

Abstract
The population persistence of schizophrenia despite associated reductions in fitness and fecundity suggests that the genetic basis of schizophrenia has a complex evolutionary history. A recent meta-analysis of schizophrenia genome-wide association studies offers novel opportunities for assessment of the evolutionary trajectories of schizophrenia-associated loci. In this study, we hypothesize that components of the genetic architecture of schizophrenia are attributable to human lineage-specific evolution. Our results suggest that schizophrenia-associated loci enrich in genes near previously identified human accelerated regions (HARs). Specifically, we find that genes near HARs conserved in nonhuman primates (pHARs) are enriched for schizophrenia-associated loci, and that pHAR-associated schizophrenia genes are under stronger selective pressure than other schizophrenia genes and other pHAR-associated genes. We further evaluate pHAR-associated schizophrenia genes in regulatory network contexts to investigate associated molecular functions and mechanisms. We find that pHAR-associated schizophrenia genes significantly enrich in a GABA-related coexpression module that was previously found to be differentially regulated in schizophrenia affected individuals versus healthy controls. In another two independent networks constructed from gene expression profiles from prefrontal cortex samples, we find that pHAR-associated schizophrenia genes are located in more central positions and their average path lengths to the other nodes are significantly shorter than those of other schizophrenia genes. Together, our results suggest that HARs are associated with potentially important functional roles in the genetic architecture of schizophrenia.
Keywords: schizophrenia, human accelerated evolution, networks, GWAS
Introduction
Schizophrenia is a severe and debilitating mental disorder that is typically characterized by symptoms such as hallucinations, delusions, and lack of concentration. Its onset usually occurs in early adulthood and is often associated with decreased reproductive success and increased mortality. Despite the evident reduction of fitness, schizophrenia affects approximately 1% of adults worldwide and is considered a fairly common mental disorder. In addition, the heritability of schizophrenia is approximately 70%, placing it among the most heritable mental disorders (Sullivan et al. 2003; van Dongen and Boomsma 2013). The prevalence and high heritability together with the fact that schizophrenia traits are not observed in other species gives rise to the hypothesis that the origin of the disease could be linked to evolutionary trajectories underlying human-specific traits such as the development of language or the disproportionately high consumption of energy by the human brain (Burns 2004; Preuss 2012). It has been theorized that mutations beneficial to human cognitive abilities might have been favored by natural selection but also predispose the risk of schizophrenia (Crow 1997, 2008). Some studies offer empirical evidence that some genes associated with schizophrenia have undergone positive selection (Crespi et al. 2007; Lo et al. 2007). However, those studies were focused on a limited number of genes.
Recent findings from genetic associations studies of psychiatric diseases offer new opportunities to reexamine the evolutionary trajectories of schizophrenia genetics. The Psychiatric Genomics Consortium (PGC) recently performed a meta-analysis combining genotype data from multiple schizophrenia genome-wide association (GWA) studies. The combined meta-analysis cohort included 36,989 schizophrenia cases and 113,075 controls, representing the largest GWA study ever performed on any psychiatric diseases (Schizophrenia Working Group of the Psychiatric Genomics Consortium 2014). Due to the exceptionally large sample size, they identified 83 new loci that passed genome-wide significance in addition to previously identified loci, providing novel insights to schizophrenia etiology. The GWA summary statistics offer a more comprehensive and quantitative representation of the genetic architecture underlying schizophrenia compared with previous candidate gene studies.
As schizophrenia traits appear to be human-specific it has been hypothesized that schizophrenia or its underlying genetic risk factors may be the result of human-specific brain evolution (Ogawa and Vallender 2014). Human accelerated regions (HARs) are genomic segments that are highly conserved among nonhuman species but experienced accelerated substitutions in the human genome (Pollard, Salama, King, et al. 2006). Thus, HARs can serve as genomic markers for human-specific evolution. Previous studies have demonstrated that HARs are mostly noncoding and likely to be regulatory elements responsible for human-specific traits. For example, Pollard et al. identified 202 HARs through comparative genomics between human and nonhuman mammals and found that the most significant HAR (named HAR1A) to be a novel long-noncoding RNA expressed in the development of human neocortex (Pollard, Salama, King, et al. 2006; Pollard, Salama, Lambert, et al. 2006). Other studies also have shown that genomic regions enriched with human-specific substitutions tend to be involved in regulation of nervous system development and other developmental processes (Prabhakar et al. 2006; Xu et al. 2013). Capra et al. (2013) assembled the HARs identified from different studies and predicted that at least 30% of the HARs were human developmental enhancers, and experimentally validated enhancer activity for 24 of 29 tested HARs.
Given HARs’ unique sequence characteristics and potential influence on human-specific traits, we hypothesize that HARs may have contributed to the emergence of psychiatric diseases such as schizophrenia. Tolosa et al. (2008) examined HAR1A in a candidate region case-control study of schizophrenia patients and found no evidence of significant association between HAR1A and the disease. However, to our knowledge, HARs have not been systematically examined in any psychiatric diseases. The recent results from the PGC meta-analysis provide a novel opportunity to investigate systematically the role of HARs in schizophrenia.
In this study, we aim to perform a genome-wide evolutionary assessment of the overlap between schizophrenia-associated loci and HARs. We consider three different types of HARs, namely, HARs based on conservation of nonhuman mammals (mHAR), HARs based on conservation of nonhuman primates (pHAR), and primate accelerated regions (PARs) based on conservation of nonprimate mammals (PAR). We find that schizophrenia loci are strongly enriched in genes near the pHARs, moderately enriched in genes near the PARs, but not enriched in genes near the mHARs. We show that pHAR-associated schizophrenia genes are under stronger selective pressure compared with the other schizophrenia genes. We then demonstrate that pHAR-associated schizophrenia genes have unique topological organization in two independent regulatory networks constructed from samples of human prefrontal cortex, and are enriched in a coexpression module that is dysregulated in schizophrenia. Finally, we provide evidence that schizophrenia loci are enriched in genes that have experienced human-specific expression shifts compared with other primates in the brain and specifically the cerebellum but not in other organs such as kidney and liver. These findings suggest that human-specific evolutionary changes may have contributed to the genetic architecture underlying schizophrenia traits in modern human populations.
Results
Schizophrenia-Associated Loci Harbor Highly Significant Number of Genes Near pHARs
To test whether the HARs or PARs are involved in schizophrenia, we first impute genomic intervals associated with schizophrenia based on the clumped GWAS single nucleotide polymorphisms (SNPs) from the meta-analysis by PGC (see Materials and Methods). The median length of the imputed intervals is approximately 52 kb under the threshold of nominal P-value < 1e-2. Because most HARs and PARs are short (<1,000 bp) and evidence suggests that HARs tend to be regulatory elements that influence the transcriptional activity of nearby genes (Pollard, Salama, King, et al. 2006; Capra et al. 2013), genes in 100 kb flanking regions are considered to be associated with the HARs or PARs (Lindblad-Toh et al. 2011). To test for enrichment of the schizophrenia GWAS regions with genes associated with HARs or PARs, we use INRICH as it accounts for confounding factors such as SNP density as well as number of overlapping genes (Lee et al. 2012; see Materials and Methods). To ensure robustness, we conduct enrichment tests using six different nominal P-value thresholds separately: P < 1e-2, P < 1e-3, P < 1e-4, P < 1e-5, P < 1e-6, and P < 1e-7. After removing nongenic intervals and merging the overlapping ones, we have 4,302, 1,676, 714, 329, 158, and 88 intervals tested under each of the P-value thresholds, separately. We find that under all of these thresholds except for P < 1e-7, the imputed schizophrenia GWAS regions are significantly enriched with pHAR-associated genes but not significantly enriched with mHAR-associated genes after stringent multiple testing correction (fig. 1). Interestingly, PAR-associated genes also show significant enrichment with the schizophrenia GWAS regions under nominal P < 1e-4 and nominal P < 1e-5, although only marginal significance is observed after multiple testing correction (fig. 1). Note that the inconsistent significance among different types of accelerated regions is not due to difference in numbers of genes covered by their respective flanking regions. In fact, the number of mHAR-associated genes is intermediate compared with the number of pHAR-associated genes and the number of PAR-associated genes (supplementary fig. S1A, Supplementary Material online). Thus, these results suggest that HARs may play an important role in regulating genes associated with schizophrenia, but not all HARs are equally important. The fact that schizophrenia GWAS regions are significantly enriched in pHAR-associated genes but not in mHAR-associated genes indicates that a significant amount of genes involved in schizophrenia tend to be regulated by genomic elements that are unique to human relative to nonhuman primates after the primates diverged from other mammals. In addition, pHARs may not be conserved in nonprimate mammals or may be different between primates and other mammals.
Fig. 1.

P-values of enrichment tests of schizophrenia GWAS regions under different nominal P-value thresholds in genes at 100 kb flanking regions of pHARs, PARs, and mHARs using INRICH. The midnight blue squares represent the highest enrichment (P < 0.001) and gray squares represent no enrichment (P > 0.1). For each enrichment test, both original P-value and multiple-testing adjusted P-value are shown. The three panels are the INRICH results using different background gene sets.
Because the HAR-associated genes are enriched in neurodevelopmental processes ([Lindblad-Toh et al. 2011; Capra et al. 2013] and results below), one may argue that the enrichment we observed when using all the human genes as background might simply indicate that both schizophrenia genes and pHAR genes are brain related. To rule out this possibility, we repeat INRICH analysis with the same data sets except that this time we use brain-expressed genes instead of all human genes as background. Specifically, we consider two sets of brain-expressed genes: genes that are expressed in the brain and genes that are highly expressed in the brain (see Materials and Methods). Both data sets yield the similar pattern of enrichment as when using all the autosomal genes as background (fig. 1). The schizophrenia GWAS regions are still significantly enriched in pHAR-associated genes but not mHAR-associated genes. In addition, compared with the marginally significant enrichment with PAR-associated genes when using all the autosomal genes as background, linkage disequilibrium (LD) intervals under the threshold P < 1e-5 show significant enrichment in PAR-associated genes after multiple testing correction when using brain-expressed genes as background.
Next, we investigate selective constraint on the HAR- and PAR-associated schizophrenia genes. Because the schizophrenia GWAS regions under the threshold P < 1e-4 exhibit the highest degree of enrichment compared with the other P-value thresholds and they cover reasonable amount of genes (supplementary fig. S1B, Supplementary Material online), we use gene sets involved in this P-value threshold for the following analyses.
pHAR-Associated Schizophrenia Genes Are Subject to Stronger Selective Pressure
If genes regulated by HARs or PARs are essential for certain biological functions in humans, these genes should be under stronger selective constraint. Human–chimpanzee pairwise nonsynonymous to synonymous substitution rate ratios (dN/dS) capture the selective pressure on genes after the two species started to diverge from their common ancestor, which is when HARs started to evolve according to how HARs are identified (Pollard, Salama, King, et al. 2006; Lindblad-Toh et al. 2011). We therefore examine human–chimpanzee pairwise dN/dS values among different sets of genes: pHAR-associated autosomal genes, mHAR-associated autosomal genes, PAR-associated autosomal genes, and all human autosomal genes. We find that while there is no significant difference between dN/dS of the three groups of AR-associated genes, both pHAR- and mHAR-associated genes have significantly lower dN/dS than all the autosomal genes (fig. 2, P = 4.2e-6 and 0.004, separately, Wilcoxon Rank-sum test), suggesting that genes regulated by pHARs and mHARs are generally under stronger selective constraint and thus implying their essential functionality. We do not observe a significant difference between the dN/dS of PAR-associated gene and all the autosomal genes probably because the dN/dS values are calculated based on human–chimpanzee pairwise comparison yet PARs represent the difference between primates and other mammals. Therefore, the human–chimpanzee pairwise dN/dS comparison does not reflect the potentially intensified selective pressure on the genes regulated by PARs.
Fig. 2.
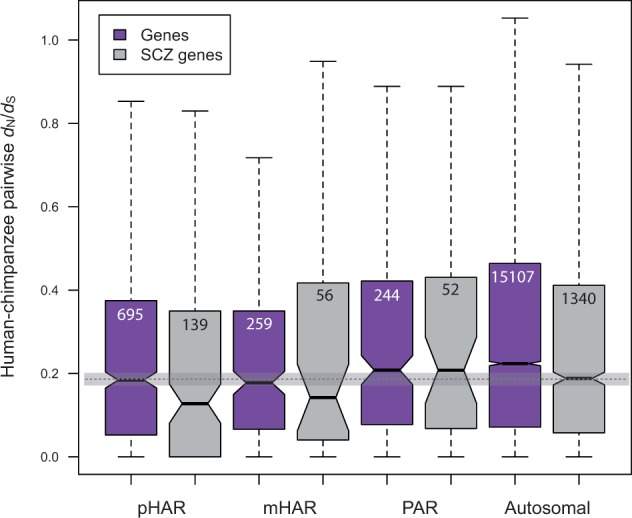
Human–chimpanzee pairwise dN/dS comparison across different sets of genes. The purple boxes represent autosomal genes and the ones that are at 100 kb flanking regions of pHARs, mHARs, and PARs. The gray boxes represent the intersection of each corresponding gene set with the schizophrenia genes with nominal P < 1e-4. Number of genes after filtering is given in the box for each category. Outliers are omitted from the plot. The horizontal shade represents median dN/dS and its confidence interval of all the schizophrenia genes (the last box).
Because we are interested in schizophrenia genes and we observe that different sets of HAR-associated genes exhibit varying degrees of enrichment with the schizophrenia LD intervals, we wonder if the pattern also holds for dN/dS comparison among different sets of schizophrenia genes. For the gene sets shown in supplementary figure S1B, Supplementary Material online, that is, under the threshold P < 1e-4, we compare the dN/dS values of HAR-associated schizophrenia gene to those of all schizophrenia genes. We find that while pHAR-associated schizophrenia genes still show significantly lower dN/dS than all the schizophrenia genes (P = 0.010, Wilcoxon Rank-sum test), mHAR-associated schizophrenia genes do not show significant difference compared with all the schizophrenia genes (fig. 2). This is consistent with the observation that the schizophrenia LD intervals are only significantly enriched in pHAR- but not mHAR-associated genes.
Interestingly, as shown in figure 2, schizophrenia genes generally have lower dN/dS than their counterparts for every pair of gene sets, that is, pHAR pair, mHAR pair, PAR pair, and autosomal pair. The difference is most pronounced between all schizophrenia genes and all autosomal genes (P = 2.24e-5, Wilcoxon Rank-sum test), suggesting that coding sequences of the schizophrenia genes are under stronger purifying selection. This is consistent with a previous study showing that most disease variants are evolutionarily more conserved than nondisease variants (Dudley et al. 2012). For the three sets of HAR or PAR genes, only pHAR-associated schizophrenia genes show significantly lower dN/dS than all the pHAR-associated genes (P = 0.044, Wilcoxon Rank-sum test).
We repeat the whole analysis using dN/dI (dI as substitution rate in introns) instead of dN/dS. The results are fairly similar. We also repeat the analysis using brain-expressed genes (supplementary fig. S2, Supplementary Material online). The patterns are generally the same except for slightly varying significance. Notably, for both data sets of brain-expressed genes, pHAR genes and schizophrenia genes still have significantly lower dN/dS than all autosomal genes. The difference of dN/dS between pHAR-associated schizophrenia genes and all the schizophrenia genes, however, becomes marginally significant for genes expressed in the brain (P = 0.069, Wilcoxon Rank-sum test) (supplementary fig. S2A, Supplementary Material online) and not significant for genes highly expressed in the brain (supplementary fig. 2B, Supplementary Material online).
Although human–chimpanzee pairwise dN/dS share the same evolutionary time frame with the HARs, these two species are closely related to each other, and therefore, the selective pressure comparisons based on these two species only may not be robust. We therefore conduct another set of comparisons using human–macaque pairwise dN/dS (supplementary fig. S3, Supplementary Material online). The resulted pattern between gene sets is similar to that using human–chimpanzee pairwise data. In fact, the statistical significance becomes stronger for almost all pairs of comparisons mentioned above. Interestingly, mHAR-associated schizophrenia genes show significantly lower human–macaque pairwise dN/dS than both all schizophrenia genes (P = 0.018, Wilcoxon Rank-sum test) and all mHAR genes (P = 0.016, Wilcoxon Rank-sum test), neither of which is significant with data from human and chimpanzee.
pHAR-Associated Schizophrenia Genes Are Enriched in a Coexpression Module that’s Dysregulated in Schizophrenia
Having found that pHAR-associated schizophrenia genes are under stronger selective pressure, we next investigate whether these genes are functionally important for schizophrenia. We use a recent weighted gene coexpression network analysis (WGCNA) in schizophrenia to test for enrichment of pHAR-associated schizophrenia genes in specific gene coexpression modules. WGCNA is a systems biology approach that aims to assign highly correlated genes into clusters (modules) based on their expression profiles (Zhang and Horvath 2005). Summarized information from each module can be used to link different modules to phenotypic traits or clinical variables. A recent analysis including 21 cases with schizophrenia and 19 control samples across four brain regions identified 32 coexpression modules, five of which showed significant differential expression between cases and controls (Roussos et al. 2012). More specifically, these modules were enriched in mitochondria, microglia, GABAergic, glutamatergic, and oligodendrocyte-related genes. Using multiple published GWA studies, they found that the neuronal (GABAergic and glutamatergic) and oligodendrocyte modules were significantly enriched for schizophrenia genetic risk variants, providing additional support for more direct involvement of these gene expression modules in schizophrenia. For each of the 32 modules, we do enrichment test for the pHAR-associated schizophrenia genes as well as all the schizophrenia genes using Fisher’s exact test (table 1). After Benjamini–Hochberg correction for multiple testing, only GABA module is significantly enriched in the pHAR-associated schizophrenia genes (multiple testing corrected P = 0.017, odds ratio = 3.83). We then perform permutation test by randomly selecting 377 genes (number of genes in the GABA module) from the gene list of the coexpression network and running Fisher’s exact test for 100,000 times. The observed odds ratio of the enrichment in the GABA module is significantly larger than those from permutation (P-value = 0.006). The GABA module, enriched with genes related to GABAergic neurons, is one of the five modules that were previously found to be significantly dysregulated in schizophrenia and enriched for schizophrenia-associated variants (Roussos et al. 2012). In comparison, the enrichment in the GABA module is not significant for all the schizophrenia genes (table 1). In fact, none of the modules has P-value smaller than 0.05 after stringent multiple testing correction for all schizophrenia genes (table 1).
Table 1.
Enrichment Test of pHAR-Associated Schizophrenia Genes and All Schizophrenia Genes in the 32 Coexpression Modules.
| Module Name | Number of Genes | Odds Ratio (95% CI) pHAR-SCZ Genes | Odds ratio (95% CI) SCZ genes |
|---|---|---|---|
| ASTRO1 | 184 | 3.05 (0.81, 8.10) | 1.34 (0.81, 2.12) |
| ASTRO2 | 90 | 0.00 (0, 5.83) | 1.04 (0.44, 2.15) |
| ASTRO3 | 40 | 0.00 (0, 13.47) | 0.59 (0.07, 2.26) |
| GABA | 377 | 3.83*** (1.79, 7.31)ψ | 1.38 (0.98, 1.89) |
| GLU1 | 466 | 2.12 (0.84, 4.51) | 1.29 (0.94, 1.73) |
| GLU2 | 42 | 3.31 (0.08, 19.70) | 1.40 (0.43, 3.53) |
| MG1 | 218 | 0.63 (0.02, 3.61) | 0.48* (0.22, 0.93) |
| MG2 | 199 | 2.10 (0.43, 6.34) | 0.76 (0.40, 1.34) |
| MG3 | 78 | 0.00 (0, 6.75) | 0.45 (0.09, 1.37) |
| MG4 | 57 | 2.43 (0.06, 14.31) | 0.82 (0.22, 2.23) |
| MG5 | 52 | 0.00 (0, 10.25) | 0.45 (0.05, 1.71) |
| MG6 | 52 | 0.00 (0, 10.25) | 0.68 (0.13, 2.09) |
| MG7 | 36 | 0.00 (0, 15.05) | 0.65 (0.08, 2.53) |
| MIT1 | 82 | 0.00 (0, 6.41) | 1.43 (0.66, 2.78) |
| MIT2 | 69 | 0.00 (0, 7.66) | 1.70 (0.78, 3.33) |
| NEU1 | 1,704 | 1.71* (1.01, 2.74) | 1.13 (0.95, 1.34) |
| NEU2 | 121 | 2.30 (0.27, 8.62) | 1.26 (0.65, 2.25) |
| NEU3 | 42 | 0.00 (0, 12.80) | 0.84 (0.17, 2.63) |
| NEU4 | 37 | 3.75 (0.09, 22.52) | 0.95 (0.19, 3.01) |
| NEU5 | 36 | 3.86 (0.09, 23.20) | 1.30 (0.34, 3.64) |
| OLIG1 | 1,228 | 1.63 (0.87, 2.82) | 1.33** (1.10, 1.60) |
| OLIG2 | 77 | 3.62 (0.43, 13.74) | 1.83 (0.91, 3.40) |
| OLIG3 | 80 | 0.00 (0, 6.57) | 0.73 (0.23, 1.78) |
| PV(+)1 | 1,288 | 2.17** (1.27, 3.52) | 1.17 (0.96, 1.41) |
| PV(+)2 | 68 | 4.10 (0.48, 15.63) | 2.25* (1.14, 4.12) |
| PV(+)3 | 66 | 2.10 (0.05, 12.28) | 1.25 (0.48, 2.72) |
| Ribosome | 551 | 1.00 (0.27, 2.63) | 0.81 (0.56, 1.12) |
| UNK1 | 782 | 1.62 (0.73, 3.18) | 1.15 (0.89, 1.46) |
| UNK2 | 159 | 0.00 (0, 3.26) | 1.03 (0.55, 1.79) |
| UNK3 | 66 | 0.00 (0, 8.01) | 1.07 (0.38, 2.45) |
| UNK4 | 57 | 0.00 (0, 9.32) | 0.62 (0.12, 1.90) |
| UNK5 | 56 | 2.48 (0.06, 14.57) | 1.05 (0.33, 2.60) |
Note.—Underlined modules are the ones that were identified to be differentially regulated in schizophrenia (Roussos et al. 2012)
*P-value < 0.05, two-sided Fisher’s exact test.
**P-value < 0.01, two-sided Fisher’s exact test.
***P-value < 0.001, two-sided Fisher’s exact test.
ψBenjamini–Hochberg corrected P-value < 0.05.
We also apply a more conservative analysis to further evaluate the enrichment of the pHAR-associated schizophrenia genes in the GABA module. Specifically, we use INRICH to test whether the schizophrenia GWAS regions that overlap with at least one pHAR gene are enriched in any of the modules. We find that among the 32 tested modules, the GABA module shows marginal significance (P = 0.064), together with two other modules also showing marginal significance: the PV(+)2 module (parvalbumin-expressing neurons, P = 0.081) and the ASTRO1 module (astrocytes, P = 0.088). However, none of them passes multiple testing correction through INRICH’s second round of permutation.
pHAR-Associated Schizophrenia Genes Are Enriched in Functional Categories Related to Synaptic Formation
To investigate functional characteristics of the schizophrenia genes that are potentially regulated by accelerated regions, we perform a series of functional enrichment tests for the following gene sets: all schizophrenia genes, pHAR-associated schizophrenia genes, PAR-associated schizophrenia genes, and mHAR-associated schizophrenia genes. As expected, the schizophrenia genes are enriched in several Gene Ontology (GO) categories related to neurological processes such as synapse and neuron projection (fig. 3A). Notably, the enrichment in calcium channel complex is consistent with previous findings that genes from this family are involved in schizophrenia (Cross-Disorder Group of the Psychiatric Genomics Consortium 2013; Schizophrenia Working Group of the Psychiatric Genomics Consortium 2014). For pHAR-associated schizophrenia genes, which is a subset of the gene set in figure 3A, show more significant GO categories (fig. 3B). Intriguingly, they are enriched in the following functional categories that do not exist in figure 3A: neuron differentiation, cell adhesion, plasma membrane, and cadherin binding, which are related to brain development and synapse formation. In contrast, mHAR-associated schizophrenia genes are not significantly enriched in any GO categories, and PAR-associated schizophrenia genes are only significantly enriched in one GO category: cell adhesion molecule binding (Benjamin–Hochberg adjusted P = 0.011). Finally, to disentangle the functional enrichment between pHAR-associated schizophrenia genes and the rest of the schizophrenia genes, we use schizophrenia genes instead of all the genes in the human genome as background to test for GO enrichment of the pHAR-associated schizophrenia genes. We find that after correcting for multiple comparisons, cell adhesion (Benjamin–Hochberg adjusted P = 0.095), biological adhesion (Benjamin–Hochberg adjusted P = 0.095), and plasma membrane (Benjamin–Hochberg adjusted P = 0.0015) are still significantly enriched (fig. 3C). This is concordant with a recent study showing that schizophrenia susceptibility is contributed by variants enriched in molecular pathways of neuronal cell adhesion and membrane scaffolding (O'Dushlaine et al. 2011).
Fig. 3.

Functional enrichment test of (A) schizophrenia genes (nominal P < 1e-4) using all the human genes as background, (B) pHAR-associated schizophrenia genes using all the human genes as background, and (C) pHAR-associated schizophrenia genes using the schizophrenia genes as background. The x axis represents fold enrichment. All the GO terms in the three figures have Benjamin–Hochberg adjusted P < 0.05 except for “cell adhesion” and “biological adhesion” in figure 3C, where they have Benjamin–Hochberg adjusted P < 0.1. BP, biological processes; CP, cellular components; MF, molecular functions.
pHAR-Associated Schizophrenia Genes Are Located in More Central Positions in Regulatory Networks of Human Prefrontal Cortex
Network structure provides a systematic view for genes’ roles in disease pathways and could also reflect their essentiality and evolutionary properties (Jeong et al. 2001; Wuchty 2004). As we have identified module enrichment and functional characteristics of the pHAR-associated schizophrenia genes, we wonder if these genes also harbor topological specialties in a network. We investigate this by constructing a coexpression network using a public data set comprising gene expression profiles of human prefrontal cortex from 220 postmortem samples (see Materials and Methods for details). The network contains 36 modules and 11,103 genes. We use eigengene-based connectivity, which is measured as the absolute value of correlation coefficient between expression profiles of a gene and the eigengene of a module (Langfelder and Horvath 2007), to examine how strongly a gene is connected to other genes in a module. We find that schizophrenia genes have a median connectivity 0.581, significantly higher than median connectivity of 0.568 of the other genes in the network (P = 0.007, Wilcoxon Rank-sum test). Similarly, pHAR genes also show significantly higher connectivity than the other genes in the network (0.600 vs. 0.568, P = 0.004, Wilcoxon Rank-sum test). But there is no significant difference of connectivity between schizophrenia genes and pHAR genes. The fact that both schizophrenia genes and pHAR genes are more strongly connected to the genes in the modules where they belong suggests their important roles in the network. Importantly, we also find that pHAR-associated schizophrenia genes show higher connectivity than other schizophrenia genes (0.615 vs. 0.579), although the significance is marginal (P = 0.068, Wilcoxon Rank-sum test). We find no significant difference of connectivity between the pHAR-associated schizophrenia genes and the pHAR genes not associated with schizophrenia (0.615 vs. 0.594, P = 0.148, Wilcoxon Rank-sum test).
To further investigate the topological difference between the pHAR-associated schizophrenia genes and other schizophrenia genes as well as other pHAR genes, we construct a Bayesian network based on gene expression profiles from another independent set of human prefrontal cortex samples (see Materials and Methods for details). The network is composed of a large connected component and several small connected components with about 7,000 genes and 8,000 edges (fig. 4). Mapping the schizophrenia genes to the network (blue and magenta nodes in fig. 4), we find that while most of them fall in the center of the largest connected component, some of them are located in the periphery of the largest component and some are located in the small components. Intriguingly, all the pHAR-associated schizophrenia genes are located in the center of the largest component and none of them falls in the small components (magenta nodes in fig. 4). We compare a series of network statistics such as centrality (degree, betweenness, and closeness), average shortest paths lengths, and eccentricity between pHAR-associated schizophrenia genes and other schizophrenia genes within this network (table 2). We find that while there is no significant difference in degree and betweenness between these two sets of genes (P > 0.1, Wilcoxon Rank-sum test), pHAR-associated schizophrenia genes have significantly greater closeness (P = 0.025, Wilcoxon Rank-sum test), significantly lower average shortest paths length (P = 0.025, Wilcoxon Rank-sum test), and significantly lower eccentricity (P = 0.001, Wilcoxon Rank-sum test) than other schizophrenia genes, indicating that pHAR-associated schizophrenia genes reside in more central positions and can reach other genes in the network more efficiently. This is consistent with our observation that pHAR-associated schizophrenia genes have higher connectivity than other schizophrenia genes to the genes in the modules from the above-mentioned coexpression network because the eigengene-based connectivity measures the connection strength between one gene to all the genes in a module. As a comparison, we also examine the same five network statistics between the pHAR-associated schizophrenia genes and the pHAR genes not associated with schizophrenia (table 2). We find that except for eccentricity, the other four network statistics have no significant difference between the two data sets. Intriguingly, pHAR-associated schizophrenia genes have significantly lower eccentricity than the pHAR genes not associated with schizophrenia (P = 0.040, Wilcoxon Rank-sum test). The smaller eccentricity of the pHAR-associated schizophrenia genes compared with other schizophrenia genes and other pHAR genes indicates their unique topological positions in the network. The eccentricity findings suggest that pHAR-associated schizophrenia genes are able to reach, and potentially influence, other genes in the network through fewer interactions compared with the other groups. A previous study also demonstrates that node eccentricity rather than degree or closeness significantly correlates with several functional data including gene expression level and gene essentiality in a yeast protein–protein interaction network (Xu et al. 2011).
Fig. 4.

The constructed Bayesian network of human prefrontal cortex in a force-directed layout. Both blue nodes and magenta nodes represent schizophrenia genes (nominal P < 1e-4) except that magenta nodes are pHAR-associated schizophrenia genes. The rest of the nodes (in green) represent other genes in the network. Although some of the blue nodes are located at the periphery of the largest connected component as well as in the small components, all the magenta nodes are in the largest connected component and virtually none of them are peripheral, suggesting that pHAR-associated schizophrenia genes are located in more central positions than other schizophrenia genes in the prefrontal cortex network.
Table 2.
Network Statistics Comparison (median and mean) between pHAR-Associated Schizophrenia Genes, pHAR Genes that Are Not Associated with Schizophrenia, and Schizophrenia Genes that Are Not Associated with the 100 kb Flanking Regions of pHARs.
| pHAR-SCZ Genes | Other pHAR Genes | Other SCZ Genes | |
|---|---|---|---|
| Degree | 2.00 (2.43) | 2.00 (2.55) | 2.00 (2.27) |
| Betweenness | 0.0003 (0.0030) | 0.0003 (0.0113) | 0.0003 (0.0045) |
| Closeness | 0.0766 (0.0756) | 0.0747 (0.0884) | 0.0735* (0.0844) |
| Average shortest path length | 13.06 (13.33) | 13.38 (13.47) | 13.61* (14.03) |
| Eccentricity | 35.00 (34.88) | 35.00* (34.94) | 36.00** (35.58) |
*P-value < 0.05, one-tailed Wilcoxon Rank-sum test comparing to pHAR-SCZ genes.
**P-value < 0.01, one-tailed Wilcoxon Rank-sum test comparing to pHAR-SCZ genes.
Discussion
In this study, we present evidence that the schizophrenia GWAS regions are enriched in genes near genomic regions that are highly conserved among nonhuman primates but experienced accelerated evolution in the human lineage (pHARs). We further demonstrate that this enrichment is not due to the fact that most genes covered by the schizophrenia GWAS regions are expressed in the brain and HAR-associated genes are involved in brain development by using brain-expressed genes as background. Similarly, the schizophrenia GWAS regions are also moderately enriched in genes near genomic regions that have accelerated in primate lineages but are conserved in nonprimate mammals (PARs). Interestingly, schizophrenia GWAS loci are not enriched in genes near genomic regions that have accelerated in humans but are conserved across nonhuman mammals (mHARs). This pattern does not seem to be attributed to the overlapping genes associated with the three types of accelerated regions. As shown in supplementary figure S1A, Supplementary Material online, the proportion of overlapping genes between pHAR and mHAR is the highest (22.9%), followed by the overlap between pHAR and PAR (9.2%) and the overlap between PAR and mHAR (8.6%). These patterns suggest that genes exclusively associated to one type of accelerated regions are more likely to contribute to the significance of enrichment. For example, CNTN4 and IGSF98, which are responsible for synaptic function and plasticity, are exclusively pHAR associated. Glutamatergic neurotransmission gene GRIN2A is exclusively PAR associated.
The results from selective pressure comparison on protein-coding sequences of the genes near accelerated regions using human–chimpanzee pairwise dN/dS values are consistent with the degree of enrichment of schizophrenia GWAS regions with the three types of accelerated regions. We first demonstrate that genes near pHARs and mHARs have significantly lower human–chimpanzee pairwise dN/dS compared with all the autosomal genes. We then show that pHAR-associated schizophrenia genes have significantly lower dN/dS than all the schizophrenia genes but mHAR-associated schizophrenia genes do not have significantly different dN/dS compared with all the schizophrenia genes, consistent with the fact that schizophrenia GWAS regions are significantly enriched in pHAR-associated genes but not in mHAR-associated genes. The evolutionary forces driving human accelerated substitutions could be either positive selection or neutral processes (Kostka et al. 2012; Hubisz and Pollard 2014). Because the reduced dN/dS values suggest stronger selective pressure on the genes near the HARs, it seems unlikely that the HARs have evolved neutrally. In fact, only a small proportion of HARs (∼15%) are driven by biased gene conversion, which is a recombination-associated neutral process (Lindblad-Toh et al. 2011). But biased gene conversion and loss of constraint can also contribute to phenotypes, especially loss-of-function. It is also worth noticing that overall the schizophrenia genes show lower dN/dS values than all the autosomal genes, suggesting that genes associated with schizophrenia are generally under stronger purifying selection. Hence, this finding is contradictory to previous studies showing that genes causing schizophrenia tend to be under positive selection (Crespi et al. 2007; Lo et al. 2007). Instead, the enrichment of schizophrenia GWAS regions in the neighborhood of pHARs suggests that regulatory elements of schizophrenia genes are likely to be under positive selection.
The results from functional enrichment test of schizophrenia genes involved in the three types of accelerated regions are also concordant with the degree of enrichment of the schizophrenia GWAS regions with the three types of accelerated regions. pHAR-associated genes show the highest enrichment with the schizophrenia GWAS regions and as a result, pHAR-associated schizophrenia genes are significantly enriched in the highest number of GO categories, among which most are neuron related. In contrast, mHAR-associated genes are not enriched in schizophrenia GWAS regions and accordingly, mHAR-associated schizophrenia genes are not enriched in any GO categories. We also used QIAGEN’s Ingenuity Pathway Analysis to detect molecular pathways that the pHAR-associated schizophrenia genes are enriched in (IPA, QIAGEN Redwood City, www.qiagen.com/ingenuity, last accessed March 11, 2015). Several signaling pathways show significance after correcting for multiple testing (supplementary table S1, Supplementary Material online), among which the most significant one is protein kinase A signaling. Interestingly, Sertoli cell junction signaling, which is essential for the spermatogenesis, is also among the top five pathways.
We demonstrate that pHAR-associated schizophrenia genes are significantly enriched in the GABA coexpression module, which is dysregulated in schizophrenia. The enrichment in the GABA module passes the stringent Benjamini–Hochberg correction when using Fisher’s exact test, but does not pass the more stringent multiple testing correction by INRICH. The reason we do not find strong signal of enrichment using INRICH could be that the test regions, that is, schizophrenia GWAS regions that overlap with pHAR genes, include other genes in addition to pHAR genes, which could weaken the statistical significance of the enrichment tests. Comparing to the other four modules that are also dysregulated in schizophrenia, GABA module is a well-replicated finding in postmortem studies of schizophrenia (Lewis et al. 1999, 2004; Lewis 2000; Hashimoto et al. 2003). In particular, studies have indicated reductions in cortical GABA content and the activity of glutamate decarboxylase 67 a GABA synthesizing enzyme. Notably, GABA-related abnormalities in schizophrenia are largely exclusive to the fast-spiking, parvalbumin-positive cortical interneurons in schizophrenia that are essential for synchronizing neuronal function and generate gamma oscillations (Lewis et al. 2005). Higher order cognitive processes that are affected in schizophrenia, like working memory depend on synchronization of neural oscillations in gamma frequency bands for optimal function. Therefore, abnormalities in GABAergic neurons will affect the generation of gamma oscillations, leading to cognitive symptoms and deficits that are present in schizophrenia.
Having seen that pHAR-associated schizophrenia genes are under stronger selective pressure and enriched in a coexpression module that’s dysregulated in schizophrenia, we hypothesize that these pHAR-associated genes may reflect the regulatory specificity in humans compared with other primates. Thus, we test whether there exists any enrichment between the pHAR-associated genes and the genes showing human-specific expression shifts relative to other primate species. We use data set from Brawand et al. (2011) where they identified groups of genes with significant human-specific expression shifts in different organs through RNA-Seq analyses of primate orthologs. Specifically, we analyze four groups of such genes, including 303 genes in the cerebellum and 4 genes in the brain (as one group), 253 genes in the kidney, 93 genes in the liver, and 628 genes in the testis. However, we find no significant enrichment of the pHAR-associated genes in any of these four groups of genes (P > 0.1, Hypergeometric test). Notably, although there are only 11 genes shared by pHAR flanking regions and the genes with expression shift in the human cerebellum and brain, two (DPYD and CACNA1C) out of these 11 genes are associated with the schizophrenia GWAS regions under nominal P < 1e-8, representing a approximately 20-fold enrichment in schizophrenia genes under this threshold. Even though we do not find evidence supporting our hypothesis, we shall not completely rule out this possibility because 1) the pHARs may regulate gene expression at specific developmental stages or in specific brain regions that are not sampled by the study by Brawand et al. (2011); and 2) the primate species used to identify pHARs and the primate species used to identify genes with human-specific expression shifts are not exactly the same. Specifically, pHARs were identified in a phylogeny including a broad range of primates including apes, Old World monkey, tarsier, and wet-nosed primates (Lindblad-Toh et al. 2011), yet genes with human-specific expression shifts were identified in a phylogeny including only apes and Old World monkey (Brawand et al. 2011).
Next, we investigate whether the schizophrenia GWAS regions are significantly enriched in any group of genes with human-specific expression shifts. Using the same approach (INRICH analysis) described previously, we find that the schizophrenia GWAS regions are significantly enriched in genes with human-specific expression shifts in the cerebellum and brain under all P-value cutoffs except for P < 1e-4. But only under the cutoff P < 1e-2 does the test pass the multiple testing correction (supplementary table S2, Supplementary Material online). In comparison, no statistical significance is found for the tests in the other organs. Note that in these INRICH analyses, instead of using all the human genes as background, we only use the primate orthologs on the autosomes (∼12,000 genes) as background. Because it is known that schizophrenia-associated loci are enriched in brain-expressed genes, the observed significance in cerebellum and brain could just mean that the tested gene set is a subset of brain-expressed genes. We therefore also test for enrichment using brain-expressed genes as background (similar to the analyses shown in fig. 1). We find that after multiple testing correction the enrichment is still significant at the P < 1e-2 cutoff (supplementary table S3, Supplementary Material online). The test is also significant at the P < 1e-7 cutoff using all the genes expressed in the brain as background (supplementary table S3, Supplementary Material online). The results thus suggest that genes whose expression has been under selection in the human cerebellum and brain are potentially involved in schizophrenia. The enrichment of the schizophrenia GWAS regions in both genes surrounding pHARs and genes with lineage-specific expression shifts in the human cerebellum and brain strongly suggests that the regulatory changes in the human genome play important roles in schizophrenia.
In conclusion, we have demonstrated that genetic loci associated with schizophrenia are enriched in genes near genomic regions that have experienced accelerated evolution in humans compared with nonhuman primates after the primate lineage diverged from mammals, that is, pHARs. We further provide evidence that these pHAR-associated schizophrenia genes are evolutionarily more conserved than other schizophrenia genes and they are enriched in a gene coexpression module that’s dysregulated in schizophrenia. Lastly, network analyses reveal that the pHAR-associated schizophrenia genes are located in more central positions than other schizophrenia genes in regulatory networks of the prefrontal cortex. We suggest that our analysis offers the first evolutionary systems understanding of the genetic architecture of schizophrenia. Although it was previously posited that the genetic basis of schizophrenia might be linked to human evolutionary trajectories due to associations with language and other human traits, our analysis offers among the first genome-wide molecular evolutionary evaluation of this hypothesis. Furthermore, the network analysis highlights specific gene regulatory network topologies that can be prioritized for further investigation into the driving molecular mechanisms of disease. While GWA studies and coexpression network analysis can identify many loci and transcripts correlated with disease status, the evolutionary informed network analysis may serve to highlight novel or prioritize opportunities for therapeutic development (Moalic et al. 2010).
Furthermore, our findings highlight the importance of taking human-specific evolution into account for interpreting genetic architectures of human-specific diseases such as schizophrenia. A recent study demonstrates that schizophrenia noncoding variants lie within enhancer sequences and affect gene expression (Roussos et al. 2014). The evidence provided suggests that integrative analysis of human-specific evolution with regulatory mechanisms of gene expression might be helpful in prioritizing genetic variants and genes associated with the etiopathogenesis of schizophrenia. With more data from GWA meta-analyses being generated, it would be interesting and valuable to examine the roles of HARs in other psychiatric diseases such as bipolar disorder and autism spectrum disorders as well as other complex diseases.
Materials and Methods
Schizophrenia GWAS Regions
Summary statistics of the clumped GWAS SNPs from the schizophrenia meta-analysis were downloaded from the PGC data repository (http://www.med.unc.edu/pgc/downloads, last accessed August 2014) (Schizophrenia Working Group of the Psychiatric Genomics Consortium 2014). In order to determine the boundaries of genomic regions harboring the nominally significant SNPs, we used genotype data from 1000 Genomes’ CEU population to estimate LD and positional distance (1000 Genomes Project Consortium 2012). After removing SNPs that are not in Hardy–Weinberg Equilibrium (P < 0.001) or have minor allele frequency less than 0.05, we used PLINK’s “LD calculations” procedure to impute LD intervals of the clumped SNPs under different nominal P-value thresholds (Purcell et al. 2007). Specifically, tagging SNPs were chosen when they are in LD with the index SNPs with r2 greater than 0.5 and they are less than 250 kb away from the index SNPs. Positions of the selected left-most and right-most tagging SNPs for an index SNP are the boundaries of its LD interval.
Human Accelerated Regions
HARs are defined to be the genomic regions that are highly conserved in nonhuman species but show signals of elevated substitution rates in syntenic regions in the human genome (Pollard, Salama, King, et al. 2006). Based on sequence conservation of mammals or vertebrates, multiple studies have identified slightly overlapping sets of HARs using different approaches (Pollard, Salama, King, et al. 2006; Prabhakar et al. 2006; Bird et al. 2007; Bush and Lahn 2008; Lindblad-Toh et al. 2011). Among these studies, Lindblad-Toh et al. (2011) contains the most comprehensive mammalian phylogeny, including six primates (human, chimpanzee, rhesus macaque, tarsier, mouse lemur, and bushbaby) and 23 nonprimate mammals. In addition to identifying HARs based on conservation of mammals (mHARs), they also identified HARs based on conservation of primates (pHARs) as well as PARs based on conservation of other mammals (Lindblad-Toh et al. 2011). These three data sets (mHARs, PARs, and pHARs) were therefore chosen as our accelerated regions. Because the accelerated regions are mostly noncoding and can potentially regulate the nearby genes, following the original article we used genes within 100 kb flanking regions for the enrichment test. Genomic coordinates based on hg18 were converted to hg19 using LiftOver tool (Kent et al. 2002).
Brain-Expressed Genes
We used the BrainSpan (http://developinghumanbrain.org, last accessed March 11, 2015) data set to define whether a gene was “brain-expressed” (Miller et al. 2014). We used the genic RPKM (reads per kilobase per million) values as supplied by the study (Gencode v10 analysis, http://download.alleninstitute.org/brainspan/RNASeq_Gencode_v10/, last accessed March 11, 2015) from the dorsolateral prefrontal cortex and hippocampus. Genes with RPKM > 1 were considered to be expressed in the brain and genes with RPKM > 5 were considered to be highly expressed in the brain. In total, we identified 11,776 autosomal genes expressed in the brain and 9,784 autosomal genes highly expressed in the brain.
Genes with Human-Specific Expression Shifts
Genes whose expression levels are consistent in all branches but one species in a phylogenetic tree are considered to have experienced natural selection on the expression levels in that particular species. Using likelihood ratio test, Brawand et al. (2011) identified a number of genes that experienced lineage-specific expression shift in different human organs by performing RNA-Seq on multiple individuals of humans, chimpanzees, bonobos, gorillas, orangutans, and macaques (Brawand et al. 2011). We chose the identified genes from five organs including brain, cerebellum, kidney, liver, and testis. We combined the genes from the brain and cerebellum together as only four genes were identified in the human brain. We did not include the genes from the heart as only 13 genes were identified in the heart, which would not give us enough power for enrichment test. The gene number for each selected organ can be found in the main text.
Testing for Enrichment of Schizophrenia GWAS Regions in HARs
To test whether the GWAS-implicated LD intervals were enriched in the 100 kb flanking regions of the HARs and PARs, we first removed the LD intervals that overlap with the broad MHC region (chr6: 25,000,000–35,000,000, assembly hg19) because this region contains a large number of genes active in immune system and thus could bias the results. We then performed rigorous enrichment test using the INRICH program (Lee et al. 2012). The tool calculates empirical P-values by randomly assigning genomic intervals along the genome for 10,000 times while matching the number and density of overlapping SNPs as well as the number of overlapping genes to those of the original LD intervals. It also corrects for multiple testing and accounts for gene length through a second round of permutation (5,000 times). The input files include: 1) list of the GWAS-implicated LD intervals, 2) list of genes flanking the HARs, 3) list of reference SNPs, that is, SNPs that were used to impute the LD intervals, and 4) list of reference genes, that is, all the human genes. Because the clumped GWAS SNPs were only available on the autosomes, all the input files were trimmed to include the autosomal data only.
Selective Pressure Comparison
Human–chimpanzee and human–macaque pairwise nonsynonymous substitution rate (dN) and synonymous substitution rate (dS) were obtained from Ensembl Biomart (Yang 2007; Vilella et al. 2009). Only the 1:1 orthologs were selected. To calculate dN/dS ratio, only the orthologs with nonzero pairwise dS were kept. We also downloaded human–chimpanzee pairwise dN/dI (dI as substitution rate of repeats in introns) ratios from the supplementary material, Supplementary Material online, of chimpanzee genome sequencing paper (Chimpanzee Sequencing and Analysis Consortium 2005).
Network Analysis
We used data set from a recent gene coexpression network analysis in cases with schizophrenia and healthy controls to test for enrichment of pHAR-associated schizophrenia genes in different coexpression modules (Roussos et al. 2012). Briefly, in this study five modules (oligodendrocyte, glutamatergic, GABAergic, microglia, and astrocytes) were found to be differentially regulated in schizophrenia among the 32 identified coexpression modules.
To examine topological specialty, we constructed another coexpression network by applying WGCNA (Zhang and Horvath 2005; Langfelder and Horvath 2008) to a previously described data set of gene expression profiles from 220 postnatal samples of the prefrontal cortex gray matter (BA46/9) from postmortem brains absent of neuropsychiatric or neuropathological diagnosis (Colantuoni et al. 2011). Thirty-six modules were identified from the analysis.
To further validate the results, we constructed a Bayesian network using a different independent prefrontal cortex data set composed of gene expression profiles from 173 samples of the dorsolateral prefrontal cortex (BA9) from postmortem brains of nondemented individuals (Zhang et al. 2013). The network was constructed using a previously described Bayesian network reconstruction algorithm implemented in the RIMBANET software package (Zhu et al. 2008, 2012). The network was then treated as undirected network and Cytoscape 3.1.1 was used for calculating node statistics and visualization (Cline et al. 2007).
Statistical Analysis
Functional enrichment tests were conducted using DAVID v6.7 (Huang et al. 2009a, 2009b) and QIAGEN’s Ingenuity Pathway Analysis (IPA, QIAGEN Redwood City, www.qiagen.com/ingenuity, last accessed March 11, 2015). All other statistical analyses were conducted using R (R Development Core Team 2008).
Supplementary Material
Supplementary material is available at Molecular Biology and Evolution online (http://www.mbe.oxfordjournals.org/).
Acknowledgments
The authors thank Douglas M. Ruderfer and Pamela Sklar for helpful comments and suggestions on the analysis. The authors would also like to thank the anonymous reviewers of this manuscript for their helpful and constructive comments. This study was supported by funding from the National Institutes of Health R01 DK098242 and U54 CA189201 to J.T.D.
References
- Genomes Project Consortium. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491:56–65. doi: 10.1038/nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird CP, Stranger BE, Liu M, Thomas DJ, Ingle CE, Beazley C, Miller W, Hurles ME, Dermitzakis ET. Fast-evolving noncoding sequences in the human genome. Genome Biol. 2007;8:R118. doi: 10.1186/gb-2007-8-6-r118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brawand D, Soumillon M, Necsulea A, Julien P, Csardi G, Harrigan P, Weier M, Liechti A, Aximu-Petri A, Kircher M, et al. The evolution of gene expression levels in mammalian organs. Nature. 2011;478:343–348. doi: 10.1038/nature10532. [DOI] [PubMed] [Google Scholar]
- Burns JK. An evolutionary theory of schizophrenia: cortical connectivity, metarepresentation, and the social brain. Behav Brain Sci. 2004;27:831–885. doi: 10.1017/s0140525x04000196. [DOI] [PubMed] [Google Scholar]
- Bush EC, Lahn BT. A genome-wide screen for noncoding elements important in primate evolution. BMC Evol Biol. 2008;8:17. doi: 10.1186/1471-2148-8-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capra JA, Erwin GD, McKinsey G, Rubenstein JL, Pollard KS. Many human accelerated regions are developmental enhancers. Philos Trans R Soc Lond B Biol Sci. 2013;368:20130025. doi: 10.1098/rstb.2013.0025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chimpanzee Sequencing and Analysis Consortium. Initial sequence of the chimpanzee genome and comparison with the human genome. Nature. 2005;437:69–87. doi: 10.1038/nature04072. [DOI] [PubMed] [Google Scholar]
- Cline MS, Smoot M, Cerami E, Kuchinsky A, Landys N, Workman C, Christmas R, Avila-Campilo I, Creech M, Gross B, et al. Integration of biological networks and gene expression data using Cytoscape. Nature Protoc. 2007;2:2366–2382. doi: 10.1038/nprot.2007.324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colantuoni C, Lipska BK, Ye T, Hyde TM, Tao R, Leek JT, Colantuoni EA, Elkahloun AG, Herman MM, Weinberger DR, et al. Temporal dynamics and genetic control of transcription in the human prefrontal cortex. Nature. 2011;478:519–523. doi: 10.1038/nature10524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crespi B, Summers K, Dorus S. Adaptive evolution of genes underlying schizophrenia. Proc R Soc B Biol Sci. 2007;274:2801–2810. doi: 10.1098/rspb.2007.0876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross-Disorder Group of the Psychiatric Genomics Consortium. Identification of risk loci with shared effects on five major psychiatric disorders: a genome-wide analysis. Lancet. 2013;381:1371–1379. doi: 10.1016/S0140-6736(12)62129-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crow TJ. Is schizophrenia the price that Homo sapiens pays for language? Schizophr Res. 1997;28:127–141. doi: 10.1016/s0920-9964(97)00110-2. [DOI] [PubMed] [Google Scholar]
- Crow TJ. The ‘big bang’ theory of the origin of psychosis and the faculty of language. Schizophr Res. 2008;102:31–52. doi: 10.1016/j.schres.2008.03.010. [DOI] [PubMed] [Google Scholar]
- Dudley JT, Chen R, Sanderford M, Butte AJ, Kumar S. Evolutionary meta-analysis of association studies reveals ancient constraints affecting disease marker discovery. Mol Biol Evol. 2012;29:2087–2094. doi: 10.1093/molbev/mss079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto T, Volk DW, Eggan SM, Mirnics K, Pierri JN, Sun Z, Sampson AR, Lewis DA. Gene expression deficits in a subclass of GABA neurons in the prefrontal cortex of subjects with schizophrenia. J Neurosci. 2003;23:6315–6326. doi: 10.1523/JNEUROSCI.23-15-06315.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang DW, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009a;37:1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009b;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- Hubisz MJ, Pollard KS. Exploring the genesis and functions of Human Accelerated Regions sheds light on their role in human evolution. Curr Opin Genet Dev. 2014;29:15–21. doi: 10.1016/j.gde.2014.07.005. [DOI] [PubMed] [Google Scholar]
- Jeong H, Mason SP, Barabasi AL, Oltvai ZN. Lethality and centrality in protein networks. Nature. 2001;411:41–42. doi: 10.1038/35075138. [DOI] [PubMed] [Google Scholar]
- Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, Haussler D. The human genome browser at UCSC. Genome Res. 2002;12:996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostka D, Hubisz MJ, Siepel A, Pollard KS. The role of GC-biased gene conversion in shaping the fastest evolving regions of the human genome. Mol Biol Evol. 2012;29:1047–1057. doi: 10.1093/molbev/msr279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langfelder P, Horvath S. Eigengene networks for studying the relationships between co-expression modules. BMC Syst Biol. 2007;1:54. doi: 10.1186/1752-0509-1-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 2008;9:559. doi: 10.1186/1471-2105-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee PH, O'Dushlaine C, Thomas B, Purcell SM. INRICH: interval-based enrichment analysis for genome-wide association studies. Bioinformatics. 2012;28:1797–1799. doi: 10.1093/bioinformatics/bts191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis DA. GABAergic local circuit neurons and prefrontal corticaldysfunction in schizophrenia. Brain Res Brain Res Rev. 2000;31:270–276. doi: 10.1016/s0165-0173(99)00042-9. [DOI] [PubMed] [Google Scholar]
- Lewis DA, Hashimoto T, Volk DW. Cortical inhibitory neurons and schizophrenia. Nat Rev Neurosci. 2005;6:312–324. doi: 10.1038/nrn1648. [DOI] [PubMed] [Google Scholar]
- Lewis DA, Pierri JN, Volk DW, Melchitzky DS, Woo TU. Altered GABA neurotransmission and prefrontal cortical dysfunction in schizophrenia. Biol Psychiatry. 1999;46:616–626. doi: 10.1016/s0006-3223(99)00061-x. [DOI] [PubMed] [Google Scholar]
- Lewis DA, Volk DW, Hashimoto T. Selective alterations in prefrontal cortical GABA neurotransmission in schizophrenia: a novel target for the treatment of working memory dysfunction. Psychopharmacology. 2004;174:143–150. doi: 10.1007/s00213-003-1673-x. [DOI] [PubMed] [Google Scholar]
- Lindblad-Toh K, Garber M, Zuk O, Lin MF, Parker BJ, Washietl S, Kheradpour P, Ernst J, Jordan G, Mauceli E, et al. A high-resolution map of human evolutionary constraint using 29 mammals. Nature. 2011;478:476–482. doi: 10.1038/nature10530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo WS, Xu ZW, Yu ZL, Pun FW, Ng SK, Chen JH, Tong KL, Zhao CY, Xu XJ, Tsang SY, et al. Positive selection within the schizophrenia-associated GABA(A) receptor beta(2) gene. PLoS One. 2007;2:e462. doi: 10.1371/journal.pone.0000462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JA, Ding SL, Sunkin SM, Smith KA, Ng L, Szafer A, Ebbert A, Riley ZL, Royall JJ, Aiona K, et al. Transcriptional landscape of the prenatal human brain. Nature. 2014;508:199–206. doi: 10.1038/nature13185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moalic JM, Le Strat Y, Lepagnol-Bestel AM, Ramoz N, Loe-Mie Y, Maussion G, Gorwood P, Simonneau M. Primate-accelerated evolutionary genes: novel routes to drug discovery in psychiatric disorders. Curr Med Chem. 2010;17:1300–1316. doi: 10.2174/092986710790936338. [DOI] [PubMed] [Google Scholar]
- O'Dushlaine C, Kenny E, Heron E, Donohoe G, Gill M, Morris D, International Schizophrenia Consortium, Corvin A. Molecular pathways involved in neuronal cell adhesion and membrane scaffolding contribute to schizophrenia and bipolar disorder susceptibility. Mol Psychiatry. 2011;16:286–292. doi: 10.1038/mp.2010.7. [DOI] [PubMed] [Google Scholar]
- Ogawa LM, Vallender EJ. Evolutionary conservation in genes underlying human psychiatric disorders. Front Hum Neurosci. 2014;8:283. doi: 10.3389/fnhum.2014.00283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard KS, Salama SR, King B, Kern AD, Dreszer T, Katzman S, Siepel A, Pedersen JS, Bejerano G, Baertsch R, et al. Forces shaping the fastest evolving regions in the human genome. PLoS Genet. 2006;2:e168. doi: 10.1371/journal.pgen.0020168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard KS, Salama SR, Lambert N, Lambot MA, Coppens S, Pedersen JS, Katzman S, King B, Onodera C, Siepel A, et al. An RNA gene expressed during cortical development evolved rapidly in humans. Nature. 2006;443:167–172. doi: 10.1038/nature05113. [DOI] [PubMed] [Google Scholar]
- Prabhakar S, Noonan JP, Paabo S, Rubin EM. Accelerated evolution of conserved noncoding sequences in humans. Science. 2006;314:786. doi: 10.1126/science.1130738. [DOI] [PubMed] [Google Scholar]
- Preuss TM. Human brain evolution: from gene discovery to phenotype discovery. Proc Natl Acad Sci U S A. 2012;109(Suppl 1):10709–10716. doi: 10.1073/pnas.1201894109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Development Core Team. Vienna Austria: R Foundation for Statistical Computing; 2008. R: a language and environment for statistical computing. ISBN 3-900051-07-0. Available from: http://www.R-project.org. [Google Scholar]
- Roussos P, Katsel P, Davis KL, Siever LJ, Haroutunian V. A system-level transcriptomic analysis of schizophrenia using postmortem brain tissue samples. Arch Gen Psychiatry. 2012;69:1205–1215. doi: 10.1001/archgenpsychiatry.2012.704. [DOI] [PubMed] [Google Scholar]
- Roussos P, Mitchell AC, Voloudakis G, Fullard JF, Pothula VM, Tsang J, Stahl EA, Georgakopoulos A, Ruderfer DM, Charney A, et al. A role for noncoding variation in schizophrenia. Cell Rep. 2014;9:1417–1429. doi: 10.1016/j.celrep.2014.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schizophrenia Working Group of the Psychiatric Genomics Consortium. Biological insights from 108 schizophrenia-associated genetic loci. Nature. 2014;511:421–427. doi: 10.1038/nature13595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan PF, Kendler KS, Neale MC. Schizophrenia as a complex trait: evidence from a meta-analysis of twin studies. Arch Gen Psychiatry. 2003;60:1187–1192. doi: 10.1001/archpsyc.60.12.1187. [DOI] [PubMed] [Google Scholar]
- Tolosa A, Sanjuan J, Leal C, Costas J, Molto MD, de Frutos R. Rapid evolving RNA gene HAR1A and schizophrenia. Schizophr Res. 2008;99:370–372. doi: 10.1016/j.schres.2007.10.011. [DOI] [PubMed] [Google Scholar]
- van Dongen J, Boomsma DI. The evolutionary paradox and the missing heritability of schizophrenia. Am J Med Genet B Neuropsychiatr Genet. 2013;162B:122–136. doi: 10.1002/ajmg.b.32135. [DOI] [PubMed] [Google Scholar]
- Vilella AJ, Severin J, Ureta-Vidal A, Heng L, Durbin R, Birney E. EnsemblCompara GeneTrees: complete, duplication-aware phylogenetic trees in vertebrates. Genome Res. 2009;19:327–335. doi: 10.1101/gr.073585.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wuchty S. Evolution and topology in the yeast protein interaction network. Genome Res. 2004;14:1310–1314. doi: 10.1101/gr.2300204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu K, Bezakova I, Bunimovich L, Yi SV. Path lengths in protein-protein interaction networks and biological complexity. Proteomics. 2011;11:1857–1867. doi: 10.1002/pmic.201000684. [DOI] [PubMed] [Google Scholar]
- Xu K, Wang J, Elango N, Yi SV. The evolution of lineage-specific clusters of single nucleotide substitutions in the human genome. Mol Phylogenet Evol. 2013;69:276–285. doi: 10.1016/j.ympev.2013.06.003. [DOI] [PubMed] [Google Scholar]
- Yang ZH. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol. 2007;24:1586–1591. doi: 10.1093/molbev/msm088. [DOI] [PubMed] [Google Scholar]
- Zhang B, Gaiteri C, Bodea LG, Wang Z, McElwee J, Podtelezhnikov AA, Zhang C, Xie T, Tran L, Dobrin R, et al. Integrated systems approach identifies genetic nodes and networks in late-onset Alzheimer's disease. Cell. 2013;153:707–720. doi: 10.1016/j.cell.2013.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Horvath S. A general framework for weighted gene co-expression network analysis. Stat Appl Genet Mol Biol. 2005;4:Article17. doi: 10.2202/1544-6115.1128. [DOI] [PubMed] [Google Scholar]
- Zhu J, Sova P, Xu Q, Dombek KM, Xu EY, Vu H, Tu Z, Brem RB, Bumgarner RE, Schadt EE. Stitching together multiple data dimensions reveals interacting metabolomic and transcriptomic networks that modulate cell regulation. PLoS Biol. 2012;10:e1001301. doi: 10.1371/journal.pbio.1001301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Zhang B, Smith EN, Drees B, Brem RB, Kruglyak L, Bumgarner RE, Schadt EE. Integrating large-scale functional genomic data to dissect the complexity of yeast regulatory networks. Nat Genet. 2008;40:854–861. doi: 10.1038/ng.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.