

Abstract
The genetic architecture underlying the heritability of cardiovascular disease (CVD) is incompletely understood. Metabolomics is an emerging technology platform that has shown early success in identifying biomarkers and mechanisms of common, chronic diseases. Integration of metabolomics, genetics and other ‘omics’ platforms in a systems biology approach holds potential for elucidating novel genetic markers and mechanisms for CVD. We review important studies that have utilized metabolomic profiling in cardiometabolic diseases, approaches for integrating metabolomics with genetics and other molecular profiling platforms, and key studies showing the potential for such studies in deciphering CVD genetics, biomarkers and mechanisms.
Keywords: metabolomics, metabolism, risk factor, genomics
Metabolomics, or the profiling of intermediary metabolites, is emerging as an important technology platform for understanding of mechanisms underlying common chronic diseases such as diabetes, obesity and cardiovascular disease (CVD). Metabolomics measures chemistry, and chemistry represents an integrated readout of upstream genetic, transcriptomic, and proteomic variation.1 It is also becoming apparent that metabolomics can be integrated with these other ‘omic’ technologies to identify novel biological pathways and disease mechanisms. At the simplest level, metabolite biomarkers can be combined with genetics, other biomarkers, and/or clinical variables to provide incremental gain in diagnostic or risk prediction models. At another level, metabolites can serve as intermediate phenotypes for genetic studies. And finally, at a more biologically and analytically complex level, metabolomics can be integrated with multiple “‘omic” platforms in a systems biology approach. A major goal of systems biology is to assemble a global map of the functional relationships and interactions between physical entities in the cell (genes, proteins, metabolites, etc.), as well as a roadmap of the interaction of different tissue and organ systems for affecting physiologic homeostasis.2
The promise of metabolomics and integrated ‘omics’ analyses for chronic disease research is just beginning to be realized. Initial successes have fueled a surge in interest, but investigators need to be aware of study design, analytic and interpretative challenges. Here we review the status of this emergent area, as illustrated by provocative proof-of-principle examples.
Genetics and Metabolomics of CVD
It is well established that CVD is heritable. Prior to the Human Genome Project, hundreds of candidate gene studies were published, but few variants were consistently associated with disease and fewer still with detailed functional data. After publication of the initial draft of the human genome, many genomewide association studies (GWAS) for CVD were conducted, consistently identifying the same intergenic locus on chromosome 9p21.3 However, the effect size of this single nucleotide polymorphism (SNP) is modest and has unclear functional effects. Larger GWAS through collaborative consortia have uncovered additional variants with weaker effects, but their impact on functions related to CVD, and their overall impact on development of disease remain to be determined.
While “static” genetic biomarkers are key components of heritability, it makes biological sense that with a chronic, systemic disease like CVD with early manifestations which can begin to develop in childhood, molecular signals more “proximal” to the disease process might serve as stronger biomarkers. Certainly, proteins reflecting the diverse biological processes of CVD are commonly used in practice (i.e. hsCRP, proBNP, troponin). The strong association of CVD with broad metabolic perturbations such as hyperglycemia and hyperlipidemia suggests that a more extensive survey of metabolic variation through application of metabolomics could identify metabolites with diagnostic and mechanistic relevance.
Indeed, metabolomics has already been used with some success to identify cardiometabolic disease biomarkers. An early study uncovered differences in NMR-derived metabolite peaks in a small group of patients with coronary artery disease (CAD) compared with controls.4 Enthusiasm for the approach was tempered when a subsequent study found that the original association was likely confounded by statin medication use and subject gender5, emphasizing the importance of evaluating medications, comorbidities, diet, and other confounders. Fortunately, several subsequent studies have emerged that include validation cohorts identifying biomarkers strongly associated with CVD and contributing conditions including insulin resistance, diabetes, and inflammation.
For example, our group has demonstrated association of a cluster of branched chain amino acids (BCAA) and related metabolites with insulin resistance.6–8 Similar association has been observed in the Framingham Heart Study9, and a subsequent study made the important observation that baseline BCAA levels predicted future development of type 2 diabetes.10 Baseline BCAA levels also predict improvement in insulin resistance with weight loss11, and BCAA levels are lower in metabolically healthy versus metabolically unwell overweight/obese individuals.12 Interestingly, BCAA levels decrease more dramatically in response to bariatric surgery compared to a dietary intervention, even when matched for amount of weight loss, consistent with the greater impact of the surgical intervention on glucose homeostasis.13 Finally, levels of BCAA and related metabolites are associated with CAD independent of type 2 diabetes mellitus.14, 15 The potential mechanistic implication of these findings is illustrated by feeding studies in rats, in which supplementation of high fat diets with BCAA promoted insulin resistance despite lesser weight gain compared to rats fed unsupplemented high fat diet.7
More recently, metabolomic studies have identified additional biomarkers of obesity and diabetes. One study demonstrated a strong association of 2-aminoadipic acid levels and incident diabetes that remained significant when corrected for BCAA levels.16 2-aminoadipic acid is a lysine metabolite. The reasons for its association with new-onset diabetes are not known, but interestingly, this metabolite was shown to increase insulin secretion in rodent and human islet cells. The authors suggest that elevated levels of 2-aminoadipic acid in the prediabetic state could cause hyperinsulinism, leading to insulin resistance. Further studies are required to test this mechanism. Another recent study identified β-aminoisobutyric acid (BAIBA) as a metabolite secreted from myocytes with forced expression of the transcriptional coactivator PGC-1α.17 BAIBA levels were found to increase in response to exercise and BAIBA infusion was found to induce the appearance of brown fat. These studies suggest that BAIBA can act as a small molecule myokine that increases energy expenditure and participates in exercise-induced protection from metabolic diseases.17 Interestingly, one potential source of BAIBA is valine metabolism, but it can also be generated by degradation of thymine. BAIBA levels in obese and insulin resistant states have not been investigated, and such studies will be required in order to fully understand its relationship to metabolic function.
In CVD itself, several biomarkers have emerged from metabolomics efforts, some with mechanistic implications. An early study capitalized on a human model of “planned MI”: plasma was collected at several time points before and after alcohol septal ablation performed for management of hypertrophic cardiomyopathy, a surrogate model for spontaneous MI.18 Several metabolites changed in blood, including a signature consisting of aconitic acid, hypoxanthine, trimethylamine N-oxide and threonine, with metabolites increasing as early as 10 minutes after initiation of MI and earlier than traditional markers (i.e. troponin). While the pathophysiology of the planned MI model differs from that of “spontaneous” MI, levels of this same set of metabolites were also different in patients with spontaneous MI as compared with patients undergoing angiography who were not having MI. Thus, these metabolites may serve as earlier and more sensitive markers of MI.18
Metabolomic profiling has also identified markers predicting incident CVD events, a phenotype where clinical prediction models are incomplete. Using targeted mass spectrometry-based metabolic profiling in baseline blood samples from 314 patients with CAD, we found that a cluster of short-chain dicarboxylacylcarnitines (SCDA) discriminated the 74 individuals who went on to suffer death or MI, with over a twofold increased risk of CVD events for every 1 unit increase in levels of the SCDA metabolic signature.1 These results were validated in a case-control cohort (N=129)1, in a sequential cohort of 2023 individuals15 and in patients undergoing coronary artery bypass grafting (Figure 1a and 1b).19 While many studies show independent association of biomarkers (i.e. after adjusting for relevant clinical variables), we demonstrated that SCDA metabolites added incremental predictive capabilities on top of clinical models. Specifically, 27% of individuals who would have classified as intermediate risk from a robust model including 23 clinical variables, were correctly reclassified into low or high risk by inclusion of metabolites (Figure 1c).15 Such analyses of incremental discrimination/prediction are important in assessing the significance of ‘omic biomarkers and will facilitate the translation of these biomarkers into clinical practice. Interestingly, little is known about the source of SCDA metabolites and how they link to CVD at a mechanistic level, but studies are underway in both areas.
Figure 1.

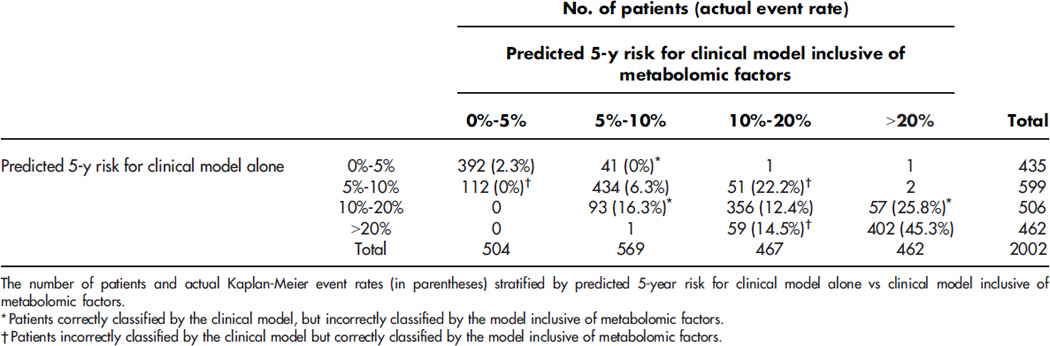
Short-chain dicarboxylacylcarnitine levels predict cardiovascular events. Unadjusted (a) and adjusted (b) Kaplan-Meier curves demonstrating increasing risk of death with higher baseline levels of SCDA metabolites; (c) risk reclassification analyses showing incremental risk prediction to 23 variable clinical model (net reclassification index [NRI] 8.8%). Reproduced with permission.15
Interest has emerged on the role of the gut microbiome in development of chronic cardiometabolic diseases. For example, using nontargeted metabolomics, higher circulating levels of trimethylamine N-oxide (TMAO), choline and betaine were observed in individuals suffering CVD events.20 The dietary lipid phosphatidylcholine is the primary dietary source of choline, and catabolism of betaine and choline by intestinal microbes leads to TMAO production. Dietary supplementation with these metabolites in mice promoted atherosclerosis, and ablation of the microbiota with antibiotics prevented the effect of dietary choline in enhancing atherosclerosis. Consistent with these results, antibiotic treatment caused TMAO levels in humans to decrease in response to an oral phosphatidylcholine challenge.21 Moreover, TMAO levels were higher in humans after L-carnitine ingestion (a trimethylamine abundant in red meat). Carnitine can also serve as a TMAO precursor via microbial metabolism.22 Interestingly, different proportions of bacterial taxa were present in the feces of vegetarians as compared to omnivores, and several of these taxa were associated with plasma TMAO concentrations. This suggests that diet could modulate intestinal microbiota composition and concomitantly, the ability to synthesize TMA and TMAO from dietary L-carnitine, thus providing a mechanistic link between diet, the gut microbiome and atherosclerosis.22
Several studies have also linked the microbiome to metabolic diseases. A recent study of twins discordant for obesity involving transplantation of fecal material into germ-free mice revealed that mice harboring the transplanted microbiota from the obese twins had higher circulating BCAA levels and higher muscle acylcarnitine levels, an indicator of muscle insulin resistance.23 Remarkably, transcriptomic analysis of the microbiota revealed induction of the entire pathway of BCAA biosynthesis in the obese microbes (this pathway is present in bacteria but not in mammals), and metabolomic analysis of the obese microbiota demonstrated higher BCAA production.23 While the relationship between microbial metabolite synthesis/production and changes in host metabolism requires further study, these new findings highlight the importance of the interactions between microbial and host genetics and metabolism.
A more integrated ‘omics approach might also contribute to better understanding of the genetic architecture underlying CVD heritability. While large CVD genetics meta-analyses are able to identify statistically significant genetic variants, they have primarily relied on “lumping” of CVD phenotypes, often resulting in a low effect size for the variants. Such studies are certainly vital for elucidating the polygenic nature of CVD, but there is also a need for parallel evaluations that “split” CVD into more discrete intermediate phenotypes. Metabolomic profiling holds great potential for enabling such phenotyping by reporting on subclinical cellular dysfunction that may not be embodied in more traditional measures. Metabolic profiling could also identify pathophysiologic changes at earlier time points in the CVD temporal continuum.
‘Omics Technologies and the Concept of Systems Biology
Advances in our understanding of disease pathophysiology, normal human physiology, and environmental influences on both have been driven recently by application of high-throughput molecular technologies (Figure 2).24 It is now possible to measure millions of genetic variants in thousands of samples in a short period of time (GWAS). Next generation sequencing allows analysis of the entire exome or genome, with many centers having the ability to perform studies on hundreds to thousands of samples, in contrast to the handful of individuals sequenced for the initial draft of the Human Genome Project. Sequencing technologies have also advanced the scientific community’s ability to measure a larger and more comprehensive number of RNA transcripts and microRNAs. Technologies are likewise evolving for high-throughput epigenetic profiling. These tools are only recently being applied to cardiometabolic diseases, but show great promise for shedding light on gene/environment interactions. Finally, in parallel, steady advances in nuclear magnetic resonance (NMR) and mass spectrometry (MS) methods have enabled more accurate and comprehensive profiling of metabolites and proteins in tissues and blood.
Figure 2.

Visual representation of ‘omics technologies available for integrated analyses. Molecular changes reflected in these markers, in combination with environmental influences, result in the health or disease phenotype. Modified with permission.24
The concept of systems biology moves beyond studying individual molecules or single reactions, integrating orthogonal data from diverse biological datasets including the genes, epigenetic modifications, RNAs, proteins, metabolites, environmental inputs, clinical variables and other factors, providing an analytic snapshot into normal and dysregulated biological function. The molecular phenotypes can be tested for relationships to each other and/or to clinical traits/diseases. These concepts are particularly relevant to cardiometabolic diseases with their dynamic temporal nature and multiple, varying environmental inputs. Such a systems biology approach may help the field to overcome the somewhat disappointing lack of clarity about genetic architecture of CVD derived from studies of ‘static’ DNA variation in isolation. It seems reasonable to postulate that clear understanding of the genetic underpinnings of CVD pathophysiology will benefit from a more holistic analysis of gene expression, proteomic and metabolic consequences of altered gene expression, and the role of incremental clinical/environmental inputs.
Additionally, while individual genetic loci identified from GWAS of disease endpoints can be tested mechanistically using traditional molecular biology experiments, this approach is difficult given the modest effects of the identified variants and the uncertainty surrounding the specific gene or gene modifier that is driving the specific SNP association. Systems biology approaches can be useful in this regard, enabling analysis of molecular interactions in the context of multiple genetic polymorphisms influencing traits and disease25, a model more directly relevant to the common, complex cardiometabolic diseases being studied. While these molecular pathway approaches will not replace mechanistic experiments, they are complementary and hypothesis-generating.
Applications of Integrated Metabolomic-Genetic Analysis
Metabolomics and genetics can be integrated for different purposes including: (1) improving clinical risk models; (2) evaluating metabolites associated with a single genotype to generate hypotheses for mechanisms underlying a specific SNP/gene; (3) using metabolites as phenotypes for genetic studies (i.e. GWAS); and (4) more unbiased, hypothesis-generating studies integrating metabolomics with genetics, transcriptomics, proteomics and other ‘omics to identify pathways of disease. While this latter systems biology approach with large orthogonal datasets may prove to be the most powerful for identification of novel mechanisms of cardiometabolic disease, integration of metabolomics with genomics at a simpler level has already led to useful advances as now reviewed.
One powerful feature of a multi-omics approach is that each molecular platform provides measures of different biological inputs to disease, meaning that their contribution to disease models can be orthogonal, thereby providing incremental discriminative/predictive capability. An early example of this approach demonstrated that inclusion of several protein-based biomarkers incrementally improved risk prediction for acute coronary syndrome.26 Another study showed the potential power of an integrated genetic risk score inclusive of SNPs for predicting CVD events, with the genotype score modestly improving risk reclassification incrementally to a clinical prediction model.27 In the newer metabolomics arena, investigators have sought to establish independent association (i.e. adjusting statistically for potential confounders) but not always incremental association (i.e. of metabolites with disease in models that account first for clinical factors). In the hopes of translating biomarkers into clinical practice, it is important to assess not just independent but also incremental association. For example, SCDA metabolites have been shown to add incremental predictive capability on top of a robust clinical model inclusive of 23 clinical variables with a net classification index (NRI) of 8.8%.15
Integration of genetics and metabolomics can also be useful in the context of a focused pathway of interest. The initial studies identifying TMAO linked host metabolism with microbiome metabolism. The same group has now demonstrated that two flavin monoxygenase family members oxidize trimethylamine (TMA) to TMAO. They further show that the FMO3 gene contributes to variations in TMAO levels in mice, that there is a relationship between variation in FMO3 expression and plasma TMAO levels, and that mice that express variants in this gene have increased susceptibility to atherosclerosis.28 Metabolomics can also help inform functional annotation for single candidate SNPs/genes. TCF7L2 SNPs have been shown to be associated with type 2 diabetes, apparently due to deficiencies in insulin secretion, although the molecular mechanism of β-cell dysfunction remains unknown.29 Metabolomic profiling has revealed alterations in phospholipid metabolism in response to glucose tolerance testing in individuals with the risk TCF7L2 genotype. The authors conclude that these results may reflect a genotype-mediated link to early metabolic abnormalities that occur before the development of impaired glucose tolerance.29
A similar approach can be employed in model organisms. Metabolomic profiling in mice with gene deletions resulting in inactivation of xanthine oxidoreductase identified, in addition to the expected derangements in purine metabolism, dysregulation of several other pathways including pyrimidine, nicotinamide, tryptophan, and phospholipid metabolism30, demonstrating the power of metabolomics for systematic assessment of direct and indirect consequences of gene mutations. Proteomics and metabolomics were combined in mice with transgenic manipulation of protein kinase C epsilon (PKCε), providing evidence for a role of PKCε in modulating cardiac glucose metabolism.31 In another study, metabolomic profiling of hearts from VEGF-B transgenic mice (which exhibit cardiac hypertrophy without cardiomyopathy) revealed apparent mitochondrial lipotoxicity, suggesting that VEGF-B regulates lipid metabolism, a heretofore unrecognized function for this angiogenic growth factor.32
Metabolites can also be used as phenotypes (“mQTLs”) for genetic evaluations by serving as intermediate early reporters on the temporal continuum of CVD development. Further, metabolites are more closely related to genes of interest, serving as intermediates between genes and clinical endpoints, and thus mapping metabolites has potential for identification of genetic variants with stronger effect sizes than seen with mapping of CVD per se.33 Moreover, the pathway in which the metabolite plays a role may provide insight into the underlying biological mechanism responsible for the development of the associated disease. Concordantly, metabolite levels have been shown to be heritable.34,35
An early study integrated metabolomics with GWAS in a small cohort of 284 participants from the KORA study.36 Using individual metabolites and ratios of metabolite concentrations (as proxies for enzymatic activity), they found that SNPs explained up to 28% of the observed variance in metabolite levels. Four of the most significant SNPs were within genes encoding enzymes in the pathway of the corresponding metabolite.36 In a GWAS of six plasma polyunsaturated fatty acids (PUFAs) in 1075 participants in the InCHIANTI study of aging, genomewide associations were found in a region of chromosome 11 encoding three fatty acid desaturases (FADS1, FADS2, and FADS3), providing further evidence that genetic variation contributes to variation in plasma fatty acid levels.37 Another study integrating GWAS with 163 metabolites in a larger sample of 1809 KORA subjects found eight genetic loci meeting genomewide significance, with most of the loci again located in or near enzyme or solute carrier coding genes involved in processing of the associated metabolite (Figure 3).38 For example, SNPs in ACADM were associated with C12/C10 acylcarnitine ratio; the enzyme encoded by this gene catalyzes the initial reaction in the beta oxidation of C4 to C12 straight-chain acyl coAs, and rare functional coding mutations in ACADM cause an inborn error of metabolism (MCAD deficiency). This suggests that common SNPs in genes that cause rare Mendelian diseases may lead to a less severe and potentially subclinical phenotype that could only be discovered by mapping the metabolite itself. Several subsequent studies combining GWAS with metabolomics have been published (Table 1). It is important to note that although some of the identified SNPs have been associated with disease phenotypes in different cohorts, these studies have not shown that metabolite-associated SNPs also associate with disease in the same cohort. In fact, many of the aforementioned studies were not performed in disease-bearing cohorts, reducing the power for “triangulating” metabolic, genetic and disease associations.
Figure 3.

Manhattan plot of GWAS of metabolites from the KORA study. Displayed is the strength of association with metabolite concentrations (top; p<10−7 in red) and concentration ratios (bottom; p<10−9 in red). Reproduced with permission.38
Table 1.
GWAS studies of metabolites
| Cohort | Platform | N | Results | Significance | Adjustment for: 1) Multiple comparisons 2) Clinical variables |
|---|---|---|---|---|---|
| KORA population-based cohort38 | Targeted tandem MS, 163 metabolites | 2231 | 9 replicating loci (FADS1, ELOVL2, ACADS ACADM, ACADL, SPTLC3, ETFDH SLC16A9, PLEKHH1) | p=3×10−24 – 6.5×10−179 | 1) Bonferroni for SNPs and metabolic combinations (p<3.64×10−12) 2) None |
| InCHIANTI study on aging37 | GC, 6 polyunsaturated unsaturated fatty acids | 1075 | FADS1-3, ELOVL2 | p=1.1×10−6 – 6.0×10−46 | 1) Bonferroni at level of SNPs (p<1×10−7) 2) Sex, age, age2 |
| Prostate cancer, Swedish men39 | Nontargeted UPLC-MS, 6138 molecular features | 893 | 7 replicating loci (PYROXD2, FADS1, PON1 CYP4F2, UGT1A8, ACADL, LIPC); pathway analysis reflected enrichment of genes with acyl-coA dehydrogenase activity | p=8.8×10−13 – 3.4×10−60 | 1) Bonferroni (p<2.44×10−11) 2) None |
| Finnish individuals40 | Nontargeted NMR, 117 metabolites | 8330 | 31 loci (including SLC1A4, PPM1K, F12 SLC25A1, GCKR, G6PC2, CPT1A, PCSK9 ANGPTL3, LPL, ABCA1, FADS1-3, LIPC CETP, LIPG, LDLR, APOE, PLTP). Metabolites showed heritability. | p=8.7×10−11 – 2.5×10−58 | 1) Bonferroni (p<2.31×10−10) 2) Age, sex, 10 genetic principal components |
| Two European cohorts41 | Nontargeted NMR in urine and plasma, 526 metabolite peaks | 211 | 3 replicating loci (PYROXD2, NAT8, AGXT2) | p=8.6×10−11 – 2.8×10−23 | 1) Permutation based procedure constraining genome-wide false discovery probability to be <0.001 for each metabolite GWAS 2) Age, gender |
| KORA population-based cohort36 | Targeted tandem MS, 363 metabolites | 284 | No individual metabolite genomewide significant; 4 loci genomewide significant for metabolite ratios (FADS1, LIPC, SCAD MCAD) | p=2.0×10−9 for best single metabolite; p=10−21 – 10−16 for metabolite ratios | 1) Bonferroni (p<1.33×10−9) 2) None |
| European cohorts42 | Targeted electrospray ionization tandem MS, 33 sphingolipids | 4400 | 5 loci with strongest associations in/near 7 genes involved in ceramide biosynthesis and trafficking (SPTLC3, LASS4, SGPP1, ATP10D FADS1-3); SNPs in 3 loci, but not necessarily the same SNPs (ATP10D, FADS3 and SPTLC3) were also associated with myocardial infarction in different cohorts | Lowest p=9.1×10−66 | 1) Bonferroni at level of SNPs (p<7.2×10−8) 2) Age, sex |
| German cohort43 | Nontargeted LC/GC tandem MS, 517 metabolic traits | 1768 | 34 loci with strongest results for 7 loci (SLC22A2, COMT, CYP3A5, CYP2C18, GBA3 UGT3A1, rs12413935); also overlaid metabolic networks to generate hypotheses for unknown compounds and performed experimental validation of those compounds. | p=1.5×10−10 – 2.2×10−281 | 1) Bonferroni (p<1.6×10−10) 2) Age, gender |
| SHIP and KORA population-based studies44 | Nontargeted NMR in urine, 59 metabolites | 2893 | 5 loci validated (SLC7A9, NAT2, SLC6A20 AGXT2, WDR66) | p=2.3×10−13 – 3.2×10−75 | 1) Bonferroni (p<4.5×10−11) 2) Age |
| KORA population-based and Twins UK studies45 | Nontargeted UPLC and GC tandem MS, >250 metabolites | 2820 | 37 loci (including NAT8, GCKR, ABO, ACADS ACADM, UGT1A, CPS1, ELOVL2); 30 of these mapped to protein biochemically linked to associated metabolites; 15 associated with disease endpoints from previous studies in other cohorts | p=1.4×10−12 – 4.4×10−305 | 1) Bonferroni (p<2.0×10−12) 2) Age, gender, family structure |
| Meta-analysis of 5 European family-based studies46 | Targeted tandem MS, 153 phospho- and sphingolipids | 4034 | 35 loci (including FADS1-2-3, PAQR9 AGPAT1, PKD2L1, PDXDC1, PLD2, APOE PNLIPRP2, ABDH3, APOA1, ELOVL2, LIPC APOE); 3 associated with disease phenotypes in other cohorts (FADS1-2-3, AGPAT1 APOA1) | p=4.9×10−8 – 9.9×10−204 | 1) Bonferroni (p<2.2×10−9) 2) Familial relatedness |
| Cardiovascular Risk in Young Finns Study (YFS) and Northern Finland Birth Cohort 1966 (NFBC66)47 | Nontargeted NMR in serum, 130 metabolite measures | 6608 | 34 loci (including PCSK9, APOB, GCKR ELOVL2, LPL, ABCA1, FADS1-2-3, CETP); SERPINA1 and AQP9 showed eQTL, upregulated in atherosclerotic plaques | p=3.9×10−9 – 3.9×10−264 | 1) Metabolic networks used as traits; significance set at p<4.5×10−9 2) None |
| KORA population-based and Twins UK studies48 | LC- and GC-tandem MS in plasma/serum, 529 metabolites | 7824 | 145 loci (including NAT8, LIPC, ANGPTL3, FMO3, ELOVL2 FADS1, CETP, CPS1, APOE, PPM1K and 84 new loci); integrated with gene expression; created web-based resources for data mining and results visualization | p=5.2×10−9 – 6.2×10−860 | 1) Bonferroni (p<1.03×10−10 for individual metabolites; p<5.08×10−13 for metabolite ratios) 2) Age, sex |
MS: mass spectrometry; GC: gas chromatography; NMR: nuclear magnetic resonance; UPLC: ultra-high performance liquid chromatography; LC: liquid chromatography.
Integrated Metabolomics-Genetics: Systems Biology Examples
Integrated approaches have been applied in model organism studies, both for focused hypothesis testing and for hypothesis generation. As an example of the latter, a study integrating metabolomics, transcriptomics and genetics in liver samples from an F2 intercross between a diabetes-resistant and diabetes-susceptible mouse strain connected variations in metabolites and transcripts to regions of the genome and constructed associative networks controlling liver metabolic processes (Figure 4a).34 Based on advanced computational analysis of this multi-omic data set, a causal network linking variations in glutamate to regulation of the key gluconeogenic enzyme PEPCK was identified, and importantly, experimentally validated by showing that glutamate induced expression of PEPCK and other genes in the network (Figure 4b).34 Studies of this nature serve as proof-of-principle for use of systems biology in identification of plausible and testable metabolic control networks.
Figure 4.


Genetic networks of liver metabolism revealed by integration of metabolic and transcriptional profiling. Results from a study of F2 intercross between diabetes-resistant C57BL/6 leptinob/ob and diabetes-susceptible BTBR leptinob/ob mouse strains: (a) metabolic quantitative trait loci (mQTL) mapping identifying genetic hotspots for metabolite regulation; and (b) causal network analysis links gene expression and metabolic changes in the context of glutamate metabolism. Reproduced with permission.34
In humans, using network analysis from multiple ‘omics platforms in a large population-based cohort from Finland, the authors elucidated functional effects of a lipid signaling module composed of a set of highly correlated genes with a prominent role in regulating the levels of 80 metabolites, and providing new links between inflammation, metabolism and adiposity.49 Such systems biology approaches may be useful even in small sample sizes. As an extreme example, an “integrative Personal Omics Profile [iPOP]” was performed on a single individual that integrated ‘omics over multiple time points over a 14 month period.50 Whole genome sequencing identified a genetic variant predisposing the individual to increased risk of type 2 diabetes and monitoring of glucose and HbA1c levels subsequently revealed the onset of the disease despite a normal body mass index. Integrated molecular profiles changed concordant with respiratory infections, showing dynamic molecular changes in response to disease. Such studies not only aid in identification of novel mechanisms of disease pathogenesis, but perhaps project future approaches to personalized medicine.
Analytic and Bioinformatic Considerations
Analysis of millions of data points per single study subject poses unique challenges in ‘omic sciences. In some instances, traditional techniques can be employed. For example, analysis of individual analytes in GWAS or metabolomic studies is facilitated by adjustment for multiple comparisons. However, approaches that explicitly incorporate the collinearity and multidimensionality of the complex data structure, take biological pathway information into account, and analyze patterns or networks within the data, may have greater statistical power and enable better mechanistic hypothesis generation. Such approaches can be unsupervised (i.e. variation within the molecular data only) or supervised (i.e. variations in the molecular data and the disease state). Analytic approaches employed include factor analysis, hierarchical clustering algorithms, Gaussian graphical modeling, and pathway- and network-based analyses that can integrate data from disparate ‘omic platforms and identify molecular interactions and pathways. Programs have been developed to aid in data visualization, statistical analysis and bioinformatic annotation and interpretation. These programs often incorporate information from publically available databases and utilize pathway and network statistical analysis techniques.
Concurrently, the metabolomics community is making advances in metabolite annotation, nomenclature and cataloguing including the Human Metabolome Project and the LIPID Metabolites and Pathways Strategy (LIPID MAPS). One issue that remains to be overcome involves methods of quantification of metabolites. Some laboratories, including ours, emphasize “targeted” approaches that make use of extensive libraries of stable isotope-labeled standards.7,34 For example, when measuring amino acids, known quantities of multiple stable isotope-labeled amino acids are added to the sample, and the concentration of the native analyte is calculated by reference to its cognate internal standard. In contrast, non-targeted methods that report analytes as relative peak areas unreferenced to specific internal standards are often used, even in epidemiological studies. It is important to note that correlation or association do not define directionality of a molecular relationship. Statistical methods for assessing cause-and-effect such as Mendelian randomization are becoming increasingly utilized, but do not replace the ultimate need for testing of predictions emanating from statistical analyses through biological experiments.
Study Design, Challenges and Future Directions
While the studies we have detailed highlight the great promise of combining high-throughput molecular data, many considerations need to be addressed to enable dissection of the signal-to-noise in such data, avoid type 1 and type 2 errors, and ensure accurate data interpretation. Investigators must carefully decide on a study design from among various options including population-based cohorts, disease case-controls studies, or evaluation of “extremes” of a clinical or molecular trait. Statistical power should be considered, particularly in the context of large numbers of molecular biomarkers measured in smaller sample sizes. The depth of molecular profiling needs to be weighed against the quantitative precision (or lack thereof) of the measurements. Analytic strategies for multidimensional data reduction, pathway/network analysis, and adjustment for multiple comparisons should be delineated early in study implementation. Built into study design should be plans for laboratory validation of molecular targets, replication in other cohorts, and experimental validation of pathways identified from statistical analysis. For example, the association between BCAA, short-chain dicarboxylacylcarnitines, and betaine-derived metabolites and cardiometabolic diseases has been validated in independent cohorts, as have several of the mQTL identified in metabolic GWAS, but other findings reviewed herein have not. Other challenges being tackled by the scientific community include a need for a common nomenclature for metabolomics, standardization across molecular platforms, and development of more robust analytic techniques for the “large p, small n” issue (i.e. thousands-millions of molecular data points for each sample, but a relatively small number of samples). Systems biology is collaborative and multi-disciplinary by nature, but fails if study teams are lacking in individuals with the relevant expertise and ability to converse across fields. Finally, the availability of relevant biological samples with well-annotated clinical phenotypes is vital to maximize the “signal-to-noise” ratio.
In the future we can anticipate the need to further refine analytic and bioinformatics approaches to accommodate even more high-dimensional datasets. Molecular ‘omic datasets will be enhanced by integration with more detailed “environomes”, “exposomes”, and “phenomes”. Research on the role of the gut microbiome in regulation of host metabolism, hormonal milieu and inflammatory tone is exploding, and a complete molecular profile will soon come to include information about gut microbiome composition, genetics and metabolism. Technologies are also evolving to enable high-throughput molecular phenotyping on single cells, facilitating mechanistic studies in heterogeneous tissues. Perhaps most importantly, ‘omic analyses of the future will integrate all of these tools to define cause-and-effect mechanisms, and to guide experimental validation of identified pathways.
In conclusion, while investigators should be careful about analytic and bioinformatics challenges, integrated metabolomic-genetic analyses and systems biology approaches hold great potential for furthering our understanding of biomarkers and mechanisms of health and disease and moving the scientific community closer to an eventual goal of more personalized medicine.
Acknowledgements
The authors thank their colleagues in the Duke Molecular Physiology Institute for their critical contributions to the work cited from our group.
Funding Sources: Work cited in this article was supported by NIH grants HL095987 (Shah) and P01-DK58398 (Newgard), and by a sponsored research agreement from Pfizer.
Footnotes
Conflict of Interest Disclosures: Both authors hold a patent on a related finding. Dr. Newgard is a member of the Pfizer CVMED scientific advisory board.
References
- 1.Shah SH, Bain JR, Muehlbauer MJ, Stevens RD, Crosslin DR, Haynes C, et al. Association of a peripheral blood metabolic profile with coronary artery disease and risk of subsequent cardiovascular events. Circ Cardiovasc Genet. 2010;3:207–214. doi: 10.1161/CIRCGENETICS.109.852814. [DOI] [PubMed] [Google Scholar]
- 2.Kitano H. Systems biology: A brief overview. Science. 2002;295:1662–1664. doi: 10.1126/science.1069492. [DOI] [PubMed] [Google Scholar]
- 3.Helgadottir A, Thorleifsson G, Manolescu A, Gretarsdottir S, Blondal T, Jonasdottir A, et al. A common variant on chromosome 9p21 affects the risk of myocardial infarction. Science. 2007;316:1491–1493. doi: 10.1126/science.1142842. [DOI] [PubMed] [Google Scholar]
- 4.Brindle JT, Antti H, Holmes E, Tranter G, Nicholson JK, Bethell HW, et al. Rapid and noninvasive diagnosis of the presence and severity of coronary heart disease using 1h–nmr-based metabonomics. Nat Med. 2002;8:1439–1444. doi: 10.1038/nm1202-802. [DOI] [PubMed] [Google Scholar]
- 5.Kirschenlohr HL, Griffin JL, Clarke SC, Rhydwen R, Grace AA, Schofield PM, et al. Proton nmr analysis of plasma is a weak predictor of coronary artery disease. Nat Med. 2006;12:705–710. doi: 10.1038/nm1432. [DOI] [PubMed] [Google Scholar]
- 6.Huffman KM, Shah SH, Stevens RD, Bain JR, Muehlbauer M, Slentz CA, et al. Relationships between circulating metabolic intermediates and insulin action in overweight to obese, inactive men and women. Diabetes Care. 2009;32:1678–1683. doi: 10.2337/dc08-2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Newgard CB, An J, Bain JR, Muehlbauer MJ, Stevens RD, Lien LF, et al. A branched-chain amino acid-related metabolic signature that differentiates obese and lean humans and contributes to insulin resistance. Cell Metab. 2009;9:311–326. doi: 10.1016/j.cmet.2009.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tai ES, Tan ML, Stevens RD, Low YL, Muehlbauer MJ, Goh DL, et al. Insulin resistance is associated with a metabolic profile of altered protein metabolism in chinese and asian-indian men. Diabetologia. 2010;53:757–767. doi: 10.1007/s00125-009-1637-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shaham O, Wei R, Wang TJ, Ricciardi C, Lewis GD, Vasan RS, et al. Metabolic profiling of the human response to a glucose challenge reveals distinct axes of insulin sensitivity. Mol Syst Biol. 2008;4:214. doi: 10.1038/msb.2008.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang TJ, Larson MG, Vasan RS, Cheng S, Rhee EP, McCabe E, et al. Metabolite profiles and the risk of developing diabetes. Nat Med. 2011;17:448–453. doi: 10.1038/nm.2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shah SH, Crosslin DR, Haynes CS, Nelson S, Turer CB, Stevens RD, et al. Branched-chain amino acid levels are associated with improvement in insulin resistance with weight loss. Diabetologia. 2012;55:321–330. doi: 10.1007/s00125-011-2356-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Batch BC, Shah SH, Newgard CB, Turer CB, Haynes C, Bain JR, et al. Branched chain amino acids are novel biomarkers for discrimination of metabolic wellness. Metabolism. 2013;62:961–969. doi: 10.1016/j.metabol.2013.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Laferrere B, Reilly D, Arias S, Swerdlow N, Gorroochurn P, Bawa B, et al. Differential metabolic impact of gastric bypass surgery versus dietary intervention in obese diabetic subjects despite identical weight loss. Sci Transl Med. 2011;3:80re82. doi: 10.1126/scitranslmed.3002043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bhattacharya S, Granger CB, Craig D, Haynes C, Bain J, Stevens RD, et al. Validation of the association between a branched chain amino acid metabolite profile and extremes of coronary artery disease in patients referred for cardiac catheterization. Atherosclerosis. 2014;232:191–196. doi: 10.1016/j.atherosclerosis.2013.10.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shah SH, Sun JL, Stevens RD, Bain JR, Muehlbauer MJ, Pieper KS, et al. Baseline metabolomic profiles predict cardiovascular events in patients at risk for coronary artery disease. Am Heart J. 2012;163:844–850. e841. doi: 10.1016/j.ahj.2012.02.005. [DOI] [PubMed] [Google Scholar]
- 16.Wang TJ, Ngo D, Psychogios N, Dejam A, Larson MG, Vasan RS, et al. 2-aminoadipic acid is a biomarker for diabetes risk. J Clin Invest. 2013;123:4309–4317. doi: 10.1172/JCI64801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Roberts LD, Bostrom P, O'Sullivan JF, Schinzel RT, Lewis GD, Dejam A, et al. Beta-aminoisobutyric acid induces browning of white fat and hepatic beta-oxidation and is inversely correlated with cardiometabolic risk factors. Cell Metab. 2014;19:96–108. doi: 10.1016/j.cmet.2013.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lewis GD, Wei R, Liu E, Yang E, Shi X, Martinovic M, et al. Metabolite profiling of blood from individuals undergoing planned myocardial infarction reveals early markers of myocardial injury. J Clin Invest. 2008;118:3503–3512. doi: 10.1172/JCI35111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shah AA, Craig DM, Sebek JK, Haynes C, Stevens RC, Muehlbauer MJ, et al. Metabolic profiles predict adverse events after coronary artery bypass grafting. J Thorac Cardiovasc Surg. 2012;143:873–878. doi: 10.1016/j.jtcvs.2011.09.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang Z, Klipfell E, Bennett BJ, Koeth R, Levison BS, Dugar B, et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature. 2011;472:57–63. doi: 10.1038/nature09922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tang WH, Wang Z, Levison BS, Koeth RA, Britt EB, Fu X, et al. Intestinal microbial metabolism of phosphatidylcholine and cardiovascular risk. N Engl J Med. 2013;368:1575–1584. doi: 10.1056/NEJMoa1109400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Koeth RA, Wang Z, Levison BS, Buffa JA, Org E, Sheehy BT, et al. Intestinal microbiota metabolism of l-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat Med. 2013;19:576–585. doi: 10.1038/nm.3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ridaura VK, Faith JJ, Rey FE, Cheng J, Duncan AE, Kau AL, et al. Gut microbiota from twins discordant for obesity modulate metabolism in mice. Science. 2013;341:1241214. doi: 10.1126/science.1241214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lewis GD, Asnani A, Gerszten RE. Application of metabolomics to cardiovascular biomarker and pathway discovery. J Am Coll Cardiol. 2008;52:117–123. doi: 10.1016/j.jacc.2008.03.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Civelek M, Lusis AJ. Systems genetics approaches to understand complex traits. Nat Rev Genet. 2014;15:34–48. doi: 10.1038/nrg3575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sabatine MS, Morrow DA, de Lemos JA, Gibson CM, Murphy SA, Rifai N, et al. Multimarker approach to risk stratification in non-st elevation acute coronary syndromes: Simultaneous assessment of troponin i, c-reactive protein, and b-type natriuretic peptide. Circulation. 2002;105:1760–1763. doi: 10.1161/01.cir.0000015464.18023.0a. [DOI] [PubMed] [Google Scholar]
- 27.Kathiresan S, Melander O, Anevski D, Guiducci C, Burtt NP, Roos C, et al. Polymorphisms associated with cholesterol and risk of cardiovascular events. N Engl J Med. 2008;358:1240–1249. doi: 10.1056/NEJMoa0706728. [DOI] [PubMed] [Google Scholar]
- 28.Bennett BJ, de Aguiar Vallim TQ, Wang Z, Shih DM, Meng Y, Gregory J, et al. Trimethylamine-n-oxide, a metabolite associated with atherosclerosis, exhibits complex genetic and dietary regulation. Cell Metab. 2013;17:49–60. doi: 10.1016/j.cmet.2012.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Then C, Wahl S, Kirchhofer A, Grallert H, Krug S, Kastenmuller G, et al. Plasma metabolomics reveal alterations of sphingo- and glycerophospholipid levels in non-diabetic carriers of the transcription factor 7-like 2 polymorphism rs7903146. PLoS One. 2013;8:e78430. doi: 10.1371/journal.pone.0078430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen Q, Park HC, Goligorsky MS, Chander P, Fischer SM, Gross SS. Untargeted plasma metabolite profiling reveals the broad systemic consequences of xanthine oxidoreductase inactivation in mice. PLoS One. 2012;7:e37149. doi: 10.1371/journal.pone.0037149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mayr M, Liem D, Zhang J, Li X, Avliyakulov NK, Yang JI, et al. Proteomic and metabolomic analysis of cardioprotection: Interplay between protein kinase c epsilon and delta in regulating glucose metabolism of murine hearts. J Mol Cell Cardiol. 2009;46:268–277. doi: 10.1016/j.yjmcc.2008.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Karpanen T, Bry M, Ollila HM, Seppanen-Laakso T, Liimatta E, Leskinen H, et al. Overexpression of vascular endothelial growth factor-b in mouse heart alters cardiac lipid metabolism and induces myocardial hypertrophy. Circ Res. 2008;103:1018–1026. doi: 10.1161/CIRCRESAHA.108.178459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Suhre K, Gieger C. Genetic variation in metabolic phenotypes: Study designs and applications. Nat Rev Genet. 2012;13:759–769. doi: 10.1038/nrg3314. [DOI] [PubMed] [Google Scholar]
- 34.Ferrara CT, Wang P, Neto EC, Stevens RD, Bain JR, Wenner BR, et al. Genetic networks of liver metabolism revealed by integration of metabolic and transcriptional profiling. PLoS Genet. 2008;4:e1000034. doi: 10.1371/journal.pgen.1000034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shah SH, Hauser ER, Bain JR, Muehlbauer MJ, Haynes C, Stevens RD, et al. High heritability of metabolomic profiles in families burdened with premature cardiovascular disease. Mol Syst Biol. 2009;5:258. doi: 10.1038/msb.2009.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gieger C, Geistlinger L, Altmaier E, Hrabe de Angelis M, Kronenberg F, Meitinger T, et al. Genetics meets metabolomics: A genome-wide association study of metabolite profiles in human serum. PLoS Genet. 2008;4:e1000282. doi: 10.1371/journal.pgen.1000282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tanaka T, Shen J, Abecasis GR, Kisialiou A, Ordovas JM, Guralnik JM, et al. Genome-wide association study of plasma polyunsaturated fatty acids in the inchianti study. PLoS Genet. 2009;5:e1000338. doi: 10.1371/journal.pgen.1000338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Illig T, Gieger C, Zhai G, Romisch-Margl W, Wang-Sattler R, Prehn C, et al. A genome-wide perspective of genetic variation in human metabolism. Nat Genet. 2010;42:137–141. doi: 10.1038/ng.507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hong MG, Karlsson R, Magnusson PK, Lewis MR, Isaacs W, Zheng LS, et al. A genome-wide assessment of variability in human serum metabolism. Hum Mutat. 2013;34:515–524. doi: 10.1002/humu.22267. [DOI] [PubMed] [Google Scholar]
- 40.Kettunen J, Tukiainen T, Sarin AP, Ortega-Alonso A, Tikkanen E, Lyytikainen LP, et al. Genome-wide association study identifies multiple loci influencing human serum metabolite levels. Nat Genet. 2012;44:269–276. doi: 10.1038/ng.1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nicholson G, Rantalainen M, Li JV, Maher AD, Malmodin D, Ahmadi KR, et al. A genome-wide metabolic qtl analysis in europeans implicates two loci shaped by recent positive selection. PLoS Genet. 2011;7:e1002270. doi: 10.1371/journal.pgen.1002270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hicks AA, Pramstaller PP, Johansson A, Vitart V, Rudan I, Ugocsai P, et al. Genetic determinants of circulating sphingolipid concentrations in european populations. PLoS Genet. 2009;5:e1000672. doi: 10.1371/journal.pgen.1000672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Krumsiek J, Suhre K, Evans AM, Mitchell MW, Mohney RP, Milburn MV, et al. Mining the unknown: A systems approach to metabolite identification combining genetic and metabolic information. PLoS Genet. 2012;8:e1003005. doi: 10.1371/journal.pgen.1003005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Suhre K, Wallaschofski H, Raffler J, Friedrich N, Haring R, Michael K, et al. A genome-wide association study of metabolic traits in human urine. Nat Genet. 2011;43:565–569. doi: 10.1038/ng.837. [DOI] [PubMed] [Google Scholar]
- 45.Suhre K, Shin SY, Petersen AK, Mohney RP, Meredith D, Wagele B, et al. Human metabolic individuality in biomedical and pharmaceutical research. Nature. 2011;477:54–60. doi: 10.1038/nature10354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Demirkan A, van Duijn CM, Ugocsai P, Isaacs A, Pramstaller PP, Liebisch G, et al. Genome-wide association study identifies novel loci associated with circulating phospho- and sphingolipid concentrations. PLoS Genet. 2012;8:e1002490. doi: 10.1371/journal.pgen.1002490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Inouye M, Ripatti S, Kettunen J, Lyytikainen LP, Oksala N, Laurila PP, et al. Novel loci for metabolic networks and multi-tissue expression studies reveal genes for atherosclerosis. PLoS Genet. 2012;8:e1002907. doi: 10.1371/journal.pgen.1002907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shin SY, Fauman EB, Petersen AK, Krumsiek J, Santos R, Huang J, et al. An atlas of genetic influences on human blood metabolites. Nat Genet. 2014;46:543–550. doi: 10.1038/ng.2982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Inouye M, Kettunen J, Soininen P, Silander K, Ripatti S, Kumpula LS, et al. Metabonomic, transcriptomic, and genomic variation of a population cohort. Mol Syst Biol. 2010;6:441. doi: 10.1038/msb.2010.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chen R, Mias GI, Li-Pook-Than J, Jiang L, Lam HY, Chen R, et al. Personal omics profiling reveals dynamic molecular and medical phenotypes. Cell. 2012;148:1293–1307. doi: 10.1016/j.cell.2012.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]