

Abstract
An overflow of regulatory RNAs (sRNAs) was identified in a wide range of bacteria. We designed and implemented a new resource for the hundreds of sRNAs identified in Staphylococci, with primary focus on the human pathogen Staphylococcus aureus. The “Staphylococcal Regulatory RNA Database” (SRD, http://srd.genouest.org/) compiled all published data in a single interface including genetic locations, sequences and other features. SRD proposes novel and simplified identifiers for Staphylococcal regulatory RNAs (srn) based on the sRNA's genetic location in S. aureus strain N315 which served as a reference. From a set of 894 sequences and after an in-depth cleaning, SRD provides a list of 575 srn exempt of redundant sequences. For each sRNA, their experimental support(s) is provided, allowing the user to individually assess their validity and significance. RNA-seq analysis performed on strains N315, NCTC8325, and Newman allowed us to provide further details, upgrade the initial annotation, and identified 159 RNA-seq independent transcribed sRNAs. The lists of 575 and 159 sRNAs sequences were used to predict the number and location of srns in 18 S. aureus strains and 10 other Staphylococci. A comparison of the srn contents within 32 Staphylococcal genomes revealed a poor conservation between species. In addition, sRNA structure predictions obtained with MFold are accessible. A BLAST server and the intaRNA program, which is dedicated to target prediction, were implemented. SRD is the first sRNA database centered on a genus; it is a user-friendly and scalable device with the possibility to submit new sequences that should spread in the literature.
Keywords: small regulatory RNAs, bacteria, Staphylococcus aureus, database, RNA-seq, sRNA identification, sRNA targets
INTRODUCTION
In the recent years, a plethora of regulatory RNAs (sRNAs) were identified in diverse bacterial genomes, including several human pathogens. sRNAs enable bacteria to induce efficient and prompt physiological feedbacks to adjust on their environments, and also to establish infection (Caldelari et al. 2013). Mechanistically, sRNAs intervene on transcription, mRNA turnover, and/or translation of target genes. sRNAs proceed in gene expression regulations, from transcription initiation to translation control and protein activity (Storz et al. 2011). The majority of sRNAs characterized up to now act by pairings with target mRNAs, either encoded on the opposite strand (cis-encoded) or transcribed apart from their targets (trans-encoded). Some have been shown to encode small peptides. While most of the bacterial sRNAs were originally studied in E. coli and other Gram-negative bacteria (Mizuno et al. 1984), a recent outburst of sRNAs was identified in Gram-positive bacteria (Brantl and Brückner 2014), including the major human pathogen Staphylococcus aureus (Fechter et al. 2014).
Staphylococcus aureus is an opportunistic pathogen that has sophisticated regulatory tracks to rapidly and efficiently adapt its growth in response to its disparate habitats and hosts. Several groups have shown experimentally that S. aureus express many sRNAs, delivered from the core genome, mobile and accessory elements (Guillet et al. 2013; Tomasini et al. 2014). They include several predicted riboswitches (cis-acting regulatory mRNA leader sequences), many cis-encoded antisense RNAs, several trans-encoded sRNAs (Romilly et al. 2012) with some containing small open reading frames that were shown to be expressed (Sayed et al. 2012; Pinel-Marie et al. 2014). However, and for the most part, their functions and mechanisms are unexplored yet. For the few sRNAs with associated functions, some detect bacterial density, modify cell surface properties for host immune escape, adjust central metabolism for optimal growth, regulate the expression of virulence factors, influence antibiotic resistance (Lalaouna et al. 2014), trigger cell death, and encodes toxins (Felden et al. 2011; Guillet et al. 2013).
The systematic and recent use of high-throughput RNA-sequencing technologies substantially raised the number of sRNAs sequences identified per bacterial species. To cope with that plethora of sRNAs, several databases have emerged from generalists to more specialize. Generalist databases such as fRNAdb (Kin et al. 2007) or NONCODE (Xie et al. 2014) focus exclusively on eukaryotes, while Rfam 11.0 (Burge et al. 2013) partitions RNAs data into families for both the eukaryotes and prokaryotes. RNAdb was originally designed for mammalian noncoding RNAs but was officially retired in June 2012 (Pang et al. 2007). On the other hand, there are databases specifically devoted to the bacterial kingdom. sRNAMap is a repository for the microbial genomes but Gram-positive bacteria, and therefore Staphylococci, are absent from this browser (Huang et al. 2009). sRNATarbase (Cao et al. 2010) was implemented to provide a list of sRNA targets, but is out of date for Staphylococci. Recently, two bacterial sRNA databases were developed: (i) sRNAdb, which focused exclusively on Gram-positive bacteria (Pischimarov et al. 2012) but provides only 39 sRNA sequences for S. aureus; (ii) BSRD is a generalist bacterial sRNA database with >700 species included (Li et al. 2013). Finally, RNAspace.org is a platform devoted to the prediction, annotation, and analysis of noncoding RNA but does not provide data set (Cros et al. 2011). In S. aureus and more generally in the Staphylococcal genus, a unified sRNA nomenclature is lacking, while many redundancies, as single sequence described under several IDs, and potential misannotated sRNAs (e.g., repeated sequences, mRNA leader or trailer sequences) would require an in-depth manual cleaning.
Therefore, there is an urgent need for additional sRNA databases focusing on a bacterial genus to provide an accurate and simple list of sRNAs. Here, we report a Staphylococcus Regulatory RNA Database (SRD, http://srd.genouest.org/) which compiles, after an in-depth scrubbing all the sRNA sequences identified so far, with a primary focus on the human pathogen S. aureus as a reference. Starting from a large set of sRNA sequences, SRD proposes a new and simple nomenclature together with individual functional, structural, and phylogenetic information and predictions. It provides a unified repository based on additional RNA-seq data analysis.
RESULTS
Construction of a database encompassing the Staphylococcal regulatory RNAs
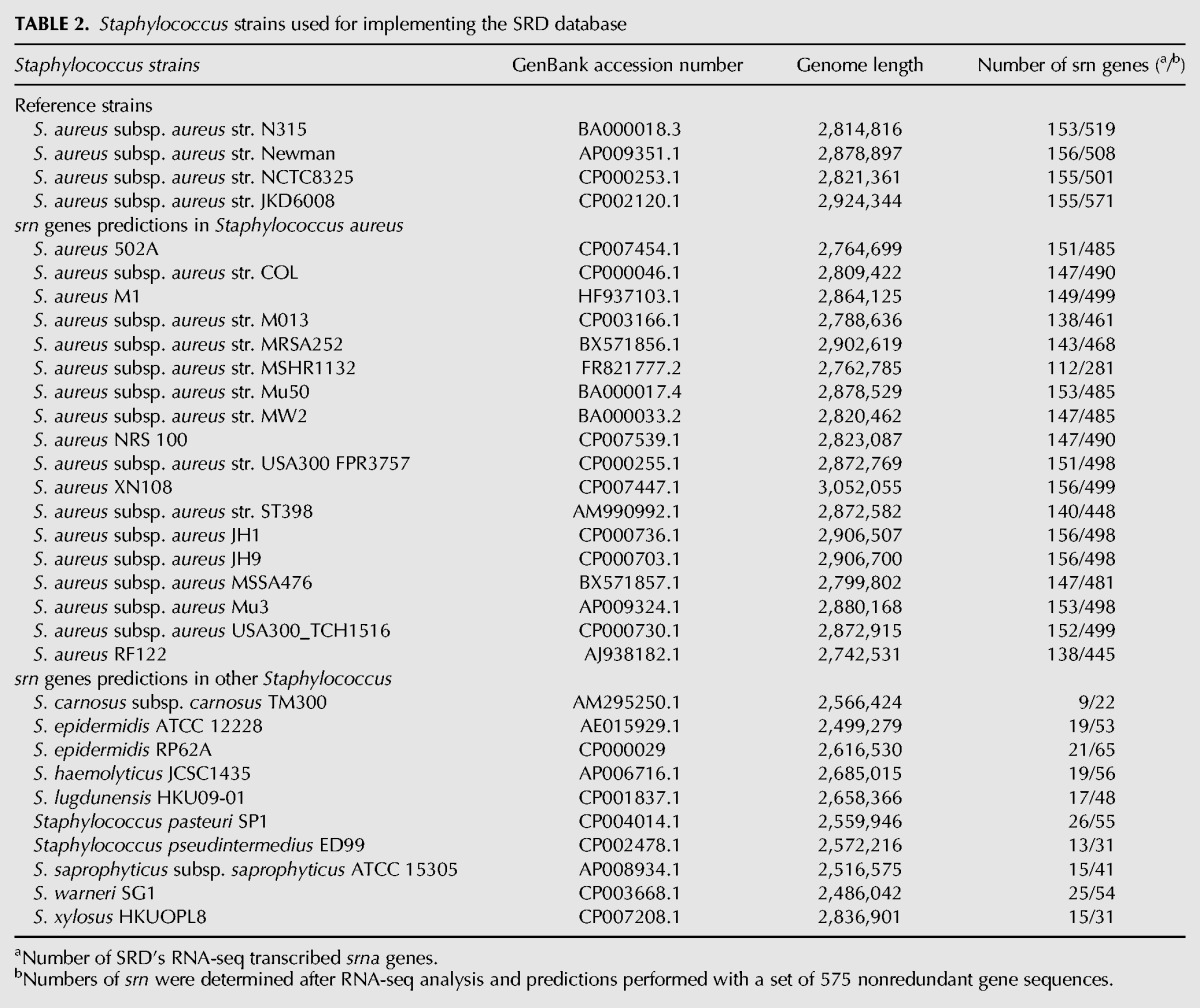
Staphylococcal sRNAs were identified and studied principally in several strains of S. aureus (Tomasini et al. 2014). The chronological discovery of the Staphylococcal sRNAs expressed in S. aureus is listed in Table 1. Those RNAs were identified by combining diverse experimental and bioinformatics approaches (Novick et al. 1989, 1993; Pichon and Felden 2005; Anderson et al. 2006; Roberts et al. 2006; Marchais et al. 2009; Nielsen et al. 2011; Morrison et al. 2012; Xue et al. 2014) including the use of Next-Generation RNA-Sequencing technologies (Geissmann et al. 2009; Abu-Qatouseh et al. 2010; Beaume et al. 2010; Bohn et al. 2010; Howden et al. 2013). A total of 894 sequences transcribed as sRNA were compiled from the literature (Fig. 1; Supplemental Data S1). We then focused on the following extensively studied and completed S. aureus genomes: N315, Newman, NCTC8325, and JKD6008 (Table 2). The BLAST program was used to locate the coordinates of each sRNA gene in any of the four genomes. Some sequences appeared, as previously suggested (Beaume et al. 2010; Howden et al. 2013), to be repeated onto the genomes, that led to an increase in the total number of sRNA sequences collected. Therefore, sequences identified as DNA repeated sequences by these authors were removed after confirming the initial statements using Blast (Supplemental Data S2). In addition, sequences located in CDSs, rRNAs, tRNAs, or spacers within the four genomes as well as the RNA sequences flanking the genes transcribed as ribosomes (reads overlapping with the ribosomes or within the intergenic regions of ribosomes) were discarded (Liu et al. 2009) to generate a first data set of 773 sequences. A significant number of redundant sequences annotated as a single sRNA could be retrieved under other names. This data set included, among others, the sau, rsa, jkdsRNA, teg, and spr genes. As an example, up to five other different gene IDs were identified for rsaE (RsaON_Sau20_Teg92_IGR6_sRNA183). Therefore, we manually cured this data set to provide a list of 575 sRNA genes exempted of redundancy.
TABLE 1.
Sequential identification of regulatory RNAs expressed in Staphylococcus aureus
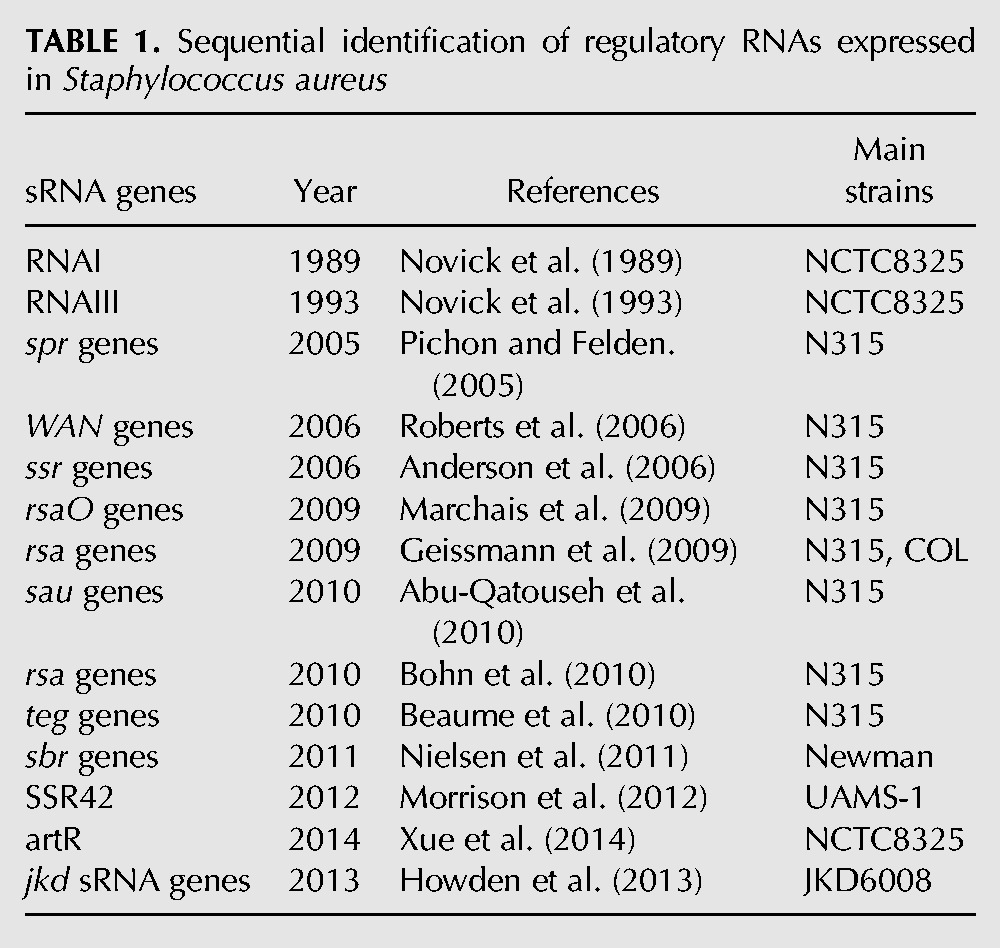
FIGURE 1.

Overview of the SRD's inputs and outputs.
TABLE 2.
Staphylococcus strains used for implementing the SRD database
Need and proposal for a novel and simplified identifier
The recent outburst in sRNAs led to spreading a large confusion in the actual number of sRNA genes and for communications as a single sRNA sequence can harbor multiple IDs. To cope with that, we assigned novel and simple identifier that clarifies the actual repertoire of S. aureus sRNAs. The genome of N315 (Kuroda et al. 2001) was used as a reference as many sRNA sequences were identified in this strain, with some experimentally validated (Pichon and Felden 2005; Beaume et al. 2010; Chabelskaya et al. 2010). Each srna gene was assigned with a srn (staphylococcus ribonucleotide) gene identifier. The srn gene identifiers were numbered based on their genetic location onto the genome of S. aureus N315 strain and starting from the origin of replication and therefore do not reflect their transcriptional level. srn numbers were assigned by increments of 10 to anticipate the identification of new sRNAs in the upcoming future which would be also numbered based on their genomic locations. When a srn gene is present in other strains it keeps the srn number assigned onto the N315 genome. When a srn identified in N315 is present in multiple copies in N315 or in another strain, the number of copies is provided (srn_1930.1, srn_1930.2, and srn 1930.3), unless experimental evidence indicated that they should be considered as distinct sRNAs (Supplemental Data S3, S4). srn identified in other strains and absent in N315 strain will be assigned with numbers starting from srn_9000 (srn_9480 formerly known as sRNA334). There were only four sRNAs (4.5 S, 6S, tmRNA, and RNase P RNA) for which a srn identifier was not generated, based on their extensive nomenclature, sequence, and functional conservation beyond Staphylococcal genomes (Supplemental Data S5). Therefore, regulatory RNAs such as riboswitches, RNAIII, or RsaE that are pioneered and/or conserved within the genus were also assigned with a srn identifier similar to what was done with the JKD6008 strain (Howden et al. 2013). However, a column entitled “most common name” is present on the website to avoid confusion when dealing with already well-described sRNAs. Additional information, including all other previously published names is provided with the list of srn on the SRD website.
Description of sRNAs in SRD
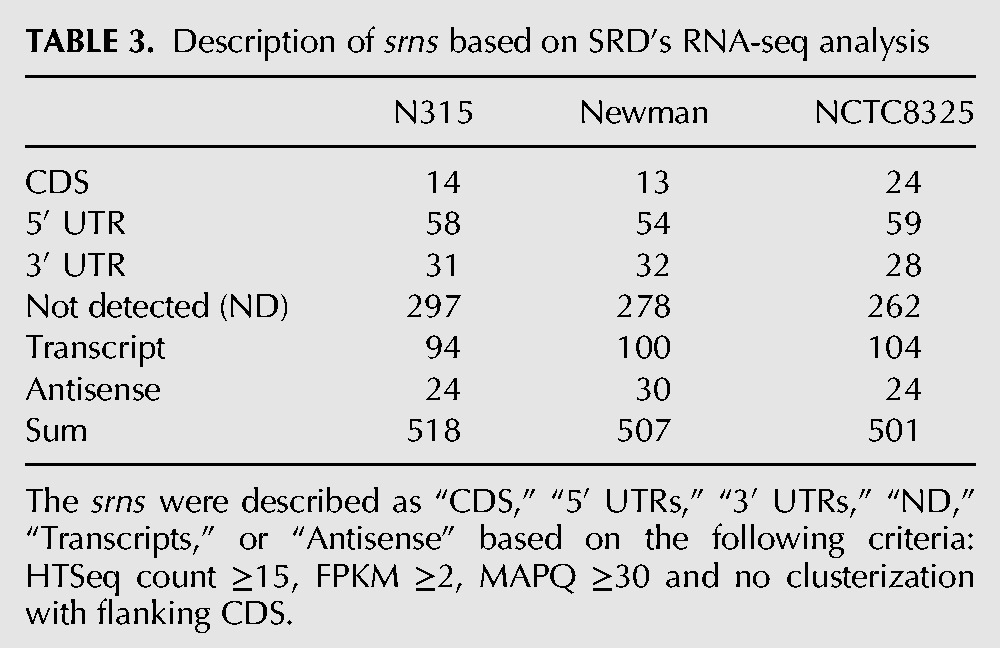
Among the 575 genomic sequences described in SRD, there are only 60 transcripts identified by multiple experimental approaches (including Northern Blot, 5′ and 3′ RACE, RT-qPCR, or RNA-seq) and a few for which their functions were characterized (Supplemental Data S3 and SRD website). Among these 60 sRNAs, 49 were described as transcripts whereas 11 were annotated as antisense sRNAs. The majority of the 575 sRNAs were identified or validated by Next-Generation RNA Sequencing (Beaume et al. 2010; Bohn et al. 2010; Lasa et al. 2011; Lioliou et al. 2012; Howden et al. 2013). Although powerful, RNA-seq, as any global approach, can lead to a substantial amount of false positive transcripts due to genomic DNA contaminations, reads mapping onto repeated sequences or/and the inaccurate detection of transcripts by bioinformatics. Some of the sRNAs previously identified by RNA-seq were of short lengths (<50 nt), mostly detected as cis-encoded antisense RNAs, including a wide antisense transcription that is not understood yet (Lasa et al. 2011). Also, some of the previously published sRNAs were already suspected as potential 5′ UTRs or 3′ UTRs (Beaume et al. 2010; Howden et al. 2013). Therefore, we searched for the presence of srn transcripts in strains N315 and Newman (data submitted to GEO with the accession number GSE64026), cultivated until the exponential phase of growth in a rich medium (BHI), and for strain NCTC8325 cultivated under 16 different growth conditions (growth phase, temperature, O2 limitations, etc.; data kindly provided by Drs. P. Bouloc and T. Rochat). Those three strains are representative as their genomes were completed and annotated (Kuroda et al. 2001; Gillaspy et al. 2006; Baba et al. 2008). Based on our initial compilation and curation, N315, Newman, and NCTC8325 are predicted to contain 518, 508, and 501 independent srn genes, respectively. Altogether, they represent a pool of 535 srn genes. RNA-seq reads were mapped at unique locations onto their respective genomes, counted using HTSeq (Anders et al. 2014) and the presence of transcripts analyzed using Artemis (Carver et al. 2012). Based on the results from HTSeq (Supplemental Data S4), an FPKM normalization (Fragment per kilobase per millions of fragments mapped) and from the visualization of reads onto the annotated genomes (Fig. 2), the srn were either described as transcripts (Fig. 2A), cis-antisense RNAs (Fig. 2B), 5′ UTR (Fig. 2C), 3′ UTR (Fig. 2D), CDS (Fig. 2E, coordinates inside an annotated gene) or not detected (Fig. 2F, ND) in the “SRD's RNA-seq evidence” column of the SRD website. A srn was considered as a transcript or cis-antisense (reads mapping onto the opposite strand of a CDS) when the HTSeq count was equal or >15 (Howden et al. 2013), the FPKM normalization >2, the mapping quality (MAPQ score in a SAM file) >30 (probability of a correct match equal to 0.999), and when the reads did not overlap with annotated genes. The UTRs were described as 5′ UTRs or 3′ UTRs when the reads are assembled with CDS and when the expression was similar to the flanking genes, as described (Yoder-Himes et al. 2009). The results of our analysis are summarized in Table 3. In N315, we identified 94 srn as transcripts, 24 as cis-antisense RNAs, 14 as CDS, 58 as 5′ UTR, 31 as 3′ UTR, and 297 were not detected. Similar results were obtained for Newman and NCTC8325 strains, respectively (see the SRD website). srn annotated as “ND” were either not detected by HTSeq count or did not meet some of the criteria retained for describing a sequence as transcript or a cis-antisense. Current Illumina sequencing kits are not perfectly adapted to the search of small bacterial transcripts and therefore it is difficult to know whether there are limits in the technology or that transcripts are absent. Forty-five of the srn that were not detected were predicted to have a length <50 nt. Ninenty-seven srn were predicted to be between 50 and 100 nt and the others were longer than 100 nt. Overall, only 159 srn were detected as transcripts or as RNA antisense under our experimental conditions (Supplemental Data S5 and http://srd.genouest.org/browsevalidated). Also, there were 24 ambiguous srn (Supplemental Data S6). These ambiguous srn presented criterion that did not allow us to add them in the list of 159 sRNA (see comments in Supplemental Data). All these results suggest that some sRNAs are expressed or detected under specific conditions that await further experimental assessment, while other srn may not be considered as independent sRNA transcriptional units or may be false positive and arise from transcriptional noise.
FIGURE 2.

Examples of the visualization of read mapping from strain Newman using Artemis. The srns are highlighted in pink. (A) Typical visualization for a srn described as transcript. (B) Example of an antisense srn. (C) Reads overlapping with a CDS and considered as a 5′ UTR. (D) srn described as a 3′ UTR. (E) srn identified within a CDS. (F) srn not detected.
TABLE 3.
Description of srns based on SRD's RNA-seq analysis
sRNA predictions in other Staphylococcal species and strains
Four S. aureus genomes were used to generate SRD. It is however important to include in a database a large number of strains and other species. Therefore, we performed srn predictions on a set of 28 strains which included 18 S. aureus subspecies and 10 other Staphylococci (Table 2). BLAST (see Materials and Methods) was used to predict the presence of srn genes using either the list of 159 srn confirmed by our RNA-seq analysis or the curated list containing 575 srn. Using the of 159 srn, this resulted in a number of predicted nucleotide sequences that ranged from 112 srn genes for Staphylococcus aureus subsp. aureus str. MSHR1132 to 156 srn genes for Staphylococcus aureus subsp. aureus str. JH1 and Staphylococcus aureus subsp. aureus str. JH9, respectively. On the opposite a reduced number of genes was predicted for the 10 other Staphylococcus species, with only 9 srn genes found for Staphylococcus carnosus subsp. carnosus str. TM300 and a maximum of 26 srn genes for Staphylococcus pasteuri SP1. Taken together these results suggest a low level of nucleotide sequence conservation for the srn genes within the genus.
Comparative analysis of the srna gene sequences
To investigate further the 159 srn gene sequences detected in the various strains a phylogenetic analysis was performed. A “phylogenomic” tree based on Staphylococcus whole-genome content (Staphylococcus tree) (Fig. 3A) and a tree based on the srn gene content were constructed (srn genes tree) (Fig. 3B). Interestingly, the tree based on the srn content showed an outstanding similar topology compared wth the tree based on the genome content and clearly differentiates Staphylococcus aureus from the other Staphylococcus species (Fig. 3). For both trees, S. aureus subsp. aureus str. MRSH 132 appeared more distant from other S. aureus subspecies. This could explain why only 112 srn genes were predicted using BLAST for that strain. A “heatmap” representation constructed using a matrix of presence/absence of srn sequences in Staphylococcus genomes showed a species clusterization similar to that of the Staphylococcus tree (Fig. 4). These results confirm a weak conservation of the 159 srn outside the aureus species. In addition, rnaseP RNA gene, 4.5S RNA gene, srn_3910 (RNAIII), and srn_2130 (rsaE) were identified in all Staphylococcus strains while the tmRNA sequences appears to be substantially degenerated only in Staphylococcus xylosus str. HKUOPL8. Regarding the S. aureus subspecies, we identified 96 srn conserved which we defined as a core sRNA set in this species.
FIGURE 3.

Phylogenetic analysis on the genome and srn content of 32 strains of the Staphylococcus genus using the Neighbor-joining algorithm. (A) Staphylococcus tree-based on genome content. (B) Staphylococcus tree-based on srn content.
FIGURE 4.

Comparative analysis through a “heatmap” cluster based on a matrix of presence (black) and absence (red) of srn sequences.
Database overview and usage
Users can access the list of srn genes through the Web interface (Fig. 5A). From the SRD home page they can (i) access the data set corresponding to the four genomes used to construct SRD, (ii) retrieve a short list of srn genes with a unified annotation, (iii) browse for predictions in other Staphylococci (described elsewhere in the text), (iv) BLAST (Altschul et al. 1990) their own sRNA sequences against the entire SRD database, or (v) search for RNA–RNA interactions using the intaRNA program (Busch et al. 2008). After entering in the webpage of one of the four curated genomes, the users will have access to the full list of srn reported so far. For each table, the new nomenclature, the genomic coordinates, the orientation, previous names, and experimental support are provided. The column “experimental evidence” was added for the community in order to have a quick overview of the srn that were identified as transcripts through RNA-seq analysis or other experimental approaches. The molecular targets and references to the experimental secondary structure information are provided within the “SRD's RNA-seq transcribed sRNA” tab of the website. In addition, the users can download the Staphylococcal genomes and the srn genes in diverse formats (“FASTA” or “gff”) suitable for subsequent RNA-seq analysis (Fig. 5B).
FIGURE 5.

Screenshots of SRD. (A) Main webpage interface to navigate and access specific features within the database. (B) Example of the presentation of srn data determined after curation of repeated and redundant sequences.
SRD specific features
Several functions have been included in the SRD website to provide an efficient device for the community working on sRNAs. BLAST and intaRNA (Altschul et al. 1990; Busch et al. 2008) were implemented in the database while external links to RNAtarget2 (Kery et al. 2014) and to RNA predator (Eggenhofer et al. 2011) are easily accessible from the webpage describing each sRNA. For BLAST comparisons, our databank includes the four initial reference genomes, the lists of validated sRNAs, and all the srn curated in the four strains. IntaRNA can be activated from the intaRNA page or directly from any srn sequences by clicking on the dedicated symbol. The users will only have to paste their mRNA sequences to see whether they may interact with each sRNA from SRD. MFold structure predictions (Zuker 2003), defined for the 575 srn RNA sequences, can be downloaded also directly from the SRD website.
Evolution of the database and future directions
SRD is a repository for the Staphylococcal sRNAs with primary focus on S. aureus. By combining an in-depth cleaning and novel RNA-seq analysis, it clarifies the status regarding the absence of a consensus nomenclature and also to the actual expanding number of individual sRNA genes. With the rising interest of the community in the field of sRNAs, the number of sRNAs detected or characterized in bacteria is predicted to constantly increase. SRD will therefore evolve, based on the identification of novel sRNAs published in the literature. In addition, researchers who would be willing to unify new discoveries under the srn nomenclature will have the possibility to submit their sRNA sequence(s) to SRD under the contact information box. SRD would allow both already published and nonpublished submission. For data that would not be already published, temporary srn numbers will be assigned and disclosed upon acceptance of the publications in peer-reviewed journals. Therefore, researchers will have the possibility to disclose their results directly with the srn nomenclature. To avoid the diffusion of a large number of genes that may not be confirmed later, researchers will be invited to describe how a new sRNA was identified. For the novel sRNAs detected by RNA-seq analysis, that could lead to false-positive sRNAs, a secure ftp link will be provided to solicit researchers to deposit their “fastq” files to check whether they meet the criteria described in the text for being annotated as sRNA transcripts.
DISCUSSION
Over the last 20 yr, the number of sequences identified as sRNAs dramatically increased in Staphylococcus aureus. However, the democratization of Next-Generation Sequencing and the absence of a consensus in the community for annotating newly identified sRNAs led to a growing confusion that become detrimental to the field. SRD compiled the existing data in a single interface, removed the repeated and or redundant sequences and proposed a novel, simplified, nomenclature. Compared with the databases on other prokaryotes sRNAs, SRD is the first sRNA database dedicated to the Staphylococcus genus. SRD assigned a single identifier for the whole genus while other sRNA databases, such as BSRD (Li et al. 2013), provide an index per strain. A unique identifier offers a substantial advantage when comparing different strains, as it should avoid the dissemination of multiple redundancies. Therefore, the data provided by SRD would have been difficult to fuse with other databases. SRD hosts a large collection of manually curated Staphylococcal sRNAs (575 srn genes) mostly exempted of repeated sequences and of redundancies. Furthermore, coupled with an SRD's RNA-seq analysis, a list of 159 RNA-seq transcribed srn is provided that includes all the 60 previously experimentally supported sRNAs. This suggests that the SRD's criteria used to describe the srn transcripts that combined previously published cut offs (Yoder-Himes et al. 2009; Howden et al. 2013) with high mapping quality scores and FPKM normalization (to prevent the detection of incorrectly mapped reads or of long transcripts that do not arise from transcriptional noise) were relevant. However, among the nearly 300 srn genes not transcribed under the physiological conditions tested, some may be later annotated as sRNA transcripts once additional data and work will provide transcriptional evidence. From this set of 159 srn genes, predictions were issued by sequence similarity to identify homologous genes in other Staphylococcal species. A comparison of the SRD's predictions with the BSRD entries for Staphylococcus strains shows that a genus-specific database is relevant. Indeed, in BSRD there is a huge discrepancy in the number of sRNAs sequences being available within the S. aureus subps. aureus species. While 154 sRNAs were listed for strain N315, mostly based on the work of Beaume et al. (2010), there were <60 sequences inventoried by similarity for other Staphylococcal strains (Li et al. 2013). All BSRD S. aureus entries are present in our database, and the predictions performed within the genus from a set of 575 srn genes (versus 8248 genes for BSRD), led to a larger set of predicted genes in SRD (Table 4). The comparative analysis performed for the srn content confirmed a low level of conservation within the genus and therefore in the bacterial kingdom. The weak sequence conservation at the DNA level in comparison with the protein level is therefore a serious limitation for retrieving genes, and therefore sRNAs, between species (Konstantinidis and Tiedje 2007; Sentausa and Fournier 2013). To our knowledge, there is no recognized standard to assign identifiers or nomenclature to sRNAs. Therefore, to not favor anyone name (spr, rsa, sau, teg, and others) we created the srn identifier. In addition, a most common name was assigned based on the chronological discovery of experimental confirmation to avoid the community to name some of the already very well-described sRNAs under a srn identifier while using the database. However, for the sRNAs that were only described by RNA-seq, the community is encouraged to use the srn identifier in an effort for unifying the work done on the sRNAs so far. We believe that SRD will help the scientific community working on Staphylococcal sRNA identification, function, and biology. SRD provides a simple and unified sRNA resource, a detailed annotation for each sRNA, a direct access to various RNA and genomic analysis tools. Finally, it shall encourage the community to participate in an effort to submitting new Staphylococcus sRNAs in SRD and to develop other genus specific sRNA databases that should be an essential extension to the generalist sRNA databases.
TABLE 4.
Comparison between BSRD and SRD number of entries

MATERIALS AND METHODS
Bacterial strains and growth conditions
Staphylococcus aureus strains Newman and N315 were grown in liquid Brain Heart Infusion broth (BHI, Oxoid) and Tryptone Soya Broth (TSB, Oxoid) at 37°C, under agitation.
RNA extraction
Overnight cultures of S. aureus were diluted to an OD600 nm of 0.1 into fresh BHI broth and cultured for 5 h at 37°C at 160 rpm. Cells were harvested by centrifugation at 15,000g for 30 sec and pellets washed with 500 µL of cold lysis buffer (20 mM sodium acetate, 1 mM EDTA, 0.5% SDS at pH 5.5). Cells were broken out using acid treated glass beads (Sigma) in the presence of phenol (pH 4) in an FP120 FastPrep cell disruptor (MP Biomedicals) for 30 sec at power 6.5. Lysates were centrifuged for 5 min at 16,000g at 4°C. Total RNAs were extracted with phenol/chloroform and precipitated overnight. The RNA samples were treated with DNase I, Amplification Grade (Invitrogen). The absence of DNA contamination was checked by qPCR in an Applied Biosystems instrument. The integrity of each RNA preparation was verified on a “Bioanalyzer” (Agilent).
cDNA Library construction and Illumina RNA-seq
Ribosomal RNAs were depleted using the Ribo-Zero Magnetic Kit (Epicentre) and following manufacturer's recommendations. Stranded cDNAs libraries were prepared using the NEBNext Ultra Directional RNA Library Prep Kit for Illumina (New England Biolabs). The concentration, quality, and purity of the libraries were determined on a BioAnalyzer (Agilent), a Qubit fluorometer (Invitrogen), and a Nanodrop (Thermo Scientific). Libraries were pooled and sequenced on an Illumina Hiseq 1500 instrument following the manufacturer's recommendations and using the rapid run mode for 200 cycles in paired-end.
Read mapping and visualization
The genome sequences and annotation files were obtained from NCBI (ftp://ftp.ncbi.nlm.nih.gov/genomes/Bacteria/). A “fastqc” report was performed prior mapping the data onto the appropriate genome. RNA-seq reads were mapped using Tophat2 and BWA (Li and Durbin 2010; Kim et al. 2013) with initial settings modified to allow the mapping of stranded library of an average mean distance between mates of 250 (for Tophat2) and to allow the alignment of a read to a single location with no mismatch (Tophat2 and BWA). BAM files were converted into SAM files and filtered on bitwise flag values (Li et al. 2009) to select properly paired reads. SAM files were then converted to BAM files, sorted by query, and counted by HTSeq count (Anders et al. 2014) for stranded library with the mode union. BAM files were visualized using the Artemis program (Carver et al. 2012).
Data set building and comparative analysis
The 32 genomes of Staphylococcus used in this study were downloaded from GenBank (Table 2). The published sRNA sequences were extracted from their respective genomes to construct a pool of sRNA. This pool was used to predict the srn genes in reference genomes. Then a pool of SRD's RNA-seq transcribed sRNA was used to predict srn genes in 28 Staphylococcus genomes. The predictions were done using “BLASTN” with a cutoff E-value <1 × 10−20, percentage similarity >80% and an alignment length >60 nt of the query length. The 32 Staphylococcus genomes and the predicted srn genes sequences were aligned using Muscle aligner implemented in Mauve software (Darling et al. 2010). Mauve alignment generated a genome content matrix for which the identity scores range between 0 and 1, where 0 indicates that no identical homologous nucleotides were found, and 1 indicates that every homologous nucleotide was identical. A matrix based on the srn gene content was generated (the similarity between two species is defined as the number of genes that they have in common divided by the total number of srn genes) (Snel et al. 1999, 2005; Huson and Bryant 2006). The genome content based matrix and the srn gene content based matrix were then used to construct, respectively, a Staphylococcus “phylogenomic” tree-based on genome content and a Staphylococcus tree-based on srn content, using Neighbor-joining algorithm in the package SplitsTree4 (Huson and Bryant 2006). A “heatmap” clusterization was constructed using a matrix based on presence and absence of srn genes using the R package (http://www.r-project.org/).
Database design
The web server has been designed in PHP with the “Symfony” framework (http://symfony.com). It includes a set of scripts that automatically parse the raw input files (srn, genomes), fill in a MySQL database for each set of genome/srn, and build some templates for all input predictions. Those scripts allow the addition of other genomes easily by simply adding the new files to the directory structure. They will update the existing information and insert the new ones. The website menus are automatically adapted to the list of analyzed genomes, removing the need to modify the website when new information is added (new genomes, new predictions, etc.). The scripts also (i) extract the FASTA sequence of each srn (to prefill IntaRNA form for example), (ii) execute MFold to create the pdf files containing the structure, and (iii) prepare a blast index for all genomes and set of srn.
A srn distribution is proposed in the web interface with pan and zoom options. This distribution is displayed using the D3js library (http://d3js.org/).
DATA DEPOSITION
The data discussed in this publication have been deposited in NCBI's Gene Expression Omnibus (Edgar et al. 2002) and are accessible through GEO Series accession number GSE64026 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE64026).
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
This research was supported by a Marie Curie International Incoming Fellowship (project 621959—SarHyb) within the 7th European Community Framework Programme and by the region Bretagne grant SAD 2013—SARS 8254. T.M. is a recipient of a fellowship from the Direction Générale pour l'Armement. We thank Dr. Philippe Bouloc and Dr. Tatiana (Orsay—Université Paris Sud) Rochat for providing a BAM file combining 16 growth conditions for strain NCTC8325. We are most grateful to the “plate-forme Génomique Santé” Biogenouest Genomics, Biosit core facility for its technical support and to the CNRS-UPMC ABiMS bioinformatics platform (http://abims.sb-roscoff.fr) for providing computational resources and support. This work has benefited from the facilities and expertise of the high-throughput sequencing platform of IMAGIF (Centre de Recherche de Gif—www.imagif.cnrs.fr).
Footnotes
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.049346.114.
REFERENCES
- Abu-Qatouseh LF, Chinni SV, Seggewiss J, Proctor RA, Brosius J, Rozhdestvensky TS, Peters G, von Eiff C, Becker K 2010. Identification of differentially expressed small non-protein-coding RNAs in Staphylococcus aureus displaying both the normal and the small-colony variant phenotype. J Mol Med (Berl) 88: 565–575. [DOI] [PubMed] [Google Scholar]
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ 1990. Basic local alignment search tool. J Mol Biol 215: 403–410. [DOI] [PubMed] [Google Scholar]
- Anders S, Pyl PT, Huber W 2014. HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics 31: 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson KL, Roberts C, Disz T, Vonstein V, Hwang K, Overbeek R, Olson PD, Projan SJ, Dunman PM 2006. Characterization of the Staphylococcus aureus heat shock, cold shock, stringent, and SOS responses and their effects on log-phase mRNA turnover. J Bacteriol 188: 6739–6756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baba T, Bae T, Schneewind O, Takeuchi F, Hiramatsu K 2008. Genome sequence of Staphylococcus aureus strain Newman and comparative analysis of staphylococcal genomes: polymorphism and evolution of two major pathogenicity islands. J Bacteriol 190: 300–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaume M, Hernandez D, Farinelli L, Deluen C, Linder P, Gaspin C, Romby P, Schrenzel J, Francois P 2010. Cartography of methicillin-resistant S. aureus transcripts: detection, orientation and temporal expression during growth phase and stress conditions. PLoS One 5: e10725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohn C, Rigoulay C, Chabelskaya S, Sharma CM, Marchais A, Skorski P, Borezée-Durant E, Barbet R, Jacquet E, Jacq A, et al. 2010. Experimental discovery of small RNAs in Staphylococcus aureus reveals a riboregulator of central metabolism. Nucleic Acids Res 38: 6620–6636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brantl S, Brückner R 2014. Small regulatory RNAs from low-GC Gram-positive bacteria. RNA Biol 11: 443–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burge SW, Daub J, Eberhardt R, Tate J, Barquist L, Nawrocki EP, Eddy SR, Gardner PP, Bateman A 2013. Rfam 11.0: 10 years of RNA families. Nucleic Acids Res 41: D226–D232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busch A, Richter AS, Backofen R 2008. IntaRNA: efficient prediction of bacterial sRNA targets incorporating target site accessibility and seed regions. Bioinformatics 24: 2849–2856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caldelari I, Chao Y, Romby P, Vogel J 2013. RNA-mediated regulation in pathogenic bacteria. Cold Spring Harb Perspect Med 3: a010298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Y, Wu J, Liu Q, Zhao Y, Ying X, Cha L, Wang L, Li W 2010. sRNATarBase: a comprehensive database of bacterial sRNA targets verified by experiments. RNA 16: 2051–2057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carver T, Harris SR, Berriman M, Parkhill J, McQuillan JA 2012. Artemis: an integrated platform for visualization and analysis of high-throughput sequence-based experimental data. Bioinformatics 28: 464–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chabelskaya S, Gaillot O, Felden B 2010. A Staphylococcus aureus small RNA is required for bacterial virulence and regulates the expression of an immune-evasion molecule. PLoS Pathog 6: e1000927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cros MJ, de Monte A, Mariette J, Bardou P, Grenier-Boley B, Gautheret D, Touzet H, Gaspin C 2011. RNAspace.org: an integrated environment for the prediction, annotation, and analysis of ncRNA. RNA 17: 1947–1956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darling AE, Mau B, Perna NT 2010. progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS One 5: e11147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar R, Domrachev M, Lash AE 2002. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res 30: 207–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eggenhofer F, Tafer H, Stadler PF, Hofacker IL 2011. RNApredator: fast accessibility-based prediction of sRNA targets. Nucleic Acids Res 39: W149–W154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fechter P, Caldelari I, Lioliou E, Romby P 2014. Novel aspects of RNA regulation in Staphylococcus aureus. FEBS Lett 588: 2523–2529. [DOI] [PubMed] [Google Scholar]
- Felden B, Vandenesch F, Bouloc P, Romby P 2011. The Staphylococcus aureus RNome and its commitment to virulence. PLoS Pathog 7: e1002006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geissmann T, Chevalier C, Cros MJ, Boisset S, Fechter P, Noirot C, Schrenzel J, Francois P, Vandenesch F, Gaspin C, et al. 2009. A search for small noncoding RNAs in Staphylococcus aureus reveals a conserved sequence motif for regulation. Nucleic Acids Res 37: 7239–7257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillaspy AF, Worrell V, Orvis J, Roe BA, Dyer DW, Landollo JJ 2006. Staphylococcus aureus NCTC8325 genome. In Gram-positive pathogens (ed. Fischetti V, et al. ), pp. 381–412 ASM Press, Washington, DC. [Google Scholar]
- Guillet J, Hallier M, Felden B 2013. Emerging functions for the Staphylococcus aureus RNome. PLoS Pathog 9: e1003767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howden BP, Beaume M, Harrison PF, Hernandez D, Schrenzel J, Seemann T, Francois P, Stinear TP 2013. Analysis of the small RNA transcriptional response in multidrug-resistant Staphylococcus aureus after antimicrobial exposure. Antimicrob Agents Chemother 57: 3864–3874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang HY, Chang HY, Chou CH, Tseng CP, Ho SY, Yang CD, Ju YW, Huang HD 2009. sRNAMap: genomic maps for small non-coding RNAs, their regulators and their targets in microbial genomes. Nucleic Acids Res 37: D150–D154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huson DH, Bryant D 2006. Application of phylogenetic networks in evolutionary studies. Mol Biol Evol 23: 254–267. [DOI] [PubMed] [Google Scholar]
- Kery MB, Feldman M, Livny J, Tjaden B 2014. TargetRNA2: identifying targets of small regulatory RNAs in bacteria. Nucleic Acids Res 42: W124–W129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL 2013. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 14: R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kin T, Yamada K, Terai G, Okida H, Yoshinari Y, Ono Y, Kojima A, Kimura Y, Komori T, Asai K 2007. fRNAdb: a platform for mining/annotating functional RNA candidates from non-coding RNA sequences. Nucleic Acids Res 35: D145–D148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konstantinidis KT, Tiedje JM 2007. Prokaryotic taxonomy and phylogeny in the genomic era: advancements and challenges ahead. Curr Opin Microbiol 10: 504–509. [DOI] [PubMed] [Google Scholar]
- Kuroda M, Ohta T, Uchiyama I, Baba T, Yuzawa H, Kobayashi I, Cui L, Oguchi A, Aoki K, Nagai Y, et al. 2001. Whole genome sequencing of meticillin-resistant Staphylococcus aureus. Lancet 357: 1225–1240. [DOI] [PubMed] [Google Scholar]
- Lalaouna D, Eyraud A, Chabelskaya S, Felden B, Massé E 2014. Regulatory RNAs involved in bacterial antibiotic resistance. PLoS Pathog 10: e1004299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasa I, Toledo-Arana A, Dobin A, Villanueva M, de los Mozos IR, Vergara-Irigaray M, Segura V, Fagegaltier D, Penades JR, Valle J, et al. 2011. Genome-wide antisense transcription drives mRNA processing in bacteria. Proc Natl Acad Sci 108: 20172–20177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R 2010. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26: 589–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R; 1000 Genome Project Data Processing Subgroup 2009. The sequence alignment/map format and SAMtools. Bioinformatics 25: 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Huang D, Cheung MK, Nong W, Huang Q, Kwan HS 2013. BSRD: a repository for bacterial small regulatory RNA. Nucleic Acids Res 41: D233–D238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lioliou E, Sharma CM, Caldelari I, Helfer AC, Fechter P, Vandenesch F, Vogel J, Romby P 2012. Global regulatory functions of the Staphylococcus aureus endoribonuclease III in gene expression. PLoS Genet 8: e1002782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu JM, Livny J, Lawrence MS, Kimball MD, Waldor MK, Camilli A 2009. Experimental discovery of sRNAs in Vibrio cholerae by direct cloning, 5S/tRNA depletion and parallel sequencing. Nucleic Acids Res 37: e46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchais A, Naville M, Bohn C, Bouloc P, Gautheret D 2009. Single-pass classification of all noncoding sequences in a bacterial genome using phylogenetic profiles. Genome Res 19: 1084–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuno T, Chou MY, Inouye M 1984. A unique mechanism regulating gene expression: translational inhibition by a complementary RNA transcript (micRNA). Proc Natl Acad Sci 81: 1966–1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison JM, Miller EW, Benson MA, Alonzo F III, Yoong P, Torres VJ, Hinrichs SH, Dunman PM 2012. Characterization of SSR42, a novel virulence factor regulatory RNA that contributes to the pathogenesis of a Staphylococcus aureus USA300 representative. J Bacteriol 194: 2924–2938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen JS, Christiansen MH, Bonde M, Gottschalk S, Frees D, Thomsen LE, Kallipolitis BH 2011. Searching for small σB-regulated genes in Staphylococcus aureus. Arch Microbiol 193: 23–24. [DOI] [PubMed] [Google Scholar]
- Novick RP, Iordanescu S, Projan SJ, Kornblum J, Edelman I 1989. pT181 plasmid replication is regulated by a countertranscript-driven transcriptional attenuator. Cell 59: 395–404. [DOI] [PubMed] [Google Scholar]
- Novick RP, Ross HF, Projan SJ, Kornblum J, Kreiswirth B, Moghazeh S 1993. Synthesis of staphylococcal virulence factors is controlled by a regulatory RNA molecule. EMBO J 12: 3967–3975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang KC, Stephen S, Dinger ME, Engström PG, Lenhard B, Mattick JS 2007. RNAdb 2.0—an expanded database of mammalian non-coding RNAs. Nucleic Acids Res 35: D178–D182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pichon C, Felden B 2005. Small RNA genes expressed from Staphylococcus aureus genomic and pathogenicity islands with specific expression among pathogenic strains. Proc Natl Acad Sci 102: 14249–14254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinel-Marie ML, Brielle R, Felden B 2014. Dual toxic peptide-coding Staphylococcus aureus RNA under antisense regulation targets host cells and bacterial rivals unequally. Cell Rep 7: 424–435. [DOI] [PubMed] [Google Scholar]
- Pischimarov J, Kuenne C, Billion A, Hemberger J, Cemič F, Chakraborty T, Hain T 2012. sRNAdb: a small non-coding RNA database for gram-positive bacteria. BMC Genomics 13: 384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts C, Anderson KL, Murphy E, Projan SJ, Mounts W, Hurlburt B, Smeltzer M, Overbeek R, Disz T, Dunman PM 2006. Characterizing the effect of the Staphylococcus aureus virulence factor regulator, SarA, on log-phase mRNA half-lives. J Bacteriol 188: 2593–2603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romilly C, Caldelari I, Parmentier D, Lioliou E, Romby P, Fechter P 2012. Current knowledge on regulatory RNAs and their machineries in Staphylococcus aureus. RNA Biol 9: 402–413. [DOI] [PubMed] [Google Scholar]
- Sayed N, Jousselin A, Felden B 2012. A cis-antisense RNA acts in trans in Staphylococcus aureus to control translation of a human cytolytic peptide. Nat Struct Mol Biol 19: 105–112. [DOI] [PubMed] [Google Scholar]
- Sentausa E, Fournier PE 2013. Advantages and limitations of genomics in prokaryotic taxonomy. Clin Microbiol Infect 19: 790–795. [DOI] [PubMed] [Google Scholar]
- Snel B, Bork P, Huynen MA 1999. Genome phylogeny based on gene content. Nat Genet 21: 108–110. [DOI] [PubMed] [Google Scholar]
- Snel B, Huynen MA, Dutilh BE 2005. Genome trees and the nature of genome evolution. Annu Rev Microbiol 59: 191–209. [DOI] [PubMed] [Google Scholar]
- Storz G, Vogel J, Wassarman KM 2011. Regulation by small RNAs in bacteria: expanding frontiers. Mol Cell 43: 880–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomasini A, Francois P, Howden BP, Fechter P, Romby P, Caldelari I 2014. The importance of regulatory RNAs in Staphylococcus aureus. Infect Genet Evol 21: 616–626. [DOI] [PubMed] [Google Scholar]
- Xie C, Yuan J, Li H, Li M, Zhao G, Bu D, Zhu W, Wu W, Chen R, Zhao Y 2014. NONCODEv4: exploring the world of long non-coding RNA genes. Nucleic Acids Res 42: D98–D103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue T, Zhang X, Sun H, Sun B 2014. ArtR, a novel sRNA of Staphylococcus aureus, regulates α-toxin expression by targeting the 5′ UTR of sarT mRNA. Med Microbiol Immunol 203: 1–12. [DOI] [PubMed] [Google Scholar]
- Yoder-Himes DR, Chain PS, Zhu Y, Wurtzel O, Rubin EM, Tiedje JM, Sorek R 2009. Mapping the Burkholderia cenocepacia niche response via high-throughput sequencing. Proc Natl Acad Sci 106: 3976–3981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuker M 2003. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res 31: 3406–3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.