantified as described in Section C2. This purification method typically yielded at least 40 nmol of DMNB-functionalized inhibitor MO.
Derivatization of MOs with the azide- or propargyl-functionalized linkers should be confirmed by mass spectrometry prior to click-chemistry conjugation. We used a Waters MicroMass ZQ liquid chromatography-mass spectrometry (LC-MS) system with a Zorbax™ SB-C18 2.1 × 30 mm column (Agilent 823700–902) and a two-solvent gradient system (solvent A: water with 0.1% formic acid; solvent B: CH3CN with 0.1% formic acid; 2–95% B in 20 min; flow rate of 0.3 mL/min). The inhibitor MO modified with the propargyl-containing, photocleavable linker typically had two masses from the same LC peak due to sample photolysis as it passed through the UV flow-cell prior to mass analysis. Accurate mass spectral data could be obtained with as little as 0.5 nmol of MO oligomer.
4. Assembly of cMOs via Azide-Alkyne Huisgen Cycloaddition
Azide-alkyne Huisgen cycloaddition, commonly referred to as ‘click chemistry’, was used to assemble cMO hairpins (Scheme 2) (Rostovtsev et al., 2002). For this reaction, the azide-functionalized 25-base MO was lyophilized to dryness, to which an equimolar amount of the propargyl-functionalized inhibitor MO dissolved in KH2PO4 buffer (pH 8) was added. The alkalinity of resulting solution was checked after mixing, and 1–2 μL of 1 M KOH was added to maintain a pH of 8 if necessary. The MO mixture was heated to 100 °C for 1 min to dissociate oligonucleotide aggregates and then cooled to room temperature. Solutions of 20 mM CuI in DMSO and 20 mM sodium ascorbate in 0.1 M KH2PO4 (pH 8) were prepared immediately prior to start of the reaction, whereas a stock solution of 20 mM tris[(1-benzyl-1H-1,2,3-triazol-4-yl)methyl]amine (TBTA) in DMSO was prepared ahead of time and stored at −20 °C between uses. The azide-alkyne coupling reaction was initiated by the addition of sodium ascorbate, TBTA, and CuI, in that order (25 μL of each 20 mM stock solution per 50 nmol-scale reaction), to the 1.5 mL microcentrifuge tube containing the solution of the two MOs. The resulting reaction mixture was briefly sonicated, protected from light with aluminum foil, and then stirred overnight with a small magnetic stir bar.
5. Purification of cMOs
After completion of the azide-alkyne coupling reaction, the stir bar was removed and consumed, insoluble catalyst was pelleted by centrifugation (16,000 g, 1 min). The supernatant was then desalted by passage through a size-exclusion column in analogy to the procedure described in Section C3. For this step we typically used one GE Life Sciences illustra NAP™-5 column per 25 nmol of MO reaction mixture according to the manufacturer’s instructions. The columns were equilibrated with water and the reaction mixture was diluted to 0.5 mL with water prior to sample loading onto the column. The crude cMO-containing eluent was lyophilized to dryness and dissolved in 100 μL of 0.02 M NaOH. The pH of the resulting solution was checked, and 1–3 μL of 1 M NaOH was added if necessary to achieve a pH greater than 12.
Our inability to resolve MO mixtures by reverse-phase and size-exclusion high-pressure liquid chromatography (HPLC) led us to develop a method for the efficient separation of cMO hairpins from unconjugated MOs using ion-exchange HPLC. Though MOs are uncharged oligomers, adenosine and thymidine bases become deprotonated (and negatively charged) at pH 11 and higher, allowing for HPLC separation with a DNAPac™ PA100 22 × 250 mm column (Dionex 043011) and a two-solvent gradient system (solvent A: 0.02 M NaOH, 1% CH3CN; solvent B: 0.02 M NaOH, 0.375 M NaClO4, 1% CH3CN). The CH3CN co-solvent was used to decrease hydrophobic interactions between MOs and the column media, while the large perchlorate anion was used to effectively compete with ionic MO/media interactions.
To purify the reaction mixture, 2 μL of the crude cMO product was first resolved on the DNAPac™ PA100 column by running a 27-min 7–50% solvent B gradient with a flow rate of 4 mL/min. As expected, MOs eluted in order of A/T base content with the cMO eluting last (Fig. 2A). MO absorbance was detected at 260 nm, and the photocleavable chromophore was detected at 347 nm. Elution time for the last unconjugated MO peak (Point X, Fig. 2A) and the beginning of the cMO hairpin peak (Point Y, Fig. 2A) was noted and converted to gradient composition (percentage of solvent B) at X and Y. An empirically derived correction factor was then subtracted from both X and Y to compensate for the solvent volume between gradient mixer and UV flow cell (7% solvent B for our HPLC system), to give the values X* and Y* (Fig. 2B). An elution method was then programmed consisting of: 7 to X*% solvent B in 5 min, X* to Y*% B in 10 min, Y* to 50% B in 1 min, and 50% B for 10 min (Fig. 2B). The 10-min pause between gradient points X* and Y* is intended to allow the elution of all unconjugated MOs, while the sharp gradient ramp from point Y* to 50% B should elute the cMO in one narrow peak. The composed method was verified using another 2 μL of the crude MO product (Fig. 2B). The unconjugated MO peak should be completely eluted from the column before the cMO hairpin is obtained as one symmetrical peak with a half-height temporal width of 30 sec or less. Gradient parameters X* and Y* were fine-tuned to meet these criteria, if needed.
Figure 2. Purification of a cMO hairpin.

(A) Chromatogram of a crude cMO mixture separated by preparative ion-exchange HPLC using a continuous solvent gradient. MO oligomers eluted in order of increasing adenosine and thymidine content, with the cMO eluting last (highlighted in gray). The dashed line represents gradient composition (% solvent B), and the retention time and gradient composition associated with points X and Y are noted. (B) Chromatogram of the same crude cMO mixture separated by preparative ion-exchange HPLC using a step-wise solvent gradient and empirically derived transition points X* and Y*. The cMO eluted as a symmetric peak with a half-height temporal width of approximately 20 sec. (C) Chromatogram of the cMO isolated by the step-wise solvent gradient in (B) and analyzed by analytical ion-exchange HPLC using a continuous solvent gradient. The cMO purity was determined to be 98.8% and its molecular mass was confirmed by LCMS.
Once a separation method was developed, up to 50 nmol of the MO mixture was loaded onto the column using a 100-μL injection loop. It is important to turn off the UV-VIS detection system during this purification step as UV light readily photolyzes the cMO hairpin. Fractions were collected every 15 sec starting at the 16-min point in the HPLC run, with cMOs typically eluting in 2–3 fractions. cMO-containing fractions were identified by UV absorbance and buffered by addition of 40 μL of 1 M NH4OAc (pH 5). After each fraction was confirmed to have a final pH of less than 7, they were combined and lyophilized to dryness. Failure to buffer these fractions resulted in linker cleavage via base-catalyzed elimination of the triazole group.
Purified cMOs were redissolved in 500 μL of water and desalted using one illustra NAP™-5 column (GE Life Sciences 17–0853) according to the manufacturer’s instructions. The columns were equilibrated with water prior to sample loading. The cMO-containing eluent was lyophilized to dryness, dissolved in 50 μL of water, and centrifuged (16,000 g, 2 min) to remove any particulates derived from size-exclusion medium. The cMO was then precipitated with 400 μL of acetone, vortexed, and centrifuged (16,000 g, 20 min, 4 °C). The precipitated cMO usually appeared as an oil in the microcentrifuge tubes that was difficult to see, and care was taken to remove most of the acetone-containing supernatent without touching the walls of the microcentrifuge tube. The cMO precipitate was then washed with 100 μL of CH3CN, briefly sonicated, and centrifuged (16,000 g, 5 min, 4 °C). The CH3CN was aspirated and the off-white cMO pellet was lyophilized for 10 min to remove trace solvent. The cMO pellet was then dissolved in 100 μL of water, heated at 100 °C for 30 sec to dissociate aggregates, cooled to room temperature, and cMO concentration was determined by the 265-nm absorbance of an aliquot dissolved in 0.1 M HCl. This purification typically yielded 5–10 nmol of pure cMO. The cMO solution should be centrifuged again (16,000g, 20 min) at room temperature to pellet insoluble particulates that can clog microinjection needles. Supernatant containing cMO was then transferred into an O-ring-capped 1.5 mL microcentrifuge tube, reduced in volume to 20 μL to facilitate the preparation of injection solutions, and stored in the dark at −20 °C. Brief exposures of the cMO solution to ambient light did not cause detectable photolysis of the cMO linker.
Prior to its use in vivo, cMO mass and purity should be confirmed by LC-MS and ion-exchange HPLC. LC-MS was performed as described in Section C4. Ion-exchange HPLC was performed using a DNAPac™ PA200 4 × 250 mm column (Dionex 063000) using a gradient of 7–50% solvent B in 27 min with a flow rate of 1.2 mL/min, using the two-solvent system described in Section C5. At least 0.3 nmol of cMO should be used for HPLC analysis, and a greater then 98% purity should be observed (Fig. 2C).
III. Synthesis of cFD
Since cFD is no longer commercially available, we prepared this reagent using a commercially available carboxymethylnitrobenzyl (CMNB)-caged fluorescein NHS ester (Invitrogen C20050) and 10-kDa aminodextran (Invitrogen D1860) (Scheme 3). Approximately 3.5–5 mg of the aminodextran dissolved in 500 μL 0.1 M Na2B4O7 buffer (pH 8.5) was added to 1 mg of caged fluorescein in its Invitrogen-supplied, tinted microcentrifuge tube, and the reaction mixture was vortexed overnight. The resulting caged fluorescein dextran was separated from unreacted caged fluorescein using a Zeba Desalt spin column (Pierce 89889) according to the manufacturer’s instructions. The yellow-colored eluent was lyophilized to dryness, weighed, dissolved in water to make a 1% (w/v) stock solution, and stored at −20 °C in 2-μL aliquots to avoid detrimental freeze-thaws. Spectroscopic analysis indicated an average loading of 2.5 caged fluorescein molecules per 1 molecule of dextran. The 1% cFD solution was typically diluted 10–20 fold for microinjection into zebrafish embryos.
Scheme 3. cFD synthesis.
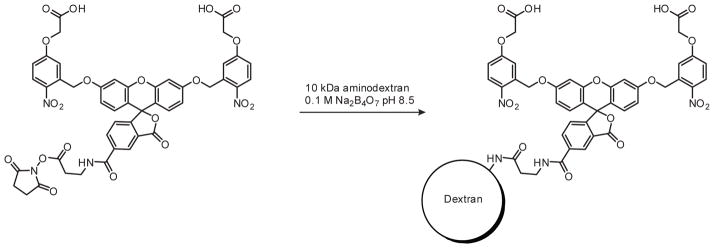
cFD was obtained in 68% yield after purification, with an average loading of 2.5 caged fluorescein molecules per 1 molecule of amminodextran.
IV. Microinjection of Caged Reagents
Injection solutions of caged reagents were prepared in Milli-Q water containing 0.1% phenol red and 100 mM KCl. cMO microinjection solutions (with or without cFD) were heated to 100 °C for 15 sec to dissociate MO aggregates and centrifuged (16,000g, 2 min, 4 °C). As with conventional MOs, cMOs and cFD were microinjected either into the animal cell or the yolk prior to 8-cell stage using previously described procedures (Ekker, 2004).
Accurate microinjection dosing is essential for the successful use of cMOs, as overdosing will recapitulate weak morphant phenotypes from hybridization between the cMO hairpin and RNA. Therefore, O-ring capped microcentrifuge tubes should always be used for handling injection solutions to avoid changes in concentration due to evaporation. The microinjection volume should also be calibrated to deliver accurate cMO doses. We recommend quantifying injection volume by the mineral oil droplet method (Nüsslein-Volhard and Dahm, 2002). In this procedure, mineral oil is placed in a glass depression slide and droplets of reagent-containing injection solution are microinjected into the oil. A stereoscope equipped with a micrometer is then used to measure the diameter of each spherical droplet, allowing the injection volume to calculated. We have not observed photolysis of cMOs or cFD from the incandescent light source on the microinjection stereoscope, though uncaging can occur with more sensitive photocleavable groups, such as the BHQ chromophore in our two-photon-compatible cMOs. To minimize compound photolysis during the microinjection procedure, the stereoscope stage can be covered with UV light-absorbing plastic, such as the 3M™ Sun Control window film.
V. Global Photoactivation of Caged Reagents
A. Embryo Mounting and Irradiation
Appropriate UV irradiation protocols are critical for reproducible photoactivation of caged reagents. The light source must emit photons of the correct wavelength to drive the photochemical reaction over the threshold activation energy. Light must also have sufficient irradiance (intensity) to attain a high probability of collision between photons and all photo-reactive molecules, thereby completing the photochemical reaction within seconds while minimizing UV damage. To achieve these conditions, we typically performed global irradiations on a Leica DM4500B upright compound epifluorescent microscope equipped with an HBO 100W mercury short-arc lamp, an A4 filter cube (360 ± 40 nm; Leica 11513874) and a 20x water-immersion objective (0.5 numerical aperture (NA); Leica 506147). We have found that irradiation with a fluorescent stereoscope (such as the Leica MZFLIII or M205FA) or a handheld UV lamp does not provide enough light intensity for efficient photoactivation. Uniform global photoactivation of caged molecules is also difficult to achieve on a confocal microscope due to the limited depth of field and short dwell time as the laser beam scans across the sample.
We have also observed that embryo placement and orientation significantly influences uncaging efficiencies. To facilitate zebrafish embryo irradiation, we typically immobilized chorionated embryos in agarose microinjection templates, orienting them on a stereoscope so that the animal cells face upward. The embryos were then irradiated one at a time on the Leica DM4500B microscope (Fig. 3A). We used microinjection templates with individual, evenly spaced wells as opposed to troughs, leaving an empty well between embryos to avoid inadvertent exposure to scattered UV light. When globally irradiating blastula-stage embryos, some rotated out of the desired animal pole orientation and had to be re-oriented on the stereoscope. Irradiating mis-oriented blastula may result in the bottom-most cells (those furthest away from the light beam) receiving lower levels of UV light due to shielding by the overlying cells. This effect was observed in embryos microinjected with mRNA encoding the Kaede protein, which undergoes a green-to-red fluorescence photoconversion upon exposure to 360-nm light (Ando et al., 2002). When such embryos were irradiated from the anterior at 7 hpf, the cells below the ‘equator’ plane of the embryo had less photo-converted Kaede protein then cells above the equator (Fig. 3B). However, when the same embryo was further irradiated laterally, red photoconverted Kaede protein was present evenly throughout the embryo (Fig. 3C). Uniform irradiation is therefore paramount to achieving consistent global photoactivation.
Figure 3. Global irradiation of zebrafish embryos.
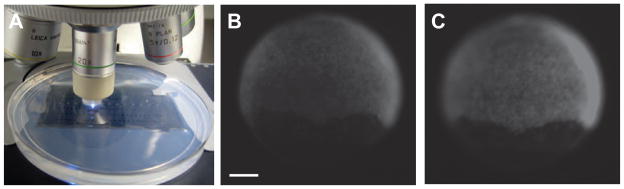
(A) Whole-embryo irradiation of zebrafish mounted in an agarose template using a Leica DM4500B upright fluorescence microscope equipped with a 20x, 0.5 NA water-immersion objective. (B) An embryo injected with mRNA encoding the photoconvertible protein Kaede and globally irradiated from the animal pole for 10 sec at 7 hpf. Fluorescence imaging revealed more Kaede photoconversion at the animal pole then at the margin. (C) The same embryo globally irradiated laterally, as mounted in (B), for an additional 10 sec, resulting in even distribution of photoconverted Kaede protein. Scale bar: 200 μm.
B. Optimization of UV Light Duration Using cFD
Since the CMNB caging group in cFD is chemically similar to the DMNB moiety in the cMO linker (Goeldner and Givens, 2005; Kobayashi et al., 2007), cFD can be used to estimate the irradiation duration that most efficiently photoconverts the cMO on a epifluorescence microscope system equipped with a specific set of illumination systems, optical filters, and objectives. In such experiments, cFD-microinjected embryos were globally irradiated at approximately 2 hpf as described above. UV light intensity was kept at 100% while irradiation duration was incrementally increased until maximum fluorescence intensity is achieved (all fluorescein was uncaged). With our 20x, 0.50 NA water-immersion objective, fluorescein signal intensity reached saturation after a 10-sec irradiation. Experimental parameters for other objectives can be estimated using the following equation describing the relationship between light intensity, NA, and magnification (Eq. 2).
| Eq. 2 |
Therefore a 20x lens with a lower NA (such as 0.40) produces UV light with 36% less intensity and disproportionally longer exposures may be required, possibly up to 20 sec. The calibration of UV irradiation duration should therefore be performed with each epifluorescence microscope prior to its use for cMO uncaging. Typical irradiation durations are in the 10–30 sec range when the microscope is capable of producing UV light with sufficient intensity to readily uncage cFD and cMO reagents.
C. Minimization of UV Phototoxicity
UV light can be damaging to zebrafish embryos, especially during blastula stages. Once the sufficient UV light exposure for cFD photoactivation was determined (10 sec with our microscope system), embryos microinjected with cFD were globally irradiated as above at 2.5 hpf and raised to 1 day post fertilization (dpf). This dose of UV light was adequately tolerated as 67% of the embryos appeared to develop normally (Fig. 4A–B, E). However, embryos globally irradiated for 15 sec or longer at 2.5 hpf had increasingly more gastrulation delays, necrotic tissues, and death by 1 dpf, indicating potential phototoxicity (Fig. 4C–E). In contrast, irradiation of wildtype embryos at 2.5 hpf or cFD-injected embryos at 4 hpf for 20 sec did not produce any noticeable toxicity (Fig. 4F). It is therefore preferable to perform global irradiations at the gastrula stage or later as blastula-stage embryos are more susceptible to UV damage. Irradiation procedures that minimize phototoxicity should therefore be established empirically on each individual microscope system prior to using caged reagents for biological studies. These assessments should also be conducted with embryos microinjected with cFD and/or cMO, as the uncaging reaction itself can be associated with some cytotoxicity (Fig. 4F).
Figure 4. UV light-induced toxicity in zebrafish embryos.

Representative micrographs of embryos microinjected with 2 nL of a 0.05% cFD solution and irradiated for 0 (A), 10 (B), 15 (C), and 20 (D) sec at 2.5 hpf. Embryos in (C) and (D) have morphological defects resulting from UV light damage. Scale bar: 200 μm. (E) Levels of embryonic toxicity and lethality by 1 dpf associated with different durations of UV irradiation at 2.5 hpf. (F) Levels of embryonic toxicity and lethality by 1 dpf associated with global, 20-sec UV irradiation. Wildtype and gastrula-stage, cFD-injected embryos were less susceptible to UV light damage than blastula-stage, cFD-injected embryos.
VI. Titration of cMO Dose for Optimum Dynamic Range
Since accurate reagent dosing is essential for cMO efficacy, it is important to perform a careful titration with the 25-base targeting MO to find the minimum effective dose. For example, in preparation for our studies with a ntla cMO, we systematically assess the embryonic phenotypes associated with different doses of the conventional ntla MO. Embryos lacking the ntla T-box transcription factor lack the notochord, are posteriorly truncated, and exhibit U-shaped rather than V-shaped somites (Halpern et al., 1993; Schulte-Merker et al., 1994). Mutants and morphants lacking ntla function also exhibit ectopic medial floor plate, a ventral region of the developing spinal cord, and it is believed that ntla acts as a transcriptional switch between notochord and medial floor plate cell fates (Amacher et al., 2002; Halpern et al., 1997). Phenotypes resulting from loss of ntla function can be categorized into four classes according to their severity: class I = a fully penetrant ntla mutant phenotype characterized by no notochord, U-shaped somites, and a lack of posterior structures; class II = no notochord, U-shaped somites, and some posterior somites; class III = incompletely vacuolated notochord, V-shaped somites, and a shortened anterior-posterior axis; and class IV = wildtype phenotype (Ouyang et al., 2009). These phenotypes can be recapitulated by microinjecting varying doses of the conventional ntla MO (GACTTGAGGCAGACATATTTCCGAT), and class I phenotype was found to require a minimum dose of 115 fmol/embryo (Fig. 5).
Figure 5. Classification of ntla morphant phenotypes.

(A) Morphology of phenotype classes I-IV at 1 dpf. Somitic (s), medial floor plate (mfp), notochord (nc), and yolk extension (ye) tissues are labeled. Scale bars: top panels, 200 μm; bottom panels, 50 μm. 115 fmol is equal to 1 ng of ntla MO. (B) Phenotypic distribution associated with different embryonic doses of the ntla MO. (C) Immunoblots showing Ntla protein levels in 10-hpf zebrafish embryos microinjected at the one-cell stage with various doses of the ntla MO. β-Actin levels are shown as a loading control. Adapted with permission (Ouyang et al., 2009; Copyright 2009, American Chemical Society).
The minimum dose of the ntla MO that produced class I phenotype (115 fmol/embryo) served as a starting point for titration of the ntla cMO. Following microinjection of the ntla cMO, half of the embryos were globally irradiated for 10 sec at 2 hpf as described in Section 5. At 1 dpf, the irradiated embryos had a mixture of class I and class II ntla phenotypes, whereas the unirradiated embryos appeared wildtype (class IV) (Fig. 6). The lack of fully restored MO activity upon uncaging was not surprising, as photochemical reactions do not typically go to completion due to competing, inactivating side-reactions. When the ntla cMO dose was increased to 230 fmol/embryo, the desired activity profile was achieved, with global irradiation producing predominately class I embryos (Fig. 6). Higher doses of ntla cMO resulted in appearance of class III phenotypes in unirradiated embryos (Fig. 6). The 230 fmol/embryo dose was therefore used along with a 10-sec irradiation protocol for all subsequent studies.
Figure 6. Dose-dependent activity of the ntla cMO.
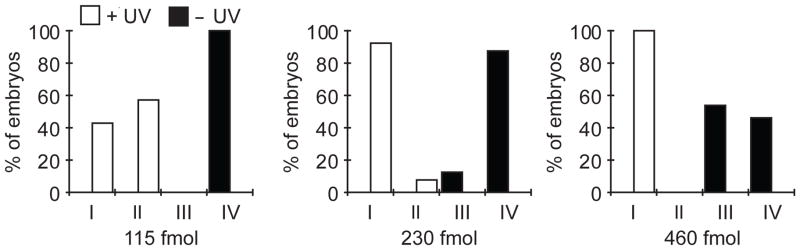
Global irradiations were performed at 2.5 hpf and phenotypes were scored at 1 dpf according to the phenotype classes described in Figure 5.
VII. Localized Photoactivation of Caged Reagents
A. Spatially Restricted UV Illumination Using a Photomask
Localized photoactivation can be performed on a compound epifluorescence microscope by reducing the size of the illumination field diaphragm. For example, our Leica DM4500B equipped with a 20× 0.5 NA water-immersion objective described in Section 5A can illuminate regions as small as a 100 μm-diameter circle or a 200 × 300 μm rectangle using its adjustable diaphragm. The spatial limits of this irradiation can also be visually confirmed using either cFD or the photoconvertible protein Kaede (Fig. 7A–B). Irradiation of smaller features or more complex patterns can be achieved using higher magnification objectives and/or micromirror array systems such as the Mosaic Digital Illumination System (Photonic Instruments).
Figure 7. Localized irradiation of zebrafish embryos.
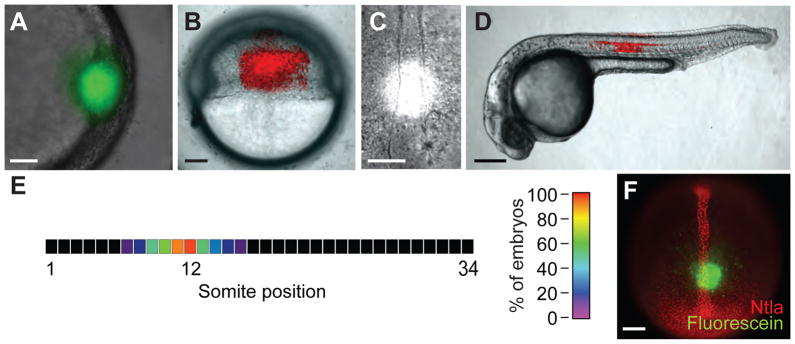
(A) Embryos injected with 2 nL of a 0.05% cFD solution were irradiated within the shield at 6 hpf for 10 sec using a circular photomask (100-μm diameter). As expected, a circular region of green fluorescence was immediately apparent in the targeted region. (B) Embryos injected with 50 pg of Kaede mRNA and irradiated laterally at 6 hpf for 10 sec using a rectangular photomask (200 × 300 μm). A rectangular region of red fluorescence was immediately observed in the targeted region. (C) Brightfield micrograph of a 10-hpf embryo undergoing UV irradiation through a circular, 100-μm-diameter photomask positioned 100 μm above the posterior end of the chordamesoderm. Grid overlays using Metamorph® software are not shown. (D) Embryos injected with 50 pg of Kaede mRNA and locally irradiated as in (C). A red fluorescent region of notochord and floor plate cells centered around the twelfth somite was visible at 1 dpf. (E) Heat map demonstrating the precision with which zebrafish embryos can be locally irradiated as described in (C). The average location of red fluorescent notochord cells along the anterior-posterior axis resulting from the targeted irradiation of 10-hpf embryos is shown (n = 18 embryos). (F) Fluorescence micrograph of a 10-hpf embryo injected with cFD, irradiate as described in (C), immediately fixed with paraformaldehyde, and immunostained with anti-Ntla and anti-fluorescein antibodies. A circular region of uncaged fluorescein was detected within the Ntla-expressing chordamesoderm, 100 μm anterior to the tailbud. Scale bars: A and C, 50 μm; B, 100 μm; D, 200 μm; F, 100 μm.
Precise irradiation targeting is a prerequisite for reproducible photoactivation experiments, requiring methods for efficiently immobilizing of live embryos and targeting specific morphological features with UV light. To mount and orient gastrulation-stage zebrafish we used the same agarose microinjection template as that described in Section 5 for our global irradiation studies and kept the embryos in their chorions. Segmentation-stage embryos were dechorinated and placed in agarose templates cast with 0.018 inch-wide channels, whereas pharyngula-stage larva were immobilized in MegaMounts (https://wiki.med.harvard.edu/SysBio/Megason/MegaMounts) and treated with tricaine. Irradiating specific tissues in a consistent manner was then achieved by using the combined brightfield and fluorescence illumination mode on the Leica DM4500B microscope. For example, to irradiate a 100 μm -diameter circular region within the chordamesoderm in 10 hpf embryos, a 50 × 50 μm grid was digitally overlayed onto the live preview window using MetaMorph® software, grid lines were aligned to the center of the fluorescent mask, and immobilized embryos oriented using the grid lines to enable irradiation of the chordamesoderm lying 100 μm from the posterior boundary (Fig. 7C). When 18 Kaede-expressing embryos were irradiated in this manner, by 1 dpf they displayed a region of red-fluorescent notochord cells tightly clustered within the same position along the anterior-posterior axis (Fig. 7D–E).
B. Determination of Protein Levels in Targeted Tissues
Since cMOs inhibit the splicing or translation of their targeted RNAs, the rate by which the encoded protein is degraded will determine the time point when gene function is actually lost after cMO photoactivation. For this reason, accurately interpreting cMO-induced phenotypes requires an assessment of protein levels for the targeted gene in the irradiated cells. This can be accomplished by immunodetection technologies, using antibodies that recognize the protein of interest and an anti-fluorescein antibody (Roche 1426320) that specifically binds to uncaged cFD. For example, when embryos microinjected with cFD and irradiated as described in Section 7A were immediately fixed and immunostained, a 100 μm-diameter circular staining pattern was apparent, marking an irradiated region within the Ntla-expressing chordamesoderm (Fig. 7F). The immunostaining procedure used an anti-Ntla polyclonal antibody (1:1000 dilution) (Schulte-Merker et al., 1994) and a monoclonal anti-fluorescein antibody (1:200 dilution), as well as their corresponding secondary antibodies (anti-rabbit Alexa-Fluor 594 and anti-mouse Alexa-Fluor 488, each at a 1:200 dilution) according to standard whole-mount immunostaining protocols (Nüsslein-Volhard and Dahm, 2002). Identical irradiations were performed on embryos co-injected with the ntla cMO and cFD, followed by fixation and immunostaining at various time points. These collective experiments revealed the time frame in which Ntla levels are significantly diminished in the irradiated cells, allowing phenotypes associated with ntla cMO activation to be linked to loss of Ntla function.
VIII. Conclusion
The protocols for cMO design, synthesis, and application described in this chapter are intended to facilitate the use of these reverse-genetic tools in zebrafish and other organisms. The cMO technology is broadly applicable to the embryonic transcriptome, as illustrated by our development of cMOs targeting several patterning genes. In addition, the methods employed for global or spatially restricted cMO photoactivation could be generally applied to emerging optogenetic systems.
While cMOs exemplify the potential of synthetic reagents for in vivo studies, it is worth discussing some of the challenges that remain in the implementation of this technology. These include both practical and experimental limitations. For example, although the synthetic and purification procedures for cMO preparation are relatively straightforward and robust, they involve techniques and equipment that are not common in most biological laboratories. Further simplification of cMO design, synthesis, and purification procedures—or perhaps more ideally, commercialization of the cMO technology—would help promote the use of these reagents by the developmental biology community.
We have also experienced difficulty in caging certain MOs. In some cases this appears to be due to cytotoxicity associated with the cMO oligonucleotide, which is typically 10–13 bases longer than conventional MOs. For instance, we previously attempted to generate a spt cMO, using a 25-base targeting MO that exhibited a narrow range of effective doses; partial spt mutant phenotypes were obtained at a dose of 3 ng/embryo and embryonic toxicity was observed at higher MO concentrations (Ouyang et al., 2009). Tethering the inhibitory oligomer to the spt MO increased rather than mitigated reagent toxicity, and photoactivation of the spt cMO did not fully recapitulate the developmental defects induced by the conventional MO. Consistent with these results, quantitative models of cMO activity suggest that as the MO dose required for gene silencing increases, the functionally equivalent dose of photoactivated cMO rises disproportionately, as the inhibitory MO released by cMO photolysis can begin to interfere with RNA hybridization by the targeting MO (Ouyang et al., 2009). The greater potential for off-target effects associated with cMO hairpins may also limit their utility in studying larval gene function, since relatively large amounts of MO are frequently microinjected in these experiments to compensate for reagent dilution during later developmental stages.
MOs that potentiate UV light-induced toxicity are also not amenable to our caging strategy, as demonstrated by our attempts to generate a cMO against notch1a (I. A. Shestopalov and J. K. Chen, unpublished observations). When we injected the notch1a cMO and globally irradiated the embryos at 2.5 hpf, we observed severe UV-dependent defects during blastula formation and gastrulation that were not found in conventional notch1a morphants. Interestingly, morphologically identical defects occurred in embryos injected with the conventional notch1a MO and globally irradiated at 2.5 hpf, indicating that the targeting MO itself can increase embryonic sensitivity to UV light damage.
New strategies for cMO design and synthesis could help overcome these challenges. Approaches that minimize the cMOs cytotoxicity and the potential for UV light damage would be an important advances. In principle, this could be achieved by minimizing or eliminating the inhibitory MO portion of these reagents, replacing the nitrobenzyl-based photocleavable group with chromophores that undergo photochemical reactions with lower doses of UV light or at less damaging wavelength, devising methods that obviate the need to utilize cMOs at doses two-fold greater than that of targeting MO, and/or utilizing new oligonucleotide scaffolds as a less toxic substitute for MOs (Shestopalov and Chen, 2010). The utility of cMOs as functional genomic probes would also benefit from the development of new uncaging technologies. For example, enzymatically triggered cMOs could be used in combination with transgenic organisms to allow MO activation with a spatial precision and three-dimensional complexity that would be difficult to achieve with photomasks or even micromirror array-based illumination. Reversible control of MO function would similarly enable gene silencing with greater temporal dexterity, facilitating studies of how genes dynamically regulate embryonic patterning.
Achieving these advances will require a collaborative effort by chemists and developmental biologists, as well as scientists trained in both disciplines. Given the amenability of zebrafish to optical technologies, transgenesis, and chemical perturbations and the burgeoning use of molecular probes in zebrafish studies, we anticipate that the zebrafish community will play a leading role in these scientific explorations. Just as synthetic reagents such as MOs have transformed how we study zebrafish biology, zebrafish biology can inspire new ways in which chemistry can provide insights into biological processes.
Acknowledgments
We gratefully acknowledge financial support from the NIH Director’s Pioneer Award (DP1 OD003792), the NIH/NIGMS (R01 GM072600), the March of Dimes Foundation (1-FY-08-433), and the California Institute for Regenerative Medicine (T1-0001). Some figures and schemes in this chapter have been adapted from the authors’ previous publication (Ouyang et al., 2009) with permission from the American Chemical Society.
References
- Alexander J, Rothenberg M, Henry GL, Stainier DY. casanova plays an early and essential role in endoderm formation in zebrafish. Dev Biol. 1999;215:343–57. doi: 10.1006/dbio.1999.9441. [DOI] [PubMed] [Google Scholar]
- Amacher SL, Draper BW, Summers BR, Kimmel CB. The zebrafish T-box genes no tail and spadetail are required for development of trunk and tail mesoderm and medial floor plate. Development. 2002;129:3311–23. doi: 10.1242/dev.129.14.3311. [DOI] [PubMed] [Google Scholar]
- Ando H, Furuta T, Tsien RY, Okamoto H. Photo-mediated gene activation using caged RNA/DNA in zebrafish embryos. Nat Genet. 2001;28:317–25. doi: 10.1038/ng583. [DOI] [PubMed] [Google Scholar]
- Ando R, Hama H, Yamamoto-Hino M, Mizuno H, Miyawaki A. An optical marker based on the UV-induced green-to-red photoconversion of a fluorescent protein. Proc Natl Acad Sci U S A. 2002;99:12651–6. doi: 10.1073/pnas.202320599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bill BR, Petzold AM, Clark KJ, Schimmenti LA, Ekker SC. A primer for morpholino use in zebrafish. Zebrafish. 2009;6:69–77. doi: 10.1089/zeb.2008.0555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blidner RA, Svoboda KR, Hammer RP, Monroe WT. Photoinduced RNA interference using DMNPE-caged 2′-deoxy-2′-fluoro substituted nucleic acids in vitro and in vivo. Mol Biosyst. 2008;4:431–40. doi: 10.1039/b801532e. [DOI] [PubMed] [Google Scholar]
- Cerda J, Reidenbach S, Pratzel S, Franke WW. Cadherin-catenin complexes during zebrafish oogenesis: heterotypic junctions between oocytes and follicle cells. Biol Reprod. 1999;61:692–704. doi: 10.1095/biolreprod61.3.692. [DOI] [PubMed] [Google Scholar]
- Ekker SC. Nonconventional antisense in zebrafish for functional genomics applications. Methods Cell Biol. 2004;77:121–36. doi: 10.1016/s0091-679x(04)77007-7. [DOI] [PubMed] [Google Scholar]
- Fedoryak OD, Dore TM. Brominated hydroxyquinoline as a photolabile protecting group with sensitivity to multiphoton excitation. Org Lett. 2002;4:3419–22. doi: 10.1021/ol026524g. [DOI] [PubMed] [Google Scholar]
- Gee KR, Weinberg ES, Kozlowski DJ. Caged Q-rhodamine dextran: a new photoactivated fluorescent tracer. Bioorg Med Chem Lett. 2001;11:2181–3. doi: 10.1016/s0960-894x(01)00421-8. [DOI] [PubMed] [Google Scholar]
- Goeldner M, Givens R. Dynamic studies in biology: phototriggers, photoswitches and caged biomolecules. Wiley-VCH; Weinheim: 2005. [Google Scholar]
- Gradinaru V, Zhang F, Ramakrishnan C, Mattis J, Prakash R, Diester I, Goshen I, Thompson KR, Deisseroth K. Molecular and cellular approaches for diversifying and extending optogenetics. Cell. 2010;141:154–65. doi: 10.1016/j.cell.2010.02.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halpern ME, Hatta K, Amacher SL, Talbot WS, Yan YL, Thisse B, Thisse C, Postlethwait JH, Kimmel CB. Genetic interactions in zebrafish midline development. Dev Biol. 1997;187:154–70. doi: 10.1006/dbio.1997.8605. [DOI] [PubMed] [Google Scholar]
- Halpern ME, Ho RK, Walker C, Kimmel CB. Induction of muscle pioneers and floor plate is distinguished by the zebrafish no tail mutation. Cell. 1993;75:99–111. [PubMed] [Google Scholar]
- Kobayashi T, Urano Y, Kamiya M, Ueno T, Kojima H, Nagano T. Highly activatable and rapidly releasable caged fluorescein derivatives. J Am Chem Soc. 2007;129:6696–7. doi: 10.1021/ja070376d. [DOI] [PubMed] [Google Scholar]
- Kozlowski DJ, Weinberg ES. Photoactivatable (caged) fluorescein as a cell tracer for fate mapping in the zebrafish embryo. Methods Mol Biol. 2000;135:349–55. doi: 10.1385/1-59259-685-1:349. [DOI] [PubMed] [Google Scholar]
- Lippincott-Schwartz J, Patterson GH. Fluorescent proteins for photoactivation experiments. Methods Cell Biol. 2008;85:45–61. doi: 10.1016/S0091-679X(08)85003-0. [DOI] [PubMed] [Google Scholar]
- Mably JD, Mohideen MA, Burns CG, Chen JN, Fishman MC. heart of glass regulates the concentric growth of the heart in zebrafish. Curr Biol. 2003;13:2138–47. doi: 10.1016/j.cub.2003.11.055. [DOI] [PubMed] [Google Scholar]
- Nasevicius A, Ekker SC. Effective targeted gene ‘knockdown’ in zebrafish. Nat Genet. 2000;26:216–20. doi: 10.1038/79951. [DOI] [PubMed] [Google Scholar]
- Nguyen QN, Chavli RV, Marques JT, Conrad PG, Jr, Wang D, He W, Belisle BE, Zhang A, Pastor LM, Witney FR, Morris M, Heitz F, Divita G, Williams BR, McMaster GK. Light controllable siRNAs regulate gene suppression and phenotypes in cells. Biochim Biophys Acta. 2006;1758:394–403. doi: 10.1016/j.bbamem.2006.01.003. [DOI] [PubMed] [Google Scholar]
- Nüsslein-Volhard C, Dahm R. Zebrafish, A Practical Approach. Oxford University Press; Oxford: 2002. [Google Scholar]
- Ouyang X, Chen JK. Synthetic strategies for studying embryonic development. Chem Biol. 2010;17:590–606. doi: 10.1016/j.chembiol.2010.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouyang X, Shestopalov IA, Sinha S, Zheng G, Pitt CL, Li WH, Olson AJ, Chen JK. Versatile synthesis and rational design of caged morpholinos. J Am Chem Soc. 2009;131:13255–69. doi: 10.1021/ja809933h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remy S, Tesson L, Menoret S, Usal C, Scharenberg AM, Anegon I. Zinc-finger nucleases: a powerful tool for genetic engineering of animals. Transgenic Res. 2010;19:363–71. doi: 10.1007/s11248-009-9323-7. [DOI] [PubMed] [Google Scholar]
- Rostovtsev VV, Green LG, Fokin VV, Sharpless KB. A stepwise huisgen cycloaddition process: copper(I)-catalyzed regioselective “ligation” of azides and terminal alkynes. Angew Chem Int Ed Engl. 2002;41:2596–9. doi: 10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- Schulte-Merker S, van Eeden FJ, Halpern ME, Kimmel CB, Nusslein-Volhard C. no tail (ntl) is the zebrafish homologue of the mouse T (Brachyury) gene. Development. 1994;120:1009–15. doi: 10.1242/dev.120.4.1009. [DOI] [PubMed] [Google Scholar]
- Shestopalov IA, Chen JK. Chemical technologies for probing embryonic development. Chem Soc Rev. 2008;37:1294–307. doi: 10.1039/b703023c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shestopalov IA, Chen JK. Oligonucleotide-based tools for studying zebrafish development. Zebrafish. 2010;7:31–40. doi: 10.1089/zeb.2010.0650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shestopalov IA, Sinha S, Chen JK. Light-controlled gene silencing in zebrafish embryos. Nat Chem Biol. 2007;3:650–1. doi: 10.1038/nchembio.2007.30. [DOI] [PubMed] [Google Scholar]
- Sumanas S, Lin S. Ets1-related protein is a key regulator of vasculogenesis in zebrafish. PLoS Biol. 2006;4:e10. doi: 10.1371/journal.pbio.0040010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Summerton J. Morpholino antisense oligomers: the case for an RNase H-independent structural type. Biochim Biophys Acta. 1999;1489:141–58. doi: 10.1016/s0167-4781(99)00150-5. [DOI] [PubMed] [Google Scholar]
- Talbot WS, Trevarrow B, Halpern ME, Melby AE, Farr G, Postlethwait JH, Jowett T, Kimmel CB, Kimelman D. A homeobox gene essential for zebrafish notochord development. Nature. 1995;378:150–7. doi: 10.1038/378150a0. [DOI] [PubMed] [Google Scholar]
- Tang X, Maegawa S, Weinberg ES, Dmochowski IJ. Regulating gene expression in zebrafish embryos using light-activated, negatively charged peptide nucleic acids. J Am Chem Soc. 2007;129:11000–1. doi: 10.1021/ja073723s. [DOI] [PubMed] [Google Scholar]
- Tang X, Swaminathan J, Gewirtz AM, Dmochowski IJ. Regulating gene expression in human leukemia cells using light-activated oligodeoxynucleotides. Nucleic Acids Res. 2008;36:559–69. doi: 10.1093/nar/gkm1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomasini AJ, Schuler AD, Zebala JA, Mayer AN. PhotoMorphs: a novel light-activated reagent for controlling gene expression in zebrafish. Genesis. 2009;47:736–43. doi: 10.1002/dvg.20554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young DD, Lusic H, Lively MO, Yoder JA, Deiters A. Gene silencing in mammalian cells with light-activated antisense agents. Chembiochem. 2008;9:2937–40. doi: 10.1002/cbic.200800627. [DOI] [PubMed] [Google Scholar]
- Zhao XF, Fjose A, Larsen N, Helvik JV, Drivenes O. Treatment with small interfering RNA affects the microRNA pathway and causes unspecific defects in zebrafish embryos. FEBS J. 2008;275:2177–84. doi: 10.1111/j.1742-4658.2008.06371.x. [DOI] [PubMed] [Google Scholar]