

Abstract
Fanconi anemia (FA) is a genetically and phenotypically heterogeneous disorder characterized by congenital malformations, progressive bone marrow failure, and predisposition to cancer, particularly hematological malignancies and solid tumors of the head and neck. The main role of FA proteins is in the repair of DNA interstrand crosslinks (ICLs). FA results from pathogenic variants in at least 16 distinct genes, causing genomic instability. Although the highly variable phenotype makes accurate diagnosis on the basis of clinical manifestations difficult in some patients, diagnosis based on a profound sensitivity to DNA crosslinking agents can be used to identify the pre-anemia patient as well as patients with aplastic anemia or leukemia who may or may not have the physical stigmata associated with the syndrome. Diepoxybutane (DEB) analysis is the preferred test for FA because other agents have higher rates of false-positive and false-negative results.
Keywords: Fanconi anemia, DEB test, DNA interstrand crosslink repair, genomic instability
Diagnosis of Fanconi anemia (FA) based on clinical manifestations alone is often difficult and unreliable because of the considerable overlap of the FA phenotype with that of a variety of genetic and nongenetic diseases (Table 8.7.1). It is currently considered standard of care that testing for crosslink sensitivity to rule out FA and testing for telomere length to rule out DKC should be part of the workup before treatment of pediatric acquired aplastic anemia (Williams et al., 2014).
Table 8.7.1.
Genetic and non-genetic diseases that overlap with the FA phenotype.
| Gene Name | MIM # | References | ||
|---|---|---|---|---|
| VATER/VACTERL Association | 192350 | Weaver et al., 1986 | ||
|
| ||||
| VACTREL-H syndrome | 276950 | Porteous et al., 1992 | ||
|
| ||||
| Dubowitz syndrome | 223370 | Tsukahara and Opitz, 1996 | ||
|
| ||||
| Velocardiofacial syndrome (VFCS) | 192430 | Taddei et al., 2001 | ||
|
| ||||
| Holt-Oram syndrome (HOS) | TBX5 | 142900 | Basson et al., 1999 | |
|
| ||||
| Thrombocytopenia absent radius syndrome (TAR) | RBM8A | 274000 | Albers et al., 2012 | |
|
| ||||
| Baller Gerold syndrome (BGS) | RECQL4 | 218600 | Van Maldergem et al., 2006 | |
|
| ||||
| Saethre-Chotzen syndrome (SCS) | TWIST1 | 101400 | El Ghouzzi et al., 1997 | |
|
| ||||
| Diamond-Blackfan anemia (DBA) a | RPS19a | 105650 | Draptchinskaia, 1999; Gerrard et al., 2013 | |
|
| ||||
| Shwachman-Diamond syndrome (SDS) | SBDS | 260400 | Boocock et al., 2003 | |
|
| ||||
| Nijmengen breakage syndrome (NBS1) | NBN | 251260 | Varone et al., 1998 | |
|
| ||||
| Seckel syndrome (SCKL1) | ATR | 210600 | O’Driscoll et al., 2003 | |
|
| ||||
| Dyskeratosis congenita (DKC) | DKCXb | DKC1 | 305000 | Heiss et al., 1998, Alder et al., 2013. |
| DKCA1c | TERC | 127550 | Vulliamy et al., 2004, Jongmans et al., 2012 | |
| DKCA2c | TERT | 613989 | Armanios et al., 2005 | |
| DKCA3c | TINF2 | 613990 | Savage et al., 2008 | |
| DKCB1d | NOLA3/NOP102 | 224230 | Walne et al., 2007 | |
| DKCB5d | RTEL1 | 615190 | Walne et al., 2013 | |
DBA is genetically heterogeneous. Genes include RPS19 and other ribosomal protein genes
DKC genes participate in regulation of telomere length, and have X-linked, autosomal dominant and autosomal recessive modes of inheritance (reviewed in Bessler et al., 2010).
X-linked inheritance
Autosomal recessive inheritance
Autosomal dominant inheritance
Although most FA patients exhibit abnormalities in growth parameters and skin pigmentation, approximately one third of the patients do not have any major birth defects (Giampietro et al., 1993, 1997). Extensive genetic heterogeneity has been shown in FA patients, by use of various strategies for gene identification including functional and positional cloning, and more recently by mass spectrometric analysis of isolated protein complexes (Meetei et al., 2004), candidate gene sequencing in FA patient DNA (Kim et al., 2011), and by whole exome sequencing (Bogliolo et al., 2013). Sixteen unique genes involved in crosslink repair, FANCA (MIM# 607139), FANCB (MIM# 300515), FANCC (MIM# 613899), FANCD1 (MIM# 605724)/BRCA2 (MIM# 600185), FANCD2 (MIM# 613984), FANCE (MIM# 613976), FANCF (MIM# 613897), FANCG (MIM# 602956), FANCI (MIM# 611360), FANCJ (MIM# 609054)/BRIP1 (MIM# 605882), FANCL (MIM# 608111), FANCM (MIM# 609644), FANCN (MIM# 610832)/PALB2 (MIM# 610355), FANCO (MIM# 613390)/RAD51C (MIM# 179617), FANCP (MIM# 613951)/SLX4 (MIM# 613278), FANCQ (MIM# 615272)/ERCC4 (MIM# 133520), have now been identified, and additional genes with pathogenic variants in FA or FA-like patients remain to be found. However, complementation groups A (~65%), C (~15%), and G (~10%) account for most patients with this disorder, with the frequency of patients with pathogenic variants in the other genes accounting for between 0.1% and 2%. Most FA genes are inherited in an autosomal recessive manner, although FANCB (Meetei et al, 2004) is an X-linked recessive. The 16 genes mutated in FA patients encode proteins implicated in a common pathway that co-ordinates multiple repair processes and checkpoint signaling events necessary for the accurate removal of DNA interstrand crosslink (ICL) lesions (reviewed in Kottemann and Smogorzewska, 2013). Of importance is the discovery that FANCD1/BRCA2, FANCJ/BACH1, FANCN/PALB2 and FANCO/RAD51C), cause FA when biallelic, and impart an increased risk for breast and other cancers in heterozygous carriers. Therefore, once a patient is diagnosed with FA by DEB-testing, it is then important to determine the complementation group by molecular methods (Chandrasekharappa et al. 2013; Flynn et al., 2014), in order to individualize treatment for the patient, and provide genetic counseling regarding cancer risk for family members.
FA affects approximately one in 100,000 live births. The average life expectancy is about 20 years, but some patients may not be diagnosed until the 3rd decade of life or even later. Hematopoietic progenitor cells in FA are genetically unstable, leading eventually to bone marrow failure, which is the main life-threatening problem in these patients. This defect also predisposes to an increased frequency of clonal cytogenetic abnormalities, myelodysplastic syndrome (MDS), and acute leukemia. Development of any of these conditions is associated with poorer survival. Allogeneic hematopoietic stem cell transplantation (HSCT) is the only curative treatment currently for bone marrow failure in patients with FA. However, HSCT treatment in individuals with FA is complicated partly because these individuals have an exaggerated sensitivity to cytoreduction regimens in standard use for HSCT. Thus it is essential that FA be ruled out in patients at risk prior to HSCT, so that specially designed pretransplant conditioning regimens avoiding high doses of DNA crosslinking agents can be used in these patients.
Because hypersensitivity to the clastogenic (chromosome-breaking) effect of DNA cross-linking agents provides a unique marker for the diagnosis of FA, this cellular characteristic is still utilized as a diagnostic test to identify the preanemic patient as well as the patient with aplastic anemia or leukemia who may or may not have the classic physical stigmata associated with FA. A variety of chemical agents can be used to test for DNA cross-link sensitivity. Diepoxybutane (DEB; 1,3-butadiene diepoxide) analysis is the preferred test for FA diagnosis, as it has the highest sensitivity and specificity, while other agents have higher rates of false-positive and false-negative test results (Auerbach, 1993; 2009). Following diagnosis of FA by DEB testing, molecular studies should be pursued, to identify the complementation group of the patient as well as the specific pathogenic variants. Due to the extensive phenotypic heterogeneity in FA, including heterogeneity in the degree of sensitivity to crosslinking agents in some rare cases, and phenotypic overlap with this large number of other genes, whole exome sequencing (WES) may be performed to investigate the diagnosis of a patient.
In this unit, a technique for applying the DEB test to rule out a diagnosis of FA using a peripheral blood sample from the patient is outlined (see Basic Protocol). In addition, instructions for working with DEB (see Support Protocol 1) and for staining slides for chromosome-breakage analysis (see Support Protocol 2), which is performed on unbanded metaphase preparations, are also given. A detailed method for applying the DEB test to cultured fibroblasts that grow as a monolayer attached to the bottom of the flask rather than in suspension, as is the case with peripheral blood lymphocytes, is also described (see Alternate Protocol 1). Finally, methods for using the DEB test for prenatal diagnosis of FA utilizing fetal cells obtained by chorionic villus sampling (CVS), amniocentesis, or fetal blood sampling, are also detailed (see Alternate Protocol 2).
CAUTION: Radioactive, biological, and chemical substances require special handling; see APPENDIX 2A for guidelines.
CAUTION: DEB is a potential carcinogen and precautions should be taken when handling this compound (see Support Protocol 1).
NOTE: All reagents and equipment coming into contact with live cells must be sterile.
NOTE: Refer to UNIT 1.13 for more information concerning Mendelian Inheritance in Man (MIM) accession numbers.
BASIC PROTOCOL
DIEPOXYBUTANE TEST FOR POSTNATAL DIAGNOSIS Of FANCONI ANEMIA
The preferred tissue for laboratory diagnosis of FA by DEB testing is peripheral blood. The sample is easy to obtain and to work with, and results of chromosome-breakage analysis of peripheral blood lymphocytes can be obtained within 3 to 4 days. Peripheral blood is cultured in complete RPMI medium with phytohemagglutinin (PHA). DEB, a highly reactive DNA cross-linking agent with a half-life in aqueous solution of ~4 days, is added to the cultures 24 hr later. After an additional 48 to 72 hr, ~2 cell cycles in DEB-treated medium, cultures are harvested and chromosome spreads are prepared for chromosome breakage analysis on Giemsa-stained metaphases. At a concentration of 0.1 μg/ml, DEB induces multiple chromosomal breaks and exchanges in FA cells, but has little clastogenic effect on cells from non-FA individuals.
Materials
Peripheral blood: collect in a preservative-free sodium heparin Vacutainer tube (e.g., Fisher)
Complete RPMI/15% FBS medium (APPENDIX 3G)/1% (w/v) phytohemagglutinin (PHA; Burroughs Wellcome)
PBS (APPENDIX 2D)
Diepoxybutane (1,3-butadiene diepoxide, DEB; Aldrich; store at 4°C)
1 μg/ml Colcemid (e.g., Life Technologies; store at 4°C)
0.075 M KCl, 37°C
Fixative: 3:1 (v/v) methanol/glacial acetic acid, fresh
10-ml syringe equipped with 18½-G needle
25-cm2 tissue culture flasks
15-ml sterile conical-bottom centrifuge tubes with caps
IEC clinical centrifuge or equivalent
Microscope slides (store in 70% ethanol)
Inverted microscope
Additional reagents and equipment for collection of peripheral blood lymphocytes (UNIT 4.1), tissue culture (APPENDIX 3G), preparing metaphase chromosome slides (UNIT 4.1), and karyotyping (APPENDIX 4A)
NOTE: All incubations are performed in a humidified 37°C, 5% CO2 incubator unless otherwise specified.
Set up peripheral blood cell culture
-
Drop peripheral blood (18 to 25 drops; 0.4 to 0.5 ml) from a 10-ml syringe equipped with an 18½-G needle into 25-cm2 tissue culture flasks containing 10 ml complete RPMI/15% FBS/1% PHA. Set up duplicate cultures for DEB studies for two concentrations of DEB and duplicate cultures to serve as untreated controls (a total of six flasks). When there is reason to believe the white count is low, set up from the buffy coat. Incubate cultures for 24 hr (APPENDIX 3G).
Phytohemagglutinin is used to transform lymphocytes into dividing lymphoblasts.
Prepare DEB
-
2
Aliquot PBS to four 15-ml sterile tubes: 10 ml to tube 1, 6 ml to tube 2, and 4.5 ml to tubes 3 and 4.
-
3
Just prior to addition to culture, add 10 μl stock DEB to tube 1, cap, and mix well. Transfer 4 ml from tube 1 to tube 2, cap, and mix well. Transfer 0.5 ml (500 μl) from tube 2 to tube 3, cap, and mix well. Make the final dilution of DEB by taking 0.5 ml (500 μl) from tube 3 and adding it to tube 4.
CAUTION: See Support Protocol 1 for safety guidelines for working with DEB.
-
4
Add 25 μl of the solution from tube 3 to two of the peripheral blood cultures that have been incubating 24 hr (from step 1). Add 25 μl of the solution from tube 4 to two of the peripheral blood cultures that have been incubating 24 hr (from step 1). Incubate all cultures, included the untreated control flasks, an additional 48 to 72 hr.
The final concentrations of DEB in the flasks are 0.1 μg/ml and 0.01 μg/ml respectively.
Harvest cultures
-
5
Add 2 ml of 1 μg/ml Colcemid to each 10-ml culture and incubate 20 min.
-
6
Transfer contents of each culture flask to a 15-ml conical-bottom centrifuge tube, cap, and centrifuge 10 min at ~150 × g (800 to 1000 rpm in an IEC clinical centrifuge).
-
7
Remove most of the supernatant. Resuspend each pellet in the residual supernatant by flicking the tube with finger, and carefully add 5 ml prewarmed 0.075 M KCl. Incubate 10 min in a 37°C water bath. Centrifuge 10 min at 150 × g, room temperature.
Hypotonic KCl is used to swell the cells without breaking them.
-
8
Remove the supernatant and gently add 1 ml fresh fixative without disturbing the pellet. Remove fixative immediately and repeat this step.
-
9
Immediately break up the pellet by striking the tube several times with an index finger. Quickly add fixative to resuspend the cells without getting clumps. Bring volume to 8 to 10 ml with fixative. Break up any clumps by pipetting with a Pasteur pipet. Let stand 30 min at room temperature.
-
10
Centrifuge 10 min at 150 × g, room temperature. Remove supernatant and resuspend pellet in 3 to 5 ml fixative. Let stand 10 min at room temperature. Repeat this step one or two more times either immediately, or after letting cells stand in fixative overnight at room temperature.
This procedure should be sufficient to remove red cell debris and fix the cells.
-
11
Centrifuge 10 min at 150 × g, room temperature. Remove the supernatant, break the pellet, and add enough fixative (~0.25 to 0.5 ml) to give the desired cell concentration for slide-making.
An optimal cell concentration provides a maximum number of cells in a microscope field without crowding metaphase spreads.
-
12
Drop a few drops of the cell suspension onto a clean microscope slide from a height sufficient to make well-spread metaphases (UNIT 4.1). Make at least six slides per culture.
This is the most critical part of the protocol. The technique should be practiced with cells that are not being used for diagnosis.
-
13
Examine slides on an inverted microscope to determine quantity and quality of metaphases. Stain slides with Giemsa (see Support Protocol 2).
Metaphases should be plentiful and not broken. Chromosomes should be adequately fixed and well spread.
Analyze slides for chromosome breakage
-
14
Analyze 25 Giemsa-stained metaphases from treatment with low-DEB concentration and 50 metaphases from high-concentration DEB-treated preparations (25 metaphases for cases with very high breakage; APPENDIX 4A). In cases with elevated breakage with low-DEB concentration, score 25 metaphases from untreated preparation.
Score for chromosome number and for the numbers and types of structural abnormalities. If chromosomal breakage is increased above the normal range, analyze an untreated preparation. Photograph abnormal cells for later analysis.
See Figure 8.7.1 for exemplary data, and APPENDIX 4C and ISCN (2013) for a description of and nomenclature for acquired chromosome aberrations.
-
15
Interpret results according to published values for baseline and DEB-induced chromosomal breakage in PHA-stimulated peripheral blood lymphocytes.
Table 8.7.2 presents data for baseline and treated cultures from normal and affected individuals. See Anticipated Results for more information regarding data analysis.
Figure 8.7.1.
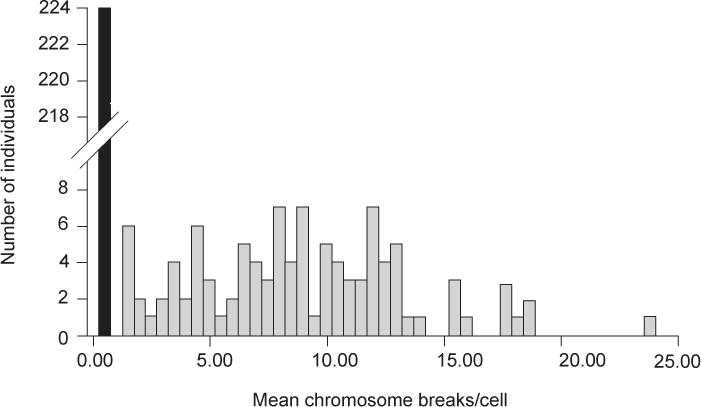
DEB-induced chromosomal breakage in peripheral blood lymphocytes from patients studied at the Rockefeller University. Black bar, DEB-insensitive (non-FA) patients; gray bars, DEB-sensitive (Fanconi anemia) patients. There is no overlap in the range for the two groups when data are expressed as mean chromosome breaks per cell. Note the wide range of breakage seen in FA cells. Reproduced from Auerbach et al. (1989) with permission of Grune & Stratton, publisher.
Table 8.7.2.
Range of Chromosomal Breakage Observed in Peripheral Blood Lymphocytes and Cultured Fetal Cells
| Tissue | Diagnosis | Mean breaks/cell
|
No. individuals tested | |
|---|---|---|---|---|
| Baseline | DEB-treateda | |||
| Peripheral blood | FA | 0.02–0.85b | 1.10–23.9 | 98 |
| Non-FA | 0.00–0.12 | 0.00–0.36 | 124 | |
| Fibroblasts | FA | 0.20–0.36 | 0.68–1.10 | 3 |
| Non-FA | 0.00–0.08 | 0.00–0.07 | 4 | |
| Amniocytes | FA | 0.18–0.45 | 0.69–0.96 | 10c |
| Non-FA | 0.00–0.12 | 0.00–0.14 | 48c | |
| Chorionic villus cells | FA | 0.30–0.46 | 1.00–1.40 | 9c |
| Non-FA | 0.00–0.10 | 0.00–0.14 | 39c | |
DEB (diepoxybutane) concentration is 0.1 μg/ml in peripheral blood cultures, and 0.01 μg/ml in fibroblast, amniotic fluid, and chorionic villus cell cultures.
Data show the range for mean breaks/cell in each category.
Number of prenatal diagnoses performed in pregnancies of couples who had a previously affected child (A.D.A., unpub. observ.).
SUPPORT PROTOCOL 1
WORKING WITH AND DISPOSING OF DIEPOXYBUTANE
DEB (1,3-butadiene diepoxide) is a potential carcinogen and precautions should be taken when handling this compound. This section describes how to work with DEB and the proper disposal of wastes.
Work space
All work with DEB must be done using a chemical fume hood or a class II Biological Safety Cabinet. The investigator should wear a laboratory coat and gloves when working with DEB. A bottle of 6 M HCl (~50 ml) should be kept at hand to use to rapidly inactivate DEB in case of spills.
Cell cultures
Cell culture work involving DEB should be done in a class II Biological Safety Cabinet. Once the cells are fixed they may be processed outside the safety cabinet. Spent medium and wash solutions should be disposed of by adding 6 M HCl and discarding as biohazardous waste. Used disposable pipets and culture flasks should be rinsed with 6 M HCl and discarded in autoclavable biohazard bags. Micropipet tips should be discarded into a small bottle of 6 M HCl.
Waste disposal
The stock solution of DEB should be disposed of as hazardous chemical waste without dilution or chemical inactivation. Disposable plasticware should be rinsed with 6 M HCl and discarded in autoclavable biohazard bags. Medium and wash solutions should be treated with 6 M HCl and disposed of as biohazardous waste.
SUPPORT PROTOCOL 2
GIEMSA STAINING FOR CHROMOSOME-BREAKAGE ANALYSIS
Giemsa stain is a histological stain with a particular affinity for nuclei and chromosomes. The procedure is simple and results in unbanded reddish-blue to purple nuclei and chromosomes.
Materials
Gurrs buffer, pH 6.8, tablets (Bio/medical Specialties)
Giemsa stain (e.g., Life Technologies)
Metaphase chromosome slide (see Basic Protocol)
Permount histological mounting medium
Coplin jars
Dissolve one Gurrs buffer, pH 6.8, tablet in 1 liter water.
-
Add 4 ml Giemsa stain to 46 ml Gurrs buffer in a staining jar.
Stain can be used for ~1 hr or to stain two groups of slides, ten slides each group. The staining solution must be prepared fresh before each use.
Stain metaphase chromosome slide 5 min.
Rinse by dipping several times in a Coplin jar containing Gurrs buffer (step 1).
Rinse slide by dipping several times in a Coplin jar containing water.
Air dry the slide.
-
Check slide for stain intensity.
Stain intensity should be sufficient so that gaps and breaks in the chromosomes appear as achromatic (nonstained) regions.
Put a coverslip on the slide using Permount histological mounting medium.
ALTERNATE PROTOCOL 1
DIEPOXYBUTANE TEST USING FIBROBLAST CULTURES
If peripheral blood is not available for testing, fibroblast cultures can be initiated from skin biopsies. Small pieces of lung or skin taken from abortuses or stillborns can also be used for testing. DEB is used at a 10-fold lower concentration for fibroblast cultures because the higher concentration is too toxic for FA fibroblasts and makes it difficult to collect sufficient metaphases for study.
Materials (also see Basic Protocol)
Tissue biopsies (e.g., full-thickness skin sample taken by a 3-mm punch)
Complete DMEM/20% and FBS (APPENDIX 3G)
0.051 M (0.38% w/v) KCl
60-mm tissue culture plates
Disposable scalpels
Additional reagents and equipment for passaging cells by trypsinization and counting cells (APPENDIX 3G)
NOTE: All incubations are performed in a humidified 37°C, 5% CO2 incubator unless otherwise specified.
Set up fibroblast culture
Place the tissue biopsy in 0.5 ml complete DMEM/20% FBS in a 60-mm tissue culture plate and cut it into 1-mm pieces with two sterile disposable scalpels.
-
Score the bottoms of at least three additional 60-mm plastic tissue culture plates by making horizontal and vertical lines with the point of a sterile scalpel. Add a few drops of complete DMEM/20% FBS to the plate with the biopsy pieces. Use a sterile Pasteur pipet to transfer 5 to 6 biopsy pieces into each 60-mm plate. Place the tissue pieces at the intersections of the scored lines.
The score lines aid in the attachment of biopsy pieces and later in locating colonies.
Incubate the plates, without any additional tissue culture medium, 15 to 30 min to allow the pieces of tissue to adhere to the plates. Add 5 ml complete DMEM/20% FBS to each plate and incubate the cultures.
-
Change the medium every 3 to 4 days without disturbing the tissue pieces. Use an inverted microscope to inspect the cultures for evidence of cell growth.
FA cells are more difficult to establish in culture than normal cells, and usually grow more slowly. It may take from a week to a month to observe good cell growth.
Subculture cells
-
5
When a large patch of fibroblasts can be seen surrounding the tissue pieces in the plate, passage the cells by treating them with 1× trypsin/EDTA solution (APPENDIX 3G), taking care not to dislodge the biopsy pieces as they will continue to grow in culture. Transfer cells to a sterile 25-cm2 tissue culture flask in complete DMEM/15% FBS.
These cells are designated as first passage. Reduce the concentration of FBS in the DMEM to 15% for this and all subsequent subcultures.
-
6
Trypsinize cells again when the first passage culture reaches confluency (APPENDIX 3G). Subculture at a 1:3 split ratio in complete DMEM/15% FBS. Proceed with DEB-testing for FA when at least three second-passage cultures reach confluency.
This culture is the second passage.
Treat cultures with DEB
-
7
Plate cells in 5 ml complete DMEM/15% FBS in 25-cm2 flasks at a density of 3 ×105 cells per flask. Incubate 24 hr. In addition to the cells being tested, normal cultured fibroblasts and FA-affected fibroblasts should be set up in parallel and handled in the same way the as test cells. This will serve as negative and positive control cultures.
Six flasks, two treated at each of two DEB concentrations (0.01 μg/ml and 0.1 μg/ml) and two untreated controls, are required for this analysis. If there are sufficient cells, make two additional flasks for repeat DEB treatment, and keep additional untreated flasks as a reserve.
-
8
Make DEB dilutions as described (see Basic Protocol, steps 2, 3 and 4).
-
9
Add 25 μl of the solution from tube 4 to 10 ml fresh complete DMEM/15% FBS. Replace the growth medium in two cultures with this DEB-containing medium (5 ml in each flask; 0.01 μg/ml final). Add 25 ul of the solution from tube 3 to 10 ml fresh complete DMEM/15% FBS. Replace the growth medium in two cultures, with this DEB-containing medium (5 ml in each flask). The final concentration of DEB in the culture is 0.1 ug/ml.
The flasks treated with a higher concentration of DEB (0.1 μg/ml final) may help with the diagnosis of affected cultures, as FA fibroblasts will usually not grow at this concentration. Affected cells may appear as if they are undergoing apoptosis; it is likely that metaphases will only be obtained if the cultures are unaffected. Even at this concentration of DEB, normal metaphases will have little increased chromosomal breakage. Additional reserve cultures with growth medium but without DEB should continue to be cultured until testing is complete, in case insufficient metaphases for analysis are obtained from the cultures in this step.
-
10
Incubate cells until the cultures are nearly confluent (~72 hr).
-
11
If cells are growing well, trypsinize cells (APPENDIX 3G) and subculture at a 1:2 or 1:3 split ratio into fresh complete DMEM/15% FBS without DEB to collect sufficient mitotic cells for cytogenetic studies. Alternatively, if cells are growing slowly, remove DEB-containing medium and replace with fresh complete medium.
Harvest cultures
-
12
Harvest cultures at the first mitotic wave after subculture (~24 to 48 hr). At 3 hr prior to harvest, add 1 ml of 1 μg/ml Colcemid to the DEB-treated cultures and the untreated controls (in 5 ml medium). Incubate 3 hr.
The timing for harvesting is determined by inspection of the cultures with an inverted microscope. Metaphase cells appear rounded.
-
13
Trypsinize cells (APPENDIX 3G). Transfer the contents of the culture flask to a sterile 15-ml centrifuge tube, cap, and centrifuge 10 min at 150 × g (800 to 1000 rpm in an IEC clinical centrifuge).
-
14
Remove most of the supernatant. Resuspend the pellet in the remaining supernatant and slowly add 5 ml of 0.051 M KCl. Incubate 10 min in a 37°C water bath. Centrifuge 10 min.
This hypotonic solution contains less KCl than that used for peripheral blood cultures. The greater hypotonicity aids in obtaining adequate swelling of fibroblasts which are larger and contain more cytoplasm than peripheral blood lymphocytes.
-
15
Fix cells and prepare slides as described (see Basic Protocol steps 9 to 12; in step 9, slowly add 3 to 5 ml of fixative).
-
16
Analyze at least 50 metaphases from high-concentration DEB, 25 cells from low-concentration DEB and 25 metaphases from untreated cultures, using each of two primary flasks in each group. Interpret results according to published values for baseline and DEB-induced chromosomal breakage in FA fibroblasts (Table 8.7.2) as well as simultaneously studied positive and negative control cultures.
ALTERNATE PROTOCOL 2
DIEPOXYBUTANE TEST FOR PRENATAL DIAGNOSIS OF FANCONI ANEMIA
FA can be diagnosed in pregnancies at risk by studying cultured trophoblast cells obtained by chorionic villus sampling (CVS) at 9 to 12 weeks of gestation, or amniotic fluid cells obtained by amniocentesis at 15 to 17 weeks. FA can also be diagnosed prenatally from a fetal blood sample using methods similar to those described previously for peripheral blood. Results with fetal blood can be obtained within 3 days from the time the sample is collected so this method is of value in pregnancies where a prenatal diagnosis could not otherwise be completed before 24 weeks.
Study of Fetal Cells Obtained by CVS or Amniocentesis
Additional Materials
Fetal cells, enough to establish cultures in two 25-cm2 flasks
Growth medium: 1:1 mixture of Chang medium (UNIT 8.4) and complete RPMI/15% FBS (APPENDIX 3G)
Additional materials and equipment for culture of chorionic villi (UNIT 8.3) and amniotic fluid (UNIT 8.4)
NOTE: All incubations are performed in a humidified 37°C, 5% CO2 incubator unless otherwise specified.
-
Establish fetal cell cultures in growth medium in 25-cm2 flasks and incubate (UNITS 8.3 & 8.4).
Chang medium facilitates growth of these cells, especially after subculture. The increased chromosomal breakage reported to be associated with this medium does not interfere with identification of affected fetuses.
Trypsinize and subculture primary cultures when flasks contain several large, rapidly growing colonies (APPENDIX 3G). Do not allow colonies to become overgrown. Use a split ratio of 1:2 or 1:3. Incubate 24 hr.
-
Replace the growth medium in at least two flasks (from two different primary cultures) with fresh growth medium containing 0.01 μg/ml of DEB, and 0.1 μg/ml of DEB in two extra flasks, if available, freshly diluted as described (see Alternate Protocol 1, steps 8 and 9).
The remaining flasks are untreated controls for baseline chromosome breakage studies.
Incubate the cultures, harvest and fix the cells, and prepare metaphase spreads as described (see Alternate Protocol 1, beginning of step 10).
Analyze a total of 50 metaphases from high-concentration DEB, 25 cells from low-concentration DEB and 25 metaphases from untreated cultures, using each of two primary flasks in each group. Interpret results according to published values for baseline and DEB-induced chromosomal breakage in FA fetal cells obtained by CVS or amniocentesis (Table 8.7.2) as well as simultaneously studied positive and negative control cultures.
Study Cells from Fetal Blood
Add ~0.25 ml heparinized fetal blood (collected in a preservative-free sodium heparin Vacutainer tube; e.g., Fisher) to a 25-cm2 flask containing 10 ml complete RPMI/15% FBS. Set up two to four flasks for baseline and DEB-treated cultures, depending on the amount of fetal blood collected.
Incubate, treat, harvest, and analyze fetal blood cultures as described (see Basic Protocol, steps 2 to 15).
COMMENTARY
Background Information
Fanconi anemia (FA) is a genetically and phenotypically heterogeneous autosomal recessive disorder defined by cellular hypersensitivity to DNA cross-linking agents (Auerbach et al., 1989; Auerbach, 1993). Clinically, FA is characterized by congenital malformations and progressive marrow failure, as well as predisposition to AML, squamous cell carcinoma, and other malignancies (Kutler et al., 2003; Wagner et al., 2003). The hypersensitivity to DNA cross-linking agents such as diepoxybutane (DEB) is used as a diagnostic test for this disease and allows the clinician to make a diagnosis of FA in patients with subtle clinical features, including those without clinically detectable congenital anomalies. Approximately one-third of FA patients do not have any obvious birth defects, but present at the time of onset of hematologic manifestations such as bone-marrow failure or acute myelogenous leukemia (Auerbach et al., 1989; Giampietro et al., 1997). These patients may manifest short stature, but this need not be remarkable. FA should be included in the differential diagnosis of all children and young adults with aplastic anemia, and the DEB test should be performed in these cases. All siblings of affected probands should also have DEB testing because a lack of concordance of phenotype in affected siblings makes clinical diagnosis unreliable even within sibships (Giampietro et al., 1993).
Delay in the diagnosis of FA may have serious consequences for the patient and family. Optimal treatment regimens for aplastic anemia depend on the etiology of the bone-marrow failure. Bone marrow or umbilical cord blood transplantation from an HLA-identical sibling is now considered the treatment of choice for FA. Prior to transplant, it is essential that potential sibling donors have DEB testing to rule out FA. Because FA patients are hypersensitive to all DNA cross-linking agents, they require a modified pretransplant conditioning regimen with a lower than usual dose of cyclophosphamide. Timely diagnosis of FA allows parents to explore their options in regard to family planning, including the conception of additional children who may be potential donors for transplant (Auerbach et al., 1990). Thus, it is now possible for FA families using preimplantation genetic diagnosis with in vitro fertilization (PGD/IVF) to select embryos for transfer that are disease free and HLA-matched to the affected child (Verlinsky et al., 2001; Grewal et al., 2004). A delay in diagnosis of FA in an affected child may result in families conceiving additional affected offspring before they are aware of the recurrence risk. Timely diagnosis is also important so that a thorough workup for FA-associated developmental disabilities such as speech, language, and motor delays are performed and the appropriate therapy implemented.
Currently, testing for DEB-induced chromosomal breakage provides the most reliable method for diagnosis of FA prenatally and postnatally (Auerbach and Wolman, 1976; Auerbach et al., 1985, 1986). The heterogeneous nature of FA makes an understanding of the correlation between genotype and phenotype important in the clinical management of FA patients. Currently, the pace of bone marrow failure and survival can be predicted for certain genotypes (Kutler et al, 2003); therefore, once FA is diagnosed, it is recommended that the gene defect be determined. This will also enable molecular diagnosis of heterozygous carriers in FA families, which cannot be done by DEB testing. The c.456+4A>T (legacy IVS4 + 4A>T) mutation in FANCC has a carrier frequency of >1/100 in the Ashkenazi Jewish population (Verlander et al., 1995). Testing for this single mutation in FANCC will be positive in ~85% of FA patients of Ashkenazi Jewish ancestry. Although 65% of FA patients are in complementation group FA-A, only two common mutations have been identified in FANCA: c.1115_1118delTTGG and c.3788_3790delTCT (Levran et al., 1997), while the rest of the mutations found in this gene are private or semiprivate. It is estimated that approximately 40% of mutations in FANCA are large genomic deletions, and that most patients are compound heterozygotes, making detection of FANCA mutations by the usual methods of exon screening from genomic DNA difficult (Flynn et al., 2014). FANCG is also characterized by many private mutations, but several ethnic-specific mutations in Korean/Japanese, Portuguese-Brazilian, French-Acadian, and Northern European FA families have been demonstrated and are easy to test with mutation-specific assays in appropriate populations (Auerbach et al., 2003).
Interstrand crosslink (ICL) repair occurs predominantly during S-phase, following replication fork stalling at the ICL, that triggers monoubiquitination of FANCD2 and FANCI, the central event of the FA pathway (reviewed in Garner and Smogorzewska, 2011). It has been suggested that immunoblotting with FANCD2 antibody to detect the presence or absence of the monoubiquitinated FANCD2-L “long” form (protein extracts from cells from FA complementation groups FA-A, -B, -C, -E, -F, -G and FA-L show only the unmodified lower FANCD2-S “short” form) be used as a diagnostic screen for FA, and as an assay to detect complementation group after transduction by cDNA-containing retroviral vectors (Shimamura et al., 2002). While this may be a useful adjunct to standard tests of hypersensitivity to the clastogenic effect of DNA cross-linking agents such as DEB, its sensitivity, specificity, and reproducibility for diagnosis are not adequate. D2 ubiquitination is normal in FA groups FA-D1/BRCA1 (Howlett et al., 2002), FA-J/BRIP1 (Levran et al., 2005), FA-N/PALB2 (Reid et al., 2007), FA-O/RAD51C (Vaz et al., 2010), FA-P/SLX4 (Kim et al., 2011), FA-Q/ERCC4/XPF (Bogliolo et al., 2013) and also in FA patients exhibiting somatic mosaicism. Analysis of chromosomal breakage after DEB treatment enables estimation of the percentage of cross-link-resistant cells, indicating the degree of mosaicism. As this may influence outcome for FA patients, it is important to apply a diagnostic test where this can be determined.
Critical Parameters and Troubleshooting
The most critical step in this protocol is slide preparation (see UNIT 4.1). Poor preparations are difficult to analyze, require additional microscopy time, and result in less accuracy in visualizing breakage. Metaphases need to be well-fixed and adequately spread, but not broken. Obtaining good preparations requires experience. Atmospheric conditions such as temperature and humidity have a significant influence on metaphase spreading, and trial and error are required to obtain optimal metaphase preparations. If necessary, additional humidity can be obtained using a humidifier or by steaming a slide over a beaker of boiling water for a minute after the cells are dropped onto the slide. The slides can then be dried 5 min on a hot plate at 60°C to obtain better spreading. If cells tend to rupture, they should be dropped on the slide more gently and additional steam or heat should not be used.
It is important to follow the safety precautions outlined in Support Protocol 1 for working with and disposing of DEB, as this compound is a potential carcinogen.
This protocol should not be performed on an occasional basis, as interpretation of results may be difficult unless the laboratory is routinely performing DEB testing to rule out Fanconi anemia. The microscopist must be experienced in chromosome-breakage analysis. Also, because of considerable individual variation in DEB sensitivity, both positive and negative controls should be studied until the laboratory staff is experienced with the performance of the DEB test. A confounding factor in the interpretation of the test results is the possible presence in the specimen of revertant cells. It is estimated that 10% to 25% of FA patients will exhibit two populations of PHA-stimulated lymphocytes, one that is sensitive to the clastogenic effects of DNA cross-linking agents and one that is resistant (Auerbach et al., 1989). This has now been shown to be the result of a somatic mutation event affecting one mutant allele in an FA patient. Several molecular mechanisms for this revertant mosaicism have now been elucidated in cultured FA-lymphoblastoid cells (LCLs; i.e., transformed B cell lines; Lo Ten Foe et al., 1997; Waisfisz et al., 1999) and even in hematopoietic stem cells from the bone marrow of a mosaic FA patient (Gregory et al., 2001). The presence of somatic revertant mosaicism may be a critical factor in interpreting DEB test results for diagnosis of FA. Normal individuals should not exhibit any metaphases with multiple chromatid breaks and exchange figures typically seen after treatment of PHA-stimulated peripheral blood cultures from FA patients with the concentration of DEB recommended in this protocol. Thus, the presence of just a few such aberrant cells should lead one to suspect FA with somatic mosaicism. The study should be repeated to confirm that the finding of revertant mosaicism was not a laboratory artifact. The percentage of DEB-resistant metaphases should be included in the laboratory report of results of tests to rule out FA, as it is useful for the clinician caring for FA patients to have information regarding somatic mosaicism. Clinical data suggest that T cell mosaicism may be a risk factor for graft rejection in recipients of unrelated marrow, and hematopoietic stem cell mosaicism may be associated with an altered “natural history” and stable hematopoietic function in the patient (Wagner et al., 2003). It is also important to be aware of any prior treatment of the test subject with chemotherapy, a compounding factor that needs to be considered. This will cause an increase in baseline breakage, but results in DEB-treated cultures should be similar to baseline. Another problem of fairly common occurrence is that of no growth of peripheral blood cultures in individuals with severe acquired aplastic anemia. This problem is seldom found in cultures from patients with genetic causes of aplastic anemia such as FA. If there are no metaphases to analyze in untreated as well as DEB-treated cultures, the test should be repeated. If the results are the same, the diagnosis is not likely to be FA. If the phenotype includes congenital malformations as well as bone marrow failure, it is recommended that an alternate tissue such as cultured fibroblasts be tested.
The reported finding that some mutations in the NBS1 gene for Nijmegen breakage syndrome can also result in a pattern of DEB-induced chromosomal breakage similar to that found in FA mosaic patients (Nakanishi et al., 2002), necessitates caution in interpretation of results of testing and the need to consider clinical symptoms in making a diagnosis of FA. As the FA pathway is further elucidated, it will ultimately be possible to identify the specific gene defect in each patient. Recent identification of rare patients with FA-like phenotypes and elevated, but lower than typical, DEB-sensitivity should be noted; this has been associated in particular with pathogenic variants in FANCO, FANP and FANCQ, as well as a few patients for whom all identified FA genes have been ruled out.
Anticipated Results
The mean baseline chromosomal breakage frequency in peripheral blood lymphocytes ranges from 0.00 to 0.05 breaks per cell in normal control individuals. The range is from 0.02 to 0.85 breaks per cell in FA patients, and from 0.00 to 0.12 in non-FA patients with some clinical features overlapping those seen in FA. Note that the baseline breakage in some FA patients does not differ from normal controls; some FA patients would be misdiagnosed if only baseline breakage studies were considered. The mean DEB-induced chromosomal breakage frequency in peripheral blood lymphocytes for normal control individuals ranges from 0.00 to 0.10 breaks per cell, for the FA patients from 1.06 to 23.9 breaks per cell, and for non-FA patients with some features of the syndrome from 0.00 to 0.36 breaks per cell. In this assay, no overlap has been observed in the range for the FA group compared with control groups (see Fig. 8.7.1 and Table 8.7.2). It is important to note that in cultures from individuals affected with FA, the percent of metaphases exhibiting the high number of chromosomal breaks and exchanges typical of FA can vary from as low as 10% to as high as 100% (see Fig. 8.7.2 for an example of such a cell). A multivariate regression analysis of data from DEB studies shows that discrimination between DEB+ and DEB− patients is best achieved by calculating breakage as breaks per cell (Auerbach et al., 1989). Also, presence of radial configurations (at two concentrations of DEB, in a clearly dosage dependent manner) is an important correlate in calling a positive result.
Figure 8.7.2.
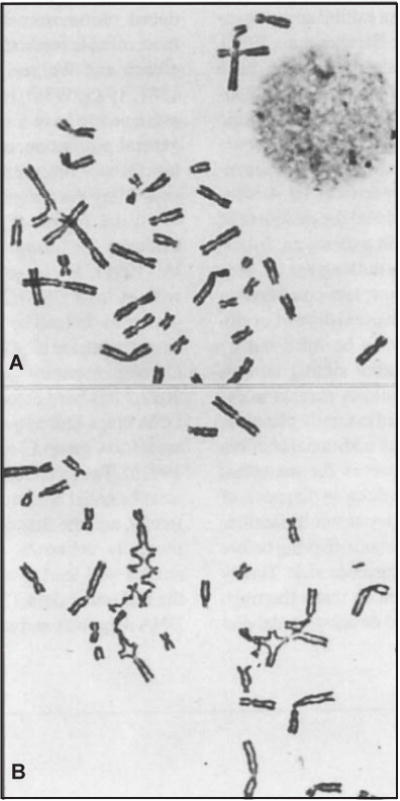
Partial metaphase spreads from Fanconi anemia lymphocytes. (A) Untreated cell showing baseline chromatid aberrations. (B) Cell exposed to 0.1 μg/ml diepoxybutane (DEB). The multiple chromatid exchange figures seen at this concentration are unique to FA cells. Reproduced from Auerbach et al. (1981) with permission of the American Academy of Pediatrics.
Table 8.7.2 can be used as a guide for the interpretation of data from the results of prenatal DEB testing using cells obtained by CVS or amniocentesis.
Time Considerations
For peripheral-blood studies (see Basic Protocol), cells are cultured for 72 to 96 hr prior to harvesting. Culture set up, preparation of DEB dilutions, and addition of DEB to cultures takes ~30 min. Harvesting takes 2½ to 3 hr. The procedure can be interrupted after the second fixation step and cells left in fixative overnight. The next morning cells can be refixed and slides made. It takes 30 to 45 min to make and grade slides. An experienced cytogeneticist should be able to perform the microscopic analysis of a case in 1 hr.
Cultured fetal cells (from CVS or amniocentesis) and cultured skin fibroblasts may take 10 to 14 days and 3 to 4 weeks in culture, respectively, before there are adequate numbers of cells to start the DEB study. Cells are then subcultured twice before harvesting; this takes an additional 4 to 9 days depending on the doubling time of the cell culture. Trypsinization and subculture steps require ~20 min each time they are repeated. Time considerations for the alternate protocols are similar to the basic protocol for peripheral blood, although it may take longer to obtain adequate fixation.
Acknowledgments
Supported in part by grant # UL1 TR000043 from the National Center for Advancing Translational Sciences (NCATS, National Institutes of Health (NIH) Clinical and Translational Science Award (CTSA) program, and the Vilcek Foundation.
Footnotes
Internet Resources
http://www.rockefeller.edu/fanconi/mutate
Fanconi Anemia Mutation Database.
Contributed by Arleen D. Auerbach, Rockefeller University, New York, New York
Literature Cited
- Albers CA, Paul DS, Schulze H, Freson K, Stephens JC, Smethurst PA, Jolley JD, Cvejic A, Kostadima M, Bertone P, Breuning MH, Debili N, Deloukas P, Favier R, Fiedler J, Hobbs CM, Huang N, Hurles ME, Kiddle G, Krapels I, Nurden P, Ruivenkamp CAL, Sambrook JG, Smith K, Stemple DL, Strauss G, Thys C, Van Geet C, Newbury-Ecob R, Ouwehand WH, Ghevaert C. Compound inheritance of a low-frequency regulatory SNP and a rare null mutation in exon-junction complex subunit RBM8A causes TAR syndrome. Nature Genet. 2012;44:435–439. doi: 10.1038/ng.1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alder JK, Parry EM, Yegnasubramanian S, Wagner CL, Lieblich LM, Auerbach R, Auerbach AD, Wheelan SJ, Armanios M. Telomere phenotypes in females with heterozygous mutations in the dyskeratosis congenita 1 (DKC1) gene. Hum Mutat. 2013;34:1481–1485. doi: 10.1002/humu.22397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armanios M, Chen JL, Chang YPC, Brodsky RA, Hawkins A, Griffin CA, Eshleman JR, Cohen AR, Chakravarti A, Hamosh A, Greider CW. Haploinsufficiency of telomerase reverse transcriptase leads to anticipation in autosomal dominant dyskeratosis congenita. Proc Nat Acad Sci. 2005;102:15960–15964. doi: 10.1073/pnas.0508124102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auerbach AD. Fanconi anemia diagnosis and the diepoxybutane (DEB) test. Exp Hematol. 1993;21:731–733. [PubMed] [Google Scholar]
- Auerbach AD. Fanconi anemia and its diagnosis. Mutat Res. 2009;668:4–10. doi: 10.1016/j.mrfmmm.2009.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auerbach AD, Wolman SR. Susceptibility of Fanconi’s anaemia fibroblasts to chromosome damage by carcinogens. Nature. 1976;261:494–496. doi: 10.1038/261494a0. [DOI] [PubMed] [Google Scholar]
- Auerbach AD, Sagi M, Adler B. Fanconi anemia: Prenatal diagnosis in 30 fetuses at risk. Pediatrics. 1985;76:794–800. [PubMed] [Google Scholar]
- Auerbach AD, Zhang M, Ghosh R, Pergament E, Verlinsky Y, Nicholas H, Boué J. Clastogen-induced chromosomal breakage as a marker for first trimester prenatal diagnosis of Fanconi anemia. Hum Genet. 1986;73:86–88. doi: 10.1007/BF00292671. [DOI] [PubMed] [Google Scholar]
- Auerbach AD, Rogatko A, Schroeder-Kurth TM. International Fanconi Anemia Registry: Relation of clinical symptoms to diepoxybutane sensitivity. Blood. 1989;73:391–396. [PubMed] [Google Scholar]
- Auerbach AD, Liu Q, Ghosh R, Pollack MS, Douglas GW, Broxmeyer HE. Prenatal identification of potential donors for umbilical cord blood transplantation for Fanconi anemia. Transfusion. 1990;30:682–687. doi: 10.1046/j.1537-2995.1990.30891020324.x. [DOI] [PubMed] [Google Scholar]
- Auerbach AD, Greenbaum J, Pujara K, Batish SD, Verlander PC, Levran O. Spectrum of sequence variation in the FANCG gene: An International Fanconi Anemia Registry (IFAR) study. Hum Mutat. 2003;21:158–168. doi: 10.1002/humu.10166. [DOI] [PubMed] [Google Scholar]
- Basson CT, Huang T, Lin RC, Bachinsky DR, Weremowicz S, Vaglio A, Bruzzone R, Quadrelli R, Lerone M, Romeo G, Silengo M, Pereira A, Krieger J, Mesquita SF, Kamisago M, Morton CC, Piermont MEM, Muller CW, Seidman JG, Seidman CE. Different TBX5 interaction in heart and limb defined by Holt-Oram syndrome mutations. Proc Natl Acad Sci. 1999;96:2919–2924. doi: 10.1073/pnas.96.6.2919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogliolo M, Schuster B, Stoepker C, Derkunt B, Su Y, Raams A, Trujillo JP, Minguillon J, Ramirez MJ, Pujol R, Casado JA, Banos R, Rio P, Knies K, Zúñiga S, Benítez J, Bueren JA, Jaspers NG, Schärer OD, de Winter JP, Schindler D, Surrallé J. Mutations in ERCC4, encoding the DNA-repair endonuclease XPF, cause Fanconi anemia. Am J Hum Genet. 2013;92:800–806. doi: 10.1016/j.ajhg.2013.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boocock GRB, Morrison JA, Popovic M, Richards N, Ellis L, Durie PR, Rommens JM. Mutations in SBDS are associated with Shwachman-Diamond syndrome. Nature Genet. 2003;33:97–101. doi: 10.1038/ng1062. [DOI] [PubMed] [Google Scholar]
- Chandrasekharappa SC, Lach FP, Kimble DC, Kamat A, Teer JK, Donovan FX, Flynn E, Sen SK, Thongthip S, Sanborn E, Smogorzewska A, Auerbach AD, Ostrander EA, NISC Comparative Sequencing Program Massively parallel sequencing, aCGH, and RNA-Seq technologies provide a comprehensive molecular diagnosis of Fanconi anemia. Blood. 2013;121:e138–148. doi: 10.1182/blood-2012-12-474585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Draptchinskaia N, Gustavsson P, Anderssson B, Pettersson M, Willig TN, Diazani I, Ball S, Tchernia G, Klar J, Matsson H, Tentler D, Mohandas N, Carlsson B, Dahl N. The gene encoding ribosomal protein S19 is mutated in Diamond-Blackfan anaemia. Nat Genet. 1999;21:169–175. doi: 10.1038/5951. [DOI] [PubMed] [Google Scholar]
- El Ghouzzi V, Le Merrer M, Perrin-Schmitt F, Lajeunie E, Benit P, Renier D, Bourgeois P, Bolcato-Bellemin AL, Munnich A, Bonaventure J. Mutations of the TWIST gene in Saethre-Chotzedn syndrome. Nat Genet. 1997;15:42–46. doi: 10.1038/ng0197-42. [DOI] [PubMed] [Google Scholar]
- Flynn EK, Kamat A, Lach FP, Donovan FX, Kimble DC, Narisu N, Sanborn E, Boulad F, Davies SM, Gillio AP, III, Harris RE, MacMillan ML, Wagner JE, Jr, Smogorzewska A, Auerbach AD, Ostrander EA, Chandrasekharappa SC. Comprehensive analysis of pathogenic deletion variants in Fanconi anemia genes. Hum Mutat. 2014 Aug 28; doi: 10.1002/humu.22680. online. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garner E, Smogorzewska A. Ubiquitylation and the Fanconi Anemia Pathway. FEBS Letters. 2011;585:2853–2860. doi: 10.1016/j.febslet.2011.04.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerrard G, Valganon M, Foong HE, Kasperaviciute D, Iskander D, Game L, Muller M, Aitman TJ, Roberts I, de la Fuente J, Foroni L, Karadimitris A. Target enrichment and high-throughput sequencing of 80 ribosomal protein genes to identify mutations associated with Diamond-Blackfan anaemia. Brit J Haemat. 2013;162:530–536. doi: 10.1111/bjh.12397. [DOI] [PubMed] [Google Scholar]
- Giampietro PF, Adler-Brecher B, Verlander PC, Pavlakis SG, Davis JG, Auerbach AD. The need for more accurate and timely diagnosis in Fanconi anemia: A report from the International Fanconi Anemia Registry. Pediatrics. 1993;91:1116–1120. [PubMed] [Google Scholar]
- Giampietro PF, Verlander PC, Davis JG, Auerbach AD. Diagnosis of Fanconi anemia in patients without congenital malformations: An International Fanconi Anemia Registry study. Am J Med Genet. 1997;68:58–61. [PubMed] [Google Scholar]
- Gregory JJ, Wagner JE, Verlander PC, Levran O, Batish SD, Eide C, Steffenhagen A, Hirsch B, Auerbach AD. Somatic mosaicism in Fanconi anemia: Evidence of genotypic reversion in lympho-hematopoietic stem cells. Proc Natl Acad Sci USA. 2001;98:2532–2537. doi: 10.1073/pnas.051609898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grewal SS, Jeffrey P, Kahn JP, MacMillan ML, Ramsay NKC, Wagner JE. Successful hematopoietic stem cell transplantation for Fanconi anemia from an unaffected HLA-genotype–identical sibling selected using preimplantation genetic diagnosis. Blood. 2004;103:1147–1151. doi: 10.1182/blood-2003-02-0587. [DOI] [PubMed] [Google Scholar]
- Heiss NS, Knight SW, Vulliamy TJ, Klauck SM, Wiemann S, Mason PJ, Poustka A, Dokal I. X-linked dyskeratosis congenita is caused by mutations in a highly conserved gene with putative nucleolar functions. Nat Genet. 1998;19:32–38. doi: 10.1038/ng0598-32. [DOI] [PubMed] [Google Scholar]
- Howlett NG, Taniguchi T, Olson S, Cox B, Waisfisz Q, De Die-Smulders C, Persky N, Grompe M, Joenje H, Pals G, Ikeda H, Fox EA, D’Andrea AD. Biallelic inactivation of BRCA2 in Fanconi anemia. Science. 2002;297:606–609. doi: 10.1126/science.1073834. [DOI] [PubMed] [Google Scholar]
- Shaffer LG, McGowan-Jordan J, Schmid M, editors. ISCN. An International System for Human Cytogenetic Nomenclature. Karger; Basel, Switzerland: 2013. [published in collaboration with Cytogenetic and Genome Research.] [Google Scholar]
- Jongmans MCJ, Verwiel ETP, Heijdra Y, Vulliamy T, Kamping EJ, Hehir-Kwa JY, Bongers EMHF, Pfundt R, van Emst L, van Leeuwen FN, van Gassen KLI, Geurts van Kessel A, Dokal I, Hoogerbrugge N, Ligtenberg MJL, Kuiper RP. Revertant somatic mosaicism by mitotic recombination in dyskeratosis congenita. Am J Hum Genet. 2012;90:426–433. doi: 10.1016/j.ajhg.2012.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y, Lach FP, Desetty R, Hanenberg H, Auerbach AD, Smogorzewska A. Mutations of the SLX4 gene in Fanconi anemia. Nat Genetics. 2011;43:142–146. doi: 10.1038/ng.750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kottemann MC, Smogorzewska A. Fanconi anaemia and the repair of Watson and Crick DNA crosslinks. Nature. 2013;493:356–363. doi: 10.1038/nature11863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kutler DI, Singh B, Satagopan J, Batish SD, Berwick M, Giampietro PF, Auerbach AD. A 20-year perspective of the International Fanconi Anemia Registry (IFAR) Blood. 2003;101:1249–1256. doi: 10.1182/blood-2002-07-2170. [DOI] [PubMed] [Google Scholar]
- Levran O, Erlich T, Magdalena N, Gregory JJ, Batish SD, Verlander PC, Auerbach AD. Sequence variation in the Fanconi anemia gene, FAA. Proc Natl Acad Sci USA. 1997;94:13051–13056. doi: 10.1073/pnas.94.24.13051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levran O, Attwooll C, Henry RT, Milton KL, Neveling K, Rio P, Batish SD, Kalb R, Velleur E, Barral S, Ott J, Petrini J, Schindler D, Hanenberg H, Auerbach AD. The BRCA1-interacting helicase BRIP1 is deficient in Fanconi anemia. Nature Genet. 2005;37:931–933. doi: 10.1038/ng1624. [DOI] [PubMed] [Google Scholar]
- Lo Ten Foe JR, Kwee ML, Rooimans MA, Oostra AB, Veerman AJ, van Weel M, Pauli RM, Shahidi NT, Dokal I, Roberts I, Altay C, Gluckman E, Gibson RA, Mathew CG, Arwert F, Joenje H. Somatic mosaicism in Fanconi anaemia: Molecular basis and clinical significance. Eur J Hum Genet. 1997;5:137–148. [PubMed] [Google Scholar]
- Meetei AR, Levitus M, Xue Y, Medhurst AL, Zwaan M, Ling C, Rooimans MA, Bier P, Hoatlin M, Pals G, de Winter JP, Wang W, Joenje H. X-linked inheritance of Fanconi anemia complementation group B. Nature Genet. 2004;36:1219–1224. doi: 10.1038/ng1458. [DOI] [PubMed] [Google Scholar]
- Nakanishi K, Taniguchi T, Ranganathan V, New HV, Moreau LA, Stotsky M, Mathew CG, Kastan MB, Weaver DT, D’Andrea AD. Interaction of FANCD2 and NBS1 in the DNA damage response. Nat Cell Biol. 2002;4:913–920. doi: 10.1038/ncb879. [DOI] [PubMed] [Google Scholar]
- O’Driscoll M, Ruiz-Perez VL, Woods CG, Jeggo PA, Goodship JA. A splicing mutation affecting expression of ataxia-telangiectasia and Rad3-related protein (ATR) results in Seckel syndrome. Nature Genet. 2003;33:497–501. doi: 10.1038/ng1129. [DOI] [PubMed] [Google Scholar]
- Porteous MEM, Cross I, Burn J. VACTERL with hydrocephalus: One end of the Fanconi anemia spectrum of anomalies. Am J Med Genet. 1992;43:1032–1034. doi: 10.1002/ajmg.1320430624. [DOI] [PubMed] [Google Scholar]
- Reid S, Schindler D, Hanenberg H, Barker K, Hanks S, Kalb R, Neveling K, Kelly P, Seal S, Freund M, Wurm M, Batish SD, Lach FP, Yetgin S, Neitzel H, Ariffin H, Tischkowitz M, Mathew CG, Auerbach AD, Rahman N. Biallelic mutations in PALB2 cause Fanconi anemia subtype FA-N and predispose to childhood cancer. Nature Genet. 2007;39:162–164. doi: 10.1038/ng1947. [DOI] [PubMed] [Google Scholar]
- Savage SA, Giri N, Baerlocher GM, Orr N, Lansdorp PM, Alter BP. TINF2, a component of the shelterin telomere protection complex, is mutated in dyskeratosis congenita. Am J Hum Genet. 2008;82:501–509. doi: 10.1016/j.ajhg.2007.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimamura A, Montes de Oca R, Svenson JL, Haining N, Moreau LA, Nathan DG, D’Andrea AD. A novel diagnostic screen for defects in the Fanconi anemia pathway. Blood. 2002;100:4649–4654. doi: 10.1182/blood-2002-05-1399. [DOI] [PubMed] [Google Scholar]
- Taddei I, Morishima M, Huynh T, Lindasy EA. Genetic factors are major determinants of phenotypic variability in a mouse model of the DiGeorge/del22q11 syndromes. Proc Natl Acad Sci. 2001;98:11428–11431. doi: 10.1073/pnas.201127298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukahara M, Opitz JM. Dubowitz syndrome: Review of 141 cases including 36 previously unreported patients. Am J Med Genet. 1996;63:277–289. doi: 10.1002/(SICI)1096-8628(19960503)63:1<277::AID-AJMG46>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- Van Maldergem L, Siitonen HA, Jalkh N, Chouery E, De Roy M, Delague V, Muenke M, Jabs EW, Cai J, Wang LL, Plon SE, Fourneau C, Kestila M, Gillerot Y, Megarbane A, Verloes A. Revisiting the craniosynostosis-radial ray hypoplasia association: Baller-Gerold syndrome caused by mutations in the RECQL4 gene. J Med Genet. 2006;43:148–152. doi: 10.1136/jmg.2005.031781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varon R, Vissinga C, Platzer M, Cerosaletti KM, Chrzanowska KH, Saar K, Beckmann G, Seemanova E, Cooper PR, Nowak NJ, Stumm M, Weemaes CMR, Gatti RA, Wilson RK, Digweed M, Rosenthal A, Sperling K, Concannon P, Reis A. Nibrin, a novel DNA double-strand break repair protein, is mutated in Nijmegen breakage syndrome. Cell. 1998;93:467–476. doi: 10.1016/s0092-8674(00)81174-5. [DOI] [PubMed] [Google Scholar]
- Vaz F, Hanenberg H, Schuster B, Barker K, Wiek C, Erven V, Neveling K, Endt D, Kesterton I, Autore F, Fraternali F, Freund M, Hartmann L, Grimwade D, Roberts RG, Schaal H, Mohammed S, Rahman N, Schindler D, Mathew CG. Mutation of the RAD51C gene in a Fanconi anemia-like disorder. Nature Genet. 2010;42:406–409. doi: 10.1038/ng.570. [DOI] [PubMed] [Google Scholar]
- Verlander PC, Kaporis AG, Liu Q, Zhang Q, Seligsohn U, Auerbach AD. Carrier frequency of the IVS4+4A>T mutation of the Fanconi anemia gene FAC in the Ashkenazi Jewish population. Blood. 1995;86:4034–4038. [PubMed] [Google Scholar]
- Verlinsky Y, Rechitsky S, Schoolcraft W, Strom C, Kuliev A. Preimplantation diagnosis for Fanconi anemia combined with HLA matching. J Am Med Assoc. 2001;285:3130–3133. doi: 10.1001/jama.285.24.3130. [DOI] [PubMed] [Google Scholar]
- Vulliamy T, Marrone A, Szydlo R, Walne A, Mason PJ, Dokal I. Disease anticipation is associated with progressive telomere shortening in families with dyskeratosis congenita due to mutations in TERC. Nature Genet. 2004;36:447–449. doi: 10.1038/ng1346. [DOI] [PubMed] [Google Scholar]
- Wagner JE, MacMillan ML, Auerbach AD. Hematopoietic cell transplantation for Fanconi anemia. In: Blume KG, Forman SJ, Appelbaum FR, editors. Thomas’ Hematopoietic Cell Transplantation. 3. Blackwell Science; Malden, MA: 2003. pp. 1483–1504. [Google Scholar]
- Waisfisz Q, Morgan NV, Savino M, de Winter JP, van Berkel CG, Hoatlin ME, Ianzano L, Gibson RA, Arwert F, Savoia A, Mathew CG, Pronk JC, Joenje H. Spontaneous functional correction of homozygous Fanconi anaemia alleles reveals novel mechanistic basis for reverse mosaicism. Nat Genet. 1999;22:379–383. doi: 10.1038/11956. [DOI] [PubMed] [Google Scholar]
- Weaver DD, Mapstone CL, Yu P. The VATER association: Analysis of 46 patients. Am J Dis Child. 1986;140:225–229. doi: 10.1001/archpedi.1986.02140170051027. [DOI] [PubMed] [Google Scholar]
- Walne AJ, Vulliamy T, Marrone A, Beswick R, Kirwan M, Masunari Y, Al Qurashi F, Aljurf M, Dokal I. Genetic heterogeneity in autosomal recessive dyskeratosis congenita with one subtype due to mutations in the telomerase-associated protein NOP10. Hum Molec Genet. 2007;16:1619–1629. doi: 10.1093/hmg/ddm111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walne AJ, Vulliamy T, Kirwan M, Plagnol V, Dokal I. Constitutional mutations in RTEL1 cause severe dyskeratosis congenita. Am J Hum Genet. 2013;92:448–453. doi: 10.1016/j.ajhg.2013.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams DA, Bennett C, Bertuch A, Bessler M, Coates T, Corey S, Dror Y, Huang J, Lipton J, Olson TS, Reiss UM, Rogers ZR, Sieff C, Vlachos A, Walkovich K, Wang W, Shimamura A. Diagnosis and Treatment of Pediatric Acquired Aplastic Anemia (AAA): An Initial Survey of the North American Pediatric Aplastic Anemia Consortium (NAPAAC) Pediatr Blood Cancer. 2014;61:869–874. doi: 10.1002/pbc.24875. [DOI] [PMC free article] [PubMed] [Google Scholar]
Key References
- Auerbach AD, Rogatko A, Schroeder-Kurth TM. 1989 See above. Presents evidence from the International Fanconi Anemia Registry showing that the DEB test is a useful discriminator for FA. [PubMed] [Google Scholar]
- Auerbach AD. Fanconi anemia diagnosis and the diepoxybutane (DEB) test. Exp Hematol. 1993;21:6. Discusses questions relating to the sensitivity and specificity of the DEB test compared to alternative tests for FA. [PubMed] [Google Scholar]
- Auerbach AD. Fanconi anemia and its diagnosis. Mutat Res. 2009;668:4–10. doi: 10.1016/j.mrfmmm.2009.01.013. Presents an overview of current knowledge regarding the varied phenotypic manifestations of FA and procedures for diagnosis based upon abnormal DNA damage responses. [DOI] [PMC free article] [PubMed] [Google Scholar]