

In a controlled, prospective, electrophysiological study, Imbach et al. demonstrate increased sleep need and excessive daytime sleepiness 6 months after traumatic brain injury. Sleep is more consolidated after brain trauma, and an increase in sleep need is associated with intracranial haemorrhage. Trauma patients underestimate their increased sleep need and sleepiness.
Keywords: traumatic brain injury, sleep, post-traumatic pleiosomnia, post-traumatic daytime sleepiness
Abstract
Post-traumatic sleep-wake disturbances are common after acute traumatic brain injury. Increased sleep need per 24 h and excessive daytime sleepiness are among the most prevalent post-traumatic sleep disorders and impair quality of life of trauma patients. Nevertheless, the relation between traumatic brain injury and sleep outcome, but also the link between post-traumatic sleep problems and clinical measures in the acute phase after traumatic brain injury has so far not been addressed in a controlled and prospective approach. We therefore performed a prospective controlled clinical study to examine (i) sleep-wake outcome after traumatic brain injury; and (ii) to screen for clinical and laboratory predictors of poor sleep-wake outcome after acute traumatic brain injury. Forty-two of 60 included patients with first-ever traumatic brain injury were available for follow-up examinations. Six months after trauma, the average sleep need per 24 h as assessed by actigraphy was markedly increased in patients as compared to controls (8.3 ± 1.1 h versus 7.1 ± 0.8 h, P < 0.0001). Objective daytime sleepiness was found in 57% of trauma patients and 19% of healthy subjects, and the average sleep latency in patients was reduced to 8.7 ± 4.6 min (12.1 ± 4.7 min in controls, P = 0.0009). Patients, but not controls, markedly underestimated both excessive sleep need and excessive daytime sleepiness when assessed only by subjective means, emphasizing the unreliability of self-assessment of increased sleep propensity in traumatic brain injury patients. At polysomnography, slow wave sleep after traumatic brain injury was more consolidated. The most important risk factor for developing increased sleep need after traumatic brain injury was the presence of an intracranial haemorrhage. In conclusion, we provide controlled and objective evidence for a direct relation between sleep-wake disturbances and traumatic brain injury, and for clinically significant underestimation of post-traumatic sleep-wake disturbances by trauma patients.
Introduction
Sleep-wake disturbances (SWD) are frequent following acute traumatic brain injury (TBI) and impair quality of life in TBI patients. Several studies highlighted the high prevalence of excessive daytime sleepiness (EDS) and increased sleep need after TBI (Guilleminault et al., 1983; Masel et al., 2001; Ouellet and Morin, 2006; Parcell et al., 2006; Baumann et al., 2007; Castriotta et al., 2007; Chaumet et al., 2008; Schreiber et al., 2008; Ponsford et al., 2013). In a recent meta-analysis of 21 clinical studies, ∼50% of TBI patients suffered from post-traumatic SWD: increased sleep need per 24 h and EDS were among the most common and disturbing post-traumatic SWD (Mathias and Alvaro, 2012). However, the reported prevalence of post-traumatic SWD varies considerably. This is because the presently available studies differ significantly regarding study design (mostly retrospective studies), assessment of sleep problems (questionnaires versus sleep laboratory examinations) and inconsistent definitions of SWD resulting in a considerable variance regarding prevalence of SWD after TBI. To address the problem of heterogeneous definitions of SWD, we recently introduced the term ‘post-traumatic pleiosomnia’ for increased sleep need following TBI (Sommerauer et al., 2013). Earlier, this symptom was referred to as post-traumatic hypersomnia, but the concept of hypersomnia comprises both increased sleep need per 24 h and EDS (American Academy of Sleep Medicine, 2005) and is therefore misleading, particularly for TBI patients who may suffer from either EDS or excessive sleep need or from both (Baumann et al., 2007).
The pathophysiology of EDS and pleiosomnia after TBI remains elusive. However, fatal TBI is associated with a partial loss of wake-promoting hypocretin (orexin)- and histamine-producing neurons (Baumann et al., 2009; Valko et al., 2014). This suggests that damage to hypothalamic neurons and to other wake-promoting neuronal populations might be associated with post-traumatic EDS and pleiosomnia, but there are no acute biomarkers to predict the evolution of these SWD. For instance, it has not been examined whether acute dysfunction in neuro-endocrine pathways involving the hypothalamic-pituitary-adrenal axis is related to the development of increased sleep propensity after TBI.
In this study, we examined the prevalence and electrophysiological characteristics of post-traumatic EDS and pleiosomnia in a prospective and controlled approach by systematically examining TBI patients 6 months after injury with both subjective and objective sleep measures in comparison to an age- and gender-matched control group. Furthermore, we tested whether sleep was more consolidated after TBI and whether TBI was associated with a relative increase in EEG power density in the delta range. These analyses were motivated by the hypothesis that increased slow-wave activity is linked to neuronal recovery (Stickgold et al., 2001; Carmichael and Chesselet, 2002; Tononi and Cirelli, 2006). As secondary endpoint, we correlated neuroimaging and laboratory measures reflecting neuronal damage in general, and more specifically dysfunction in the hypothalamic-pituitary-adrenal axis in the acute phase of TBI with the occurrence of post-traumatic SWD, to identify potential early predictors of post-traumatic EDS and pleiosomnia. The rationale to screen for hormonal disturbances in TBI patients as possible predictors of SWD was based on the hypothesis that shearing injuries may cause loss of hormone-releasing neurons in the brainstem and hypothalamus, and would possibly result in the disruption of neuro-endocrine pathways, similar to the loss of hypocretin-producing neurons after TBI (Baumann et al., 2009).
Materials and methods
The protocol for this clinical study was approved by the local ethics committee (Kantonale Ethikkommission Zurich). Written informed consent for study participation was obtained from all patients and healthy controls before participation.
Study design
This is a prospective controlled trial to examine the prevalence, severity, and natural history of SWD after TBI in relation to severity of trauma (Glasgow Coma Scale), presence of brain damage (intracranial haemorrhage), and selected neurochemical biomarkers. Patients were examined at two time points: (i) in the acute phase after TBI for clinical staging, brain CT scan and laboratory assessment; and (ii) 6 months after TBI for a detailed analysis of sleep and wakefulness (2-week actigraphy, followed by nocturnal video-polysomnography and multiple sleep latency tests, MSLT) and follow-up laboratory assessment (Fig. 1).
Figure 1.

Overview of study design and patient enrolment. Explanations for loss to follow-up are given in the results section of the manuscript.
Participants
Between July 2009 and June 2012, we screened 140 patients with acute, first-ever TBI admitted to our hospital and included 60 patients. Patients with prior TBI, other neurological or systemic diseases, drug or alcohol abuse, or psychiatric comorbidities were excluded.
For the analyses of sleep and wakefulness, we enrolled an age- and gender-matched healthy control group without prior TBI. For this purpose, we prospectively searched for controls by word-of-mouth advertising. To achieve optimal controlling of the sleep-wake outcome measures, we introduced a novel strategy for matching the control group: to achieve similar extents of sleep satiation before sleep laboratory examinations, we matched for the difference of total sleep times on actigraphy between working days and weekends/holidays, because this difference reflects chronic sleep restriction during workdays. By this additional matching, we could therefore rule out a possible bias in the assessment of sleep measures due to chronic sleep restriction. Both after screening and again after sleep-wake assessments in the sleep laboratory, participants with SWD including circadian rhythm disorders on actigraphy were excluded as controls. Furthermore, volunteers with neurological disorders, a history of SWD or psychiatric illnesses were excluded, as well as parents with young children or subjects on shift work. To get a representative rather than an artificial control sample (Mignot et al., 2006), apnoea-hypopnoea indices (AHI) from 5 to 15 on polysomnography or mean sleep latencies from 5 to 8 min on MSLT were accepted for inclusion as control if no subjective EDS was indicated by the Epworth Sleepiness Scale. Sleep laboratory examinations in controls were performed only once. Six controls were included in a previous study (Sommerauer et al., 2013).
Examinations in the acute phase after traumatic brain injury
We categorized the severity of TBI using the Glasgow Coma Scale (13–15: mild; 9–12: moderate; and 3–8: severe) on admission to the hospital. Furthermore, we performed a CT scan within the first 4 h after trauma to estimate severity and localization of brain injury.
Laboratory examinations within the first 5 days after trauma included the assessments of the hypothalamic-pituitary-adrenal axis (adrenocorticotropic hormone, cortisol, and aldosterone, adrenaline, noradrenaline, dopamine), and of serum biomarkers reflecting neural injury and indicating poor overall recovery after TBI (S100 calcium binding protein B, S100B, and neuron-specific enolase, NSE; Berger et al., 2002; Lamers et al., 2003; Vos et al., 2004). The same blood tests were performed again 6 months after TBI.
Assessment of sleep-wake disturbances 6 months after traumatic brain injury
We screened for changes in sleep habits, sleep quality, daytime vigilance and insomnia by detailed and structured interviews. Patients and controls completed validated questionnaires such as the Epworth Sleepiness Scale, the Sleep Apnoea Scale of the Sleep Disorders Questionnaire, the Ullanlinna Narcolepsy Scale and the Fatigue Severity Scale. We further investigated psychological mood, fatigue, and quality of life using reliable and widely used self-report measures, and the ‘Allgemeine Depressionsskala’ (ADS) to assess depressive symptoms (Hautzinger and Bailer, 1993).
To screen for circadian SWD and to quantify sleep need per 24 h, patients and controls were examined with sleep logs and wrist actigraphy (Actiwatch, Neurotechnology, King et al., 2005). Wrist actigraphy was recorded and analysed as described before (Cippa et al., 2013).
Thereafter, we performed overnight video-polysomnography from 11 p.m. to 7 a.m. as described previously (Baumann et al., 2007). Sleep-stage scoring was performed visually according to revised international criteria (Iber et al., 2007) and sleep stage distribution was assessed by calculating the relative time in each sleep behavioural state. For further signal processing of the raw data, we used MatLab (The MathWorks Inc. 2009). We calculated delta power in slow-wave sleep as follows: first, all 30-s epochs that were scored as non-rapid eye movement (NREM) sleep stage 3 were included. This data set was then subdivided into epochs of 5-s length. Artefacts were rejected by an automated dual algorithm based on a short-term kurtosis measure (mainly for electrode artefacts) and spectral information (e.g. movement artefacts). Fast Fourier Transformation was then applied on each 5-s epoch after multiplication by a Hann window to address the problem of edge discontinuities (zero padding was used to expand the signal in each 5-s epoch of 500 data points to a window size of 512 points). For better comparability between subjects, the calculated delta power (0.5–4.5 Hz) was normalized to the total spectral power (1–50 Hz) for each individual. To calculate a quantitative measure of sleep fragmentation, we first assessed the total number of behavioural state bouts. A behavioural state bout was defined as a consecutive series of epochs in the same behavioural state without state transitions. The resulting amount of behavioural state bouts was then divided by the total number of 5-s epochs in the same sleep stage. This sleep fragmentation index reflects therefore the relative number of behavioural state bouts per time (e.g. highly fragmented sleep with many changes in sleep-wake behavioural states will result in a high sleep fragmentation index). We chose 5-s epochs for this analysis because the artefact rejection algorithm was based on this time interval, but sleep stage classification was based on the classical 30-s epoch length (Iber et al., 2007).
Objective EDS was assessed by standardized MSLT. Briefly, the procedure entails four opportunities for sleep lasting 20 min every 2 h; subjects were told to relax, and sleep was permitted. Sleepiness was then assessed by mean sleep latencies as derived from the concurrent EEG in the absence of external alerting factors. Mean sleep latencies below 8 min were considered to represent objective EDS (American Academy of Sleep Medicine, 2005).
Statistical analyses
We used means, proportions and quartiles for descriptive comparative analysis of continuous data. TBI patients and controls were compared with unpaired two-sided t-tests and data from TBI patients at different time points with paired t-tests. More than two groups were compared by one-way ANOVA. We used χ2-statistics to compare nominal data. To compare objective and subjective EDS (i.e. comparison between MSLT versus Epworth Sleepiness Scale), we used McNemar’s test to account for marginal homogeneity (i.e. to test the null hypothesis whether subjective and objective assessment do not differ significantly). Bivariate correlation analysis was done by calculation of Pearson’s correlation coefficient to reveal significant relations between clinical characteristics or endocrine markers with sleep outcome measures.
Furthermore, we performed a subgroup analysis for the TBI patients with respect to severity of TBI (mild versus severe TBI) and compared the two groups by one-way ANOVA. For all analyses, two-tailed P-values < 0.05 were considered to be significant.
Results
Of 140 screened patients, 33 were excluded because of substance abuse or neurological, sleep–wake or psychiatric disorders prior to TBI, and 47 patients denied to participate, resulting in a cohort of 60 TBI patients (range 18–71 years, 74% male). Forty-two out of these patients with first-ever traumatic brain injury and no prior SWD were available for follow-up examinations 6 months after TBI (Fig. 1). After acute phase examinations, 18 participants were lost to follow-up for various reasons (new onset of severe TBI-unrelated disease: n = 1, work: n = 2, moved to a foreign country: n = 2, socioeconomic problems: n = 1, new onset epilepsy: n = 1, no reason given: n = 11). Except for one subject in the severe TBI group, all TBI patients had ADS scores below the cut-off of 23, indicating that all but one TBI patient did not suffer from clinical depression. Thirty-one of 42 patients with full sleep laboratory assessments had complete serum laboratory results in both the acute and the 6-month phase.
Post-traumatic pleiosomnia and excessive daytime sleepiness
Continuous actigraphy assessment over 2 weeks confirmed pleiosomnia in TBI patients: we found that TBI patients slept 1.2 h more per 24 h (8.3 ± 1.1 h, mean ± SD) than controls (7.1 ± 0.8 h, mean ± SD, t-test: P < 0.0001, Fig. 2A). On MSLT, average sleep latency was 12.1 ± 4.7 min in controls and 8.7 ± 4.6 min in TBI patients (t-test: P < 0.001, Fig. 2B). Total sleep time in polysomnography was significantly longer in TBI patients (392 ± 10 min in controls and 419 ± 7 min in TBI patients, t-test: P < 0.05, Fig. 2C). Objective EDS (defined as a mean sleep latency <8 min) was found in 57% of TBI patients as compared to 19% in healthy subjects (Fig. 3A, χ2 test: P =0.001, Cramer’s φ: 0.39), resulting in an odds ratio of 5.6 (confidence interval: 2.1–15.1) for TBI patients versus healthy controls (Fig. 3A). Other causes than TBI of increased EDS and pleiosomnia such as sleep apnoea or insufficient sleep syndrome were not present (no difference between groups, Table 1). TBI patients did not show signs of circadian dysfunction in actigraphy.
Figure 2.
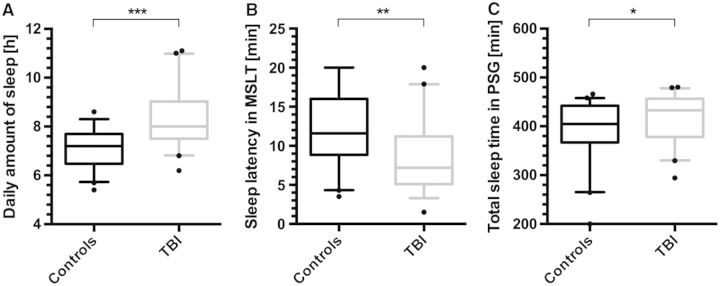
Sleep amount and sleep latency in TBI patients and healthy controls. (A) Daily hours of sleep as measured by continuous actigraphy over 2 weeks, showing an increase of 1.2 h in total amount of sleep/24 h in TBI patients as compared to the control group. (B) Mean sleep latencies on the MSLT in TBI patients are 28% lower than in healthy controls. (C) Total sleep time in 8 h-polysomnography (PSG) reveals an increased amount of sleep in TBI patients. Box plots indicate means (horizontal line), upper and lower quartiles (box) and extrema (whiskers), outliers are shown as black dots. ***P < 10−6, **P < 0.0001, *P < 0.05.
Figure 3.

Objective versus subjective EDS in TBI patients and controls. (A) Objective EDA (defined as average sleep latency <8 min on MSLT, filled bars) was found in 24/42 TBI patients (57%) as compared to 8/42 healthy subjects (19%; odds ratio: 5.6, P < 0.001). (B) Comparison of subjective EDS (based on Epworth Sleepiness Scale, open bars) and objective EDS (based on MSLT, filled bars). In healthy controls, no difference between subjective and objective measures was found, but in TBI patients, EDS was three times more prevalent when assessed objectively (McNemar objective versus subjective EDS: P > 0.99 in controls and **P < 0.0005 in TBI patients).
Table 1.
Demographic data for TBI patients and controls
| Controls | TBI | P-value | |
|---|---|---|---|
| Population characteristics | |||
| Age | 36.5 ± 13.2 | 35.5 ± 14.4 | 0.84 |
| Gender (M/F) | 31/11 | 31/11 | n.a. |
| Polysomnography | |||
| Total Sleep Time [min] | 392 ± 10 | 420 ± 7 | 0.04 |
| Sleep efficiency | 87.6 ± 2% | 87.9 ± 1.3% | 0.98 |
| Sleep latency (NREM2) [min] | 31.9 ± 6.7 | 23.7 ± 2.8 | 0.27 |
| Wake after sleep onset [min] | 11.4 ± 1.7 | 8.2 ± 1.1 | 0.17 |
| Arousal Index | 7.7 ± 0.8 | 6.0 ± 0.6 | 0.09 |
| PLMS Index | 1.3 ± 0.6 | 2.7 ± 1.2 | 0.30 |
| AHI | 3.2 ± 1 | 2.3 ± 0.5 | 0.42 |
| ODI | 3.3 ± 1 | 2.2 ± 0.4 | 0.32 |
| NREM1 | 9.1 ± 0.7 | 9.9 ± 1.2% | 0.06 |
| NREM2 | 41.2 ± 1.4 | 43.9 ± 1.3% | 0.32 |
| NREM3 | 20.2 ± 1.2 | 20.1 ± 1.0% | 0.29 |
| REM | 18.1 ± 0.9 | 18.0 ± 1.0% | 0.43 |
| WAKE | 11.4 ± 1.7 | 8.2 ± 1.1% | 0.49 |
| Actigraphy | |||
| Sleep time/24 h (actigraphy) | 7.1 ± 0.8# | 8.3 ± 1.1* | P < 0.00001 |
| Sleep time/24 h (sleep log) | 7.3 ± 1.1# | 7.5 ± 1.4* | n.s |
| Difference WD/WE | 0.4 ± 0.8 | 0.5 ± 0.8 | n.s. |
Relative amounts of sleep stages are given in percentage of total time in bed for NREM1, NREM2, NREM3, REM sleep, and wakefulness. Sleep time/24 h (actigraphy) = time asleep as measured by actigraphy recordings; Sleep time/24 h (sleep log) = time asleep per 24 h as assessed by sleep logs; Difference WD/WE = difference of daily hours of sleep between weekdays and weekends. P-values are given for comparison of controls versus TBI patients. Sleep time per 24 h as assessed objectively by actigraphy and sleep time per 24 h as reported on sleep logs did not differ in controls (#, n.s), whereas sleep times were subjectively underestimated in TBI patients (*P < 0.005, paired t-test). AHI = apnoea-hypopnoea index; PLMS = periodic limb movements during sleep; ODI = oxygen desaturation index.
Sleep state misperception after traumatic brain injury
Subjective EDS and fatigue were assessed by the Epworth Sleepiness Scale and the Fatigue Severity Scale, and subjective sleep time per 24 h by sleep logs. Although objective EDS and sleep need per day differed significantly between patients and controls, we found no significant differences of these subjective measures between the two groups (Table 2 and Fig. 3B). Similarly, when comparing the objective against subjective EDS evaluation, we found no significant differences for controls, whereas in TBI patients objective EDS was markedly more pronounced than recorded by subjective assessment (Fig. 3B, McNemar objective versus subjective: P-values > 0.99 in controls and 0.0004 in TBI patients). In other words, 8 of 42 TBI patients (19%) reported subjective EDS, but objective EDS was detected in 24 of 42 patients (57%, P < 0.0005). Similarly, sleep logs in controls showed no difference in sleep times between sleep logs and actigraphy, but in TBI patients, sleep times were underestimated by sleep logs when compared to actigraphy results (P = 0.002, Table 1).
Table 2.
Subjective sleep measures for TBI patients and controls
| TBI | CONTROLS | P-value | Stat | |
|---|---|---|---|---|
| Epworth Sleepiness Scale | 6.1 ± 3.1 | 5.6 ± 3.3 | 0.64 (n.s.) | t-test |
| FSS | 3.0 ± 1.1 | 2.7 ± 1.3 | 0.23 (n.s.) | t-test |
| Ullanlinna | 7.0 ± 3.5 | 6.2 ± 3.2 | 0.42 (n.s.) | t-test |
| SNS | 17.5 ± 17.0 | 22.6 ± 13.2 | 0.54 (n.s.) | t-test |
| Sleepiness yes/no | 16 | 10 | 0.15 (n.s.) | Chi-Square |
| SWD yes/no | 12 | 10 | 0.61 (n.s.) | Chi-Square |
| Epworth Sleepiness Scale>10 | 8 | 7 | 0.77 (n.s.) | Chi-Square |
FSS = Fatigue Severity Scale; Ullanlinna = Ullanlinna Narcolepsy Scale; SNS = Swiss Narcolepsy Scale; Sleepiness yes/no = overall subjective assessment of daytime sleepiness; SWD yes/no = overall subjective assessment of the presence of a sleep-wake disorder.
Consolidated slow-wave sleep in traumatic brain injury patients
Next, we addressed the question whether objective EDS and pleiosomnia in TBI patients are related to changes of sleep architecture or delta power of deep slow-wave sleep (NREM3) during polysomnography. Consolidation of sleep was assessed by calculation of a sleep fragmentation index based on manual sleep scoring (see ‘Materials and methods’ section). This measure represents the average number of behavioural state bouts per time (thus lower values indicate a more consolidated sleep). In TBI patients, NREM3, but also NREM1 and NREM2 sleep were significantly more consolidated (lower number of sleep bouts per time) than in controls, whereas REM sleep was not altered (Fig. 4, t-test: P = 0.002 for total effect). For the wake state, on the other hand, we observed a higher fragmentation index in TBI patients. Thus, whereas NREM sleep was more consolidated after TBI, the wake state showed a higher instability. These findings are in line with a trend towards a higher amount of normalized delta power (0.5–4.5 Hz) in TBI patients that resulted in a 22% increase in delta power (normalized delta power 0.44 in controls and 0.54 in TBI patients, t-test: P = 0.1, data not shown). Similarly, we also found less wake after sleep onset in the TBI group during polysomnography, although this finding did not reach statistical significance (Table 1). Relative sleep stage distribution showed no significant difference between TBI patients and controls (Table 1).
Figure 4.

Sleep fragmentation in TBI patients and controls. Sleep fragmentation (number of sleep bouts/time) is reduced in NREM sleep in TBI patients (grey bars) as compared to controls (black bars), whereas fragmentation of REM sleep did not differ. Wakefulness was more fragmented in TBI patients (t-test: **P < 0.005, *P < 0.05).
Severe trauma and intracranial haemorrhage as risk factors for pleiosomnia
To identify predictors or risk factors for post-traumatic SWD, we compared the sleep-wake outcome measures with clinical and imaging TBI characteristics in the acute phase. Correlation analysis revealed a strong dependence of pleiosomnia on low initial Glasgow Coma Scale, i.e. high TBI severity, and on haemorrhage as assessed by CT scans. Pearson’s bivariate correlation coefficients were R = 0.46 (P = 0.003) for pleiosomnia versus initial Glasgow Coma Scale and R = 0.42 (P = 0.005) for pleiosomnia versus presence of intracranial haemorrhage (statistically controlled for age and gender, Fig. 5A and D, full correlation not shown). Occurrence and extent of pleiosomnia were not associated with size or location of intracranial bleeding. Other clinical measures such as age or gender had no influence on sleep-wake outcome. In a subgroup analysis, the sleep amount in the haemorrhage TBI group differed significantly from controls, as well as from the non-haemorrhage TBI group (Fig. 5A and C), whereas in TBI patients without intracranial haemorrhage the increased sleep amount was not significantly different from controls. Mean sleep latency on MSLT, on the other hand, was decreased in all TBI patients and did not differ between patients with or without intracranial haemorrhage (Fig. 5B). Similarly, the subgroup analysis for low severity and high severity TBI based on initial Glasgow Coma Scale revealed again that pleiosomnia depends on TBI severity and differed between high and low grade TBI patients, whereas objective EDS was independent from the initial Glasgow Coma Scale on hospital admission (Fig. 5D–F).
Figure 5.

Pleiosomnia and excessive daytime sleepiness with respect to occurrence of intracranial haemorrhage and TBI severity. (A) Patients with intracranial haemorrhage (ICB+) had significantly more sleep per 24 h than both controls and TBI patients without bleedings (ICB−). (B) Mean sleep latencies on MSLT differed between controls and TBI patients, but no significant difference was observed between TBI patients with (ICB+) and without (ICB−) intracranial bleeding (Pearson correlation: R = −0.292, P < 0.01). (C) Patients with intracranial haemorrhage (ICB+) showed increased total sleep time on polysomnography (PSG), whereas sleep time in patients without haemorrhage (ICB−) did not differ from the control group. (D) Patients with severe TBI (Grade III, Glasgow Coma Scale ≤ 8) had more sleep per 24 h than both controls and mild TBI patients (Grade I, Glasgow Coma Scale ≥ 13). (E) Mean sleep latencies on MSLT differed between controls and TBI patients, but not between TBI patients with low and high severity trauma. (F) Patients with Grade III TBI had elevated total sleep time in polysomnography as compared to controls. (***P < 0.001, **P < 0.01 *P < 0.05 in one-way ANOVA, n = 84).
Correlation of sleep-wake outcome with acute phase laboratory markers
To find neurochemical predictors of post-traumatic sleep-wake outcome, we assessed whether sleep-wake changes 6 months after trauma were related to the presence of serum TBI markers, or to pathological levels of hypothalamo-pituitary-adrenal hormones in the acute phase after TBI. In a first step, we compared these acute laboratory measures with follow-up values at the time of sleep evaluation (6 months after TBI). Trauma markers such as NSE and S100B were elevated in the acute phase (Fig. 6A) but had no predictive value for sleep-wake outcomes. In terms of neuro-endocrine parameters, only adrenaline levels were significantly elevated (Fig. 6A). Overall cortisol levels were not changed, but we observed a change of the diurnal cortisol pattern: in the acute phase, the circadian gradient between morning and afternoon cortisol was flattened, as compared to the normal-more pronounced difference between a.m. and p.m. cortisol levels 6 months after TBI (Fig. 6B). Next, we compared these neurochemical markers with sleep-wake outcome parameters in a bivariate correlation analysis and we found that decreased morning cortisol levels in the acute phase correlated significantly with objective EDS 6 months after TBI (Fig. 6C, R = 0.88, P = 0.005, n = 10).
Figure 6.

Levels of potential biomarkers in the acute phase versus follow-up after TBI. (A) Acute phase lab values (within 5 days after TBI, grey circles) normalized to values at follow-up (6 months after TBI, dotted line) revealed increased values for TBI markers (S100) and elevated adrenaline levels (*P < 0.05). (B) Change of the diurnal cortisol pattern. The difference between a.m. (black) and p.m. (grey) cortisol levels was diminished in the acute phase (#P = 0.44 for comparison acute versus 6 months, paired two-sided t-test) as compared to 6 months after TBI (##P = 0.06 for comparison acute versus 6 months, unpaired one-sided t-test). (C) Morning cortisol levels (cortisol am) in the acute phase correlated significantly with sleepiness as measured by MSLTs 6 months after TBI (R = 0.88, P = 0.005, n = 10). S100 = S100 calcium binding protein; ACTH = adrenocorticotropic hormone; NSE = neuron-specific enolase.
Discussion
We present the first prospective and controlled clinical trial to examine EDS and pleiosomnia—i.e. excessive sleep need—in TBI patients by objective and subjective sleep measures. Six months after TBI, we found a marked increase of objective EDS and clinically relevant pleiosomnia as compared to an age- and gender-matched control group. Polysomnography revealed that NREM sleep in TBI patients is more consolidated than in controls, but the relative distribution of sleep stages is not different. The observed increase in total delta power after TBI was not significant in our sample. In contrast to these objective findings, TBI patients appear to underestimate both EDS and pleiosomnia if only subjective measures such as sleep logs or sleep questionnaires are applied.
The observed prevalence of post-traumatic EDS (57% of TBI patients) is in line with previous uncontrolled or retrospective trials (Masel et al., 2001; Baumann et al., 2007; Castriotta et al., 2007), and the observed prevalence of EDS in controls is in general agreement with earlier findings in large community-based cohort studies (Mignot et al., 2006). Our control group was not only matched for age and gender, but also for difference of total sleep times on actigraphy between working days and weekends/holidays, i.e. for overall sleep satiation before polysomnography and MSLT. By controlling for this measure, we were able to compare objective sleep-wake measures between groups without a potential bias due to chronic sleep deprivation. Furthermore, by including also subjects with sleep latencies<8 min as controls, we were able to compare TBI patients to a reliable and not overly preselected healthy reference group, because moderate objective daytime sleepiness is a frequent finding in healthy subjects (Mignot et al., 2006). By doing this, we confirm a significantly increased prevalence of EDS and of pleiosomnia and conclude—in the absence of other potential causes of SWD—that these are probably directly related to the trauma. A potential confounding factor is the occurrence of depression after TBI and development of secondary post-traumatic SWD (e.g. insomnia). However, standardized neuropsychiatric assessment revealed that only one TBI patient had scores suggestive of a clinical depression. Therefore, we conclude that EDS and pleiosomnia in this cohort of TBI patients is not caused or worsened by accompanying mood disorders.
Pleiosomnia was strongly correlated with both severe TBI as assessed with the Glasgow Coma Scale and with the presence of an intracranial haemorrhage upon CT scan in the acute phase after TBI. This indicates that trauma patients with an objective correlate of severe TBI, i.e. intracranial bleedings, are more susceptible to developing post-traumatic pleiosomnia. Other clinical parameters such as age or gender had no influence on the development of post-traumatic pleiosomnia. Furthermore, as SWDs were not associated with size or localization of bleedings, we suggest that intracranial haemorrhage itself is not the origin for poor sleep-wake outcome, but rather an indicator of the severity of the trauma. We rather hypothesize that patients with severe trauma may suffer more often from direct or secondary lesions in the hypothalamus or rostral brainstem, which are not accessible by CT scan. This might lead to the otherwise observed loss of wake-promoting neuronal systems with TBI, which could explain the occurrence of pleiosomnia. These deductions, however, lie beyond the scope of the current study. More detailed (MRI-based) neuroanatomical analyses in TBI patients are needed to investigate the occurrence and distribution of micro-lesions in the brainstem after TBI. In line with this, we recently found a significant loss of wake-promoting histaminergic cells in the hypothalamic tuberomammillary nucleus (Valko et al., 2014). On the other hand, we cannot exclude the possibility that other mechanisms that are related to intracranial haemorrhage—such as vasospasms that may affect cerebral blood flow to the midbrain—might contribute to increased sleep need (Cetas et al., 2009). However, given the fact that recordings were made 6 months after trauma, such mechanisms are likely to be less relevant.
On the other hand, we might speculate whether increased sleep need might be associated with the need for neuroplasticity after TBI. It has been shown that synchronous slow wave EEG activity is associated with neuronal plasticity or synaptic strength, both during sleep and in the awake state (Stickgold et al., 2001; Carmichael and Chesselet, 2002; Tononi and Cirelli, 2006). Our finding of more consolidated deep sleep and a tendency towards higher delta power in NREM sleep might be in favour of such a hypothesis. The percentage breakdown of sleep stages was unaltered in TBI patients as compared to healthy controls. In other words, TBI patients are in need for longer sleep times, but still have the same relative amounts of sleep stages. Altogether, our findings indicate that patients with more severe TBI and higher levels of cellular or axonal damage might be in need of longer sleep times per 24 h and for more consolidated slow-wave sleep with higher delta power. In this regard, it would be interesting to learn whether overall sleep need and slow wave sleep with increased delta power are even more accentuated early on after TBI. This question could be addressed by further studies.
We diagnosed pleiosomnia based on actigraphy recordings. It must be noted that decreased levels of activity might have led to an overestimation of sleep times per 24 h. This is why we double-checked actigraphy data with the sleep logs that have been filled by the patients during actigraphy recordings. Furthermore, sleep laboratory examinations in a controlled setting (MSLT, polysomnography; Fig. 2B and C) confirmed the occurrence of increased sleep need in TBI patients. However, maximal sleep time at polysomnography was limited to 8 h, which leads to a quantitative underestimation of sleep need per 24 h, particularly in subjects with increased sleep need (>8 h; Fig. 5). Therefore and despite the given methodological limitations, we believe that actigraphy delivers better insights into total average sleep time per 24 h.
Post-traumatic EDS, on the other hand, occurs independently of traumatic intracranial haemorrhage and trauma severity. In our population, many patients with minor head trauma still developed clinically relevant EDS. Therefore, other factors than direct neuronal damage might influence post-traumatic EDS. In line with this, analysis of neuro-endocrine markers revealed that flattened diurnal cortisol profiles and low a.m. cortisol levels correlate with the development of post-traumatic EDS, i.e. shorter sleep latency on MSLT. Impaired alertness (Chapotot et al., 1998) and awakening problems have been reported in patients with lowered cortisol levels (Dijk et al., 2012). Thus, pathological cortisol signalling seems to herald a subsequent evolution of midbrain-driven EDS. Earlier studies have suggested that midbrain structures are particularly vulnerable to TBI (Crompton, 1971a, b; Baumann et al., 2009). Considering our findings, it seems that even mild TBI can lead to a relevant disturbance in the central regulation of the hypothalamic-pituitary-adrenal axis without any further brain damage or focal neurological deficits.
However, as an important limitation, this study was not designed to account for diurnal cortisol changes. The analysis of a.m. and p.m. levels was done post hoc and therefore the correlation analysis was done only for patients in whom a.m. and p.m. cortisol was available. Therefore, no conclusion for TBI patients in general can be drawn at this point and only future systematic approaches might reveal whether or not diurnal cortisol level measurements might be predictive of post-traumatic EDS.
Finally, many TBI patients with marked increase of ‘objective’ EDS in MSLT did not report ‘subjective’ EDS as assessed by various sleep questionnaires. The same applies for increased sleep need: pleiosomnia was obvious in TBI patients as compared to controls, but sleep logs revealed similar reported sleep times in both groups. In other words: although TBI patients presented with pronounced objective EDS and pleiosomnia, the applied subjective tests failed to capture both EDS and pleiosomnia. These results indicate a strong sleep state misperception in TBI patients. The reason for this striking difference between objective and subjective sleep measures remains unclear. As this study was not designed to assess for anosognosia in TBI patients, this question should be addressed in future studies by appropriate neuropsychological assessments. From a clinical point of view, this finding poses a diagnostic challenge, as standardized and validated screening instruments for sleepiness or hypersomnia (e.g. Epworth Sleepiness Scale, sleep logs, history-taking) obviously fail to detect SWD in TBI patients. Consequently, considering these data and forensic consequences of EDS (e.g. in patients driving motor vehicles), TBI patients should be examined with sleep laboratory examinations rather than self-reported sleep measures whenever possible. As polysomnography is expensive and time-consuming, further studies might elucidate whether actigraphy and MSLT alone might be sufficient.
Acknowledgements
We thank Dr Dilek Könü and Dr Oguzkan Sürücü for their help in patient recruitment and Dr D. Noain for many helpful discussions.
Glossary
Abbreviations
- EDS
excessive daytime sleepiness
- MSLT
Multiple Sleep Latency Test
- NREM
non-rapid eye movement
- SWD
sleep-wake disturbances
- TBI
traumatic brain injury
Funding
This study was supported by the Swiss National Science Foundation (SNF, grant no. 32003B-125504), and by the Clinical Research Priority Program ‘Sleep and Health’ of the University of Zurich.
References
- American Academy of Sleep Medicine. . 2nd edn. Westchester, IL: American Academy of Sleep Disorders Association; 2005. International Classification of sleep disorders (ICSD-2), diagnostic and coding manual, 2nd edn. [Google Scholar]
- Baumann CR, Werth E, Stocker R, Ludwig S, Bassetti CL. Sleep-wake disturbances 6 months after traumatic brain injury: a prospective study. Brain. 2007;130:1873–83. doi: 10.1093/brain/awm109. [DOI] [PubMed] [Google Scholar]
- Baumann CR, Bassetti CL, Valko PO, Haybaeck J, Keller M, Clark E, et al. Loss of hypocretin (orexin) neurons with traumatic brain injury. Ann Neurol. 2009;66:555–9. doi: 10.1002/ana.21836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger RP, Pierce MC, Wisniewski SR, Adelson PD, Clark RS, Ruppel RA, et al. Neuron-specific enolase and S100B in cerebrospinal fluid after severe traumatic brain injury in infants and children. Pediatrics. 2002;109:E31. doi: 10.1542/peds.109.2.e31. [DOI] [PubMed] [Google Scholar]
- Carmichael ST, Chesselet MF. Synchronous neuronal activity is a signal for axonal sprouting after cortical lesions in the adult. J Neurosci. 2002;22:6062–70. doi: 10.1523/JNEUROSCI.22-14-06062.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castriotta RJ, Wilde MC, Lai JM, Atanasov S, Masel BE, Kuna ST. Prevalence and consequences of sleep disorders in traumatic brain injury. J Clin Sleep Med. 2007;3:349–56. [PMC free article] [PubMed] [Google Scholar]
- Cetas JS, Lee DR, Alkayed NJ, Wang R, Iliff JJ, Heinricher MM. Brainstem control of cerebral blood flow and application to acute vasospasm following experimental subarachnoid hemorrhage. Neuroscience. 2009;163:719–29. doi: 10.1016/j.neuroscience.2009.06.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cippa MA, Baumann CR, Siccoli MM, Bassetti CL, Poryazova R, Werth E. Actigraphic assessment of periodic leg movements in patients with restless legs syndrome. J Sleep Res. 2013;22:589–92. doi: 10.1111/jsr.12053. [DOI] [PubMed] [Google Scholar]
- Chaumet G, Quera-Salva MA, Macleod A, Hartley S, Taillard J, Sagaspe P, et al. Is there a link between alertness and fatigue in patients with traumatic brain injury? Neurology. 2008;71:1609–13. doi: 10.1212/01.wnl.0000334753.49193.48. [DOI] [PubMed] [Google Scholar]
- Chapotot F, Gronfier C, Jouny C, Muzet A, Brandenberger G. Cortisol secretion is related to electroencephalographic alertness in human subjects during daytime wakefulness. J Clin Endocrinol Metab. 1998;83:4263–8. doi: 10.1210/jcem.83.12.5326. [DOI] [PubMed] [Google Scholar]
- Crompton MR. Hypothalamic lesions following closed head injury. Brain. 1971a;94:165–72. doi: 10.1093/brain/94.1.165. [DOI] [PubMed] [Google Scholar]
- Crompton MR. Brainstem lesions due to closed head injury. Lancet. 1971b;1:669–73. doi: 10.1016/s0140-6736(71)92680-8. [DOI] [PubMed] [Google Scholar]
- Dijk DJ, Duffy JF, Silva EJ, Shanahan TL, Boivin DB, Czeisler CA. Amplitude reduction and phase shifts of melatonin, cortisol and other circadian rhythms after a gradual advance of sleep and light exposure in humans. PLoS One. 2012;7:e30037. doi: 10.1371/journal.pone.0030037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guilleminault C, Faull KF, Miles L, van den Hoed J. Posttraumatic excessive daytime sleepiness: a review of 20 patients. Neurology. 1983;33:1584–9. doi: 10.1212/wnl.33.12.1584. [DOI] [PubMed] [Google Scholar]
- Hautzinger M, Bailer M. Allgemeine depressions skala. Manual . Göttingen: Beltz; 1993. [Google Scholar]
- Iber C, Chesson A, Quan S, editors. 1st edn. Westchester IL: American Academy of Sleep Medicine; 2007. The AASM manual for the scoring of sleep and associated events: rules, terminology, and technical specification. [Google Scholar]
- King MA, Jaffre MO, Morrish E, Shneerson JM, Smith IE. The validation of a new actigraphy system for the measurement of periodic leg movements in sleep. Sleep Med. 2005:507–13. doi: 10.1016/j.sleep.2004.12.010. [DOI] [PubMed] [Google Scholar]
- Lamers KJ, Vos P, Verbeek MM, Rosmalen F, van Geel WJ, van Engelen BG. Protein S-100B, neuron-specific enolase (NSE), myelin basic protein (MBP) and glial fibrillary acidic protein (GFAP) in cerebrospinal fluid (CSF) and blood of neurological patients. Brain Res Bull. 2003;61:261–4. doi: 10.1016/s0361-9230(03)00089-3. [DOI] [PubMed] [Google Scholar]
- Masel BE, Scheibel RS, Kimbark T, Kuna ST. Excessive daytime sleepiness in adults with brain injuries. Arch Phys Med Rehabil. 2001;82:1526–32. doi: 10.1053/apmr.2001.26093. [DOI] [PubMed] [Google Scholar]
- Mathias JL, Alvaro PK. Prevalence of sleep disturbances, disorders, and problems following traumatic brain injury: a meta-analysis. Sleep Med. 2012;13:898–905. doi: 10.1016/j.sleep.2012.04.006. [DOI] [PubMed] [Google Scholar]
- Mignot E, Lin L, Finn L, Lopes C, Pluff K, Sundstrom ML, et al. Correlates of sleep-onset REM periods during the multiple sleep latency test in community adults. Brain. 2006;129:1609–23. doi: 10.1093/brain/awl079. [DOI] [PubMed] [Google Scholar]
- Ouellet MC, Morin CM. Subjective and objective measures of insomnia in the context of traumatic brain injury: a preliminary study. Sleep Med. 2006;7:486–97. doi: 10.1016/j.sleep.2006.03.017. [DOI] [PubMed] [Google Scholar]
- Parcell DL, Ponsford JL, Rajaratnam SM, Redman JR. Self-reported changes to nighttime sleep after traumatic brain injury. Arch Phys Med Rehabil. 2006;87:278–85. doi: 10.1016/j.apmr.2005.10.024. [DOI] [PubMed] [Google Scholar]
- Ponsford JL, Parcell DL, Sinclair KL, Roper M, Rajaratnam SM. Changes in sleep patterns following traumatic brain injury: a controlled study. Neurorehabil Neural Repair. 2013;27:613–21. doi: 10.1177/1545968313481283. [DOI] [PubMed] [Google Scholar]
- Schreiber S, Barkai G, Gur-Hartman T, Peles E, Tov N, Dolberg OT, et al. Long-lasting sleep patterns of adult patients with minor traumatic brain injury (mTBI) and non-mTBI subjects. Sleep Med. 2008;9:481–7. doi: 10.1016/j.sleep.2007.04.014. [DOI] [PubMed] [Google Scholar]
- Sommerauer M, Valko P, Werth E, Baumann CR. Excessive sleep need following traumatic brain injury: a case-control study of 36 patients. J Sleep Res. 2013;22:634–9. doi: 10.1111/jsr.12068. [DOI] [PubMed] [Google Scholar]
- Stickgold R, Hobson JA, Fosse R, Fosse M. Sleep, learning, and dreams: off-line memory reprocessing. Science. 2001;294:1052–7. doi: 10.1126/science.1063530. [DOI] [PubMed] [Google Scholar]
- Tononi G, Cirelli C. Sleep function and synaptic homeostasis. Sleep Med Rev. 2006;10:49–62. doi: 10.1016/j.smrv.2005.05.002. [DOI] [PubMed] [Google Scholar]
- Valko PO, Gavrilov Y, Yamamoto M, Finn K, Reddy H, Haybaeck J, et al. Damage to histamine and other hypothalamic neurons with traumatic brain injury. Ann Neurol. doi: 10.1002/ana.24298. Advance Access published 2014. [DOI] [PubMed] [Google Scholar]
- Vos PE, Lamers KJ, Hendriks JC, van Haaren M, Beems T, Zimmerman C, et al. Glial and neuronal proteins in serum predict outcome after severe traumatic brain injury. Neurology. 2004;62:1303–10. doi: 10.1212/01.wnl.0000120550.00643.dc. [DOI] [PubMed] [Google Scholar]