

A number of single gene disorders share clinical and MRI characteristics with multiple sclerosis and may be overlooked in the differential diagnosis. Weisfeld-Adams et al. review features that should serve as ‘red flags’, alerting clinicians to the possibility of specific Mendelian or mitochondrial disorders in patients with suspected multiple sclerosis.
Keywords: multiple sclerosis, misdiagnosis, inherited disease, mitochondrial disease
Abstract
Several single gene disorders share clinical and radiologic characteristics with multiple sclerosis and have the potential to be overlooked in the differential diagnostic evaluation of both adult and paediatric patients with multiple sclerosis. This group includes lysosomal storage disorders, various mitochondrial diseases, other neurometabolic disorders, and several other miscellaneous disorders. Recognition of a single-gene disorder as causal for a patient’s ‘multiple sclerosis-like’ phenotype is critically important for accurate direction of patient management, and evokes broader genetic counselling implications for affected families. Here we review single gene disorders that have the potential to mimic multiple sclerosis, provide an overview of clinical and investigational characteristics of each disorder, and present guidelines for when clinicians should suspect an underlying heritable disorder that requires diagnostic confirmation in a patient with a definite or probable diagnosis of multiple sclerosis.
Introduction
Multiple sclerosis is a common, chronic demyelinating neurological disease primarily affecting young adults, with a prevalence of ∼0.1% in the Caucasian population (Miller and Leary, 2007). Early, accurate diagnosis is critical to effective patient management and counselling, but assignment of an incorrect diagnosis of multiple sclerosis remains a frequent concern.
Patients with multiple sclerosis are classified according to their clinical phenotype, with ∼85% following a relapsing-remitting course (relapsing-remitting multiple sclerosis) characterized by recurrent, acute neurological deficits punctuating periods of latency or remission (Lublin and Reingold, 1996). Typical symptoms of relapses may be referable to demyelinating pathology involving the optic nerves (e.g. optic neuritis), brainstem (e.g. internuclear ophthalmoplegia) or spinal cord (e.g. partial myelitis), although non-specific symptoms referable to the cerebral hemispheres or other brain regions can also occur (Katz Sand and Lublin, 2013). Approximately 15% of patients follow a primary progressive or progressive relapsing course from disease onset, usually characterized by symptoms of progressive myelopathy (gait instability, spasticity, bladder symptoms) and cognitive impairment. Approximately half of relapsing-remitting patients develop a secondary progressive course within 20 years of disease onset (secondary progressive multiple sclerosis; Tremlett et al., 2008).
McDonald Criteria (Polman et al., 2011) for multiple sclerosis integrate data from neurological history, physical examination, and MRI appearances of the brain and cord. Diagnostic criteria and classification of multiple sclerosis subtypes have evolved in recent decades, and, although successive versions have differed in emphasis, all have required dissemination of disease in space (requiring involvement of multiple areas of the CNS) and in time (requiring ongoing disease activity over time). Unmatched oligoclonal bands are observed in the CSF of >80% of patients with multiple sclerosis and may be considered as supporting evidence, but are neither completely sensitive nor specific for multiple sclerosis. In the absence of pathognomonic clinical findings or a definitive laboratory test, the diagnosis of multiple sclerosis remains challenging in many patients, and diagnostic criteria emphasize the caveat of ‘no better explanation’ for a patient’s clinical presentation and MRI findings. If the presentation is atypical, alternative diagnoses should be explored and investigated, and patients subjected to a diligent search for clinical, imaging or laboratory ‘red flags’; in 2008, a scheme and ‘red flag’ scoring instrument was presented as a tool to be used in the differential diagnosis of multiple sclerosis (Miller et al., 2008). Patients with adolescent or adult-onset inherited disorders, as well as those with non-multiple sclerosis idiopathic inflammatory demyelinating disease entities such as neuromyelitis optica and acute disseminated encephalomyelitis, or infectious, neoplastic, or vascular disorders, can all present similarly to multiple sclerosis, resulting in misdiagnoses (Miller et al., 2008; Rudick and Miller, 2012).
The spectrum of disorders mistaken for multiple sclerosis have likely changed over time as a result of evolution of diagnostic criteria for multiple sclerosis (Solomon et al., 2012). These disorders share characteristics with multiple sclerosis but have quite different clinical courses and prognostic implications. The incidence of multiple sclerosis misdiagnosis probably varies between individual clinicians and institutions, and depends upon efforts used to investigate for other disorders in patients with ‘multiple sclerosis-like’ presentations (Engell, 1988; Solomon and Weinshenker, 2013). In a recent editorial concerned with multiple sclerosis misdiagnosis, the authors suggested that multiple sclerosis diagnosis is based on symptoms, signs and MRI findings, clinical judgement, experience, and, crucially, the capacity needed to identify an alternative diagnosis (Rudick and Miller, 2012). They also discussed over-reliance on MRI appearances in multiple sclerosis diagnosis, as well as the phenomenon of ‘cessation of critical thinking about alternative diagnoses once the multiple sclerosis diagnosis has been made’.
Multiple historical case reports describe clinical and diagnostic confusion between multiple sclerosis and various single gene disorders. Diagnosis of some of these rare disorders is likely to be subject to ascertainment bias, with classic presentations occurring during early childhood, and later-onset variants going unrecognized and conferring more subtle presentations. Several such disorders can present in adolescence or adulthood with progressive spastic paraparesis and accompanying white matter lesions on MRI, mimicking primary progressive multiple sclerosis. Less commonly patients with such disorders can display relapsing-remitting or insidiously progressive neurological symptoms. The overlap may at times result in misdiagnosis of multiple sclerosis, ultimately leading to inappropriate treatment with immunomodulatory therapies and, in certain disorders, a missed opportunity for other potentially helpful management interventions.
No comprehensive review of single-gene aetiologies of ‘multiple sclerosis-like’ presentations has been written since that by Natowicz and Bejjani (1994), 20 years ago. Since then, neuroimaging technologies have evolved unrecognizably, and have brought an abundance of knowledge regarding MRI characteristics in multiple sclerosis and other conditions. In the same period, the molecular genetic and/or biochemical basis of many ‘multiple sclerosis mimics’ has been elucidated, and much has been learned about the phenotypic spectrum of these disorders. Most inherited mimics of multiple sclerosis are now amenable to molecular genetic testing of specific genes, or interrogation of multiple genes using next-generation, whole exome, or whole genome sequencing. The current article, based on an extensive review of the literature of cases of Mendelian diseases misdiagnosed as multiple sclerosis, and the authors’ own clinical experiences, presents a scheme for neurologists and medical geneticists to follow for differential diagnosis of several single-gene disorders that may mimic multiple sclerosis and seem to meet McDonald criteria, and outlines strategies for evaluation of such patients.
Disorders of lysosomal storage and peroxisomal function
Lysosomal storage diseases are inherited metabolic disorders characterized by defective catabolism of complex molecules, including lipids, glycoproteins, and oligosaccharides, and by resultant toxic accumulation of cellular debris. Several lysosomal disorders have CNS white matter involvement in patterns that may overlap with those observed in multiple sclerosis, and attenuated forms of Krabbe disease (globoid cell leukodystrophy), metachromatic leukodystrophy, Niemann-Pick disease type C (a disorder of cholesterol trafficking), Fabry disease, and variant forms of X-linked adrenoleukodystrophy and adrenomyeloneuropathy, also have the potential to show clinical overlap with multiple sclerosis in adult patients, and are probably underdiagnosed. Several published single cases or case series in the literature report individuals with confirmed lysosomal storage disease or peroxisomal disease who were originally diagnosed with ‘suspected multiple sclerosis’ or clinically definite multiple sclerosis (Zwetsloot et al., 1992; Stöckler et al., 1993; Kazibutowska et al. 2002; Chebel et al., 2009; Godeiro-Junior et al., 2009; Carra-Dalliere et al., 2013; Weisfeld-Adams et al., 2013). The biochemical defects underlying specific lysosomal disorders and peroxisomal disorders that can potentially mimic multiple sclerosis, together with corresponding clinical and radiologic features and guidelines for diagnostic testing, are summarized in Table 1.
Table 1.
Lysosomal and peroxisomal disorders with potential to mimic multiple sclerosis
| Disorder | Gene(s); biochemical defect | Inheritance mechanism | Neurologic presentations in adult-onset disease | ‘Typical’ MRI appearances | Recommended diagnostic testing |
|---|---|---|---|---|---|
| Globoid cell leukodystrophy (Krabbe disease) |
|
AR | SP and gait dysfunction, cognitive deterioration, clumsiness; lower extremity paraesthesia and visual loss reported rarely | T2/FLAIR hyperintensities involving CS or pyramidal tracts are characteristic. Symmetric periventricular T2/FLAIR WM hyperintensity, predominantly posterior (parieto-occipital) with common involvement of the splenium of the CC; U-fibres generally spared |
|
| Metachromatic leukodystrophy |
|
AR | Neuropsychiatric symptoms with cognitive deterioration progressing to dementia; spasticity and incoordination; visual failure; dystonia; peripheral neuropathy | Symmetrical ‘sheet-like’ areas of abnormal T2/FLAIR WM periventricular hyperintensity, typically with frontal predominance, progressing to involve subcortical WM while sparing the U-fibres; variable involvement of cerebellar, thalamic or projection fibre WM in advanced disease; typically no enhancement |
|
| X-linked adrenoleukodystrophy/ adrenomyeloneuropathy |
|
X-linked | Male hemizygotes: SP with progressive gait, spastic paraparesis and bladder dysfunction; erectile dysfunction; abnormalities of proprioception and vibration sense; headache, hemiparesis and ataxia also reported; primary adrenocortical insufficiency with/without neurologic symptoms; Female heterozygotes: 20–50% eventually develop SP or mild myelopathy, often after middle age; adrenocortical insufficiency rare | In AMN, primary involvement of frontopontine or corticospinal projection fibres; many AMN patients develop cerebral involvement limited to long tracts (posterior limbs internal capsules, brain stem) or with lobar involvement similar in appearance to childhood X-ALD (frontal or parieto-occipital); peripheral enhancement common | |
| Fabry disease |
|
X-linked | Temperature-sensitive acroparasthesias usually signal clinical onset of disease; anhidrosis; recurrent/chronic abdominal pain; transient TIA/stroke episodes include hemiparesis or vertigo; chronic fatigue, anxiety and depression commond | Non-specific, multifocal, asymmetric T2/FLAIR hyperintense lesions in deep and subcortical WM; progressive WM lesion load over time due to cerebral vasculopathy; T1 hyperintensities of thalamic pulvinar are characteristic but only present in 25% of patients |
|
| Niemann-Pick disease, type C |
|
AR | Partial/generalized seizure disorder with gelastic cataplexy and vertical supranuclear gaze palsy are characteristic; also neuropsychiatric symptoms with cognitive deterioration and dementia; dystonia; dysarthria; peripheral neuropathy reported rarely | Thinning of CC with increased signal in periatrial WM; widespread GM/WM abnormalities; reduced pons:midbrain ratio; cerebellar vermian atrophy (late) |
|
| Chédiak-Higashi disease |
|
AR | Late-onset disease characterized by SP, spinocerebellar degeneration, parkinsonism, dystonia, cognitive deteriorationg | T2-FLAIR hyperintensity in periventricular WM becoming more prominent with time; global atrophy, especially spinocerebellar atrophy |
|
AMN = adrenomyeloneuropathy; AR = autosomal recessive; CC = corpus callosum; CS = corticospinal; GM = grey matter; LDL = low-density lipoprotein; NCV = nerve conduction velocities; SP = spastic paraparesis; TIA = transient ischaemic attack; VLCFA = very long chain fatty acids; WM = white matter; X-ALD = X-linked adrenoleukodystrophy.
a High prevalence of 30 kb deletion allele in Caucasian population.
b VLCFA analysis normal in 10-20% of female heterozygotes (Moser et al., 1999); VLCFA abnormalities generally correlate poorly with clinical phenotypes.
c Males only; 70% of adult males with adrenomyeloneuropathy phenotype have adrenal insufficiency.
d Concurrent evidence of cardiovascular, cerebrovascular, or renal disease common; female heterozygotes can be asymptomatic with normal life expectancy or present similarly to hemizygous males with classic disease, depending on pattern of X-inactivation.
e Full diagnostic algorithm provided in Gal et al. (2011); both plasma and leucocytes should be assayed as some mutations specifically affect trafficking of enzyme; measure of α-gal A is insufficient for detection of carrier status in females.
f Limited availability outside of research laboratories at time of submission.
g Adult-onset patients with mild or subclinical immunodeficiency; pigmentary abnormalities of skin, hair and retina common but less apparent than in infantile onset disease.
Various newborn screening initiatives for several lysosomal and peroxisomal disorders are now underway (Matern et al., 2013) and may help answer several unsolved questions relating to their natural histories and phenotypic spectra. Over 7 years of experience with newborn screening for Krabbe disease in New York State suggests that late-onset Krabbe disease is significantly more common than previously recognized. This is indicated by large numbers of asymptomatic infants with low galactosylceramidase (GALC) activity and biallelic, pathogenic GALC mutations. Specific GALC missense alleles (p.G57S, p.W115R and p.G286D) are associated with late-onset Krabbe disease (Lissens et al., 2007). Many infants with very low GALC activity on newborn screening have remained asymptomatic over several years (Wang et al., 2011) and at what age, if ever, these individuals will develop symptomatic Krabbe disease is unclear. Late-onset Krabbe disease typically presents in adolescence or adulthood with insidiously progressive paraparesis and gait disturbance which can appear similar to progressive multiple sclerosis. MRI ‘red flags’ to suggest the diagnosis of Krabbe disease over multiple sclerosis include prominent involvement of the pyramidal tracts and/or symmetric or confluent involvement of the posterior white matter, splenium of the corpus callosum, and corticospinal tracts (Fig. 1A–C; Farina et al., 2000; Sedel et al., 2008).
Figure 1.
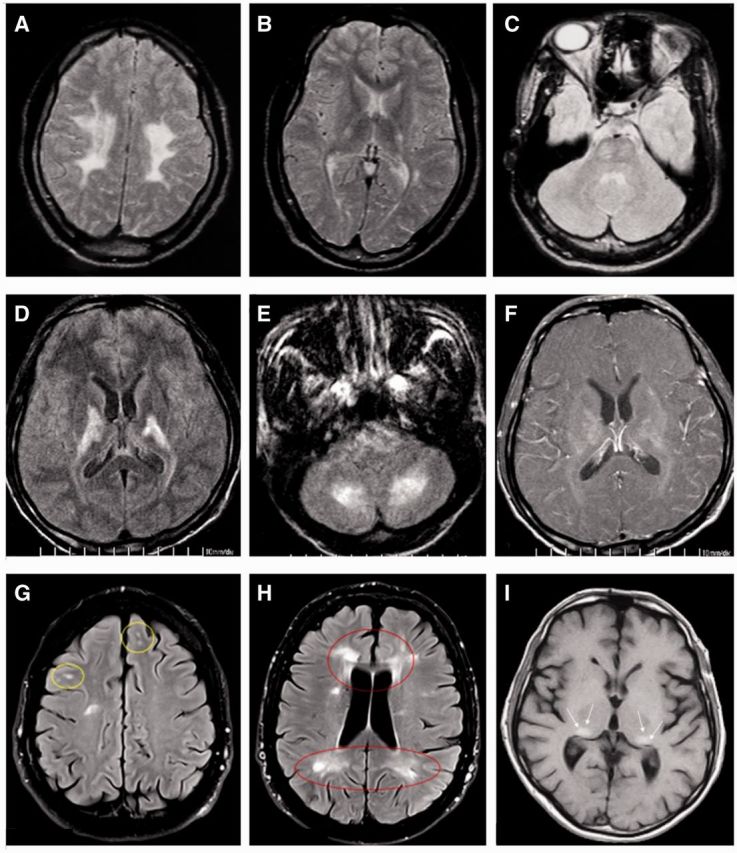
Selected magnetic resonance images from patients with adult-onset lysosomal and peroxisomal disorders. (A) Axial T2-weighted image of a 24-year-old female with Krabbe disease (globoid cell leukodystrophy) demonstrating confluent T2 hyperintensity in the bilateral centrum semiovale, centred in the fronto-parietal lobes and involving the corticospinal tracts. The patient presented with a 2-year history of worsening spastic paraparesis and symmetrical lower extremity sensory disturbance. (B and C) Axial T2 and proton-density weighted images of the same patient showing symmetric hyperintensity in the posterior limbs of the internal capsule and pons, corresponding to the corticospinal tracts. (D and E) Axial FLAIR images from a 35-year-old Taiwanese male with an 18-year history of progressive spastic paraplegia, diagnosed with adrenomyeloneuropathy. Images demonstrate asymmetric T2 hyperintensity within the internal capsules with corticospinal tract involvement, and asymmetric T2 hyperintensity in cerebellar white matter and dentate nuclei. (F) Post-contrast T1-weighted image of the same patient demonstrating patchy contrast enhancement in and around corresponding regions of signal hyperintensity (reproduced with permission, Mo et al., 2004). (G and H) Axial FLAIR images of a 64-year-old female with a confirmed diagnosis of Fabry disease and with a 40-year history of ataxia and intermittent right-sided hemiparesis and hemisensory disturbance. There is focal and confluent T2 hyperintensity in the periventricular, subcortical and deep white matter, in a pattern mimicking multiple sclerosis (reproduced with permission, Böttcher et al. 2013). Although the patient met McDonald criteria for multiple sclerosis, oligoclonal bands were absent from CSF. (I) Axial T1-weighted image of a 46-year-old male with Fabry disease demonstrating classic, intrinsic T1 hyperintensities in the pulvinar nuclei bilaterally, a classic imaging feature of Fabry disease (reproduced with permission, Burlina et al., 2008).
Based on their extensive clinical overlap with each other and with acquired neurologic conditions, it is probably reasonable to extrapolate the under-ascertainment phenomenon suggested by early experiences with newborn screening for Krabbe and Fabry diseases to other disorders including metachromatic leukodystrophy and X-linked adrenoleukodystrophy/adrenomyeloneuropathy (X-ALD/AMN). Among European patients with adult-onset metachromatic leukodystrophy, the p.I179S allele (associated with a predominant psychiatric phenotype) and p.P426L, account for the majority of ARSA alleles (Ługowska et al., 2005). Non-pathogenic pseudodeficiency alleles occur at relatively high frequencies in ARSA, and may complicate interpretation of ARSA enzymology for metachromatic leukodystrophy; the best overall initial screening test for metachromatic leukodystrophy is therefore urinary sulphatides. A complex ARSA pseudodeficiency allele (c.[1049A>G; *96A>G]) has been recognized at higher frequency among patients with multiple sclerosis compared to matched controls (Kappler et al., 1991; Baronica et al., 2011). Genotype–phenotype correlations have also been observed in adult-onset forms of Chédiak-Higashi disease, a disorder of lysosomal trafficking with associated pigmentary abnormalities, progressive spastic paraplegia and progressive spinocerebellar degeneration and leukoencephalopathy on MRI, with most cases occurring in the setting of biallelic missense mutations or in-frame deletions in LYST (Karim et al., 2002; Weisfeld-Adams et al., 2013).
Patients with the adrenomyeloneuropathy phenotype of X-linked adrenoleukodystrophy can be confused with primary progressive multiple sclerosis, and classically present with progressive spastic paraparesis and bladder disturbance in early or middle adulthood. Families with ABCD1 mutations exhibit extreme intra and interfamilial heterogeneity, and very long chain fatty acid profiles are poorly predictive of clinical phenotypes. More than 80% of female heterozygotes harbouring pathogenic mutations in ABCD1 develop adult-onset spastic paraparesis, mild myeloneuropathy, and/or peripheral neuropathy usually after 40 years of age, and have a high incidence of bladder spasticity and faecal incontinence. Prevalence of symptoms in female heterozygotes seems to correlate strongly with advancing age (Engelen et al., 2014). Cerebral disease is highly unusual in females. Most hemizygous males develop adult-onset adrenomyeloneuropathy (accounting for 40–45% of males) or childhood-onset cerebral adrenoleukodystrophy (30–35% of males), whereas a much smaller proportion of adult males develop clinical and radiological features of cerebral disease without preceding myeloneuropathy symptoms (Engelen et al., 2012). Although the prevalence of cerebral disease in males with adrenomyeloneuropathy was previously estimated at 20% within 10 years of onset (van Geel et al., 2001), a more recent Dutch study evaluating 27 males with adrenomyeloneuropathy with mean symptomatic onset at 35.3 years of age showed that >60% of patients developed evidence of cerebral demyelination a mean of 10.2 years after onset of adrenomyelomeuropathy (de Beer et al., 2014). Furthermore, survival after onset of cerebral demyelinating symptoms in these patients was limited to a mean of 3.4 ± 2.9 years, suggesting that cerebral disease in adult males, once underway, may be as devastating and as rapidly progressive as is the case for childhood-onset cerebral X-linked adrenoleukodystrophy. Head trauma and stroke may precipitate the onset of cerebral disease (Raymond et al., 2010). Several different MRI patterns have been described in adults and children; these patterns have utility in predicting clinical course and, together with extent of disease and age of onset, may be helpful in determining candidacy for haematopoietic stem cell transplantation. The classic pattern seen in childhood-onset cerebral X-linked adrenoleukodystrophy encompasses symmetric T2 hyperintensity of parieto-occipital white matter, classically periatrially (so-called ‘Pattern 1’ disease); most cases are of early-onset and are characterized by rapid progression (Loes et al., 2003). Males with the adrenomyeloneuropathy phenotype can have normal brain MRI, changes that appear in advance of clinical disease, or changes that appear several years or decades later (Loes et al., 2003; de Beer et al., 2014). Abnormalities restricted to the pyramidal tracts, or diffuse MRI abnormalities involving the cerebral hemispheres, posterior limbs of the internal capsules, brainstem and cerebellum can also occur (Fig. 1D–F; Kumar et al., 1995).
Pilot newborn screening studies for Fabry disease, which frequently presents with intermittent acroparaesthesias and abnormal white matter appearances on MRI, have also revealed an unexpectedly high incidence among male newborns (Spada et al., 2006; Inoue et al., 2013). Relapsing-remitting symptoms sharing characteristics with relapsing-remitting multiple sclerosis have been observed in Fabry disease. In a retrospective study of 187 confirmed patients with Fabry disease, 11 patients had concomitant diagnoses of clinically definite multiple sclerosis or possible multiple sclerosis; Fabry disease was diagnosed a mean of 8.2 years after the multiple sclerosis diagnosis was assigned (Böttcher et al., 2013) and some patients with ‘true’ Fabry disease had unmatched oligoclonal bands in CSF. ‘Red flag’ characteristics suggesting an alternative diagnosis of Fabry disease were suggested and included patients with asymmetric, confluent white matter T2 hyperintensity on MRI (Fig. 1G and H), normal spinal MRI, ectatic vertebrobasilar arteries, proteinuria and absence of unmatched CSF oligoclonal bands (Böttcher et al., 2013). Additionally, T1 hyperintensity in the pulvinar nuclei of the thalami, seen in 25% of patients with Fabry disease, are a specific feature of the condition (Fig. 1I; Moore et al., 2003; Burlina et al., 2008).
Further complicating the differentiation of these disorders from multiple sclerosis, unmatched oligoclonal bands have been reported in confirmed cases of Fabry disease, X-linked adrenoleukodystrophy and adrenomyeloneuropathy, metachromatic leukodystrophy, and Niemann-Pick disease type C (Böttcher et al., 2013). Clinical clues to lysosomal or peroxisomal disease in patients with ‘multiple sclerosis-like’ presentations include peripheral neuropathy (frequently observed in Krabbe disease, metachromatic leukodystrophy, and X-linked adrenoleukodystrophy spectrum disorders), extra-neurological manifestations, such as cardiac, renal and dermatological manifestations of Fabry disease, splenomegaly in Niemann-Pick disease type C and more rarely in Fabry disease, and immunodeficiency and pigmentary abnormalities in Chédiak-Higashi disease. Phenotypic heterogeneity is a well-known feature of Niemann-Pick disease type C, and data from an international patient registry demonstrated that 27% of participants experienced symptomatic onset in late adolescence or adulthood, and that a majority presented with ataxia, dysarthria, and cognitive impairment, features often seen in progressive multiple sclerosis. Many patients also had clinical features more specific to Niemann-Pick disease type C and less typical of multiple sclerosis, such as vertical supranuclear gaze palsy and gelastic cataplexy (Patterson et al., 2013).
Of all the groups of heritable disorders that have the potential to mimic multiple sclerosis, lysosomal storage disorders are likely to be among the most amenable to pharmacotherapeutic interventions that can effectively modify disease outcomes, illustrating the importance of accurate and timely diagnosis of these conditions. Available treatments for Krabbe disease, metachromatic leukodystrophy, and childhood cerebral X-linked adrenoleukodystrophy are currently limited to haematopoietic stem cell transplantation, and are generally reserved for pre- or early symptomatic disease, and outcomes have only been reported in any detail in paediatric patients (Escolar et al., 2005; Biffi et al., 2013). There are currently limited data to support the efficacy of haematopoietic stem cell transplantation in adults with cerebral X-linked adrenoleukodystrophy (Engelen et al., 2012) and other adult-onset leukodystrophies. Trials of gene therapies, stem cell therapies, or intrathecal enzyme replacement therapy are underway or in development for Krabbe disease, metachromatic leukodystrophy, and X-linked adrenoleukodystrophy, and are showing promise in animal subjects as well as in small-scale human studies (Cartier et al., 2009; Stroobants et al., 2011; Rafi et al., 2012; Patil et al., 2013).
The efficacy of systemic enzyme replacement therapy is well established for individuals with Fabry disease and significantly improves survival and quality of life (Pisani et al., 2012), reasserting the need for accurate and expedient diagnosis. In a randomized clinical trial of Niemann-Pick type C patients treated with oral miglustat, a reversible inhibitor of glycosphingolipid synthesis, miglustat successfully stabilized key neurological manifestations of the disease in paediatric and adult patients (Patterson et al., 2007). Other chemical chaperone approaches, substrate reduction therapies, and stop-codon read-through strategies have been used in several other lysosomal storage diseases or are in development (Boustany, 2013), and are beyond the scope of this review.
Mitochondrial disorders
Dysfunctional mitochondria play a central role in the pathogenesis of various neurological disorders including multiple sclerosis, Alzheimer's disease, Parkinson's disease and stroke. Mitochondrial dysfunction increases production of reactive oxygen species which may contribute to oxidative stress, potentially leading to neuronal injury, neurodegeneration and, ultimately, clinical disability (Witte et al., 2010; Friese et al., 2014). Biochemical defects in respiratory chain complex I activity have been proposed as contributory to the pathogenesis of multiple sclerosis, and the lack of complex IV activity in demyelinated axons supports axonal dysfunction as a contributor to disease progression in multiple sclerosis (Mahad et al., 2009). The capacity for remyelination and axonal repair, critical factors in delaying or preventing progressive disease, also seem to be dependent on healthy mitochondria.
Given these indicators that mitochondrial dysfunction is integral to multiple sclerosis pathology, it is not surprising that several mitochondrial respiratory chain defects, caused by defined mutations in nuclear or mitochondrial DNA (mtDNA) may present with neurologic syndromes sharing characteristics with multiple sclerosis (Iñiguez et al., 1998). Clinical features, MRI appearances and diagnostic testing strategies for these disorders are outlined in Table 2. Prominent myopathy, cardiomyopathy or cardiac conduction abnormalities, external ophthalmoplegia, early severe optic neuropathy that does not recover, seizures, cerebrovascular disease, peripheral neuropathy, pigmentary retinopathy, and diabetes mellitus may all serve as clinical clues differentiating this group of disorders from multiple sclerosis. Myoclonic epilepsy with lactic acidosis and stroke-like episodes (MELAS), myoclonic epilepsy with ragged red fibres (MERRF), and Leber’s hereditary optic neuropathy (LHON) are caused by specific mutations or structural rearrangements in mtDNA, and can present with spasticity, ataxia and optic neuropathy. Leigh disease encompasses a heterogeneous group of conditions, some of which are caused by autosomal recessively-inherited mutations in nuclear genes that are disruptive to mitochondrial function or maintenance, and some by mutations in mtDNA itself. Non-Mendelian/maternal transmission, if observed, is strong evidence for a mitochondrially-inherited condition; however, given the epidemiology of multiple sclerosis (females affected more frequently than males; increased risk of multiple sclerosis in first-degree relatives) it is not unusual to see multiple sclerosis in a patient with a similarly affected mother. Further, the absence of family history does not exclude the diagnosis of a mtDNA-encoded disorder, owing to the phenomena of incomplete penetrance and heteroplasmy in mtDNA disorders. POLG is a nuclear gene encoding polymerase gamma, a DNA polymerase in humans that effects mtDNA replication and repair.
Table 2.
Mitochondrial disorders with potential to mimic multiple sclerosis
| Disorder | Gene(s); molecular genetic pathogenesis | Inheritance mechanism | Clinical presentation in adult-onset disease | ‘Typical’ MRI appearances | Recommended diagnostic testing |
|---|---|---|---|---|---|
| Leber hereditary optic neuropathy (LHON) |
|
mitochondrial | Bilateral, painless, subacute visual failure with central field loss in young adult life, typically with more complete penetrance in males; tremor, peripheral neuropathy, myopathy, and movement disorders more common in individuals with LHON than in controls (‘LHON-plus’); females with LHON may develop MS-like illness (‘Harding disease’) | Often normal in patients with ‘pure’ LHON; multifocal WM disease mimicking demyelinating lesions of MS seen in LHON patients with apparently pure optic neuropathy as well as in ‘LHON-plus’ and Harding disease phenotypes |
|
| MELAS/MERRF |
|
mitochondrial | Seizures (prominent myoclonus in MERFF), gait instability/ataxia (often initial manifestation in MERFF), myopathy, stroke-like episodes, sensorineural deafness, dementia, PEO, hemianopsia, pigmentary retinopathy, peripheral neuropathy; growth delay, cardiomyopathy, dysrhythmia | MELAS: acute and chronic stroke-like episodes with GM/WM T2/FLAIR hyperintensity, typically involving the posterior cerebrum, thalamus, and not conforming to major vascular territories; in acute phase lesions typically do not restrict diffusion as is seen in ischaemic stroke; elevated lactate on MR spectroscopy; MERRF: multicystic or cavitating leukoencephalopathy, cerebral, cerebellar or brain stem atrophy; basal ganglia calcification/necrosis |
|
| Leigh disease |
|
AR/X-linked; mitochondrial | Spasticity, dystonia, weakness, hypo- or hyperreflexia, seizures (myoclonic or generalized tonic-clonic; infantile spasms), cerebellar ataxia, peripheral neuropathy; dysphagia, persistent vomiting, thermoregulatory dysfunction; optic atrophy, retinitis pigmentosa; liver and renal disease; cardiomyopathy | Bilateral, symmetrical T2/FLAIR hyperintense lesions in brain stem and/or basal ganglia (especially putamina) are characteristic; lesions affecting the mamillothalamic tracts, substantia nigra, medial lemniscus, medial longitudinal fasciculus, spinothalamic tracts, and cerebellum |
|
| POLG-related disease |
|
ARf | Cerebellar ataxia, sensory ataxia, axonal neuropathy, migraine (often first symptom), encephalopathy, seizures, myoclonus, PEO, myopathy, dysarthria; liver disease | ‘MS-like’ T2/FLAIR hyperintense periventricular WM lesions; gadolinium-enhancing brain and cord lesions described |
|
| Optic atrophy, type 1 |
|
AD | Some patients with extraocular features early (sensorineural deafness) or in early-mid adulthood (proximal myopathy, cerebellar or sensory ataxia, axonal sensory and/or motor neuropathy); history of insidious, usually asymmetric, decrease in visual acuity usually in childhood or early adulthood | Normal or optic atrophy; some individuals with basal ganglia calcifications, T2/FLAIR-hyperintense periventricular MS-like lesions, non-specific WM lesions, and cortical and cerebellar atrophy |
|
| Pyruvate dehydrogenase (PDH) deficiency | PDHA1 most common; also PDHB, DLAT, PDHX DLD, PDP1; assembly of pyruvate dehydrogenase enzyme complex | X-linked (PDHA1); AR (other forms) | Intermittent or episodic ataxia or other acute neurological symptoms described, especially in male patients; dystonia, seizures, developmental delay, axonal neuropathy | Leigh-like GM appearances; basal ganglia abnormalities; thin corpus callosum, cortical atrophy; MRI may be normal |
|
AD = autosomal dominant; AR = autosomal recessive; GM = grey matter; MERRF = myoclonic epilepsy with ragged red fibers; MS = multiple sclerosis; PEO = progressive external ophthalmoplegia; RNFL = retinal nerve fibre layer; RRF = ragged red fibres; WM = white matter.
a Mutations in MT-ND4 (including m.11778G>A, accounting for 70% of all LHON cases), MT-ND6 (including m.14484T>C), MT-ND1 (including m.3460G>A), MT-ND2, MT-ND4L, and MT-ND5 are known to cause LHON.
b A specific mutation in MT-TL1 (m.3243A>G) is responsible for approximately 80% of all MELAS cases; other mtDNA genes known to harbor mutations in MELAS patients include MT-ND5, MT-TC, MT-TK, MT-TV, MT-TF, MT-TQ, MT-TS1, MT-TS2, MT-TW, MT-CO1, MT-CO2, MT-CO3, MT-CYB, MT-ND1, MT-ND3, and MT-ND6. Four MT-TK mutations (m.8344A>G, m.8356T>C, m.8363G>A, and m.8361G>A) account for >90% of all individuals with a clinical diagnosis of MERRF; m.8344A>G alone accounts for >80% of cases. Rare cases of MERRF are attributable to mutations in MT-TF or MT-TP.
c Negative selection of leucocytes with high mutant mitochondria burden means that other tissues besides blood (cultured fibroblasts, skeletal muscle) should be used for mtDNA mutational analysis in patients with suspected Leigh disease, MELAS, or MERRF.
d Other nuclear genes associated with Leigh disease include NDUFV1, NDUFS1, NDUFS2, NDUFS3, NDUFS4, NDUFS7, NDUFS8, NDUFA1, NDUFA2, NDUFA10, NDUFAF2, C8orf38, C20orf7, FOXRED1, SDHA, COX10, COX15, LRPPR1, PDSS2, POLG, SUCLG1, and C12orf65. m.8993T>G and m.8993T>C (in MT-ATP6) collectively account for 10–20% of all Leigh disease. Other mtDNA genes implicated in Leigh disease include MT-TL1, MT-TK, MT-TW, MT-TV, MT-ND1, MT-ND2, MT-ND3, MT-ND4, MT-ND5, MT-ND6, and MT-CO3.
e Decreased citrulline also observed in Leigh disease patients with m.8993T>G mtDNA mutation.
f All clinical phenotypes of POLG-related disease follow AR inheritance, except PEO, which follows AD inheritance.
g Oligonucleotide array should be strongly considered if there is reasonable clinical suspicion for POLG-related disease, as microdeletions involving intragenic regions of POLG are reported and therefore relevant in a symptomatic individual with a single heterozygote pathogenic mutation.
i Normal analysis of skeletal muscle for mtDNA depletion/multiple deletions and respiratory chain enzymology does not exclude POLG-related disease.
LHON, caused by specific point mutations in mtDNA (typically at positions 3460, 11778, or 14484) encoding complex I of the respiratory chain, merits special consideration because of its particularly strong associations with multiple sclerosis, occurring with ∼50 times higher frequency in patients with multiple sclerosis than would be expected by chance (Palace, 2009), an association referred to as ‘LHON-plus’ or ‘Harding disease’. Subpopulations of multiple sclerosis patients (those with early/severe optic neuritis) appear to harbour characteristic LHON mutations at high frequencies (Leuzzi et al., 1997). Curiously, although LHON is of lower penetrance among females harbouring mtDNA mutations, the coexistence of the disorder with multiple sclerosis-like illness occurs with a similar female preponderance as is observed in isolated multiple sclerosis. Harding reported three females with LHON harbouring m.11778G>A mutations who did not have extraocular symptoms but had white matter lesions on MRI mimicking multiple sclerosis (Harding et al., 1992), and MRI appearances in LHON commonly closely mimic those observed in multiple sclerosis (Inglese et al., 2001; Lerman-Sagie et al., 2005; Küker et al., 2007; Fig. 2A–C). Unmatched oligoclonal bands have been detected in multiple patients (Palace, 2009). Some authors have proposed that specific mtDNA mutations traditionally associated with LHON are associated with and predispose to multiple sclerosis; an inflammatory process and clinical course similar to that observed in ‘LHON-plus’ has also been described in patients with MELAS (Maruyama et al., 1998), although MELAS is predominantly a grey matter disorder.
Figure 2.
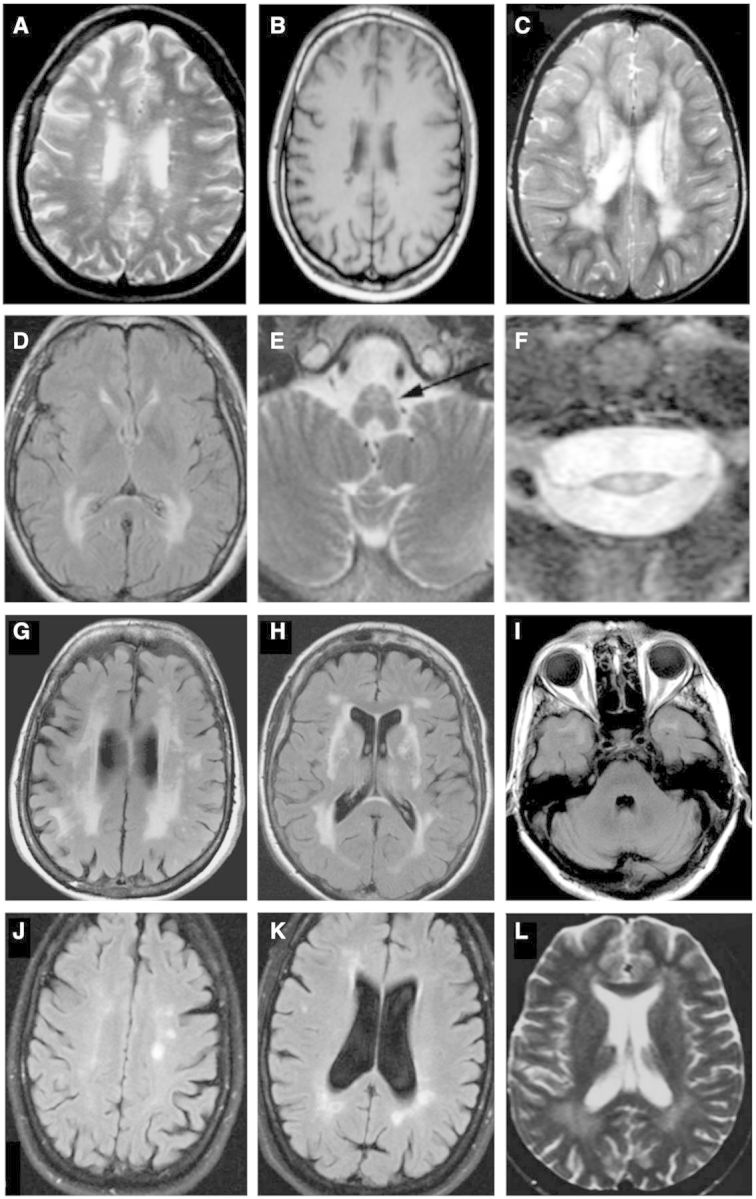
Selected magnetic resonance images from miscellaneous hereditary conditions mimicking multiple sclerosis. (A) Axial FLAIR image of a 36-year-old female with spastic paraparesis and painless visual loss, known to harbour the m.11778G>A mtDNA mutation, diagnostic of LHON-plus, also known as Harding’s disease. Numerous foci of hyperintensity of the periventricular and deep white matter strongly resemble appearances seen in multiple sclerosis (reproduced with permission, Küker et al., 2007). Four years after presentation, this patient developed bilateral optic atrophy, ataxia, and internuclear ophthalmoplegia. (B) Axial T1-weighted image of another patient with LHON showing white matter lesions with hypointense signal, indistinguishable from multiple sclerosis (reproduced with permission, Inglese et al., 2001). (C) Axial T2-weighted image demonstrating more confluent T2 hyperintense white matter disease in a third LHON patient (reproduced with permission, Lerman-Sagie et al., 2005). (D–F) Axial FLAIR and T2-weighted imaging through the brain (D), brainstem (E) and cervical cord (F) in three individual patients with adult-onset Alexander disease. The female patient in (D) presented at 43 years of age with bulbar weakness; patients (E) and (F) presented with lower extremity weakness. The images demonstrate confluent posterior predominant T2 hyperintensity in the periventricular and deep white matter (D) together with atrophy of medullary pyramids (arrow in E) and marked thinning and T2 hyperintensity in the cervical cord (F) (reproduced with permission, Pareyson et al., 2008). (G-I) Axial FLAIR imaging in a patient with CADASIL. Patchy and confluent hyperintensity in the periventricular white matter (G) with involvement of the external capsules (H) and subcortical white matter of the temporal poles (I) are characteristic. (J and K) Axial FLAIR imaging of a 49-year-old female patient with PLP1 disease (hereditary spastic paraplegia, SPG2 type) presenting with a phenotype mimicking primary progressive multiple sclerosis. She had a 10-year history of progressive spastic paraplegia, dysarthria, and ataxia. Her son had died in childhood of a previously undiagnosed severe neurodevelopmental disorder. Multifocal T2 hyperintensities are seen in the periventricular and deep white matter near the atria and occipital horns of the ventricles, together with and other scattered foci of T2 signal in the white matter, mimicking multiple sclerosis (reproduced with permission, Warshawsky et al., 2005). She had abnormal visual-evoked potentials bilaterally and CSF was positive for unmatched oligoclonal bands. (L) Axial T2-weighted image in a 22-year-old female with hereditary spastic paraplegia, SPG11 type, presenting with subacute gait disturbance, urge incontinence, and mild, subjective cognitive disturbance. Confluent T2 hyperintensity is seen in the posterior white matter together with small, scattered foci elsewhere in the brain (reproduced with permission, Romagnolo et al., 2014).
Late-onset autosomal recessive Leigh disease is clinically and genetically heterogeneous and has been associated with missense mutations in SURF1 as well as several other nuclear genes and mtDNA mutations, and is characterized by motor regression, dementia and dystonia. Although Leigh disease, like MELAS, is primarily a disorder of grey matter, its clinical course may be acute, gradually progressive, or relapsing, and there are historical reports of misdiagnosis with multiple sclerosis (Malojcic et al., 2004). Clues to Leigh disease or other mitochondrial disorders on MRI in suspected multiple sclerosis include symmetric bilateral T2 hyperintense lesions in the basal ganglia, or multifocal grey matter involvement. Common variants in two prominent mitochondrial Leigh disease genes, MT-ATP6 and MT-ND2, have been associated with multiple sclerosis (Vyshkina et al., 2008).
Mutations in POLG have also been associated with multiple sclerosis-like presentations in several patients. The clinical spectrum of POLG-related disorders comprise a continuum of overlapping phenotypes including a subset of ataxia-neuropathy disorders, which overlap with MELAS/MERRF phenotypes and presenting with optic neuritis, white matter hyperintensities, unmatched oligoclonal bands in CSF, and variable axonal polyneuropathy (Degos et al., 2014). Other POLG mutations result in infantile-onset hepatocerebral disease (Alpers-Huttenlocher syndrome) with no clinical resemblance to multiple sclerosis. Although heterozygous POLG mutations can cause ophthalmoplegia phenotypes, most other POLG-related phenotypes, including ataxia-neuropathy, are recessively inherited. Heterozygous mutations in OPA1, another nuclear gene, also cause dysfunction and misdirection of mitochondria in neural tissue; the most common phenotype is insidiously progressive childhood-onset optic nerve atrophy, although a proportion of patients have extraocular neurologic involvement including ataxia and/or spastic paraplegia. Some patients have diffuse white matter hyperintensities on MRI with prominent involvement of periventricular white matter (Yu-Wai-Man et al. 2010; Pretegiani et al., 2011). Several patients with pyruvate dehydrogenase complex deficiency, classically described as a severe infantile-onset disease, have been described with later-onset presentations including recurrent, episodic ataxia. These patients are usually males with specific missense mutations of PHDA1, a nuclear gene, and may have Leigh-like grey matter appearances and other basal ganglia changes on MRI (Barnerias et al., 2010) that are unlikely to mimic ‘typical’ MRI findings observed in multiple sclerosis.
Other metabolic leukodystrophies, leukoencephalopathies and myelopathies
‘Leukodystrophy’ refers to a group genetic disorders primarily involving myelin pathology and with prominent white matter involvement, while ‘leukoencephalopathy’ is a generic, largely radiological term encompassing many genetic (and acquired) disorders with diffuse white matter abnormalities on MRI and including some disorders where neuropathology is primarily axonal. Although many lysosomal disorders (discussed above) are classifiable as leukodystrophies, several other leukodystrophies and leukoencephalopathies potentially presenting in adulthood are not classifiable as disorders of lysosomal or peroxisomal function, or as mitochondrial disorders. Several of these disorders can present with spastic paraparesis, or with other neurological features that can mimic multiple sclerosis (Table 3). Other neurometabolic disorders that mimic multiple sclerosis and that are treatable or potentially treatable with pharmacotherapy include urea cycle disorders, acute intermittent porphyria, biotinidase deficiency, cerebrotendinous xanthomatosis, and some disorders of vitamin B12 or folate metabolism. Some patients with urea cycle disorders, acute intermittent porphyria, and some methylmalonic acidaemias may be candidates for liver transplantation.
Table 3.
Other leukodystrophies and leukoencephalopathies and neurometabolic diseases with potential to mimic multiple sclerosis
| Disorder | Gene; biochemical/cellular defect | Inheritance mechanism | Clinical presentation in adult-onset disease | ‘Typical’ MRI appearances | Recommended diagnostic testing |
|---|---|---|---|---|---|
| Urea cycle disorders |
|
AR | Progressive SP, bladder dysfunction, neurocognitive deficits, seizures; also liver disease, episodic hyperammonaemia | Multicystic WM lesions with subcortical predominance; cortical and cerebellar atrophy, demyelination, stroke-like lesions, basal ganglia calcifications |
|
| Adult polyglucosan body disease |
|
ARa | Gait disturbance, bladder spasticity, mixed UMN/LMN involvement, sensory loss predominantly in the distal lower extremities, and mild cognitive difficulties | Predominantly periventricular and to a lesser extent subcortical T2/FLAIR hyperintense WM abnormalities, esp. posterior internal capsule, pyramidal tracts, medial lemniscus of pons/medulla, cerebellar peduncles, dentate, anterior medulla; atrophy of medulla and cord |
|
| Acute intermittent porphyria |
|
AD | Acute neurovisceral attacks of abdominal pain, in some cases with mental changes, seizures, focal neurological symptoms, and peripheral neuropathy | Diffuse, patchy T2 hyperintensities in periventricular WM in some patients |
|
| Disorders of vitamin B12 (cobalamin), homocysteine, and folate metabolism |
|
AR | Tubulointerstitial renal disease, basal ganglia stroke, growth failure, optic nerve atrophy, episodic metabolic acidosis, lethargy/coma, vomiting, dehydration in mutase deficiency; myelopathy, SP, neuropsychiatric features, nystagmus, maculopathy, or optic atrophy in cblC defect; myelopathy and spasticity in MTHFR deficiency | Swelling and hyperintensity of supratentorial WM, thinning of CC in severe cases, bilateral T2 hyperintensity in dorsal columns of the spinal cord; signal abnormalities variable between individual disorders; abnormalities potentially reversible with treatment |
|
| Biotinidase deficiency |
|
AR | Affected older children and adolescents may exhibit motor weakness, SP, and decreased visual acuity; other clinical features include seizures, developmental delay, hearing loss, aloplecia, immunodeficiency and eczema; manifestations partially/completely reversible with biotin | Periventricular and subcortical WM T2 hyperintensity followed by atrophy |
|
| Cerebrotendinous xanthomatosis |
|
AR | Pyramidal and/or cerebellar signs, SP, dystonia, parkinsonism, peripheral neuropathy, neuropsychiatric symptoms, seizures; premature atherosclerosis; cataracts, xanthomas, chronic diarrhoea | Diffuse cerebral and cerebellar atrophy with variable nonspecific T2/FLAIR signal abnormalities in deep and subcortical white matter, sometimes with parieto-occipital predominance; symmetric T2 hyperintensity in the dentate nuclei and basal ganglia; brain stem and cerebellar white matter can become involved as disease progresses; decreased NAA and increased lactate on MRS |
|
AD = autosomal dominant; AR = autosomal recessive; cblC = cobalamin C defect; CC = corpus callosum; GBE = glycogen branching enzyme; HHH = hyperornithinaemia-hyperammonaemia-homocitrullinaemia syndrome; LMN = lower motor neuron; MRS = magnetic resonance spectroscopy; MTHFR = methylenetetrahydrofolate reductase; NAA = N-acetylaspartate; SP = spastic paraparesis; UMN = upper motor neuron; WM = white matter.
a Heterozygous GBE1 mutations reported in some clinically affected individuals.
Two primary urea cycle disorders (arginase deficiency and the hyperornithinaemia-hyperammonaemia-homocitrullinaemia syndrome) are known to onset with spasticity and diplegia of late-onset in rare patients. Both disorders can present with liver disease and life-threatening hyperammonaemia and are managed with dietary protein restriction and pharmacologic ammonia scavengers, and patients with diplegia may improve clinically with treatment (Prasad et al., 1997). Several historical reports attest to the potential of adult polyglucosan body disease, caused by defective glycogen branching and allelic to glycogen storage disease type IV, to be confused with multiple sclerosis. Although adult polyglucosan body disease is recessively inherited, progressive gait disturbance and cognitive decline have also been reported in heterozygotes (Paradas et al., 2014). Management for adult polyglucosan body disease is currently supportive only.
Neurologic phenotypes common to nutritional or autoimmune B12 deficiency and inherited disorders of B12 metabolism include paraesthesias, ataxia, impaired proprioception, and spasticity. Many patients also have megaloblastic anaemia. Several late-onset forms of many inherited disorders of B12 metabolism have been described, including methylmalonic acidaemia (MMA), mutase type, and some cobalamin defects, and may share characteristics with nutritional/autoimmune B12 deficiency and with multiple sclerosis. The cobalamin C defect has been reported in association with adult-onset myelopathy, neuropsychiatric disturbance and patchy white matter T2 hyperintensities on MRI, which may involve the cord (Thauvin-Robinet et al., 2008; Huemer et al., 2014). Optic atrophy presenting with acute visual loss has been reported in mutase deficiency methylmalonic acidaemia (Pinar-Sueiro et al., 2010). MTHFR deficiency, most often characterized by early-childhood clinical onset, also has late-onset forms with white matter disease on MRI and progressive myelopathic gait disturbance. Reported patients lacked a history of relapses and were negative for unmatched CSF oligoclonal bands (Haworth et al., 1993). Screening plasma B12 levels, as well as quantification of methylmalonic acid (MMA) in plasma and/or urine, are already recommended for all individuals for whom the diagnosis of progressive multiple sclerosis is considered; we also recommend additional screening of plasma homocysteine if there is any reasonable suspicion for these disorders, since B12 and methylmalonic acid will be normal in patients with MTHFR deficiency or cobalamin E or G defects.
Adolescents and adults with partial biotinidase deficiency with diffuse white matter disease and spastic paraparesis have also been described; this condition is subject to newborn screening in many countries, can be diagnosed with straightforward biochemical testing, and is treatable with oral biotin (vitamin B7) supplementation.
Cerebrotendinous xanthomatosis is an autosomal recessive lipid storage disease that is also classified as a leukodystrophy, and characterized by infantile-onset diarrhoea, cataracts, and adolescent or adult-onset tendon xanthomas and progressive neurological disease causing cerebellar signs, spasticity, dystonia, parkinsonism, and peripheral neuropathy. MRI may show diffuse cerebral and cerebellar atrophy with variable T2 abnormalities involving deep and subcortical white matter which may overlap with abnormalities seen in multiple sclerosis. When present, characteristic symmetric T2 hyperintensity in the dentate nuclei is suggestive of this disorder (De Stefano et al., 2001). Cerebrotendinous xanthomatosis is a potentially treatable condition with clinical symptoms stabilizing with administration of chenodeoxycholic acid (Yahalom et al., 2013).
Many patients with acute intermittent porphyria, an autosomal dominant defect of haem metabolism with limited penetrance, experience recurrent attacks of poorly localized abdominal pain; some patients experience acute neurologic deficits or psychiatric symptoms for which pathomechanisms are poorly characterized. Such patients may have evidence of both peripheral nerve and CNS disease. Many patients harbouring pathogenic HMBS mutations (and who have symptomatic family members) have ‘latent’ porphyria and remain asymptomatic throughout their lives. A proportion of patients have hyperintense white matter lesions on MRI that can occur in a multiple sclerosis-like distribution (Bylesjö et al., 2004). In relation to haem and iron metabolism, iron is known to accumulate in the CNS in multiple sclerosis, although the pathomechanisms for this phenomenon and its contribution to multiple sclerosis pathogenesis are incompletely understood. Iron may modify disease activity in multiple sclerosis by promoting production of pro-inflammatory mediators, mitochondrial dysfunction, and reactive oxygen species production (Williams et al., 2012). In one large multiple sclerosis cohort, there was no enrichment for mutations in HFE, the gene for haemochromatosis (Ramagopalan et al., 2008). Although other inherited disorders of metabolism of iron and other metals [including neurodegeneration with brain iron accumulation (NBIA), and Wilson disease] may rarely involve white matter, they do not typically mimic multiple sclerosis clinically and generally have distinct radiological features (van Wassenaer-van Hall et al., 1995; Hayflick et al., 2006).
Non-metabolic leukodystrophies/leukoencephalopathies and small artery disorders
Several other heritable disorders of white matter can mimic multiple sclerosis, form a heterogeneous group, and are not strictly classifiable as metabolic diseases (Table 4). Adult-onset Alexander disease is caused by heterozygous, usually missense, mutations in GFAP; both sporadic cases (with de novo mutations) and familial cases with autosomal dominant transmission and incomplete penetrance have been described (Li et al., 2006; Balbi et al., 2010). Alexander disease is characterized by fibrinoid bodies in astrocytes and classically described as an infantile-onset disorder presenting with macrocephaly, growth failure, seizures, and progressive neurological impairment. In recent years, knowledge of the clinical spectrum has expanded, with late-onset disease now thought to account for at least a quarter of all cases of Alexander disease. The clinical course of late-onset disease entails progressive gait ataxia, spasticity and pyramidal signs, bulbar signs, and autonomic features, and can mimic multiple sclerosis (Tschampa et al., 2011); unlike infantile-onset Alexander disease, a very low incidence of macrocephaly and mental retardation is observed in adult-onset disease (Pareyson et al., 2008). Thirty-five per cent of adult patients in one series had palatal myoclonus, which is only rarely seen in other disorders and may be a useful diagnostic clue (Balbi et al., 2010). Adult patients may have T2 hyperintensities in periventricular and deep white matter (Fig. 2D), and atrophy and T2 signal abnormalities of the medulla oblongata and cervical cord on MRI (Fig. 2E and F), which may also be seen in multiple sclerosis. Sixty-five to 75% of patients with adult-onset Alexander disease in two series had white matter hyperintensities on MRI in varying distributions which have potential to be mistaken for multiple sclerosis (Farina et al., 2008; Yoshida et al., 2011). MRI can be normal in some adult patients.
Table 4.
Non-metabolic leukodystrophies and leukoencephalopathies with potential to mimic multiple sclerosis
| Disorder | Gene; biochemical/cellular defect | Inheritance mechanism | Clinical presentation in adult-onset disease | ‘Typical’ MRI appearances | Recommended diagnostic testing |
|---|---|---|---|---|---|
| Adult-onset Alexander disease |
|
AD | Bulbar/pseudobulbar signs: palatal myoclonus, dysphagia, dysarthria, spasticity, hyperreflexia, ataxia, nystagmus, dysmetria, incontinence, constipation, orthostatic hypotension, seizures, diplopia | Atrophy and T2 hyperinetnsity of medulla and cervical cord in almost all patients; cerebral T2 WM hyperintensities in most, often with with frontal predominance; hyperintensity and swelling involving basal ganglia and thalami; MRI can be normal |
|
| Autosomal Dominant Leukodystrophy (ADLD) |
|
AD | Early autonomic dysfunction, cognitive impairment, pyramidal lesions, cerebellar dysfunction, typical onset age 30–50 years | Subtle to extensive symmetrical WM T2 hyperintensities with frontoparietal predominance; may also involve middle cerebellar peduncles, brainstem; typically periventricular WM is spared or less affected than other regions; atrophy of corpus callosum and brain stem | Molecular testing of LMNB1 (for common duplication first; use quantitative PCR or FISH initially) |
| Leukoencephalopathy with brainstem and spinal cord involvement and lactate elevation (LBSL) |
|
AR | Slowly progressive cerebellar ataxia, spasticity, abnormal proprioception/vibration sense, dysarthria; epilepsy and/or learning disability in some patients | Confluent or patchy T2 hyperintensities in cerebral WM, dorsal columns and lateral corticospinal tracts of cord, and medulla; relative sparing of the U-fibres; abnormalities may also be seen in corpus callosum, internal capsule, cerebellar peduncles, spinocerebellar tracts and cerebellar WM |
|
| Hereditary diffuse leukencephalopathy with spheroid cysts (HDLS) |
|
AD | Executive dysfunction with frontal lobe signs, memory decline, personality changes, motor impairment, seizures; usual age of onset 25–45 years | Bifrontal or bifrontoparietal T2/FLAIR hyperintensities in deep, subcortical, and periventricular WM, often asymmetric and patchy, becoming confluent with time; involvement of corpus callosum and corticospinal tract lesions common; brain stem atrophy and contrast enhancement in brain parenchyma usually absent |
|
| Cerebral Arteriopathy with Subcortical Infarcts and Leukoencephalopathy (CADASIL/CARASIL) |
|
AD (CADASIL) AR (CARASIL) | Progressive cerebrovascular disease from mid-adulthood, progressing to spastic paraplegia and dementia; migraine and mood disturbance | Classically T2-hyperintense WM lesions involving the temporal pole and external capsule; numerous subcortical infarcts; optic nerves and cord typically spared |
|
| ClC-2 chloride channel deficiency leukoencephalopathy |
|
AR | Cerebellar ataxia, SP, optic neuropathy; cognitive defects, headache, retinopathy | Restricted diffusion with T2 hyperintensity in midbrain cerebral peduncles, middle cerebellar peduncles, and posterior limb of internal capsule; possible WM hyperintensity more diffusely | Molecular testing of CLCN2 |
| X-linked Charcot–Marie–Tooth disease (CMTX) |
|
X-linked | Usually accompanied by demyelinating polyneuropathy with amyotrophy, distal weakness, and pes cavus; episodic or persistent ataxia and dysarthria; sensorineural hearing loss in some families; males generally more severely affected; “relapsing” events triggered by e.g. high altitude | Non-enhancing periventricular, symmetrical, confluent white matter abnormalities with posterior predominance; T1 hypointensity in splenium of CC and middle cerebellar peduncles; new CNS abnormalities may appear acutely, especially at altitude |
|
AD = autosomal dominant; AR = autosomal recessive; CC = corpus callosum; FISH = fluorescent in situ hybridization; MRS = magnetic resonance spectroscopy; NCV = nerve conduction velocity; SP = spastic paraparesis; WM = white matter.
Autosomal dominant leukodystrophy is associated with duplications of the lamin B1 (LMNB1) gene, causing lamin B1 overexpression, resultant defects in oligodendroglial differentiation, and subtotal demyelination. The disorder has been misdiagnosed as multiple sclerosis, and was originally identified as a distinct disorder in two large Irish-American kindreds sharing a common founder duplication allele (Padiath et al., 2006). Leukoencephalopathy with brainstem and cord involvement and lactate elevation (LBSL) is an autosomal recessive disorder of white matter caused by mutations in DARS2. It is characterized by slowly progressive pyramidal, cerebellar, and dorsal column dysfunction, manifesting as cerebellar ataxia and spasticity, with a predominance of lower extremity involvement, and dysarthria (Isohanni et al., 2010). Onset is typically in adolescence or early adulthood, but the disorder exhibits phenotypic heterogeneity and may be incompletely penetrant in some families (Labauge et al., 2011). MRI shows a pattern of homogeneous, often enhancing white matter disease, and involvement of brain stem and spinal tracts. Increased lactate is often present spectroscopically in abnormal white matter. One Finnish study screened for two common DARS2 mutations in 321 patients with multiple sclerosis; no enrichment for these mutations was observed (Isohanni et al., 2010). Hereditary diffuse leukoencephalopathy with spheroid cysts (HDLS) is an autosomal dominant disorder that may resemble primary progressive multiple sclerosis (Keegan et al., 2008), and is caused by mutations in CSF1R. Most cases are inherited from an affected parent, although de novo cases are also reported (Rademakers et al., 2011). This disorder typically becomes symptomatic in early adulthood and has a prominent psychiatric or cognitive impairment phenotype. White matter disease on MRI may be asymmetric and patchy and mimic multiple sclerosis, developing confluence later. Significant grey matter pathology, brainstem atrophy and cerebellar abnormalities are minimal or absent.
Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL) is a progressive, debilitating cerebrovascular disorder that typically follows a stepwise course, with symptomatic onset in the fourth or fifth decade. More than 90% of individuals with CADASIL have autosomal dominantly-inherited heterozygous mutations in NOTCH3; de novo mutations are uncommon, most individuals having an affected parent. Missense mutations located distally in the NOTCH3 transcript may confer a less specific phenotype with predisposition to age-related progression of small vessel vasculopathy-associated white matter lesions and a high incidence of progressive disability compared to controls. CARASIL, a, similar autosomal recessive disorder, shares phenotypic characteristics with CADASIL and is caused by homozygous or compound heterozygous mutations in HTRA1 (Fukutake et al., 1995). The pathologic hallmark of CADASIL is electron-dense granules in the media of arterioles that can often be identified by electron microscopic evaluation of skin biopsies; these appearances are not observed in CARASIL. On MRI, CADASIL/CARASIL have a predilection for involving subcortical white matter of the anterior temporal lobes, frontoparietal regions near the vertex, and the bilateral external capsules, but may also exhibit diffuse leukoencephalopathy that may mimic multiple sclerosis (Fig. 2G–I; O’Riordan et al., 2002).
Exome sequencing recently identified several adult patients with autosomal recessive ClC-2 chloride channel deficiency with clinical and imaging features resembling multiple sclerosis. These patients had leukoencephalopathy with MRI signal abnormality in the posterior limb of the internal capsule and middle cerebellar peduncles, and presented with variable clinical features including optic neuropathy, spasticity, and cerebellar ataxia; some patients had chorioretinopathy. Among individuals with adult-onset disease, age of onset ranged from 30–57 years. All identified patients were homozygous or compound heterozygous for CLCN2 mutations (Depienne et al., 2013). Finally, several families with X-linked Charcot–Marie–Tooth disease (CMTX) have been described in which individuals have presented with, in addition to the mixed axonal demyelinating polyneuropathy that characterizes the disease, ‘relapsing-remitting’ neurological deficits (including transient limb weakness, hemiparesis, or dysarthria) and transient CNS white matter changes often with confluence and posterior or parietal predominance (Hanemann et al., 2003; Sato et al., 2012). In some individuals these episodes seemed to be triggered by physical exertion or high altitude (Paulson et al., 2002).
Hereditary spastic paraplegias
Hereditary spastic paraplegias are a heterogenous and expanding group of disorders characterized by slowly progressive paraparesis and spasticity. Historically, these disorders have been broadly classified as ‘uncomplicated’ if neurological impairment is limited to progressive spastic paraparesis, hypertonic urinary bladder disturbance, and diminution of lower-extremity vibration sensation and proprioception. The primary pathology is axonal degeneration, which is maximal in distal corticospinal tracts and dorsal column fibres (Harding, 1993). There may be a family history of similarly affected individuals, especially in dominantly-inherited subtypes; however, absence of a family history should not exclude hereditary spastic paraplegias, as many subtypes exhibit incomplete or variable penetrance in some families, result from de novo mutations, or follow an autosomal recessive inheritance mechanism. MRI is often normal, helping to differentiate hereditary spastic paraplegia from multiple sclerosis and other conditions, but may reveal mild global atrophy, atrophy of the corpus callosum, non-specific white matter lesions, T2 hyperintensity in the internal capsule, and cord atrophy (Casali et al., 2004; Hourani et al., 2009), making exclusion of a multiple sclerosis diagnosis challenging in some cases. A recent study revealed alterations in multiple diffusion-tensor indices in many patients, suggesting widespread disruption of myelin integrity (Aghakhanyan et al., 2014). Uncomplicated autosomal dominant forms include types 3A, 4, 6, 8, 10, 13, 17, 31 and 33; types 5A, 7 and 11 are autosomal recessive, and X-linked forms have been described. While many cases of hereditary spastic paraplegia have prominent clinical features that are unlike those seen in multiple sclerosis (e.g. amyotrophy, mental retardation), others may present with dysarthria, optic disc pallor or other features that overlap with multiple sclerosis.
PLP1 disease is classified as X-linked hereditary spastic paraplegia under many schemes, with segmental duplication of Xq22 the most common molecular mechanism described. The classic phenotype of PLP1 disease is early childhood-onset Pelizaeus-Merzbacher disease in young males, although numerous reports of attenuated disease exist, particularly in female heterozygotes with adult-onset spastic paraplegia phenotypes (Hurst et al., 2006), designated as the SPG2 subtype of hereditary spastic paraplegia. SPG2 has been documented in association with mutations involving specific domains of the PLP1 protein transcript or less-conserved gene regions, and some gene truncations. Adult patients with SPG2, as well as clinically asymptomatic obligate carrier mothers of males with classical Pelizaeus-Merzbacher disease, have been described with non-specific periventricular and subependymal white matter changes on MRI, and cystic changes in the basal ganglia (Fig. 2J and K; Battini et al., 2003; Warshawsky et al., 2005; Hurst et al., 2006). A young female presenting with subacute gait disturbance and periventricular white matter lesions was recently described with biallelic pathogenic mutations in SPG11 (Fig. 2L; Romagnolo et al., 2014). Using a whole exome sequencing approach in combination with bioinformatics and functional validation, several additional putative hereditary spastic paraplegia genes were recently identified (Novarino et al., 2014), although as yet, the potential for these hereditary spastic paraplegia subtypes to mimic multiple sclerosis has not been evaluated.
Other miscellaneous Mendelian disorders of relevance to multiple sclerosis
Other Mendelian genes can harbour highly penetrant, rare variants implicated in specific disorders that overlap with multiple sclerosis. Familial haemophagocytic lymphohistiocytosis is a genetically heterogenous autosomal recessive disorder characterized by proliferation/infiltration of macrophages and T-lymphocytes; adult disease has been reported. Several reports exist of cases with severe CNS manifestations, and multiple sclerosis has been reported with higher frequency among patients harbouring PRF1 mutations and polymorphisms (Camiña-Tato et al., 2010). Neurological features may include spasticity, cranial nerve palsies, ataxia, quadriplegia and blindness. Other findings, including fever, cytopaenias, rash, lymphadenopathy, liver dysfunction, and haemophagocytosis are not universally present, and the overall picture can mimic acute disseminated encephalomyelitis as well as multiple sclerosis.
Friedreich ataxia usually results from biallelic trinucleotide expansions in FRDA, a nuclear gene, and causes degeneration of dorsal columns, spinocerebellar and pyramidal tracts, dorsal root ganglia, and peripheral nerves. On MRI, atrophy of the medulla and upper cervical cord is common, and in late onset forms, cerebellar atrophy and cerebellar white matter abnormalities become evident (Bhidayasiri et al., 2005). It and other hereditary ataxias can mimic multiple sclerosis, although this group of disorders typically have clinical characteristics that are clearly distinguishable from multiple sclerosis.
Familial Mediterranean fever (FMF), which typically presents with recurrent febrile episodes with or without abdominal pain, is caused by recessively-inherited mutations in MEFV, encoding pyrin and implicated in the regulation of the pro-inflammatory cytokine interleukin 1β. MEFV mutations and polymorphisms have been identified at higher frequencies in patients with multiple sclerosis compared to healthy control groups; it has been suggested that endothelial dysfunction in familial Mediterranean fever may disrupt blood–brain barrier integrity, an important element in the evolution of inflammatory lesions and plaques (Alpayci et al., 2012).
Other miscellaneous rare disorders merit consideration in a discussion of hereditary mimics of multiple sclerosis. Gerstmann-Straussler-Scheinker syndrome is a rare, usually autosomal dominant prion disease with a protracted course encompassing a progressive ataxia syndrome, with MRI and CSF findings suggestive of a demyelinating-inflammatory process. It has been noted that certain vascular malformation disorders (cerebral angiomatosis; von Hippel-Lindau disease) may cause progressive neurological features punctuated by acute clinical deteriorations, and radiological appearances that could potentially be confused with multiple sclerosis (Natowicz and Bejjani, 1994).
Discussion
Despite significant recent advances in the understanding of multiple sclerosis pathogenesis and revisions to diagnostic criteria, there remains no pathognomonic clinical or investigative finding for the diagnosis of multiple sclerosis. Misdiagnosis occurs (Rudick and Miller, 2012; Solomon et al., 2012; Solomon and Weinshenker, 2013) and remains a challenging problem with significant repercussions. Diagnostic criteria have been designed to increase sensitivity, resulting in lowered specificity to the extent that patients with other disorders, both inherited and acquired, have the potential to meet McDonald Criteria when ‘no better explanation’ for symptoms, signs, or investigational findings is elicited by the treating physician. This highlights the importance of expanding the diagnostic evaluation to exclude other, rarer conditions when one or more ‘red flags’ are present. Although a relapsing-remitting course can be mimicked by, for example, various mitochondrial disorders (pyruvate dehydrogenase complex deficiency, Leigh disease, MELAS) Fabry disease, and X-linked Charcot–Marie–Tooth disease, awareness of these red flags is especially important for clinicians evaluating patients meeting criteria for (or carrying tentative diagnoses of) primary progressive multiple sclerosis, as most other heritable mimics of multiple sclerosis are more likely to exhibit a progressive disease course. Patients meeting diagnostic criteria or suspected of having primary progressive multiple sclerosis are a particularly heterogenous group of patients whose presentation is typically characterized by spasticity or myelopathic signs with insidious onset, but who generally have fewer perivascular inflammatory white matter lesions on MRI than relapsing patients (Revesz et al., 1994). Overall clinical presentation and even MRI characteristics in primary progressive disease may overlap with, or be indistinguishable from, that seen in adult-onset forms of several rare Mendelian genetic conditions. Primary progressive patients are generally older than relapsing patients at onset, are more likely to be male, have a greater prevalence of motor impairment and cognitive impairment at diagnosis (Wachowius et al., 2005) and show poor response to immunomodulatory therapies (Miller and Leary, 2007). Although some genetic or inherited metabolic disorders may exhibit a relapsing course, most such conditions have slow, insidiously progressive or stepwise neurologic decline, without clear remissions. Paediatric multiple sclerosis is almost always relapsing at presentation with a prominent inflammatory component at symptomatic onset (Banwell et al. 2007; Krupp et al., 2007).
The most recent McDonald Criteria for diagnosis of multiple sclerosis (Polman et al., 2011), especially those for multiple sclerosis ‘with progression from onset’, are more sensitive and less specific than previous iterations (Polman et al., 2005) and no longer stipulate presence of oligoclonal bands as an absolute requirement. In practice, many neurologists do not routinely evaluate CSF, although it is generally recognized that CSF evaluation remains an important aid in the confirmation of multiple sclerosis, especially in atypical patients. Oligoclonal bands are themselves non-specific and incompletely sensitive; their absence does not exclude multiple sclerosis (Solomon et al., 2012) and patients with other disorders may have them, reflecting non-specific CSF inflammatory disease. The presence or absence of oligoclonal bands is therefore an imperfect discriminator, but patients whose multiple sclerosis diagnosis is already in doubt and who are negative for unmatched oligoclonal bands in CSF should be subjected to particularly rigorous scrutiny. In all types of multiple sclerosis, oligoclonal bands are of lower prevalence in non-Caucasian individuals (Link and Huang, 2006); this may be of particular relevance to primary progressive disease, where a higher percentage of patients are non-Caucasian (Naismith et al., 2006). CSF protein may be elevated in multiple sclerosis, but is also of poor specificity and sensitivity.
Several ‘red flag’ clinical, paraclinical and MRI characteristics of a patient’s presentation and disease course should prompt careful consideration of specific Mendelian genetic or mitochondrial disorders in the differential diagnosis of an individual presenting with a multiple sclerosis-like illness and are outlined in Table 5. These ‘flags’, together with family history, patient age, sex and ethnicity (specific inherited disorders occur with high prevalent in specific populations) should be routinely considered in the evaluation of such patients. In addition to disorder-specific features outlined in earlier sections, clinical red flags include absent ocular involvement, absent callosal lesions (Dawson’s fingers) on MRI, absent cord lesions on MRI, positive family history of (unexplained) neurologic symptoms, early or very late (>70 years) age of onset, presence of unexplained non-CNS pathology, and absence of clinical remissions (Rudick et al., 1986; Miller et al., 2008). Although collectively these red flags have some utility, the discriminating power of each ‘flag’ individually is low, as illustrated by presence of one or more flags in many ‘true’ multiple sclerosis patients. Primary progressive patients in particular always fulfill ‘absence of clinical remission’, which in part explains additional uncertainty that often accompanies a primary progressive multiple sclerosis diagnosis. With specific reference to alternative inherited disorders, ‘family history of multiple sclerosis’, ‘early age of onset’ and ‘presence of non-CNS disease’ have been suggested as flags, but again, each ‘flag’ individually has poor discriminatory power; many patients with ‘true’ multiple sclerosis have affected family members, and a clear genomic basis for multiple sclerosis is now well established. In a collaborative genome-wide association study (GWAS) involving almost 30 000 patients with multiple sclerosis and ∼50 000 controls (Beecham et al., 2013), 110 low penetrance susceptibility variants were established at 103 loci. Patients with autosomal recessive disorders (or incompletely penetrant autosomal dominant disorders) frequently lack any discernible family history. ‘Early age of onset’ is also a poor discriminator; the phenomenon of paediatric multiple sclerosis is well established, as is, increasingly, the recognition of attenuated or adult-onset inherited conditions exhibiting overlap with multiple sclerosis. For females with X-linked disorders (PLP1 disease/SPG2, Fabry disease, X-linked adrenoleukodystrophy, X-linked Charcot–Marie–Tooth disease), disease severity and age of onset may be determined by the pattern of X-inactivation.
Table 5.
Fifty-six ‘red flag’ clinical, laboratory, and radiologic characteristics raising suspicion for Mendelian diseases in patients evaluated for multiple sclerosis or with ‘multiple sclerosis-like’ phenotypes
| Disease characteristic | Red Flaga | Examples of alternative inherited disorders |
|---|---|---|
| Clinical neurologic or neuro-ophthalmologic features | ||
| Peripheral neuropathy/delayed NCV | Major | CMTX, KD, MLD, X-ALD/AMN, cblC defect, PLP1 disease, CTX, mito disease, POLG, APBD |
| Myopathy | Major | Mito disease, POLG |
| Amyotrophy | Major | Specific HSP subtypes |
| Retinopathy | Major | Mito disease, cblC defect |
| Prominent cerebrovascular disease | Major | CADASIL, FD |
| Prominent neuropsychiatric disease/dementia | Intermediate | MLD, WD, HDLS, CTX, cblC defect |
| Isolated, progressive or intermittent ataxia | Intermediate | FA, spinocerebellar ataxia, CTX, PDH deficiency |
| Apparently isolated myelopathy | Minor | X-ALD/AMN, cblC defect |
| Early, severe optic nerve involvement | NA | LHON, X-ALD, OPA1 disease, CLCN2 disease |
| Prominent dystonia/parkinsonism | NA | NPC, CHD, MLC1 disease, CTX, NBIA, WD, APBD, Leigh disease and other mito disease |
| Prominent bulbar symptoms | NA | AOAD, KD |
| Prominent epilepsy | NA | MLD, cblC defect, POLG, MELAS/MERFF, other mito disease |
| Palatal myoclonus | NA | AOAD |
| Hearing loss | NA | FD, NPC, Leigh disease, other mito disease |
| Vertical supranuclear gaze palsy (VSGP) | NA | NPC |
| Gelastic cataplexy | NA | NPC |
| Tongue hemiatrophy | NA | KD |
| Relapsing-remitting neurological symptoms or stroke-like episodes | NA | PDH deficiency, MELAS/MERFF, CMTX; cblC defect, MTHFR deficiency, CADASIL, FD |
| Gender discrepancy in clinical neurological phenotype or more severely affected brothers/male maternal relatives (implying X-linked disease) | NA | FD, X-ALD/AMN, PLP1 disease/SPG2; CMTX, PDH deficiency |
| Extra-neurologic features | ||
| Renal disease | Major | FD, MMA |
| Cutaneous angiokeratomas | Major | FD |
| Liver disease | NA | UCD, mito disease, POLG, NPC, WD |
| Cardiomyopathy | NA | FD, mito disease |
| Tendon xanthomas | NA | CTX |
| Early cataract/lenticular opacity | NA | FD, WD, CTX |
| Symptomatic anaemia | NA | WD, MMA/cobalaminopathies |
| Adrenocortical insufficiency | NA | X-ALD/AMN |
| Recurrent abdominal pain | NA | AIP, FMF, FD |
| Chronic diarrhoea | NA | CTX, mito disease |
| Laboratory findings | ||
| Elevated lactate, plasma | Major | mtDNA disorders, POLG, PDH deficiency; MMA |
| Elevated CK, plasma | NA | Mito disease, POLG |
| Elevated mean corpuscular volume | NA | MMA/cobalaminopathies |
| Haemolytic anaemia | NA | WD |
| Persistently elevated methylmalonic acid, plasma/urine (with normal B12) | NA | MMA/cobalaminopathies |
| Hyperhomocysteinaemia and/or homocystinuria | NA | cblC defect; MTHFR deficiency |
| Neuroimaging appearances | ||
| Pattern/distribution of white matter involvement | ||
| Frontal (anterior) predominance | NA | MLD, HDLS, ADLD |
| Parieto-occipital (posterior) predominance | NA | X-ALD, KD, CTX |
| Periventricular predominance | NA | APBD |
| Symmetric, confluent white matter abnormalities | Intermediate | MLD, SPG11, CMTX, other leukodystrophies and leukoencephalopathies |
| Anterior temporal lobe, external capsule | NA | CADASIL/CARASIL |
| Sparing of the U-fibres | NA | MLD, CXT, APBD, Krabbe |
| Early involvement of U-fibres | NA | MTHFR deficiency, mito disease, APBD |
| Sparing of the corpus callosum | NA | APBD, MLC1 disease |
| Corticospinal/pyramidal tract involvement | NA | KD, HDLS, X-ALD/AMN, mito disease |
| Other findings | ||
| Cerebellar/brainstem signal abnormality | NA | AMN, CTX, AOAD, ADLD, POLG, APBD |
| Gadolinium contrast enhancement | NA | X-ALD/AMN, POLG, AOAD |
| T2 hyperintensities of dentate nucleus | Major | CTX, WD, mito disease |
| T1 hyperintensities of pulvinar | Major | FD |
| Symmetric T2 hyperintensity of basal ganglia | Major | Leigh disease, other mito disease; CTX |
| Calcification on CT | Intermediate | Mito disease |
| Elevated white matter lactate on MRS | Intermediate | Mito disease, LBSL, X-ALD |
| Cystic change or degeneration of white matter | NA | PLP1 disease/SPG2, CLC2, mito disease |
| ‘Eye of the tiger’ appearance to GP | NA | NBIA |
| Prominent brainstem/upper cord atrophy | NA | AOAD |
| Prominent cerebellar atrophy | NA | CHD, NPC, spinocerebellar ataxia |
| Cord hyperintensity | NA | cblC defect, MTHFR deficiency, CTX, mito disease, B12/folate deficiencies, AOAD, LHON |
aMajor, intermediate, and minor red flags as determined by Miller et al., 2008 (NA, not addressed).
ADLD = autosomal dominant leukodystrophy; AIP = acute intermittent porphyria; AMN = adrenomyeloneuropathy; AOAD = adult-onset Alexander disease; APBD = adult polyglucosan body disease; cblC = cobalamin C defect; CARASIL = cerebral autosomal recessive arteriopathy with subcortical infarcts and leukoencephalopathy; CHD = Chédiak-Higashi disease; CK = creatine kinase; CLC2 = CLC2 disease; CMTX = X-linked Charcot–Marie–Tooth disease; CTX = cerebrotendinous xanthomatosis; FD = Fabry disease; FMF = familial Mediterranean fever; GP = globus pallidus; HDLS = hereditary diffuse leukoencephalopathy with spheroids; KD = Krabbe disease; LBSL = leukoencephalopathy with brain stem involvement and lactate elevation; MLD = metachromatic leukodystrophy; MMA = methylmalonic acadaemia; MRS = magnetic resonance spectroscopy; MTHFR = methylenetetrahydrofolate reductase; NBIA = neurodegeneration with brain iron accumulation; NCV = nerve conduction velocity; NPC = Niemann-Pick disease type C; PDH = pyruvate dehydrogenase deficiency; POLG = polymerase gamma-related disease; UCD = urea cycle disorders; X-ALD = X-linked adrenoleukodystrophy.
For the patient who is ‘rediagnosed’ with an inherited disorder, there are several key practical implications: (i) availability of therapeutic interventions for some conditions; (ii) anticipation of natural history of the newly diagnosed disorder, including involvement of other tissues and organs, and expected prognosis; (iii) genetic counselling and/or testing opportunities for relatives of the proband; and (iv) avoidance of risks associated with the erroneous multiple sclerosis diagnosis including side effects of immunomodulatory therapies. For clinicians and researchers, more accurate and timely diagnosis of multiple sclerosis, and improved characterization of patient cohorts, reduces the risk of spurious conclusions from clinical or basic research. In the years ahead, clinically-applicable sequencing technologies are expected to become even more comprehensive, and less expensive, and many rare Mendelian and mitochondrial disorders appear destined to become more accessible to novel therapeutics, some of which are already in development or in clinical trials. For these reasons it is now more important—and more realistic—than ever to accurately recognize rare genetic disorders that masquerade with multiple sclerosis-like phenotypes.
Funding
Dr Honce receives research funding from Biogen Idec. Dr Weisfeld-Adams, Dr Katz Sand and Dr Lublin have no relevant funding or financial conflicts of interest to disclose.
Glossary
Abbreviations
- CADASIL
cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy
- LHON
Leber’s hereditary optic neuropathy
- MELAS
myoclonic epilepsy with lactic acidosis
References
- Aghakhanyan G, Martinuzzi A, Frijia F, Vavla M, Hlavata H, Baratto A, et al. Brain white matter involvement in hereditary spastic paraplegias: analysis with multiple diffusion tensor indices. Am J Neuroradiol. 2014;35:1533–8. doi: 10.3174/ajnr.A3897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alpayci M, Bozan N, Erdem S, Gunes M, Erden M. The possible underlying pathophysiologic mechanisms for development of multiple sclerosis in familial Mediterranean fever. Med Hypotheses. 2012;78:717–20. doi: 10.1016/j.mehy.2012.02.017. [DOI] [PubMed] [Google Scholar]
- Balbi P, Salvini S, Fundarò C, Frazzitta G, Maestri R, Mosah D, et al. The clinical spectrum of late-onset Alexander disease; a systematic literature review. [Review] J Neurol. 2010;257:1955–62. doi: 10.1007/s00415-010-5706-1. [DOI] [PubMed] [Google Scholar]
- Banwell B, Ghezzi A, Bar-Or A, Mikaeloff Y, Tardieu M. Multiple sclerosis in children: clinical diagnosis, therapeutic strategies and future directions. [Review] Lancet Neurol. 2007;6:887–902. doi: 10.1016/S1474-4422(07)70242-9. [DOI] [PubMed] [Google Scholar]
- Barnerias C, Saudubray JM, Touati G, De Lonlay P, Dulac O, Ponsot G, et al. Pyruvate dehydrogenase complex deficiency: four neurological phenotypes with differing pathogenesis. Dev Med Child Neurol. 2010;52:e1–9. doi: 10.1111/j.1469-8749.2009.03541.x. [DOI] [PubMed] [Google Scholar]
- Baronica KB, Mlinac K, Ozretić D, Vladić A, Bognar SK. Arylsulfatase A gene polymorphisms in relapsing remitting multiple sclerosis: genotype-phenotype correlation and estimation of disease progression. Coll Antropol. 2011;35(Suppl 1):11–16. [PubMed] [Google Scholar]
- Battini R, Bianchi MC, Boespflug-Tanguy O, Tosetti M, Bonanni P, Canapicchi R, et al. Unusual clinical and magnetic resonance imaging findings in a family with proteolipid protein gene mutation. Arch Neurol. 2003;60:268–72. doi: 10.1001/archneur.60.2.268. [DOI] [PubMed] [Google Scholar]
- Beecham AH, Patsopoulos NA, Xifara DK, Davis MF, Kemppinen A, Cotsapas C, et al. International Multiple Sclerosis Genetics Consortium (IMSGC). Analysis of immune-related loci identifies 48 new susceptibility variants for multiple sclerosis. Nat Genet. 2013;45:1353–60. doi: 10.1038/ng.2770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhidayasiri R, Perlman SL, Pulst SM, Geschwind DH. Late-onset Friedreich ataxia: phenotypic analysis, magnetic resonance imaging findings, and review of the literature. Arch Neurol. 2005;62:1865–9. doi: 10.1001/archneur.62.12.1865. [DOI] [PubMed] [Google Scholar]
- Biffi A, Montini E, Lorioli L, Cesani M, Fumagalli F, Plati T, et al. Lentiviral hematopoietic stem cell gene therapy benefits metachromatic leukodystrophy. Science. 2013;341:1233158. doi: 10.1126/science.1233158. [DOI] [PubMed] [Google Scholar]
- Böttcher T, Rolfs A, Tanislav C, Bitsch A, Köhler W, Gaedeke J, et al. Fabry disease – Underestimated in the differential diagnosis of multiple sclerosis? PLoS One. 2013;8:e7189. doi: 10.1371/journal.pone.0071894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boustany RM. Lysosomal storage diseases – the horizon expands. [Review] Nat Rev Neurol. 2013;9:583–98. doi: 10.1038/nrneurol.2013.163. [DOI] [PubMed] [Google Scholar]
- Burlina AP, Manara R, Caillaud C, Laissy JP, Severino M, Klein I, et al. The pulvinar sign: frequency and clinical correlations in Fabry disease. J Neurol. 2008;255:738–44. doi: 10.1007/s00415-008-0786-x. [DOI] [PubMed] [Google Scholar]
- Bylesjö I, Brekke OL, Prytz J, Skjeflo T, Salvesen R. Brain magnetic resonance imaging white matter lesions and cerebrospinal fluid findings in patients with acute intermittent porphyria. Eur Neurol. 2004;51:1–5. doi: 10.1159/000074909. [DOI] [PubMed] [Google Scholar]
- Camiña-Tato M, Morcillo-Suárez C, Bustamante MF, Ortega I, Navarro A, Muntasell A, et al. Gender-associated differences of perforin polymorphisms in the susceptibility to multiple sclerosis. J Immunol. 2010;185:5392–404. doi: 10.4049/jimmunol.1000102. [DOI] [PubMed] [Google Scholar]
- Carra-Dalliere C, Scherer C, Ayrignac X, Menjot de Champfleur N, Bellesme C, Labauge P, et al. Adult-onset cerebral X-linked adrenoleukodystrophy with major contrast-enhancement mimicking acquired disease. Clin Neurol Neurosurg. 2013;115:1906–7. doi: 10.1016/j.clineuro.2013.05.006. [DOI] [PubMed] [Google Scholar]
- Cartier N, Hacein-Bey-Abina S, Bartholomae CC, Bougnères P, Schmidt M, Kalle CV, et al. Hematopoietic stem cell gene therapy with a lentiviral vector in X-linked adrenoleukodystrophy. Science. 2009;326:818–23. doi: 10.1126/science.1171242. [DOI] [PubMed] [Google Scholar]
- Casali C, Valente EM, Bertini E, Montagna G, Criscuolo C, De Michele G, et al. Clinical and genetic studies in hereditary spastic paraplegia with thin corpus callosum. Neurology. 2004;62:262–8. doi: 10.1212/wnl.62.2.262. [DOI] [PubMed] [Google Scholar]
- Chebel S, Barboura I, Boughammoura-Bouatay A, Ammar M, Ferchichi S, Miled A, et al. Adult-type metachromatic leukodystrophy mimicking multiple sclerosis. Can J Neurol Sci. 2009;36:521–3. doi: 10.1017/s0317167100007940. [DOI] [PubMed] [Google Scholar]
- De Beer M, Engelen M, van Geel BM. Frequent occurrence of cerebral demyelination in adrenomyeloneuropathy. Neurology. 2014;83:2227–31. doi: 10.1212/WNL.0000000000001074. [DOI] [PubMed] [Google Scholar]
- De Stefano N, Dotti MT, Mortilla M, Federico A. Magnetic resonance imaging and spectroscopic changes in brains of patients with cerebrotendinous xanthomatosis. Brain. 2001;124:121–31. doi: 10.1093/brain/124.1.121. [DOI] [PubMed] [Google Scholar]
- Degos B, Laforet P, Jardel C, Sedel F, Jossay-Winter M, Romero NB, et al. POLG mutations associated with relapsing-remitting neurological events. J Clin Neurosci. 2014;21:186–8. doi: 10.1016/j.jocn.2013.03.019. [DOI] [PubMed] [Google Scholar]
- Depienne C, Bugiani M, Dupuits C, Galanaud D, Touitou V, Postma N, et al. Brain white matter oedema due to ClC-2 chlroride channel deficiency: an observational analytical study. Lancet Neurol. 2013;12:659–68. doi: 10.1016/S1474-4422(13)70053-X. [DOI] [PubMed] [Google Scholar]
- Engelen M, Kemp S, de Visser M, van Geel BM, Wanders RJ, Aubourg P, et al. X-linked adrenoleukodystrophy (X-ALD): clinical presentation and guidelines for diagnosis, follow-up, and management. Orphanet J Rare Dis. 2012;7:51. doi: 10.1186/1750-1172-7-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelen M, Barbier M, Dijkstra IM, Schür R, de Bie RM, Verhamme C, et al. X-linked adrenoleukodystrophy in women: a cross-sectional cohort study. Brain. 2014;137(Pt 3):693–706. doi: 10.1093/brain/awt361. [DOI] [PubMed] [Google Scholar]
- Engell T. A clinic-pathoanatomical study of multiple sclerosis diagnosis. [Review] Acta Neurol Scand. 1988;78:39–44. doi: 10.1111/j.1600-0404.1988.tb03616.x. [DOI] [PubMed] [Google Scholar]
- Escolar ML, Poe MD, Provenzale JM, Richards KC, Allison J, Wood S, et al. Transplantation of umbilical-cord blood in babies with infantile Krabbe's disease. N Engl J Med. 2005;352:2069–81. doi: 10.1056/NEJMoa042604. [DOI] [PubMed] [Google Scholar]
- Farina L, Bizzi A, Finocchiaro G, Pareyson D, Sghirlanzoni A, Bertagnolio B, et al. MR Imaging and Proton MR Spectroscopy in Adult Krabbe Disease. AJNR Am J Neuroradiol. 2000;21:1478–82. [PMC free article] [PubMed] [Google Scholar]
- Farina L, Pareyson D, Minati L, Ceccherini I, Chiapparini L, Romano S, et al. Can MR imaging diagnose adult-onset alexander disease? AJNR Am J Neuroradiol. 2008;29:1190–6. doi: 10.3174/ajnr.A1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friese MA, Schattling B, Fugger L. Mechanisms of neurodegeneration and axonal dysfunction in multiple sclerosis. [Review] Nat Rev Neurol. 2014;10:225–38. doi: 10.1038/nrneurol.2014.37. [DOI] [PubMed] [Google Scholar]
- Fukutake T, Hirayama K. Familial young-adult-onset arteriosclerotic leukoencephalopathy with alopecia and lumbago without arterial hypertension. Eur Neurol. 1995;35:69–79. doi: 10.1159/000117096. [DOI] [PubMed] [Google Scholar]
- Gal A, Hughes DA, Winchester B. Toward a consensus in the laboratory disgnostics of Fabry disease - recommendations of a European expert group. J Inherit Metab Dis. 2011;34(2):509–14. doi: 10.1007/s10545-010-9261-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godeiro-Junior C, Felicio AC, Benites V, Chieia MA, Oliveira AS. Late-onset hexosaminidase A deficiency mimicking primary lateral sclerosis. Arq Neuropsiquiatr. 2009;67:105–6. doi: 10.1590/s0004-282x2009000100024. [DOI] [PubMed] [Google Scholar]
- Hanemann CO, Bergmann C, Senderek J, Zerres K, Sperfeld AD. Transient, recurrent, white matter lesions in X-linked Charcot-Marie-Tooth disease with novel Connexin 32 mutation. Arch Neurol. 2003;60:605–9. doi: 10.1001/archneur.60.4.605. [DOI] [PubMed] [Google Scholar]
- Harding AE, Sweeney MG, Miller DH, Mumford CJ, Kellar-Wood H, Menard D, et al. Occurrence of a multiple sclerosis-like illness in women who have a Leber’s hereditary optic neuropathy mutation. Brain. 1992;115:979–89. doi: 10.1093/brain/115.4.979. [DOI] [PubMed] [Google Scholar]
- Harding AE. Hereditary spastic paraplegias. [Review] Semin Neurol. 1993;13:333–6. doi: 10.1055/s-2008-1041143. [DOI] [PubMed] [Google Scholar]
- Haworth JC, Dilling LA, Surtees RAH, Seargeant LE, Lue-Shing H, Cooper BA, et al. Symptomatic and asymptomatic methylenetetrahydrofolate reductase deficiency in two adult brothers. Am J Med Genet. 1993;45:572–6. doi: 10.1002/ajmg.1320450510. [DOI] [PubMed] [Google Scholar]
- Hayflick SJ, Hartman M, Coryell J, Gitschier J, Rowley J. Brain MRI in neurodegeneration with brain iron accumulation with and without PANK2 mutations. AJNR Am J Neuroradiol. 2006;27:1230–3. [PMC free article] [PubMed] [Google Scholar]
- Hourani R, El-Hajj T, Barada WH, Hourani M, Yamout BI. MR imaging findings in autosomal recessive hereditary spastic paraplegia. Am J Neuroradiol. 2009;30:936–40. doi: 10.3174/ajnr.A1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huemer M, Scholl-Bürgi S, Hadaya K, Kern I, Beer R, Seppi K, et al. Three new cases of late-onset cblC defect and review of the literature illustrating when to consider inborn errors of metabolism beyond infancy. Orphanet J Rare Dis. 2014;9:161. doi: 10.1186/s13023-014-0161-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurst S, Garbern J, Trepanier A, Gow A. Quantifying the carrier female phenotype in Pelizaeus-Merzbacher disease. Genet Med. 2006;8:371–8. doi: 10.1097/01.gim.0000223551.95862.c3. [DOI] [PubMed] [Google Scholar]
- Iñiguez C, Arenas J, Montoya J, Mostacero E, Lopez del Val J, Morales F. Mitochondrial respiratory chain deficiency may present as multiple sclerosis. Neurologia. 1998;13:199–203. [PubMed] [Google Scholar]
- Inglese M, Rovaris M, Bianchi S, Mantia LL, Mancardi GL, Ghezzi A, et al. Magnetic resonance imaging, magnetization transfer imaging, and diffusion weighted imaging correlates of optic nerve, brain and cervical cord damage in Leber’s hereditary optic neuropathy. J Neurol Neurosurg Psychiatry. 2001;70:444–9. doi: 10.1136/jnnp.70.4.444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue T, Hattori K, Ihara K, Ishii A, Nakamura K, Hirose S. Newborn screening for Fabry disease in Japan: prevalence and genotypes of Fabry disease in a pilot study. J Hum Genet. 2013;58:548–52. doi: 10.1038/jhg.2013.48. [DOI] [PubMed] [Google Scholar]
- Isohanni P, Linnankivi T, Buzkova J, Lönnqvist T, Pihko H, Valanne L, et al. DARS2 mutations in mitochondrial leucoencephalopathy and multiple sclerosis. J Med Genet. 2010;47:66–70. doi: 10.1136/jmg.2009.068221. [DOI] [PubMed] [Google Scholar]
- Kappler J, Pötter W, Gieselmann V, Kiessling W, Friedl W, Propping P. Phenotypic consequences of low arylsulfatase A genotypes (ASAp/ASAp and ASA-/ASAp): does there exist an association with multiple sclerosis? Dev Neurosci. 1991;13:228–31. doi: 10.1159/000112165. [DOI] [PubMed] [Google Scholar]
- Karim MA, Suzuki K, Fukai K, Oh J, Nagle DL, Moore KJ, et al. Apparent genotype-phenotype correlation in childhood, adolescent, and adult Chédiak-Higashi syndrome. Am J Med Genet. 2002;108:16–22. [PubMed] [Google Scholar]
- Katz Sand IB, Lublin FD. Diagnosis and differential diagnosis of multiple sclerosis. [Review] Continuum (Minneap Minn) 2013;19:922–43. doi: 10.1212/01.CON.0000433290.15468.21. [DOI] [PubMed] [Google Scholar]
- Kazibutowska Z, Bal A, Gołba A, Ługowska A. Metachromatic leukodystrophy in adult patient initially diagnosed as multiple sclerosis. Neurol Neurochir Pol. 2002;36:1209–19. [PubMed] [Google Scholar]
- Keegan BM, Giannini C, Parisi JE, Lucchinetti CF, Boeve BF, Josephs KA. Sporadic adult-onset leukoencephalopathy with neuroaxonal spheroids mimicking cerebral MS. Neurology. 2008;70:1128–33. doi: 10.1212/01.wnl.0000304045.99153.8f. [DOI] [PubMed] [Google Scholar]
- Krupp L, Banwell B, Tenembaum S International Pediatric MS Study Group. Consensus definitions proposed for pediatric multiple sclerosis and related disorders. Neurology. 2007;68(Suppl 2):S7–12. doi: 10.1212/01.wnl.0000259422.44235.a8. [DOI] [PubMed] [Google Scholar]
- Küker W, Weir A, Quaghebeur G, Palace J. White matter changes in Leber’s hereditary optic neuropathy: MRI findings. Eur J Neurol. 2007;14:591–3. doi: 10.1111/j.1468-1331.2007.01757.x. [DOI] [PubMed] [Google Scholar]
- Kumar AJ, Kohler W, Kruse B, Naidu S, Bergin A, Edwin D, et al. MR findings in adult-onset adrenoleukodystrophy. Am J Neuroradiol. 1995;16:1227–37. [PMC free article] [PubMed] [Google Scholar]
- Labauge P, Dorboz I, Eymard-Pierre E, Dereeper O, Boespflug-Tanguy O. Clinically asymptomatic adult patient with extensive LBSL MRI pattern and DARS2 mutations. J Neurol. 2011;258:335–7. doi: 10.1007/s00415-010-5755-5. [DOI] [PubMed] [Google Scholar]
- Lerman-Sagie T, Leshinsky-Silver E, Watemberg N, Luckman Y, Lev D. White matter involvement in mitochondrial diseases. Mol Genet Metab. 2005;84:127–36. doi: 10.1016/j.ymgme.2004.09.008. [DOI] [PubMed] [Google Scholar]
- Leuzzi V, Carducci C, Lenza M, Salvetti M, Ristori G, Di Giovanni S, et al. LHON mutations in Italian patients affected by multiple sclerosis. Acta Neurol Scand. 1997;96:145–8. doi: 10.1111/j.1600-0404.1997.tb00257.x. [DOI] [PubMed] [Google Scholar]
- Li R, Johnson AB, Salomons GS, van der Knaap MS, Rodriguez D, Boespflug-Tanguy O, et al. Propensity for paternal inheritance of de novo mutations in Alexander disease. Hum Genet. 2006;119:137–44. doi: 10.1007/s00439-005-0116-7. [DOI] [PubMed] [Google Scholar]
- Link H, Huang YM. Oligoclonal bands in multiple sclerosis cerebrospinal fluid: an update on methodology and clinical usefulness. [Review] J Neuroimmunol. 2006;180:17–28. doi: 10.1016/j.jneuroim.2006.07.006. [DOI] [PubMed] [Google Scholar]
- Lissens W, Arena A, Seneca S, Rafi M, Sorge G, Liebaers I, et al. A single mutation in the GALC gene is responsible for the majority of late onset Krabbe disease patients in the Catania (Sicily, Italy) region. Hum Mutat. 2007;28:742. doi: 10.1002/humu.9500. [DOI] [PubMed] [Google Scholar]
- Loes DJ, Fatemi A, Melhem ER, Gupte N, Bezman L, Moser HW, et al. Analysis of MRI patterns aids prediction of progression in X-linked adrenoleukodystrophy. Neurology. 2003;61:369–74. doi: 10.1212/01.wnl.0000079050.91337.83. [DOI] [PubMed] [Google Scholar]
- Lublin FD, Reingold SC. Defining the clinical course of multiple sclerosis: results of an international survey. National Multiple Sclerosis Society (USA) Advisory Committee on Clinical Trials of New Agents in Multiple Sclerosis. Neurology. 1996;46:907–11. doi: 10.1212/wnl.46.4.907. [DOI] [PubMed] [Google Scholar]
- Ługowska A, Berger J, Tylki-Szymanska A, Löschl B, Molzer B, Zobel M, et al. Molecular and phenotypic characteristics of metachromatic leukodystrophy patients from Poland. Clin Genet. 2005;68:48–54. doi: 10.1111/j.1399-0004.2005.00451.x. [DOI] [PubMed] [Google Scholar]
- Mahad DJ, Ziabreva I, Campbell G, Lax N, White K, Hanson PS, et al. Mitochondrial changes within axons in multiple sclerosis. Brain. 2009;132:1161–74. doi: 10.1093/brain/awp046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malojcic B, Brinar V, Poser C, Djakovic V. An adult case of Leigh disease. Clin Neurol Neurosurg. 2004;106:237–40. doi: 10.1016/j.clineuro.2004.02.028. [DOI] [PubMed] [Google Scholar]
- Maruyama S, Yamada T, Ishimoto Y, Hara H, Taniwaki T, Kira J. A case of MELAS showing CSF pleocytosis associated with stroke-like episodes. Rinsho Shinkeigaku. 1998;38:641–4. [PubMed] [Google Scholar]
- Matern D, Oglesbee D, Tortorelli S. Newborn screening for lysosomal storage disorders and other neuronopathic conditions. [Review] Dev Disabil Res Rev. 2013;17:247–53. doi: 10.1002/ddrr.1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller DH, Leary SM. Primary progressive multiple sclerosis. [Review] Lancet Neurol. 2007;6:903–12. doi: 10.1016/S1474-4422(07)70243-0. [DOI] [PubMed] [Google Scholar]
- Miller DH, Weinshenker BG, Filippi M, Banwell BL, Cohen JA, Freedman MS, et al. Differential diagnosis of suspected multiple sclerosis: a consensus approach. Mult Scler. 2008;14:1157–74. doi: 10.1177/1352458508096878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mo YH, Chen YF, Liu HM. Adrenomyeloneuropathy, a dynamic progressive disorder: brain magnetic resonance imaging in two cases. Neuroradiology. 2004;46:296–300. doi: 10.1007/s00234-003-1096-8. [DOI] [PubMed] [Google Scholar]
- Moore DF, Ye F, Schiffmann R, Butman JA. Increased Signal Intensity in the Pulvinar on T1-Weighted Images: A Pathognomonic MR Imaging Sign of Fabry Disease. AJNR Am J Neuroradiol. 2003;24:1096–101. [PMC free article] [PubMed] [Google Scholar]
- Naismith RT, Trinkaus K. Cross AH. Phenotype and prognosis in African-Americans with multiple sclerosis: a retrospective chart review. Mult Scler. 2006;12:775–81. doi: 10.1177/1352458506070923. [DOI] [PubMed] [Google Scholar]
- Natowicz MR, Bejjani B. Genetic disorders that masquerade as multiple sclerosis. [Review] Am J Med Genet. 1994;49:149–69. doi: 10.1002/ajmg.1320490202. [DOI] [PubMed] [Google Scholar]
- Novarino G, Fenstermaker AG, Zaki MS, Hofree M, Silhavy JL, Heiberg AD, et al. Exome sequencing links corticospinal motor neuron disease to common neurodegenerative disorders. Science. 2014;343:506–11. doi: 10.1126/science.1247363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Riordan S, Nor AM, Hutchinson M. CADASIL imitating multiple sclerosis: the importance of MRI markers. Mult Scler. 2002;8:430–2. doi: 10.1191/1352458502ms834oa. [DOI] [PubMed] [Google Scholar]
- Palace J. Multiple sclerosis associated with Leber’s hereditary optic neuropathy. J Neurol Sci. 2009;286:24–7. doi: 10.1016/j.jns.2009.09.009. [DOI] [PubMed] [Google Scholar]
- Padiath QS, Saigoh K, Schiffmann R, Asahara H, Yamada T, Koeppen A, et al. Lamin B1 duplications cause autosomal dominant leukodystrophy. Nat Genet. 2006;38:1114–23. doi: 10.1038/ng1872. [DOI] [PubMed] [Google Scholar]
- Paradas C, Akman HO, Ionete C, Lau H, Riskind PN, Jones DE, et al. Branching enzyme deficiency: expanding the clinical spectrum. JAMA Neurol. 2014;71:41–7. doi: 10.1001/jamaneurol.2013.4888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pareyson D, Fancellu R, Mariotti C, Romano S, Salmaggi A, carella F, et al. Adult-onset Alexander disease: a series of eleven unrelated cases with review of the literature. Brain. 2008;131:2321–31. doi: 10.1093/brain/awn178. [DOI] [PubMed] [Google Scholar]
- Patil SA, Maegawa GH. Developing therapeutic approaches for metachromatic leukodystrophy. [Review] Drug Des Devel Ther. 2013;7:729–45. doi: 10.2147/DDDT.S15467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson MC, Vecchio D, Prady H, Abel L, Wraith JE. Miglustat for treatment of Niemann-Pick C disease: a randomised controlled study. Lancet Neurol. 2007;6:765–72. doi: 10.1016/S1474-4422(07)70194-1. [DOI] [PubMed] [Google Scholar]
- Patterson MC, Mengel E, Wijburg FA, Muller A, Schwierin B, Drevon H, et al. Disease and patient characteristics in NP-C patients: findings from an international disease registry. Orphanet J Rare Dis. 2013;8:12. doi: 10.1186/1750-1172-8-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulson HL, Garbern JY, Hoban TF, Krajewski KM, Lewis RA, Fischbeck KH, et al. Transient central nervous system white matter abnormality in X-linked Charcot-Marie-Tooth disease. Ann Neurol. 2002;52:429–34. doi: 10.1002/ana.10305. [DOI] [PubMed] [Google Scholar]
- Pinar-Sueiro S, Martínez-Fernández R, Lage-Medina S, Aldamiz-Echevarria L, Vecino E. Optic neuropathy in methylmalonic acidemia: the role of neuroprotection. J Inherit Metab Dis. 2010;33(Suppl 3):S199–203. doi: 10.1007/s10545-010-9084-8. [DOI] [PubMed] [Google Scholar]
- Pisani A, Visciano B, Roux GD, Sabbatini M, Porto C, Parenti G, et al. Enzyme replacement therapy in patients with Fabry disease: state of the art and review of the literature. [Review] Mol Genet Metab. 2012;107:267–75. doi: 10.1016/j.ymgme.2012.08.003. [DOI] [PubMed] [Google Scholar]
- Polman CH, Reingold SC, Edan G, Filippi M, Hartung HP, Kappos L, et al. Diagnostic criteria for multiple sclerosis: 2005 revisions to the “McDonald Criteria”. Ann Neurol. 2005;58:840–6. doi: 10.1002/ana.20703. [DOI] [PubMed] [Google Scholar]
- Polman CH, Reingold SC, Banwell B, Clanet M, Cohen JA, Filippi M, et al. Diagnostic criteria for multiple sclerosis: 2010 revisions to the McDonald criteria. Ann Neurol. 2011;69:292–302. doi: 10.1002/ana.22366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasad AN, Breen JC, Ampola MG, Rosman NP. Argininemia: a treatable genetic cause of progressive spastic diplegia simulating cerebral palsy: case reports and literature review. [Review] J Child Neurol. 1997;12:301–9. doi: 10.1177/088307389701200502. [DOI] [PubMed] [Google Scholar]
- Pretegiani E, Rufa A, Gallus GN, Cardaioli E, Malandrini A, Federico A. Spastic paraplegia in ‘dominant optic atrophy plus’ phenotype due to OPA1 mutation. Brain. 2011;134:e195. doi: 10.1093/brain/awr101. [DOI] [PubMed] [Google Scholar]
- Rademakers R, Baker M, Nicholson AM, Rutherford NJ, Finch N, Soto-Ortolaza A, et al. Mutations in the colony stimulating factor 1 receptor (CSF1R) gene cause hereditary diffuse leukoencephalopathy with spheroids. Nat Genet. 2011;44:200–5. doi: 10.1038/ng.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rafi MA, Rao HZ, Luzi P, Curtis MT, Wenger DA. Extended normal life after AAVrh10-mediated gene therapy in the mouse model of Krabbe disease. Mol Ther. 2012;20:2031–42. doi: 10.1038/mt.2012.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramagopalan SV, Cukjati M, Cernilec M, DeLuca GC, Dyment DA, Degenhardt A, et al. Mutations in the hemochromatosis gene and the clinical outcome of multiple sclerosis. J Neuroimmunol. 2008;203:104–7. doi: 10.1016/j.jneuroim.2008.06.036. [DOI] [PubMed] [Google Scholar]
- Raymond GV, Seidman R, Monteith TS, Kolodny E, Sathe S, Mahmood A, et al. Head trauma can initiate the onset of adrenoleukodystrophy. J Neurol Sci. 2010;290:70–4. doi: 10.1016/j.jns.2009.11.005. [DOI] [PubMed] [Google Scholar]
- Revesz T, Kidd D, Thompson AJ, Barnard RO, McDonald WI. A comparison of the pathology of primary and secondary progressive multiple sclerosis. Brain. 1994;117:759–65. doi: 10.1093/brain/117.4.759. [DOI] [PubMed] [Google Scholar]
- Romagnolo A, Masera S, Mattioda A, Superti G, Santorelli FM, Mongini T, et al. Atypical hereditary spastic paraplegia mimicking multiple sclerosis associated with a novel SPG11 mutation. Eur J Neurol. 2014;21:e14–15. doi: 10.1111/ene.12297. [DOI] [PubMed] [Google Scholar]
- Rudick RA, Schiffer RB, Schwetz KM, Herndon RM. Multiple sclerosis: the problem of incorrect diagnosis. [Review] Arch Neurol. 1986;43:578–83. doi: 10.1001/archneur.1986.00520060042015. [DOI] [PubMed] [Google Scholar]
- Rudick RA, Miller AE. Multiple sclerosis or multiple possibilities: the continuing problem of misdiagnosis. [Review] Neurology. 2012;78:1904–6. doi: 10.1212/WNL.0b013e318259e2e2. [DOI] [PubMed] [Google Scholar]
- Sato K, Kubo S, Fujii H, Okamoto M, Takahashi K, Takamatsu K, et al. Diffusion tensor imaging and magnetic resonance spectroscopy of transient cerebral white matter lesions in X-linked Charcot-Marie-Tooth disease. J Neurol Sci. 2012;316:178–80. doi: 10.1016/j.jns.2012.01.017. [DOI] [PubMed] [Google Scholar]
- Sedel F, Tourbah A, Fontaine B, Lubetski C, Baumann N, Saudubray JM, et al. Leukoencephalopathies associated with inborn errors of metabolism in adults. [Review] J Inherit Metab Dis. 2008;31:295–307. doi: 10.1007/s10545-008-0778-0. [DOI] [PubMed] [Google Scholar]
- Solomon AJ, Klein EP, Bourdette D. “Undiagnosing” multiple sclerosis: the challenge of misdiagnosis in MS. [Review] Neurology. 2012;78:1886–91. doi: 10.1212/WNL.0b013e318259e1b2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solomon AJ, Weinshenker BG. Misdiagnosis of multiple sclerosis: frequency, causes, effects, and prevention. [Review] Curr Neurol Neurosci Rep. 2013;13:403. doi: 10.1007/s11910-013-0403-y. [DOI] [PubMed] [Google Scholar]
- Spada M, Pagliardini S, Yasuda M, Tukel T, Thiagarajan G, Sakuraba H, et al. High incidence of later-onset Fabry disease revealed by newborn screening. Am J Hum Genet. 2006;79:31–40. doi: 10.1086/504601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stöckler S, Millner M, Molzer B, Ebner F, Körner E, Moser HW. Multiple sclerosis-like syndrome in a woman heterozygous for adrenoleukodystrophy. Eur Neurol. 1993;33:390–2. doi: 10.1159/000116978. [DOI] [PubMed] [Google Scholar]
- Stroobants S, Gerlach D, Matthes F, Hartmann D, Fogh J, Gieselmann V, et al. Intracerebroventricular enzyme infusion corrects central nervous system pathology and dysfunction in a mouse model of metachromatic leukodystrophy. Hum Mol Genet. 2011;20:2760–9. doi: 10.1093/hmg/ddr175. [DOI] [PubMed] [Google Scholar]
- Thauvin-Robinet C, Roze E, Couvreur G, Horellou MH, Sedel F, Grabli D, et al. The adolescent and adult form of cobalamin C disease: clinical and molecular spectrum. J Neurol Neurosurg Psychiatry. 2008;79:725–8. doi: 10.1136/jnnp.2007.133025. [DOI] [PubMed] [Google Scholar]
- Tremlett H, Zhao Y, Devonshire V. Natural history of secondary progressive multiple sclerosis. [Review] Mult Scler. 2008;14:314–24. doi: 10.1177/1352458507084264. [DOI] [PubMed] [Google Scholar]
- Tschampa HJ, Greschus S, Vinahl M, Urbach H, Mueller MM, Gerding WM. MS-like presentation of Alexander disease with multifocal lesions and oligoclonal bands. J Neurol. 2011;258:935–7. doi: 10.1007/s00415-010-5846-3. [DOI] [PubMed] [Google Scholar]
- Van Geel BM, Bezman L, Loes DJ, Moser HW, Raymond GV. Evolution of phenotypes in adult male patients with X-linked adrenoleukodystrophy. Ann Neurol. 2001;49:186–94. doi: 10.1002/1531-8249(20010201)49:2<186::aid-ana38>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- Van Wassenaer-van Hall HN, van den Heuvel AG, Jansen GH, Hoogenraad TU, Mali WP. Cranial MR in Wilson disease: abnormal white matter in extrapyramidal and pyramidal tracts. Am J Neuroradiol. 1995;16:2021–7. [PMC free article] [PubMed] [Google Scholar]
- Vyshkina T, Sylvester A, Sadiq S, Bonilla E, Canter JA, Perl A, et al. Association of common mitochondrial DNA variants with multiple sclerosis and systemic lupus erythematosus. Clin Immunol. 2008;129:31–5. doi: 10.1016/j.clim.2008.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wachowius U, Talley M, Silver N, Heinze HJ, Sailer M. Cognitive impairment in primary and secondary progressive multiple sclerosis. J Clin Exp Neuropsychol. 2005;27:65–77. doi: 10.1080/138033990513645. [DOI] [PubMed] [Google Scholar]
- Wang RY, Bodamer OA, Watson MS, Wilcox WR ACMG Work Group on Diagnostic Confirmation of Lysosomal Storage Diseases. Lysosomal storage diseases: diagnostic confirmation and management of presymptomatic individuals. Genet Med. 2011;13:457–84. doi: 10.1097/GIM.0b013e318211a7e1. [DOI] [PubMed] [Google Scholar]
- Warshawsky I, Rudick RA, Staugaitis SM, Natowicz MR. Primary progressive multiple sclerosis as a phenotype of a PLP1 gene mutation. Ann Neurol. 2005;58:470–3. doi: 10.1002/ana.20601. [DOI] [PubMed] [Google Scholar]
- Weisfeld-Adams JD, Mehta L, Rucker JC, Dembitzer FR, Szporn A, Lublin FD, et al. Atypical Chédiak-Higashi syndrome with attenuated phenotype: three adult siblings homozygous for a novel LYST deletion and with neurodegenerative disease. Orphanet J Rare Dis. 2013;8:46. doi: 10.1186/1750-1172-8-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams R, Buchheit CL, Berman NE, LeVine SM. Pathogenic implications of iron accumulation in multiple sclerosis. [Review] J Neurochem. 2012;120:7–25. doi: 10.1111/j.1471-4159.2011.07536.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witte ME, Guerts JJ, de Vries HE, van der Valk P, van Horssen J. Mitochondrial dysfunction: a potential link between neuroinflammation and neurodegeneration. [Review] Mitochondrion. 2010;10:411–18. doi: 10.1016/j.mito.2010.05.014. [DOI] [PubMed] [Google Scholar]
- Yahalom G, Tsabari R, Molshatzki N, Ephraty L, Cohen H, Hassin-Baer S. Neurological outcome in cerebrotendinous xanthomatosis treated with chenodeoxycholic acid: early versus late diagnosis. Clin Neuropharmacol. 2013;36:78–83. doi: 10.1097/WNF.0b013e318288076a. [DOI] [PubMed] [Google Scholar]
- Yoshida T, Sasayama H, Mizuta I, Okamoto Y, Yoshida M, Riku Y, et al. Glial fibrillary acidic protein mutations in adult-onset Alexander disease: clinical features observed in 12 Japanese patients. Acta Neurol Scand. 2011;124:104–8. doi: 10.1111/j.1600-0404.2010.01427.x. [DOI] [PubMed] [Google Scholar]
- Yu-Wai-Man P, Griffiths PG, Gorman GS, Lourenco CM, Wright AF, Auer-Grumbach M, et al. Multi-system neurological disease is common in patients with OPA1 mutations. Brain. 2010;133:771–86. doi: 10.1093/brain/awq007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwetsloot CP, Padberg GW, van Seters AP, Maaswinkel-Mooy PD, Onkenhout W. Adult adrenoleukodystrophy: the clinical spectrum in a large Dutch family. J Neurol. 1992;239:107–11. doi: 10.1007/BF00862985. [DOI] [PubMed] [Google Scholar]