

Abstract
Rationale
Diabetes is associated with cardiac fibrosis. Matricellular proteins are induced in fibrotic conditions and modulate fibrogenic and angiogenic responses by regulating growth factor signaling.
Objective
To test the hypothesis that the prototypical matricellular protein thrombospondin (TSP)-1, a potent angiostatic molecule and crucial activator of TGF-β, may play a key role in remodeling of the diabetic heart.
Methods and results
Obese diabetic db/db mice exhibited marked myocardial TSP-1 upregulation in the interstitial and perivascular space. In order to study the role of TSP-1 in remodeling of the diabetic heart we generated and characterized db/db TSP-1 null (dbTSP) mice. TSP-1 disruption did not significantly affect weight gain and metabolic function in db/db animals. When compared with db/db animals, dbTSP mice had increased left ventricular dilation associated with mild non-progressive systolic dysfunction. Chamber dilation in dbTSP mice was associated with decreased myocardial collagen content and accentuated Matrix Metalloproteinase (MMP)-2 and -9 activity. TSP-1 disruption did not affect inflammatory gene expression and activation of TGF-β/Smad signaling in the db/db myocardium. In cardiac fibroblasts populating collagen pads, TSP-1 incorporation into the matrix did not activate TGF-β responses, but inhibited leptin-induced MMP-2 activation. TSP-1 disruption abrogated age-associated capillary rarefaction in db/db mice, attenuating myocardial upregulation of angiopoietin-2, a mediator that induces vascular regression. In vitro, TSP-1 stimulation increased macrophage, but not endothelial cell, angiopoietin-2 synthesis.
Conclusions
TSP-1 upregulation in the diabetic heart prevents chamber dilation by exerting matrix-preserving actions on cardiac fibroblasts and mediates capillary rarefaction through effects that may involve angiopoietin-2 upregulation.
Keywords: diabetic cardiomyopathy, remodeling, matricellular gene, fibrosis
INTRODUCTION
Diabetes and obesity are associated with increased susceptibility to cardiovascular disease 1, 2. Data derived from the Framingham study suggest that diabetic men have a 2.4-fold increase in the incidence of heart failure; the risk of heart failure is even higher (5.1-fold increase) in diabetic women 3. The increased prevalence of heart failure in diabetes is only in part due to the increased risk of atherosclerotic coronary disease and its complications. Diabetics also develop a unique cardiomyopathic condition, termed “diabetic cardiomyopathy” 4, 5, 6 that is independent of coronary artery disease. Diabetic cardiomyopathy is characterized by extensive fibrotic changes, expansion of the cardiac interstitium, 7 and profound alterations in the cardiac interstitial matrix 8 leading to increased myocardial stiffness and development of diastolic dysfunction 9. Despite its significance, the pathophysiologic basis of cardiac fibrosis in diabetes remains poorly understood.
Tissue fibrosis requires the dynamic participation of the extracellular matrix and is regulated by a family of structurally unrelated macromolecules called matricellular proteins 10. Matricellular proteins are generally not expressed in the normal heart, but are markedly upregulated in the remodeling myocardium, and through binding to structural matrix proteins serve as molecular bridges between the matrix and the cells, transducing or modulating growth factor signals 11, 12, 13, 14. Thrombospondin (TSP)-1 is a prototypical matricellular protein that is not part of the normal cardiac matrix network, but is upregulated in cardiac remodeling due to myocardial infarction 15 or pressure overload 16. In the pressure-overloaded heart, TSP-1 modulates fibroblast phenotype by activating Transforming Growth Factor (TGF)-β and preserves the matrix by inhibiting matrix metalloproteinase (MMP) activity 16. In addition to its pro-fibrotic and matrix-preserving actions, TSP-1 is also a potent angiostatic mediator 17, 18 that promotes endothelial cell apoptosis through activation of a CD36/p59fyn/p38 Mitogen-Activate Protein Kinase (MAPK) pathway 19.
Experimental and clinical studies have demonstrated that obesity and diabetes are associated with marked upregulation of TSP-1 in the adipose tissue and in the cardiovascular system 20, 21. In adipose tissue harvested from obese patients, TSP-1 expression was markedly increased and was strongly associated with insulin resistance and inflammatory activity 21. Moreover, in obese diabetic Zucker rats, TSP-1 protein expression was markedly upregulated in the vascular adventitia and in the cardiac interstitium 20; TSP-1 induction in diabetic vessels was associated with reduced density of vasa vasorum. In vitro, hyperglycemia potently upregulated TSP-1 synthesis; high glucose levels induced a 30-fold increase in TSP-1 expression by isolated endothelial cells, smooth muscle cells and fibroblasts 20.
We hypothesized that, through its pro-fibrotic and angiostatic properties, TSP-1 may play an important role in remodeling of the diabetic heart. Incorporation of TSP-1 in the interstitial matrix of the diabetic myocardium may locally activate TGF-β signaling, inducing fibroblast activation and promoting fibrotic remodeling. Moreover, through its potent angiostatic properties, TSP-1 may be responsible for the impaired angiogenic responses in the diabetic heart. In order to test this hypothesis we used the db/db mouse, a model of genetic obesity and diabetes due to central leptin resistance that is associated with progressive cardiac fibrosis 22. We demonstrate marked TSP-1 upregulation in db/db hearts predominantly localized in perivascular and interstitial areas. Through the generation and systematic study of db/db TSP-1 null mice, we found that TSP-1 loss attenuates diabetes-associated cardiac fibrosis and enhances myocardial protease activity, without significantly improving diastolic function. In contrast, loss of the matrix-preserving actions of TSP-1 resulted in mild left ventricular dilation and modest non-progressive systolic dysfunction. In vitro, TSP-1 incorporation in collagen gels did not activate latent TGF-β, but exerted matrix-preserving actions by inhibiting leptin-induced MMP activation. Assessment of the cardiac vasculature demonstrated that TSP-1 loss attenuated diabetes-associated vascular rarefaction, documenting that TSP-1 upregulation is an important angiostatic mediator in the diabetic myocardium. Our in vivo and in vitro studies demonstrated that TSP-1 induction in the diabetic heart upregulates angiopoietin-2 expression, suggesting a novel TSP-1-mediated angiostatic pathway.
METHODS
(Detailed methodology is provided in the online supplement)
Generation of db/db TSP-1 −/− mice
Animal studies were approved by the Institutional Animal Care and Use Committees at Albert Einstein College of Medicine and at Baylor College of Medicine. Leprdm/+ on a C57BL6J background (db/+) were purchased from Jackson Laboratories (Bar Harbor, ME, USA). The TSP-1 −/− mice (on a C57BL6J background) were obtained from our own colony 16, 15, 23. TSP-1-deficient db/db mice were generated by crossing db/+ mice and TSP-1 null animals. Genotyping was performed through established PCR protocols.
Assessment of adiposity
Lean and fat tissue mass was measured in db/db (male, n=9; female, n=7) and dbTSP mice (male, n=11; female, n=5) at 2mo and 4mo of age by dual-energy x-ray absorptiometry (DEXA).
Assessment of metabolic profile
Animals used for assessment of metabolic parameters in the serum (n=5 per group) were euthanized at 2 months of age and blood was collected by aortic puncture. Plasma glucose, insulin, cholesterol, leptin, adiponectin, triglyceride, free fatty acid (FFA) and glycerol levels were assessed at the Baylor College of Medicine Diabetes and Endocrinology Research Center Core.
Echocardiography
For echocardiographic analysis db/db (n=14) and dbTSP mice (n=19) were imaged at 4, 6, 8 and 12 months of age using a Vevo770 system (Visualsonics, Toronto, Canada).
Assessment of contractile function and reserve in isolated perfused mouse hearts
WT (n=7), db/db (n=10) and dbTSP mice (n=14) were used for functional analysis in an isolated perfused heart system. Hearts were perfused in the Langendorff mode and subjected to dobutamine infusion to assess inotropic and lusitropic responses.
Immunohistochemistry and histology
For histopathological analysis zinc/formalin-fixed, paraffin-embedded sections were obtained from WT, db/db and dbTSP hearts at 6 and 12mo of age. Collagen fibers were identified by Picrosirius Red staining. For visualization of myocardial capillary network, CD31 immunohistochemistry was performed using the rat anti-mouse CD31 antibody (BD Pharmingen) as previously described 24. In order to identify cardiac arterioles, immunohistochemistry with an anti-α-SMA antibody (Sigma) was performed as previously described 24. TSP-1 immunostaining was performed using a mouse monoclonal antibody to TSP-1 (NeoMarkers). Assessment of capillary and arteriolar density was performed. Capillary and arteriolar densities were expressed as vessels /mm2.
RNA extraction and quantitative real-time PCR
Gene expression in hearts and cultured fibroblasts was assessed using quantitative PCR. The sequences of primers used in the study are listed in Online Table I.
Protein extraction and western blotting
Protein was isolated from whole WT, TSP-1 KO, db and dbTSP hearts (n=8/group) and Western blotting was performed as previously described.
Hydroxyproline assay
To assess collagen deposition in WT, db/db and dbTSP hearts (n=8 per group) we used a previously described method 25 that utilizes hydroxyproline content as surrogate measure of collagen.
Zymography
MMP activity in murine WT, db and dbTSP hearts (n=8 per group) was assessed by gelatin zymography as previously described 26.
Isolation and stimulation of cardiac fibroblasts in collagen pads
WT mouse cardiac fibroblasts were isolated by enzymatic digestion as previously described 25. In order to study the effects of matrix-bound TSP-1 on cardiac fibroblasts an in vitro assay of cardiac fibroblasts populating collagen pads in the presence or absence of TSP-1, leptin and active or latent TGF-β was used as previously described 25, 27. At the end of the experiment, gel area was assessed using Image Lab 3.0 software to measure the contractile activity of fibroblasts. Subsequently, each collagen pad was divided in half and used for analysis of MMP activity by gelatin zymography and for gene expression studies by qPCR.
Isolation and stimulation of mouse splenic macrophages
Splenic macrophages were isolated from WT mice using immunomagnetic bead sorting for CD11b as previously described28.
Stimulation of mouse cardiac microvascular endothelial cells (MCMECs)
MCMECs were purchased from Cell Biologics and were stimulated with TSP-1 in order to assess expression of angiogenesis-related genes.
Statistical analysis
Data are expressed as mean ± SEM. Statistical analysis was performed using unpaired, 2-tailed Student’s t test or non-parametric t-test for non-Gaussian distributions (Mann-Whitney) and 1-way ANOVA with Tukey’s multiple comparison test or non-parametric ANOVA (Kruskal-Wallis) for non-Gaussian distributions. Statistical analyses were performed using Prism 6.0 software. P<0.05 was considered to be significant. Mortality was compared using the log rank test.
RESULTS
1. Cardiac fibrosis in db/db mice is associated with TSP-1 upregulation
Obese diabetic db/db mice exhibited cardiac fibrosis. At 2 months of age, db/db animals had higher myocardial collagen I mRNA expression than lean WT animals (Figure 1A). At 6 months of age myocardial collagen content, measured through a hydroxyproline biochemical assay, was significantly higher in db/db animals (Figure 1B). Sirius red staining identified collagen fibers in the myocardium showing expansion of the cardiac interstitium and extensive deposition of collagen in db/db mouse hearts (Figure 1C–D). Cardiac fibrosis in db/db animals was associated with significant TSP-1 mRNA upregulation (Figure 1E). Immunohistochemical staining demonstrated low levels of TSP-1 immunoreactivity in WT mice (Figure 1F); in contrast, db/db animals had intense TSP-1 staining in perivascular and interstitial areas (Figure 1G). TSP-1 KO mice were used as negative controls and had no myocardial staining for TSP-1 (Figure 1H).
Figure 1. Cardiac fibrosis in the db/db myocardium is associated with TSP-1 upregulation.

A–D: db/db mice had significant fibrotic remodeling of the myocardium. qPCR analysis showed marked upregulation of collagen I mRNA in db/db hearts at 2 months of age (A). A hydroxyproline biochemical assay showed that 6-month old db/db mice had significantly higher myocardial collagen content when compared with age-matched WT animals (B). Sirius red staining shows increased deposition of collagen in the cardiac interstitium of db/db animals (D), when compared with lean WT mice (C). E: qPCR shows that TSP-1 mRNA expression was markedly increased in db/db hearts. F–G: Immunohistochemical staining of 6 month-old mouse hearts showed weak immunoreactivity in lean WT mice (F). In contrast, in the db/db myocardium, intense TSP-1 staining was noted (G) and was predominantly localized in perivascular and interstitial areas (arrows). TSP-1 KO mice showed negligible staining for TSP-1 (H) (*p<0.05 vs. WT; **p<0.01 vs. WT). Scale bar: 50μm.
2. Weight gain and adiposity in db/db TSP-1 −/− mice
In order to study the role of TSP-1 in fibrotic remodeling of the db/db heart we generated db/db TSP-1 −/− mice (dbTSP). During a 12 month follow-up, mortality was comparable between db/db and dbTSP mice (Figure 2A). In db/db mice, TSP-1 loss was associated with an increase in body weight at 4 months of age that was statistically significant only for male mice; in contrast, mouse length was comparable between groups (Figure 2B–C). DEXA analysis showed that in db/db mice, TSP-1 disruption did not affect lean weight (Figure 2D), but resulted in a modest increase in total fat weight (Figure 2E) and abdominal fat weight (Figure 2F) in both male and female animals. However, no gender-specific differences in weight and fat gain were noted between 2 and 4 months of age (Online Table II) and the percentage of total and abdominal fat content in db/db animals was not affected by TSP-1 loss (Figure 2G–H).
Figure 2. Effects of TSP-1 loss on survival, weight gain and adiposity of db/db mice.

A. db/db and db/db TSP-1 KO mice had comparable survival curves after 12 months of follow-up. B. TSP-1 loss did not affect mouse size (length). C. TSP-1 loss had subtle gender-specific effects on weight gain in db/db mice. At 4 months of age, male db/db TSP-1 KO animals (dbT) had significantly increased body weight; however, in female mice no statistically significant difference was noted between db/db and db/db TSP-1 −/− animals. D. Lean weight was comparable between groups. E–F. At 4 months of age TSP-1 loss was associated with increased fat weight in male db/db mice and with higher abdominal fat weight in both male and female animals. G–H. However, fat percent content was comparable between db/db and db/db TSP-1 null mice. (*p<0.05 and **p<0.01 vs. corresponding db/db mice).
3. Effects of TSP-1 loss on the metabolic profile in db/db mice
Next, we examined whether TSP-1 loss had an effect on the development of metabolic dysfunction in db/db mice. At 2 months of age, db/db and dbTSP mice had comparable plasma glucose and insulin levels; HOMA IR was also not significantly different between groups (Figure 3A–C). TSP-1 disruption in db/db animals was associated with a significant increase in plasma cholesterol levels (Figure 3D). However, plasma glycerol, FFA and triglycerides were not significantly affected by TSP-1 loss (Figure 3E–G). Plasma levels of the adipokines leptin and adiponectin were also comparable between db/db and dbTSP mice (Figure 3H–I).
Figure 3. Effects of TSP-1 loss on the metabolic profile in db/db mice.

Serum glucose, insulin, HOMA IR, glycerol, triglyceride, FFA, leptin and adiponectin levels were comparable between db/db and dbTSP animals at 2 months of age (A–C, E–I). However, TSP-1 loss was associated with an increase in serum cholesterol levels (D). (*p<0.05 vs. db/db mice).
4. dbTSP mice exhibit non-progressive ventricular dilation accompanied by modest systolic dysfunction
Because cardiac TSP-1 upregulation may play a role in remodeling of the diabetic heart, we examined the effects of TSP-1 loss on function and geometry of the db/db heart. As they age, db/db mice exhibit marked cardiac hypertrophy in the absence of significant systolic dysfunction. At 4–12 months of age, dbTSP mice had increased left ventricular dimensions and volumes when compared with db/db animals (Figure 4A–B, Online Table II), suggesting that TSP-1 absence induces chamber dilation. At 4 months of age, dbTSP mice had mildly depressed systolic function, evidenced by a reduction in ejection fraction (Figure 4C, Online Table II). However, systolic dysfunction in dbTSP mice was not progressive; at later timepoints, ejection fraction was not affected by TSP-1 loss. dbTSP animals also had lower LV mass than db/db mice and reduced anterior and posterior wall thickness, suggesting attenuated cardiac hypertrophy (Figure 4D–F).
Figure 4. TSP-1 loss in db/db mice results in dilative cardiac remodeling and decreased left ventricular wall thickness.

Echocardiographic imaging showed that db/db TSP-1 null mice (dbTSP) had increased Left Ventricular End-Diastolic Diameter (LVEDD) (A) and Left Ventricular End-Diastolic Volume (LVEDV) (B) when compared with db/db animals (db), suggesting increased dilative remodeling (*p<0.05, **p<0.01 vs. age-matched db/db mice; db/db, n=14; dbTSP, n=19). C. At 4 months of age, systolic function was mildly depressed in dbTSP mice; however, loss of TSP-1 did not cause prolonged or progressive systolic ventricular dysfunction. D–F. TSP-1 absence in db/db animals attenuated cardiac hypertrophy leading to reductions in left ventricular mass (D), anterior wall end-diastolic thickness (AWT, E) and posterior wall end-diastolic thickness (PWT, F). G. At 6 months of age, db/db mice had significantly increased left atrial area when compared with WT animals (**p<0.01). TSP-1 loss significantly abrogated the increase in LA size observed in diabetic animals H. Mitral inflow Doppler echocardiography showed that db/db animal had a significant reduction in the E:A ratio suggesting impaired relaxation (*p<0.05 vs. WT). TSP-1 loss did not affect the E:A ratio in lean or obese mice. I–L. Assessment of left ventricular contractile reserve in a Langendorff model demonstrated that at 6 months of age, obese diabetic db/db mice and WT lean animals had no difference in systolic function, assessed through measurements of dp/dtmax (I) and LVDP (J), both at baseline and after infusion of dobutamine. TSP-1 loss did not significantly affect systolic function in db/db mice; only trends towards reduced LVDP and dp/dtmax were observed with low-dose dobutamine infusion. However, TSP-1 loss in db/db mice significantly delayed the peak systolic response after dobutamine infusion (K). L. Diastolic function, measured through assessment of −dp/dtmin was not significantly affected by TSP-1 loss (**p<0.01 vs. baseline −dp/dtmin).
5. When compared with db/db hearts, dbTSP hearts exhibit delayed responses to dobutamine
We studied the effects of TSP-1 loss on contractile reserve in dobutamine-stimulated isolated working hearts. Lean WT and TSP-1 −/− mouse hearts had comparable systolic and diastolic function at baseline and upon stimulation with dobutamine (Online Table III). At 6 months of age db/db mice and dbTSP animals had no evidence of systolic dysfunction; baseline dp/dtmax and LVDP were comparable between WT, db/db and dbTSP hearts (Figure 4G–H). During dobutamine infusion, db/db mice and WT animals had comparable increases in systolic contractile function. Upon infusion of low-dose dobutamine (7.5 ng/min) dbTSP hearts exhibited a trend towards a blunted contractile response, evidenced by lower dp/dtmax and LVDP (Figure 4I–J). Moreover, with high doses of dobutamine infusion (37.5 ng/min-75 ng/min), dbTSP hearts had a marked temporal delay in mounting a peak contractile response (Figure 4K).
6. Effects of TSP-1 loss on the development of diastolic dysfunction in the db/db heart
Diastolic function was compared between WT, db/db and dbTSP mice by assessing left atrial size, by performing mitral inflow Doppler echocardiography and through hemodynamic assessment of the isolated perfused heart. At 6 months of age, db/db mice had increased left atrial size in comparison to WT animals. TSP-1 loss in db/db animals was associated with a reduction in left atrial dimensions (Figure 4G). Mitral inflow Doppler echocardiography showed that, when compared with WT mice, db/db animals had a reduced E:A ratio; however, TSP-1 loss did not significantly affect the E:A ratio in db/db mice (Figure 4H). Assessment of diastolic function in isolated perfused hearts showed that −dp/dtmax was comparable between db/db and dbTSP animals (Figure 4L). Thus, TSP-1 absence was not associated with consistent evidence of altered diastolic function in diabetic animals.
7. dbTSP mice exhibit reduced myocardial collagen content and increased MMP activation
Next, we explored the basis for dilative remodeling in dbTSP hearts. Because the extracellular matrix plays a key role in determining cardiac geometry, we studied the effects of TSP-1 loss on cardiac matrix metabolism. Using a hydroxyproline assay we demonstrated that db/db hearts have increased myocardial collagen deposition; TSP-1 loss significantly reduced collagen content in db/db animals (Figure 5A). Reduced collagen content in dbTSP hearts was not due to lower collagen mRNA synthesis (Figure 5B), but was associated with marked increases in MMP activity. dbTSP mice had a 2-fold increase in MMP-2 activity and a 5-fold increase in MMP-9 activity when compared with db/db animals (Figure 5C–E). Increased protease activity in the absence of TSP-1 was not associated with increased MMP mRNA or protein levels. qPCR showed that myocardial MMP-2 and MMP-9 mRNA levels were comparable between db/db and dbTSP mice (Figure 5F–G). Moreover, western blotting demonstrated that db/db and dbTSP mice had comparable myocardial MMP-2 and MMP-9 protein levels (Online Figure I).
Figure 5. Dilative remodeling of the TSP-1-deficient db/db heart is associated with decreased collagen content and enhanced MMP activity.

A. Assessment of collagen content using a hydroxyproline biochemical assay demonstrated that TSP-1 loss significantly reduced collagen content in 6 month-old db/db animals (*p<0.05 vs. db/db; db/db, n=8/group). B. Reduced collagen content was not associated with attenuated collagen mRNA levels; myocardial collagen I mRNA levels were higher in dbTSP hearts. C–E. Zymography showed that TSP-1 absence resulted in significantly increased MMP-2 and MMP-9 activity in db/db animals (*p<0.05 vs. WT, **p<0.01 vs. WT; ^^p<0.01 vs. db/db). F–G. Enhanced MMP activity in dbTSP hearts was not associated with increased MMP-2 or MMP-9 mRNA levels.
8. Dilation and increased matrix remodeling in dbTSP mice is not due to impaired TGF-β activation
TGF-β promotes matrix preservation and critically regulates cardiac remodeling 29. Because TSP-1 is an essential activator of TGF-β, we hypothesized that the effects of TSP-1 disruption in db/db hearts may be due to impaired TGF-β signaling. When compared with WT animals, db/db mice had significantly increased myocardial expression of p-Smad2 and p-Smad3, suggesting activation of the TGF-β/Smad signaling pathway. p-Smad2 and p-Smad3 expression was comparable between db/db and dbTSP mice (Figure 6A–C), suggesting that TSP-1 loss does not affect activation of canonical TGF-β signaling. Moreover, db/db and dbTSP mice had comparable expression of TGF-β1 mRNA in the myocardium (Figure 6D).
Figure 6. TSP-1 deficiency does not affect myocardial inflammation and activation of TGF-β/Smad signaling in the db/db myocardium.
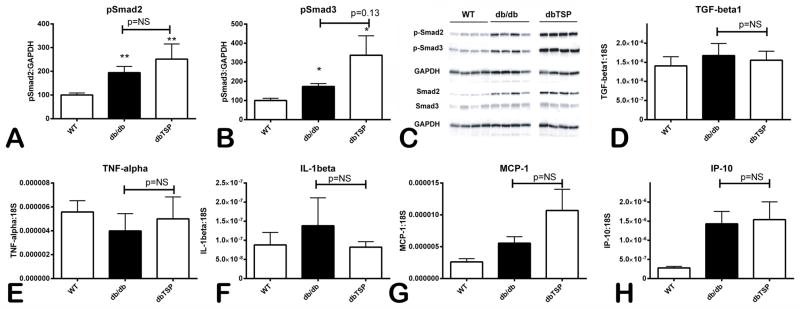
A–C. Western blotting showed that myocardial expression of phosphorylated Smad2 (A) and phosphorylated Smad3 (B) was significantly increased in db/db animals when compared with lean WT mice. db/db mice lacking TSP-1 (dbTSP) had comparable myocardial p-Smad2 levels and showed a trend towards increased pSmad3 expression when compared with db/db mice. Moreover, db/db and dbTSP mice had comparable myocardial mRNA expression of the cytokines TGF-β (D), TNF-α (E), IL-1β (F) and the chemokines MCP-1/CCL2 (G) and IP-10/CXCL10 (H) (n=8 mice per group).
9. Dilation of the dbTSP heart is not due to accentuated inflammation
Pro-inflammatory mediators, such as IL-1β and TNF-α, mediate cardiac dilation and dysfunction by inducing and activating MMPs. Because TSP-1 downmodulates pro-inflammatory responses, we examined whether dilation and accentuated matrix metabolism in dbTSP hearts is due to increased inflammatory activity. Myocardial expression of the pro-inflammatory cytokines TNF-α and IL-1β (Figure 6E–F), and of the chemokines MCP-1 and IP-10 (Figure 6G–H) was comparable between db/db and dbTSP mice suggesting that TSP-1 loss does not affect inflammatory activity in db/db mice.
10. Cardiac fibroblasts harvested from dbTSP mice exhibit attenuated matrix-synthetic capacity
In order to explore the basis for accentuated matrix degrading activity in the absence of TSP-1, we isolated cardiac fibroblasts from db/db and dbTSP hearts and compared baseline expression of genes associated with matrix synthesis and metabolism. dbTSP fibroblasts had reduced collagen I and III mRNA expression and showed trends for increased expression of MMP-2 and MMP-3 mRNA (Figure 7A–D). TIMP-1 and -2 mRNA expression was comparable between groups (Figure 7E). Altered expression of genes associated with matrix metabolism in dbTSP cardiac fibroblasts was not associated with impaired activation of TGF-β/Smad signaling. Western blotting demonstrated that p-Smad2 expression was comparable between fibroblasts isolated from db/db and dbTSP hearts (Figure 7F).
Figure 7. Matrix-preserving effects of TSP-1 in isolated cardiac fibroblasts.

Cardiac fibroblasts isolated from db/db TSP-1 null hearts had reduced collagen I (A) and III (B) mRNA expression and showed trends towards increased MMP-2 (C) and MMP-3 (D) synthesis when compared with fibroblasts harvested from db/db hearts. In contrast, TIMP-1 mRNA synthesis (E) and expression of p-Smad2 protein (F) was comparable between db/db and dbTSP fibroblasts. G. In fibroblasts populating collagen pads incorporation of TSP-1 into the matrix did not activate TGF-β responses. Active TGF-β (aTGF) induced marked gel contraction in fibroblast-populated pads, reflecting myofibroblast transdifferentiation. In contrast, TSP-1 incorporation into the pad failed to increase the effects of latent TGF-β (lTGFbeta) on gel contraction suggesting that, in this model, TSP-1 cannot activate TGF-β. H. Leptin stimulation modestly, but significantly, increased gel contraction in fibroblast-populated pads. TSP-1 did not affect the effects of leptin on gel contraction. I. In fibroblast-populated collagen pads, TSP-1 incorporation into the matrix abrogated the stimulatory effects of leptin on MMP-2 activity. J–L. The inhibitory actions of TSP-1 on MMP-2 activation were not due to modulation of MMP-2 (J), MT1-MMP (K) and TIMP-2 (L) synthesis (n=6 per experimental condition).
11. When bound to the matrix, TSP-1 does not activate TGF-β responses, but enhances matrix preservation, inhibiting leptin-induced MMP activation
As a matricellular protein TSP-1 modulates cellular phenotype when bound to the structural matrix. Accordingly, we tested the hypothesis that incorporation of TSP-1 into the matrix may induce a matrix-preserving phenotype in cardiac fibroblasts using a cell biological assay of fibroblast-populated collagen pads in the presence or absence of TSP-1. Because TSP-1 is considered an essential activator of TGF-β, we examined whether incorporation of TSP-1 into the matrix promotes TGF-β-mediated actions in fibroblasts stimulated with latent TGF-β. Active, but not latent, TGF-β induced gel contraction in fibroblast-populated collagen pads (Figure 7G), reflecting the prominent effects of the growth factor on myofibroblast transdifferentiation. Incorporation of TSP-1 into the matrix had no effect on gel contraction, both in the presence or absence of latent TGF-β (Figure 7G), suggesting that TSP-1 exerts no significant TGF-β–activating effects in this model. On the other hand, leptin stimulation induced modest, but significant, gel contraction in the presence, or absence, of TSP-1 (Figure 7H). Zymography demonstrated that leptin markedly enhanced MMP-2 activity in fibroblast-populated collagen pads (Figure 7I). When incorporated into the matrix, TSP-1 attenuated the matrix-degrading effects of leptin. The effects of leptin and TSP-1 on MMP-2 activity were not due to modulation of MMP-2 mRNA levels (Figure 7J). Because MT1-MMP and TIMP-2 are implicated in MMP-2 activation we examined whether they are involved in mediating the inhibitory effects of TSP-1 on MMP-2 activity. We found that leptin and TSP-1 had no effects on fibroblast-derived MT1-MMP and TIMP-2 mRNA synthesis (Figure 7K–L).
12. TSP-1 loss abrogates capillary, but not arteriolar, rarefaction in db/db mice
Through its potent angiostatic effects, TSP-1 may be responsible for impaired angiogenesis in the diabetic heart. In order to study the effects of TSP-1 loss on the microvascular network in diabetic hearts we compared vascular density between db/db and dbTSP animals. At 6 and 12 months of age, db/db mice exhibited significantly lower capillary and arteriolar density when compared with WT animals (Figure 8A–B). TSP-1 disruption attenuated capillary rarefaction in db/db animals, but had no significant effect on arteriolar density (Figure 8A–E).
Figure 8. TSP-1 upregulates angiopoietin-2 and mediates capillary rarefaction in the db/db myocardium.

A–C. Myocardial capillaries were identified using staining with the endothelial cell marker CD31. Representative images of stained sections from 6mo-old WT (A), db/db (B) and dbTSP (C) hearts are shown. D. Quantitative analysis demonstrated that db/db hearts exhibited significant reduction in vascular density at 6 and 12 months of age (^^p<0.01 vs. corresponding WT mice). dbTSP mice were partially protected from microvascular loss (**p<0.01 vs. db/db). E. Myocardial arterioles were identified using α-SMA staining. Quantitative analysis showed that db/db mice had reduced myocardial arteriolar density when compared with WT animals (*p<0.05 vs. WT). However, TSP-1 absence did not affect the progression of arteriolar loss in db/db mice (6 mo group: WT, n=7; db/db, n=12; dbTSP, n=5 – 12 mo group: WT, n=10; db/db, n=12; dbTSP, n=5). F–G: TSP-1 mediates angiopoietin-2 upregulation in the db/db heart. Western blotting was used to measure myocardial angiopoietin-2 expression levels in 6 month-old (F) and 12-month-old (G) animals. At 12 months of age, db/db mice exhibited marked myocardial angiopoietin-2 upregulation (*p<0.05 vs. WT). db/db mice lacking TSP-1 had significantly reduced myocardial angiopoietin-2 expression when compared with age-matched db/db animals (^p<0.05, ^^p<0.01 vs. db/db). H. In splenic macrophages TSP-1 stimulation for 4h significantly upregulated angiopoietin-2 mRNA synthesis (*p<0.05 vs. control n=6). I. Cardiac fibroblasts harvested from db/db mice had significantly increased angiopoietin-2 mRNA expression levels; TSP-1 loss abrogated the increase in angiopoietin-2 synthesis (**p<0.01 vs. WT; ^p<0.05 vs. db/db, n=6). Scalebar: 40μm.
13. TSP-1 mediates myocardial angiopoietin-2 upregulation in db/db mice
Angiogenesis is regulated through dynamic interactions between Vascular Endothelial Growth Factor (VEGF) and angiopoietins30. Because diabetes is associated with overexpression of angiopoietin-2, a mediator that promotes vascular regression31, 32 we examined whether attenuation of capillary rarefaction in dbTSP mice is associated with reduced angiopoietin-2 expression. Western blotting showed that 12 month-old db/db mice had significantly higher myocardial angiopoietin-2 expression than WT animals (Figure 8G). TSP-1 absence markedly reduced myocardial Angpt-2 expression in 6 and 12-month old animals (Figure 8F–G). In contrast, TSP-1 loss did not affect myocardial VEGF protein expression (Online Figure II).
14. TSP-1 stimulation upregulates angiopoietin-2 expression in macrophages
We next examined whether TSP-1 directly induces angiopoietin-2 expression. In isolated mouse macrophages TSP-1 stimulation increased angiopoietin-2 mRNA synthesis (Figure 8H). Moreover, cardiac fibroblasts harvested from db/db mice showed increased angiopoietin-2 mRNA expression; the increase in angiopoietin-2 levels was abrogated in cells isolated from dbTSP hearts (Figure 8I). In contrast, TSP-1 had no effect on macrophage-derived VEGF-A mRNA synthesis and TSP-1 loss did not affect VEGF-A mRNA expression by db/db cardiac fibroblasts. Mouse cardiac microvascular endothelial cells constitutively expressed VEGF-A mRNA, but did not express angiopoietin-2 in the presence or absence of TSP-1 (Online Figure III).
DISCUSSION
Our study provides the first direct evidence supporting the role of the prototypical matricellular protein TSP-1 in matrix remodeling and regulation of angiogenesis in the diabetic heart. We report several novel observations: i) Diabetic cardiac fibrosis is associated with marked TSP-1 upregulation and deposition in the cardiac interstitium. ii) In the diabetic heart TSP-1 exerts matrix-preserving actions preventing dilative remodeling. iii) The matrix-preserving effects of TSP-1 appear to be due to direct actions on MMP activation and not to activation of TGF-β signaling. iv) TSP-1 induction in the diabetic heart upregulates angiopoietin-2 expression, exerting potent angiostatic actions and mediating rarefaction of the capillary network.
The development of interstitial and perivascular fibrosis is a consistent characteristic of diabetic cardiomyopathy6. In non-hypertensive patients with familial diabetes, increased collagen deposition was noted in the cardiac perivascular and interstitial space33. Fibrotic cardiac remodeling in diabetic subjects has important functional implications and may be involved in the pathogenesis of diastolic dysfunction. In patients with early type 2 diabetes, collagen synthesis, assessed through measurement of serum levels of the carboxy-terminal propeptide of procollagen type I, correlated with the severity of diastolic impairment34. Animal models of type 2 diabetes also exhibit significant cardiac fibrosis. Using qPCR, histochemical staining and a hydroxyproline biochemical assay, we found that obese diabetic db/db mice exhibit a marked increase in myocardial collagen content (Figure 1). Collagen deposition in the db/db heart is associated with marked TSP-1 upregulation. We localized TSP-1 immunoreactivity in vascular cells and in perivascular areas suggesting that endothelial cells and pericytes may be important sources of TSP-1 in the diabetic myocardium (Figure 1). Hyperglycemia is a potent inducer of TSP-1 in both endothelial and vascular smooth muscle cells35, and may mediate TSP-1 upregulation in the db/db heart. Glucose-mediated activation of the hexosamine pathway induces TSP-1 upregulation in vascular smooth muscle cells36. A similar mechanism for glucose-induced TSP-1 upregulation is operative in endothelial cells37; however, the transcription factors activated by high glucose appear to be cell-type specific35.
What is the function of TSP-1 in the diabetic myocardium? Systematic assessment of cardiac dimensions and function in db/db TSP-1 null mice demonstrated that TSP-1 plays an important role in preservation of the geometry of the diabetic heart. As they age, db/db mice develop cardiac hypertrophy with preserved left ventricular chamber dimensions. TSP-1 loss in db/db mice was associated with a 20% increase in LVEDV, reflecting significant dilative remodeling of the ventricle. Dilation of the left ventricle in dbTSP mice was associated with modest, non-progressive systolic dysfunction and impairment of contractile reserve (Figure 4). Dilation and dysfunction of the dbTSP heart was associated with significant loss of myocardial collagen and increased MMP activity (Figure 5). Taken together these findings suggest that loss of TSP-1 deprives the db/db myocardium from an important matrix-preserving signal leading to geometric remodeling of the ventricle.
TSP-1 may exert matrix-preserving actions through several distinct pathways. First, as a potent activator of TGF-β38, TSP-1 may enhance growth factor activity in the cardiac interstitium, promoting matrix deposition and increasing expression of protease inhibitors, such as TIMP-1. In order to test this hypothesis we measured activation of the pro-fibrotic, canonical TGF-β/Smad pathway by assessing Smad2/3 phosphorylation in the myocardium. db/db mice had markedly increased myocardial p-Smad2 and p-Smad3 expression; however, TSP-1 disruption did not affect TGF-β/Smad activation. Moreover, in an in vitro model using collagen pads populated with cardiac fibroblasts, incorporation of TSP-1 in the collagenous matrix did not stimulate gel contraction and did not upregulate TGF-β-inducible genes in cells stimulated with latent TGF-β. These observations suggested that the matrix-preserving actions of TSP-1 in the remodeling diabetic heart are not due to TGF-β activating effects. Second, TSP-1 may preserve the matrix by attenuating the inflammatory response, thus preventing cytokine-induced protease synthesis and activation. However, TSP-1 absence in db/db mice did not affect myocardial cytokine and chemokine synthesis suggesting that overactive inflammation is not responsible for the increased chamber dilation in db/db TSP-1 knockout hearts. Third, TSP-1 may directly inhibit MMP activity39. TSP-1 loss in db/db mice was associated with a 2-fold increase in myocardial MMP-2 activity and a 5-fold increase in MMP-9 activity (Figure 5). In vitro, TSP-1 incorporation into the matrix inhibited leptin-induced MMP-2 activation; the inhibitory effects of TSP-1 on MMP activity were not due to transcriptional regulation of MMPs (Figure 6). Thus, our in vivo and in vitro observations suggest that the matrix-preserving actions of TSP-1 in the diabetic heart are likely due to direct inhibition of protease activity and may not involve activation of growth factors that stimulate matrix synthesis, such as TGF-β.
In addition to its effects in matrix metabolism, TSP-1 is also involved in regulation of myocardial angiogenesis in diabetic mice. In both human patients and in experimental animal models, diabetes is associated with impaired angiogenesis. Diabetic patients exhibit attenuated angiogenic responses to myocardial ischemia40 and have poor development of collateral coronary vessels41. Moreover, microvascular rarefaction is a characteristic of the diabetic heart and may be responsible for development of contractile dysfunction and for increased susceptibility to myocardial ischemia42,43. The molecular signals responsible for impaired angiogenesis in diabetic subjects remain poorly understood; animal model studies and investigations in human patients have produced somewhat conflicting results. In diabetic animals, expression of angiogenic growth factors (such as HIF-1α and VEGF) was reduced, suggesting that diabetes-associated impairment in the angiogenic response may be due to attenuation of growth factor synthesis40. In contrast, in human patients with coronary disease and type 2 diabetes, myocardial VEGF levels were increased; however, VEGF receptor phosphorylation was reduced and downstream myocardial VEGF signaling was impaired contributing to reduced angiogenesis44. A potential mechanism of impaired angiogenesis in diabetes may involve activation of angiostatic pathways. Our findings suggest that upregulation of the angiostatic matricellular protein TSP-1 is in part responsible for the age-related rarefaction of the cardiac microvascular network in db/db hearts (Figure 8). Published evidence suggests that the antiangiogenic effects of TSP-1 may involve several distinct biological processes45. First, TSP-1 may antagonize VEGF signaling through direct binding and inhibition of its release from the matrix and via inhibition of VEGF receptor phosphorylation46. Second, TSP-1 may induce endothelial cell apoptosis through a CD36/fyn-mediated pathway19. Third, TSP-1 may directly suppress endothelial cell cycle progression in a CD36-independent manner, or inhibit endothelial cell migration through interactions involving β1 integrins47. Fourth, TSP-1-induced suppression of nitric oxide signaling may also be involved in activation of angiostatic signaling48 in endothelial cells. Our experiments identified angiopoietin-2 induction, as a new mechanism that may be implicated in the angiostatic actions of TSP-1 in the diabetic myocardium. Angiopoietin-2 potently regulates angiogenesis in a context-dependent manner. Ang-2 promotes endothelial cell death if endogenous VEGF activity is inhibited49 and promotes vascular regression by inhibiting pericyte recruitment50. We show that angiopoietin-2 protein is upregulated in db/db hearts; loss of TSP-1 is associated with marked reduction of angiopoietin-2 levels (Figure 8). Thus, the angiostatic effects of TSP-1 in the diabetic myocardium may be in part due to upregulation of angiopoietin-2 expression, in an environment where VEGF signaling is also attenuated (Online Figure II). Our in vitro experiments demonstrated cell-specific stimulatory effects of TSP-1 on angiopoietin-2 expression. TSP-1 induced angiopoietin-2 synthesis in macrophages, but not in cardiac microvascular endothelial cells (Figure 8).
In addition to its role in tissue remodeling, angiogenesis and fibrosis, TSP-1 has been recently identified as an adipokine that is highly expressed in fat tissue harvested from obese insulin-resistant subjects and is associated with adipose inflammation21. Our recent work demonstrated that TSP-1 null mice have attenuated fat accumulation and reduced weight gain when fed a high-fat diet or a high-carbohydrate low-fat diet51 and suggested effects of TSP-1 in promoting adipose tissue inflammation and adipocyte proliferation. Li and co-workers also showed that TSP-1 null mice fed a high fat diet exhibit reduced adipose inflammation and improved insulin sensitivity52. In contrast, Olerud and coworkers found that TSP-1 null mice were glucose intolerant due to islet dysfunction; reconstitution of the TGF-βactivating effects of TSP-1 prevented the development of insulin resistance in TSP-1 −/− mice 53. In our current study, db/db mice that lacked TSP-1 had only subtle alterations in adipose tissue gain and in metabolic function when compared with db/db animals. The percentage of total and abdominal fat, glucose, insulin levels and HOMA IR were not significantly affected by TSP-1 loss; however db/db TSP-1 KO mice had higher serum cholesterol levels than db/db animals. The differences in the findings between investigations may reflect the distinct mechanisms of obesity in various experimental models. db/db mice develop morbid obesity and severe metabolic dysfunction due to central nervous system leptin resistance that results in hyperphagia and rapid weight gain; the very high caloric intake in these animals may induce metabolic dysfunction regardless of the presence or absence of TSP-1. An alternative possibility is that any effects of TSP-1 on metabolic function may require intact leptin signaling.
Conclusions
TSP-1 does not play a significant role in weight gain and metabolic dysfunction in db/db mice, but is highly induced in the db/db myocardium where it exerts matrix-preserving and angiostatic actions. TSP-1-mediated matrix preservation protects the diabetic heart from dilation by locally inhibiting MMP activity; however, dilative remodeling in the absence of TSP-1 is not progressive and is associated with mild systolic dysfunction. In addition to its effects in maintaining chamber geometry, TSP-1 exerts potent angiostatic actions mediating vascular rarefaction in the aging diabetic heart. Because the anti-angiogenic effects of TSP-1 are mediated through binding of specific angiostatic domains with CD36 or CD4754,45, selective inhibition of the angiostatic effects of TSP-1 may be a promising approach to correct impaired angiogenic responses in the diabetic myocardium.
Supplementary Material
NOVELTY AND SIGNIFICANCE.
What is known?
Diabetics develop diastolic heart failure associated with a fibrotic cardiomyopathy and exhibit impaired myocardial angiogenesis.
Matricellular proteins are critically involved in fibrotic and angiogenic responses by modulating growth factor and cytokine signaling.
The matricellular protein Thrombospondin-1 is markedly upregulated by hyperglycemia and may play an important role in remodeling of the diabetic heart by modulating growth factor signaling, by inhibiting protease activation and by exerting angiostatic actions.
What new information does this article contribute?
Obese diabetic db/db mice exhibit marked myocardial upregulation of Thrombospondin-1, localized in interstitial and perivascular areas.
Thrombospondin-1 upregulation in the diabetic myocardium increases myocardial collagen content, but also preserves chamber geometry and systolic function in the diabetic heart by inhibiting matrix metalloproteinase activation.
Thrombospondin-1 induction in the diabetic heart mediates age-related capillary rarefaction by upregulating expression of angiopoietin-2.
The matricellular protein Thrombospondin (TSP)-1 is markedly induced in diabetes and may be implicated in fibrotic remodeling of diabetic hearts. Moreover, through its potent angiostatic actions, TSP-1 may be responsible for impaired angiogenesis in the diabetic myocardium. Obese diabetic db/db mice exhibit marked TSP-1 upregulation in the myocardium. In order to investigate the role of TSP-1 in diabetic cardiomyopathy we generated db/db TSP-1 null mice. Our findings show for the first time that: a) loss of TSP-1 in db/db mice does not significantly affect weight gain and metabolic function, b) TSP-1 disruption in db/db animals results in chamber enlargement and modest systolic dysfunction, associated with reduced collagen content and enhanced matrix metalloproteinase activity in the myocardium, c) The effects of TSP-1 on the matrix are not due to activation of Transforming Growth Factor-β responses, but may reflect inhibition of Matrix Metalloproteinase activity, d) TSP-1 mediates age-associated capillary rarefaction in the diabetic heart by upregulating expression of angiopoietin-2, a mediator that induces vascular regression. Our findings identify TSP-1-mediated angiopoietin-2 induction as a crucial mechanism responsible for impaired preservation of the cardiac microvascular network in diabetes. Inhibition of the angiostatic actions of TSP-1 may have therapeutic potential in diabetic heart disease.
Acknowledgments
FUNDING SOURCES: Supported by NIH grants R01HL76246, R01HL85440, T32HL007675 and the Wilf Family Cardiovascular Research Institute.
NON-STANDARD ABBREVIATIONS AND ACRONYMS
- TSP-1
thrombospondin-1
- TGF-β
transforming growth factor-β
- MMP
matrix metalloproteinase
- MAPK
mitogen-activated protein kinase
- DEXA
dual energy X-ray absorptiometry
- FFA
free fatty acids
- α-SMA
α-smooth muscle actin
- LVDP
left ventricular developed pressure
- TNF-α
tumor necrosis factor-α
- IL-1β
interleukin-1β
- MCP-1
monocyte chemoattractant protein-1
- IP-10
interferon-γ-inducible protein-10
- Smad
small mothers against decapendaplegic
- MT1-MMP
membrane type matrix metalloproteinase-1
- TIMP
tissue inhibitor of metalloproteinases
- VEGF
vascular endothelial growth factor
- HIF
hypoxia-inducible factor
Footnotes
DISCLOSURES: None.
References
- 1.Marwick TH. Diabetic heart disease. Heart. 2006;92:296–300. doi: 10.1136/hrt.2005.067231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Abel ED, Litwin SE, Sweeney G. Cardiac remodeling in obesity. Physiol Rev. 2008;88:389–419. doi: 10.1152/physrev.00017.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kannel WB, Hjortland M, Castelli WP. Role of diabetes in congestive heart failure: the Framingham study. Am J Cardiol. 1974;34:29–34. doi: 10.1016/0002-9149(74)90089-7. [DOI] [PubMed] [Google Scholar]
- 4.Rubler S, Dlugash J, Yuceoglu YZ, Kumral T, Branwood AW, Grishman A. New type of cardiomyopathy associated with diabetic glomerulosclerosis. Am J Cardiol. 1972;30:595–602. doi: 10.1016/0002-9149(72)90595-4. [DOI] [PubMed] [Google Scholar]
- 5.Asbun J, Villarreal FJ. The pathogenesis of myocardial fibrosis in the setting of diabetic cardiomyopathy. J Am Coll Cardiol. 2006;47:693–700. doi: 10.1016/j.jacc.2005.09.050. [DOI] [PubMed] [Google Scholar]
- 6.Boudina S, Abel ED. Diabetic cardiomyopathy, causes and effects. Rev Endocr Metab Disord. 2010;11:31–39. doi: 10.1007/s11154-010-9131-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fischer VW, Barner HB, Larose LS. Pathomorphologic aspects of muscular tissue in diabetes mellitus. Hum Pathol. 1984;15:1127–1136. doi: 10.1016/s0046-8177(84)80307-x. [DOI] [PubMed] [Google Scholar]
- 8.Law B, Fowlkes V, Goldsmith JG, Carver W, Goldsmith EC. Diabetes-induced alterations in the extracellular matrix and their impact on myocardial function. Microsc Microanal. 2012;18:22–34. doi: 10.1017/S1431927611012256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Uusitupa MI, Mustonen JN, Airaksinen KE. Diabetic heart muscle disease. Ann Med. 1990;22:377–386. doi: 10.3109/07853899009147274. [DOI] [PubMed] [Google Scholar]
- 10.Bornstein P. Matricellular proteins: an overview. J Cell Commun Signal. 2009;3:163–165. doi: 10.1007/s12079-009-0069-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schellings MW, Pinto YM, Heymans S. Matricellular proteins in the heart: possible role during stress and remodeling. Cardiovasc Res. 2004;64:24–31. doi: 10.1016/j.cardiores.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 12.Frangogiannis NG. Matricellular proteins in cardiac adaptation and disease. Physiol Rev. 2012;92:635–688. doi: 10.1152/physrev.00008.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Goldsmith EC, Bradshaw AD, Spinale FG. Contributory Pathways Leading to Myocardial Fibrosis - Moving Beyond Collagen Expression. Am J Physiol Cell Physiol. 2012 doi: 10.1152/ajpcell.00347.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jugdutt BI, Jelani A, Palaniyappan A, Idikio H, Uweira RE, Menon V, Jugdutt CE. Aging-related early changes in markers of ventricular and matrix remodeling after reperfused ST-segment elevation myocardial infarction in the canine model: effect of early therapy with an angiotensin II type 1 receptor blocker. Circulation. 2010;122:341–351. doi: 10.1161/CIRCULATIONAHA.110.948190. [DOI] [PubMed] [Google Scholar]
- 15.Frangogiannis NG, Ren G, Dewald O, Zymek P, Haudek S, Koerting A, Winkelmann K, Michael LH, Lawler J, Entman ML. The critical role of endogenous Thrombospondin (TSP)-1 in preventing expansion of healing myocardial infarcts. Circulation. 2005;111:2935–2942. doi: 10.1161/CIRCULATIONAHA.104.510354. [DOI] [PubMed] [Google Scholar]
- 16.Xia Y, Dobaczewski M, Gonzalez-Quesada C, Chen W, Biernacka A, Li N, Lee DW, Frangogiannis NG. Endogenous thrombospondin 1 protects the pressure-overloaded myocardium by modulating fibroblast phenotype and matrix metabolism. Hypertension. 2011;58:902–911. doi: 10.1161/HYPERTENSIONAHA.111.175323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ren B, Yee KO, Lawler J, Khosravi-Far R. Regulation of tumor angiogenesis by thrombospondin-1. Biochim Biophys Acta. 2006;1765:178–188. doi: 10.1016/j.bbcan.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 18.Iruela-Arispe ML, Lombardo M, Krutzsch HC, Lawler J, Roberts DD. Inhibition of angiogenesis by thrombospondin-1 is mediated by 2 independent regions within the type 1 repeats. Circulation. 1999;100:1423–1431. doi: 10.1161/01.cir.100.13.1423. [DOI] [PubMed] [Google Scholar]
- 19.Jimenez B, Volpert OV, Crawford SE, Febbraio M, Silverstein RL, Bouck N. Signals leading to apoptosis-dependent inhibition of neovascularization by thrombospondin-1. Nat Med. 2000;6:41–48. doi: 10.1038/71517. [DOI] [PubMed] [Google Scholar]
- 20.Stenina OI, Krukovets I, Wang K, Zhou Z, Forudi F, Penn MS, Topol EJ, Plow EF. Increased expression of thrombospondin-1 in vessel wall of diabetic Zucker rat. Circulation. 2003;107:3209–3215. doi: 10.1161/01.CIR.0000074223.56882.97. [DOI] [PubMed] [Google Scholar]
- 21.Varma V, Yao-Borengasser A, Bodles AM, Rasouli N, Phanavanh B, Nolen GT, Kern EM, Nagarajan R, Spencer HJ, 3rd, Lee MJ, Fried SK, McGehee RE, Jr, Peterson CA, Kern PA. Thrombospondin-1 is an adipokine associated with obesity, adipose inflammation, and insulin resistance. Diabetes. 2008;57:432–439. doi: 10.2337/db07-0840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Park CW, Kim HW, Lim JH, Yoo KD, Chung S, Shin SJ, Chung HW, Lee SJ, Chae CB, Kim YS, Chang YS. Vascular endothelial growth factor inhibition by dRK6 causes endothelial apoptosis, fibrosis, and inflammation in the heart via the Akt/eNOS axis in db/db mice. Diabetes. 2009;58:2666–2676. doi: 10.2337/db09-0136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lawler J, Sunday M, Thibert V, Duquette M, George EL, Rayburn H, Hynes RO. Thrombospondin-1 is required for normal murine pulmonary homeostasis and its absence causes pneumonia. J Clin Invest. 1998;101:982–992. doi: 10.1172/JCI1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zymek P, Bujak M, Chatila K, Cieslak A, Thakker G, Entman ML, Frangogiannis NG. The role of platelet-derived growth factor signaling in healing myocardial infarcts. J Am Coll Cardiol. 2006;48:2315–2323. doi: 10.1016/j.jacc.2006.07.060. [DOI] [PubMed] [Google Scholar]
- 25.Dobaczewski M, Bujak M, Li N, Gonzalez-Quesada C, Mendoza LH, Wang XF, Frangogiannis NG. Smad3 signaling critically regulates fibroblast phenotype and function in healing myocardial infarction. Circ Res. 2010;107:418–428. doi: 10.1161/CIRCRESAHA.109.216101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dobaczewski M, Xia Y, Bujak M, Gonzalez-Quesada C, Frangogiannis NG. CCR5 signaling suppresses inflammation and reduces adverse remodeling of the infarcted heart, mediating recruitment of regulatory T cells. Am J Pathol. 2010;176:2177–2187. doi: 10.2353/ajpath.2010.090759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bujak M, Dobaczewski M, Gonzalez-Quesada C, Xia Y, Leucker T, Zymek P, Veeranna V, Tager AM, Luster AD, Frangogiannis NG. Induction of the CXC chemokine interferon-gamma-inducible protein 10 regulates the reparative response following myocardial infarction. Circ Res. 2009;105:973–983. doi: 10.1161/CIRCRESAHA.109.199471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen W, Saxena A, Li N, Sun J, Gupta A, Lee DW, Tian Q, Dobaczewski M, Frangogiannis NG. Endogenous IRAK-M attenuates postinfarction remodeling through effects on macrophages and fibroblasts. Arterioscler Thromb Vasc Biol. 2012;32:2598–2608. doi: 10.1161/ATVBAHA.112.300310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dobaczewski M, Chen W, Frangogiannis NG. Transforming growth factor (TGF)-beta signaling in cardiac remodeling. J Mol Cell Cardiol. 2011;51:600–606. doi: 10.1016/j.yjmcc.2010.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Holash J, Maisonpierre PC, Compton D, Boland P, Alexander CR, Zagzag D, Yancopoulos GD, Wiegand SJ. Vessel cooption, regression, and growth in tumors mediated by angiopoietins and VEGF. Science. 1999;284:1994–1998. doi: 10.1126/science.284.5422.1994. [DOI] [PubMed] [Google Scholar]
- 31.Feng Y, vom Hagen F, Pfister F, Djokic S, Hoffmann S, Back W, Wagner P, Lin J, Deutsch U, Hammes HP. Impaired pericyte recruitment and abnormal retinal angiogenesis as a result of angiopoietin-2 overexpression. Thromb Haemost. 2007;97:99–108. [PubMed] [Google Scholar]
- 32.Cao Y, Sonveaux P, Liu S, Zhao Y, Mi J, Clary BM, Li CY, Kontos CD, Dewhirst MW. Systemic overexpression of angiopoietin-2 promotes tumor microvessel regression and inhibits angiogenesis and tumor growth. Cancer Res. 2007;67:3835–3844. doi: 10.1158/0008-5472.CAN-06-4056. [DOI] [PubMed] [Google Scholar]
- 33.Regan TJ, Lyons MM, Ahmed SS, Levinson GE, Oldewurtel HA, Ahmad MR, Haider B. Evidence for cardiomyopathy in familial diabetes mellitus. J Clin Invest. 1977;60:884–899. doi: 10.1172/JCI108843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ihm SH, Youn HJ, Shin DI, Jang SW, Park CS, Kim PJ, Kim HY, Chang K, Seung KB, Kim JH, Choi KB. Serum carboxy-terminal propeptide of type I procollagen (PIP) is a marker of diastolic dysfunction in patients with early type 2 diabetes mellitus. Int J Cardiol. 2007;122:e36–38. doi: 10.1016/j.ijcard.2007.07.057. [DOI] [PubMed] [Google Scholar]
- 35.Raman P, Harry C, Weber M, Krukovets I, Stenina OI. A novel transcriptional mechanism of cell type-specific regulation of vascular gene expression by glucose. Arterioscler Thromb Vasc Biol. 2011;31:634–642. doi: 10.1161/ATVBAHA.110.219675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Raman P, Krukovets I, Marinic TE, Bornstein P, Stenina OI. Glycosylation mediates up-regulation of a potent antiangiogenic and proatherogenic protein, thrombospondin-1, by glucose in vascular smooth muscle cells. J Biol Chem. 2007;282:5704–5714. doi: 10.1074/jbc.M610965200. [DOI] [PubMed] [Google Scholar]
- 37.Dabir P, Marinic TE, Krukovets I, Stenina OI. Aryl hydrocarbon receptor is activated by glucose and regulates the thrombospondin-1 gene promoter in endothelial cells. Circ Res. 2008;102:1558–1565. doi: 10.1161/CIRCRESAHA.108.176990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Crawford SE, Stellmach V, Murphy-Ullrich JE, Ribeiro SM, Lawler J, Hynes RO, Boivin GP, Bouck N. Thrombospondin-1 is a major activator of TGF-beta1 in vivo. Cell. 1998;93:1159–1170. doi: 10.1016/s0092-8674(00)81460-9. [DOI] [PubMed] [Google Scholar]
- 39.Bein K, Simons M. Thrombospondin type 1 repeats interact with matrix metalloproteinase 2. Regulation of metalloproteinase activity. J Biol Chem. 2000;275:32167–32173. doi: 10.1074/jbc.M003834200. [DOI] [PubMed] [Google Scholar]
- 40.Boodhwani M, Sellke FW. Therapeutic angiogenesis in diabetes and hypercholesterolemia: influence of oxidative stress. Antioxid Redox Signal. 2009;11:1945–1959. doi: 10.1089/ars.2009.2439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Abaci A, Oguzhan A, Kahraman S, Eryol NK, Unal S, Arinc H, Ergin A. Effect of diabetes mellitus on formation of coronary collateral vessels. Circulation. 1999;99:2239–2242. doi: 10.1161/01.cir.99.17.2239. [DOI] [PubMed] [Google Scholar]
- 42.Chen JX, Zeng H, Reese J, Aschner JL, Meyrick B. Overexpression of angiopoietin-2 impairs myocardial angiogenesis and exacerbates cardiac fibrosis in the diabetic db/db mouse model. Am J Physiol Heart Circ Physiol. 2011;302:H1003–1012. doi: 10.1152/ajpheart.00866.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schiekofer S, Galasso G, Sato K, Kraus BJ, Walsh K. Impaired revascularization in a mouse model of type 2 diabetes is associated with dysregulation of a complex angiogenic-regulatory network. Arterioscler Thromb Vasc Biol. 2005;25:1603–1609. doi: 10.1161/01.ATV.0000171994.89106.ca. [DOI] [PubMed] [Google Scholar]
- 44.Sasso FC, Torella D, Carbonara O, Ellison GM, Torella M, Scardone M, Marra C, Nasti R, Marfella R, Cozzolino D, Indolfi C, Cotrufo M, Torella R, Salvatore T. Increased vascular endothelial growth factor expression but impaired vascular endothelial growth factor receptor signaling in the myocardium of type 2 diabetic patients with chronic coronary heart disease. J Am Coll Cardiol. 2005;46:827–834. doi: 10.1016/j.jacc.2005.06.007. [DOI] [PubMed] [Google Scholar]
- 45.Lawler PR, Lawler J. Molecular basis for the regulation of angiogenesis by thrombospondin-1 and -2. Cold Spring Harb Perspect Med. 2012;2:a006627. doi: 10.1101/cshperspect.a006627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang X, Kazerounian S, Duquette M, Perruzzi C, Nagy JA, Dvorak HF, Parangi S, Lawler J. Thrombospondin-1 modulates vascular endothelial growth factor activity at the receptor level. Faseb J. 2009;23:3368–3376. doi: 10.1096/fj.09-131649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Short SM, Derrien A, Narsimhan RP, Lawler J, Ingber DE, Zetter BR. Inhibition of endothelial cell migration by thrombospondin-1 type-1 repeats is mediated by beta1 integrins. J Cell Biol. 2005;168:643–653. doi: 10.1083/jcb.200407060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Isenberg JS, Martin-Manso G, Maxhimer JB, Roberts DD. Regulation of nitric oxide signalling by thrombospondin 1: implications for anti-angiogenic therapies. Nat Rev Cancer. 2009;9:182–194. doi: 10.1038/nrc2561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lobov IB, Brooks PC, Lang RA. Angiopoietin-2 displays VEGF-dependent modulation of capillary structure and endothelial cell survival in vivo. Proc Natl Acad Sci U S A. 2002;99:11205–11210. doi: 10.1073/pnas.172161899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fagiani E, Lorentz P, Kopfstein L, Christofori G. Angiopoietin-1 and -2 exert antagonistic functions in tumor angiogenesis, yet both induce lymphangiogenesis. Cancer Res. 2011;71:5717–5727. doi: 10.1158/0008-5472.CAN-10-4635. [DOI] [PubMed] [Google Scholar]
- 51.Kong P, Gonzalez-Quesada C, Li N, Cavalera M, Lee DW, Frangogiannis NG. Thrombospondin-1 regulates adiposity and metabolic dysfunction in diet-induced obesity enhancing adipose inflammation and stimulating adipocyte proliferation. Am J Physiol Endocrinol Metab. 2013;305:E439–450. doi: 10.1152/ajpendo.00006.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li Y, Tong X, Rumala C, Clemons K, Wang S. Thrombospondin1 deficiency reduces obesity-associated inflammation and improves insulin sensitivity in a diet-induced obese mouse model. PLoS One. 2011;6:e26656. doi: 10.1371/journal.pone.0026656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Olerud J, Mokhtari D, Johansson M, Christoffersson G, Lawler J, Welsh N, Carlsson PO. Thrombospondin-1: an islet endothelial cell signal of importance for beta-cell function. Diabetes. 2011;60:1946–1954. doi: 10.2337/db10-0277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Henkin J, Volpert OV. Therapies using anti-angiogenic peptide mimetics of thrombospondin-1. Expert Opin Ther Targets. 2011;15:1369–1386. doi: 10.1517/14728222.2011.640319. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.