

Abstract
Identification of microRNAs (miRNAs) is an important step toward understanding post-transcriptional gene regulation and miRNA-related pathology. Difficulties in identifying miRNAs through experimental techniques combined with the huge amount of data from new sequencing technologies have made in silico discrimination of bona fide miRNA precursors from non-miRNA hairpin-like structures an important topic in bioinformatics. Among various techniques developed for this classification problem, machine learning approaches have proved to be the most promising. However these approaches require the use of training data, which is problematic due to an imbalance in the number of miRNAs (positive data) and non-miRNAs (negative data), which leads to a degradation of their performance. In order to address this issue, we present an ensemble method that uses a boosting technique with support vector machine components to deal with imbalanced training data. Classification is performed following a feature selection on 187 novel and existing features. The algorithm, miRBoost, performed better in comparison with state-of-the-art methods on imbalanced human and cross-species data. It also showed the highest ability among the tested methods for discovering novel miRNA precursors. In addition, miRBoost was over 1400 times faster than the second most accurate tool tested and was significantly faster than most of the other tools. miRBoost thus provides a good compromise between prediction efficiency and execution time, making it highly suitable for use in genome-wide miRNA precursor prediction. The software miRBoost is available on our web server http://EvryRNA.ibisc.univ-evry.fr.
Keywords: microRNA prediction, classification, boosting, support vector machine (SVM), imbalanced data
INTRODUCTION
MicroRNAs (miRNAs) are small single stranded noncoding RNAs (21–22 nt) found in eukaryotic cells. MiRNAs function by regulating gene expression either by translational inhibition or message degradation, and thus are key to many biological processes. Dysregulation of miRNAs is known to cause a wide range of diseases (miR2Disease database Jiang et al. 2009) such as hereditary progressive hearing loss (Mencía et al. 2009), growth and skeleton defects (de Pontual et al. 2011), various cancers (He et al. 2005; Mraz et al. 2009), heart diseases (Thum et al. 2007), and Alzheimer's disease (Maes et al. 2009). Mature miRNAs are made from precursors (pre-miRNAs) of ∼90 nt in length, characterized by a hairpin-like structure. More than 18,000 miRNAs have been discovered in ∼140 species, of which >1500 are in Homo sapiens (Kozomara and Griffiths-Jones 2011). However, recent studies reveal that a large number of miRNAs have yet to be discovered (van Rooij 2011). The identification of novel miRNAs from genomes is thus of key importance for both biological and medical sciences. Novel miRNAs are difficult to detect in cells with experimental techniques due to their small size and low abundance (Lagos-Quintana et al. 2001; Lai et al. 2003). In silico prediction is therefore useful for identifying potential pre-miRNAs, which can be subsequently validated experimentally. Several methods have been recently developed to detect pre-miRNAs, including comparative genomics, homology-based, and ab initio approaches. Comparative genomics approaches use multiple alignments of sequences to compare genomes of related species for detection of conserved pre-miRNAs. Such approaches are RNAmicro (Hertel and Stadler 2006), miRFinder (Huang et al. 2007), miRSeeker (Lai et al. 2003), MiRScan (Lim et al. 2003), and miRRim (Terai et al. 2007). Homology-based methods exploit information from homologous sequences and structures to identify new pre-miRNAs that are homologous to existing ones, as proposed in ERPIN (Legendre et al. 2004) and miRAlign (Wang et al. 2005). Nonetheless, it is unlikely that comparative genomics and homology-based methods would work efficiently when a new candidate sequence without a known homolog or cross-species sequence conservation is studied.
The ab initio approaches, which may help to avoid this issue, can be classified into three categories. The first, considered as completely ab initio, searches for potential pre-miRNAs occurring in genomes via intrinsic properties of sequence and structure of pre-miRNAs, as in CID-miRNA (Tyagi et al. 2008) and miRNAFold (Tempel and Tahi 2012). The second category predicts potential pre-miRNAs considering additional information, for example, the positions or neighbors of a given sequence in a genomic sequence, as in miR-abela (Sewer et al. 2005) and MIReNA (Mathelier and Carbone 2010). These two categories are applied as a rough filter for pre-miRNA candidates, which might be subsequently refined using other techniques.
Following the primary filters for pre-miRNA candidates, the third category classifies these as real or pseudo pre-miRNAs. Among different techniques developed for this classification problem, machine learning approaches have demonstrated to be the most promising. Several machine learning techniques have been applied to deal with the classification of pre-miRNAs, such as genetic programming (miRPred, Brameier and Wiuf 2007), random forests (MiPred, Jiang et al. 2007), random walk (miRank, Xu et al. 2008), Bayesian networks (BayesMiRNAfinder, Yousef et al. 2006), kernel density estimator (mir-KDE, Chang et al. 2008), and hidden Markov models (CSHMM, Agarwal et al. 2010; proMIR, Nam et al. 2005).
Besides these above-mentioned methods, support vector machines (SVM) (Vapnik 1998) have been widely applied for classification in bioinformatics, information retrieval, computer vision, etc. The SVM defines a separating hyperplane that divides the space into two sides by maximizing the margin or the distance from the hyperplane to the closest samples. A number of computational tools using SVM have been implemented to identify pre-miRNAs, such as: miPred (Ng and Mishra 2007), miRPara (Wu et al. 2011), and triplet-SVM (Xue et al. 2005).
Nevertheless, as the number of determined non-miRNAs is much higher than that of identified pre-miRNAs, we are faced with an imbalance in the training data. The traditional learning-based classifiers, such as standard SVM, which aim to achieve the highest accuracy for the whole set of samples, are not suitable to deal with imbalanced learning tasks as they tend to classify all given samples into the more prevalent class in the training data (Wu and Chang 2003). Hence, a majority of candidate sequences would be predicted as non-miRNAs. Several kinds of modifications have been included in SVM approaches to deal with imbalanced data sets. Morik et al. (1999) presented an SVM model in which the cost factors for positive and negative examples were distinguished. Lewis et al. (2004) described a thresholding strategy while Li and Shawe-Taylor (2003) introduced an SVM with uneven margins. The parameter of uneven margins represents the ratio of the negative margins to the positive margins of the SVM classifier and is equal to 1 in the standard SVM. For an imbalanced data set with a few positive samples and many negative ones, it would be beneficial to use larger margins for the positive ones than for the negative ones. Over-sampling and under-sampling techniques have also been implemented in Ling and Li (1998), Japkowicz (2000), and Chawla et al. (2002). Certain ensemble methods, with no aims to treat this problem, showed a good performance in dealing with imbalanced data, such as bagging (Breiman 1996) and boosting (Schapire 1990), in which boosting was empirically shown to perform better when the data do not have much noise (Bauer and Kohavi 1999; Opitz and Maclin 1999).
A few methods have been recently developed to overcome the imbalance issue of pre-miRNAs, but all of them are unusable on large data sets because of their speed or their unavailability. Such methods include microPred (Batuwita and Palade 2009), MiRenSVM (Ding et al. 2010), mirExplorer (Guan et al. 2011), HeteroMirPred (Lertampaiporn et al. 2013), and HuntMi (Gudys et al. 2013).
In this paper, we introduce miRBoost, which uses the boosting method, combined with weakened SVM component classifiers, for dealing with imbalanced training data in ab initio pre-miRNA classification. The principle is to have a sequence of SVM classifiers, where each classifier is applied on the subset of data that were not well classified. This technique not only has shown good performance in classifying imbalanced data, but is also advantageous in terms of execution time.
miRBoost takes a set of sequence candidates as input and classifies each of them as a pre-miRNA or not. The classification of pre-miRNAs is based on a set of features or parameters that characterize a given sequence. These features should not only be distinctive for the classification task but should also be independent between them. A feature that is irrelevant to discrimination will reduce the predictive capability (Dash and Liu 1997). Meanwhile, different features may provide a similar discriminative power and the removal of redundancy in such a set of features may improve the performance in terms of execution time as well as in prediction accuracy. However, the selection of helpful features utilized for pre-miRNA identification is still in question (Stepanowsky et al. 2012). In this study, we extracted appropriate features from 62 novel features that we developed and 125 existing ones from the literature using search-based techniques that find highly scored low-dimensional projections of the data.
We evaluated the method on human and cross-species data. The positive data sets were taken from miRBase (version 18) (Kozomara and Griffiths-Jones 2011), followed by a refinement. For the negative ones, firstly, we generated, from exonic regions of protein-coding genes, sequences that could be folded into hairpin-like structures and verify several pre-miRNA characteristics. Secondly, we took the noncoding RNAs that are not pre-miRNAs from different databases. The evaluation was conducted through a fivefold cross-validation and a prediction on novel pre-miRNA and non-miRNA sequences. With the boosting technique, using the appropriately selected features, our method has shown favorable performance in comparison with state-of-the-art tools, on both prediction results as well as on running time. The software miRBoost is available on our web server http://EvryRNA.ibisc.univ-evry.fr.
The paper is organized as follows. In the two next sections, we present and discuss the results obtained with miRBoost compared with several existing pre-miRNA classification methods, in terms of classification performance, sensitivity and specificity in predicting new pre-miRNAs, and execution time. Then, we describe the materials and methods, including the human and cross-species training and prediction data sets we use, our feature selection process, and the algorithm. The existing tools that we compared with miRBoost are detailed afterwards.
RESULTS
Experimental setup
We measure the performance of miRBoost on one data set from human and one from cross-species. The data sets and the feature selection process are described below in the Materials and Methods.
miRBoost was compared with several existing computational tools of pre-miRNA classification in order to evaluate its performance: CSHMM (Agarwal et al. 2010) and triplet-SVM (Xue et al. 2005), which do not take into account the imbalance problem; HeteroMirPred (Lertampaiporn et al. 2013), microPred (Batuwita and Palade 2009), MiPred (Jiang et al. 2007), and mirExplorer (Guan et al. 2011), which deal with imbalanced data; and MIReNA (Mathelier and Carbone 2010), which does not apply machine learning techniques for classification. These tools are described in more detail in the Materials and Methods.
We mainly used sensitivity, specificity and g-mean to measure the performance. We denote TP, FP, TN, FN as the numbers of true positive, false positive, true negative, and false negative predictions, respectively. Sensitivity SE = TP/(TP + FN) measures the fraction of pre-miRNAs correctly classified. Specificity SP = TN/(TN + FP) measures the fraction of non-miRNAs correctly classified. g-mean, which is usually used for evaluating classifiers on imbalanced data, is the geometric mean sensitivity and specificity . A high g-mean signifies a high value for both sensitivity and specificity simultaneously. Other measures are also reported in Supplemental Data 1.
Classification performance
We conducted a fivefold cross-validation to evaluate the classification performance of miRBoost. miRBoost predicts each of the five subsamples with the model trained on the combination of four remaining subsamples, using the features identified on this training set via our feature selection process, for human and cross-species data, respectively. These cross-validated results were compared to those obtained with other existing pre-miRNA classification methods. As the cross-validation requires the models built on the different data subsets, we considered, for this comparison, CSHMM, triplet-SVM, and microPred, the only machine learning methods that allow retraining their model, and also MIReNA, which is not based on machine learning.
ROC spaces for the classification results of miRBoost and the existing tools on human and cross-species are given in Figure 1. As shown, miRBoost gives the best compromise between sensitivity and specificity, i.e., the ability to simultaneously predict pre-miRNAs and reject non pre-miRNAs. In both cases of human and cross-species, the score (1-specificity, sensitivity) of miRBoost is always the closest to (0,1). This is also proved by the highest g-mean of 0.90 and other measures achieved with miRBoost (see Supplemental Data 1). microPred, which is the only method that manages to solve the imbalanced data issue tested here, performs slightly better in specificity and slightly worse in sensitivity in comparison with miRBoost, yielding a lower g-mean for both data sets (0.87 and 0.89). Meanwhile, CSHMM, triplet-SVM, and MIReNA show considerably lower classification ability, with g-mean from 0.69 to 0.87 for human, and from 0.64 to 0.86 for cross-species. A table giving the results obtained with miRBoost and those tools, expressed in several other measures, is provided in Supplemental Data 1.
FIGURE 1.
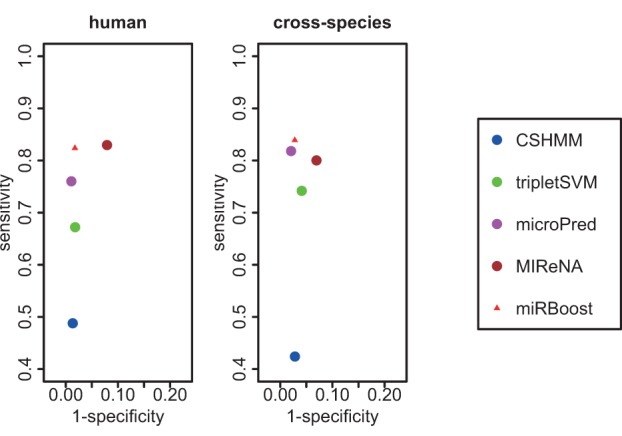
ROC space for cross-validated classification results of miRBoost and the other methods on human and cross-species data.
Predictive sensitivity and specificity on new sequences
We evaluated the predictive sensitivity of miRBoost and the other methods on 690 novel pre-miRNAs from the difference of miRBase versions 19 and 20 and version 18, and the predictive specificity on 8246 non-miRNA sequences (see “Data sets” section). We applied the model trained on the whole data set of cross-species for the prediction, with 18 features identified on this set through our feature selection process (see Pre-miRNA Feature Selection). Similarly, the models of CSHMM, triplet-SVM, and microPred were trained on that whole data set. We also compared with MiPred, HeteroMirPred, and mirExplorer, for which we used the provided models, as they do not allow retraining of their models with other data. The predicting results are given in Figure 2 and Supplemental Data 1.
FIGURE 2.
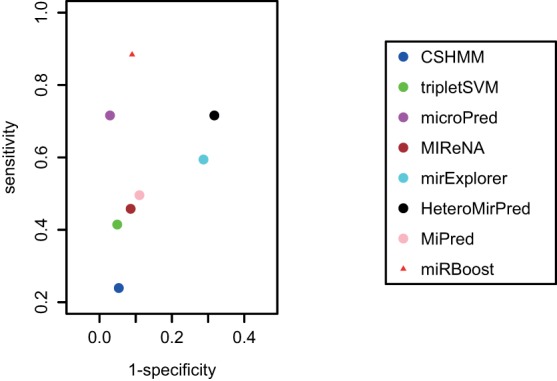
ROC space for prediction results of miRBoost and the other methods on new data.
miRBoost predicts 610 (88.4%) sequences as pre-miRNAs and rejects 7504 (91.0%) non-miRNA sequences. Our method provides the best equilibrium between sensitivity and specificity, with a g-mean of 0.90, which is almost 7% higher than the second best method, microPred. Most of the other methods tested are far from producing a correct prediction. CSHMM, triplet-SVM, MIReNA, and MiPred reject not only most of non-miRNA sequences (89.0%–95.1%) but also a large number of pre-miRNAs (50.4%–76.1%). HeteroMirPred produces a fair equilibrium between sensitivity and specificity, yet both are low, giving a g-mean of 0.70. Our results show that miRBoost can considerably prevail over the other approaches in predicting novel pre-miRNAs.
Running time
Due to the ever-increasing amount of data coming from new sequencing technologies, execution time has become an important factor in evaluating computational prediction tools. In Table 1, we present the running time of miRBoost and the other tools (except for mirExplorer, which could not be installed on our Linux machine) for predicting the 690 novel pre-miRNAs and the 8246 non-miRNA sequences considered above.
TABLE 1.
Comparison of miRBoost and other existing tools running time in classifying 690 pre-miRNAs and 8246 non-miRNAs

miRBoost is the only method that performs well the classification, when dealing with the imbalanced data issue, in a reasonable amount of time (2 min 29 sec). This compares with several hours for related methods such as microPred, MiPred, and HeteroMirPred. Although microPred shows similar results to miRBoost in classification ability, it takes over 1400 times longer than miRBoost to achieve this. miRBoost is also 10 times faster than CSHMM. triplet-SVM and MIReNA seem to be very rapid (three times faster than miRBoost), yet their prediction performance is much lower than that of miRBoost.
All experiments were performed on a Linux machine with four six-core processors Intel Xeon X5680 of 3.33 GHz and 20 GB of RAM, except for mirExplorer, where the tests were performed on a Windows machine with Core 2 Duo Intel E8400 of 3.00 GHz and 4 GB of RAM. None of the programs were executed in parallel mode.
Selected features
We carried out two different processes of feature selection. First, feature sets were determined through the fivefold cross-validation. We validated miRBoost on each of the five subsamples with the features selected on the set of the four others, on human and cross-species data, respectively. Second, we selected upstream a feature set on each whole data set of human and cross-species. Table 2 shows the number of features selected in these cross-validated training sets and whole data sets (see Supplemental Data 2 for more details).
TABLE 2.
Number of selected features for each training set
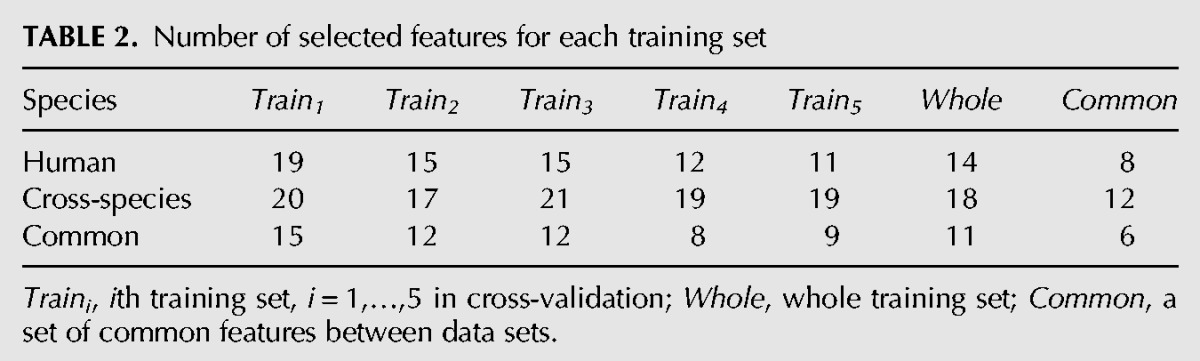
We find eight common features for human among the whole data set and the five training sets from the cross-validation process: maximum number of consecutive G's in the longest exact stem, folding free energy of the longest nonexact stem, imbalance of G + A with regard to C + U in the longest nonexact stem, maximum number of consecutive C's in the hairpin, maximum number of consecutive G's in the hairpin, folding free energy adjusted by the hairpin size, average folding free energy of the exact stems, and size of bulges. These features can be considered as the properties that characterize human pre-miRNAs.
A similar conservation is observed in cross-species data with 12 common features: folding free energy of the longest nonexact stem, maximum number of consecutive G's in the longest nonexact stem, percentage of dinucleotides CC and GA, maximum number of consecutive C's in the hairpin, maximum number of consecutive G's in the hairpin, percentage of G–U pairs, folding free energy adjusted by the hairpin size, percentage of paired U, average folding free energy of the exact stems, percentage of triplets unpaired–unpaired A–paired (denoted as A••(), and size of bulges. These 12 features are thus presumed characteristics for cross-species pre-miRNAs.
As shown in Table 2, six features are common for the two data sets: folding free energy of the longest nonexact stem, maximum number of consecutive C's in the hairpin, maximum number of consecutive G's in the hairpin, folding free energy adjusted by the hairpin size, average folding free energy of the exact stems, and size of bulges. The feature selection processes on the two data sets also show a good correlation with >50% of features conserved in each pair of corresponding training sets, illustrating the fitness of our strategy in the selection of features.
DISCUSSION
Robustness regarding selected features
Classification performance is usually affected by the features used for classification. To verify the robustness of miRBoost regarding the selected features, we performed a cross validation using 14 features for human and 18 features for cross-species, which were identified upstream. The results obtained, compared with those obtained when the features were determined through fivefold cross validation (as described in “Classification Performance” section), are equivalent for cross-species, and very slightly different for human (see Supplemental Data 1). This indicates the robustness of miRBoost with the selected features.
Consistent features
As shown in the Results, six features characterizing pre-miRNAs are always selected and are common to all data sets. Among these features, four belong to the 62 new features that we have defined: maximum number of consecutive C's in the hairpin, maximum number of consecutive G's in the hairpin, folding free energy adjusted by the hairpin size, and folding free energy of all exact stems over number of paired nucleotides in the hairpin. The two other features are related to the folding free energy of the longest nonexact stem and the size of all bulges.
We analyzed the six features on the whole cross-species data. 94.9% and 96.6% of pre-miRNAs contain from 2 to 5 (mostly 2–3) consecutive C's and G's in the hairpin, respectively. These two features concern GC-content and low-complexity regions, and may thus suggest related properties on pre-miRNA composition. With regard to the folding free energy adjusted by the hairpin size, the average for pre-miRNAs is −44.91 kcal/mol, which is much lower than the average of −31.11 for non-miRNAs. For the folding free energy of all exact stems, pre-miRNAs have an average of −0.8, whereas non-miRNAs have −0.7, per base pair. In pre-miRNAs, the folding free energy of the longest nonexact stem ranges from −96.6 to −4.6 kcal/mol, with an average of −32.03. This energy has a much higher average of −15.77 in non-miRNAs, ranging from −77.2 to −4.0 kcal/mol. It is noteworthy that the values of those features must be moderate for a valid pre-miRNA. If the folding free energy is too high or there is no consecutive C's and G's in the hairpin, the secondary structure is thermodynamically unstable. On the contrary, if the folding free energy is too low or the hairpin contains several consecutive C's and G's, the secondary structure is so stable that the RNA-induced silencing complex (RISC) might hardly cleave the two complementary strains of the hairpin to create the miRNA. Finally, 96.8% of pre-miRNAs have a total bulge size of 0 or 1, which represents the high symmetry between the two strands. It suggests that pre-miRNAs should not contain large asymmetric bulges, as the latter increase the folding free energy and degrade the hairpin structure stability.
Here we can make two remarks. Firstly, those features are not only restricted to the hairpin secondary structure, but also related to the nucleotide composition, and thus show that consecutive nucleotides such as microsatellites (short tandem repeats) could be inappropriate for the pre-miRNA structure. Secondly, folding free energies on the exact stems and the longest nonexact stem, as well as size of the bulges, which we initially exploited in miRNAFold (Tempel and Tahi 2012), are confirmed in this work to be significantly important to the hairpin structure.
Effect of the boosting technique on miRBoost performance
To show how the boosting technique improves the classification results in dealing with imbalanced data, we tested the standard SVM technique without boosting. The SVM model was trained on the whole cross-species data set. We considered the 18 selected features for cross-species mentioned above. The model parameters were optimized with the intrinsic cross-validation protocol of LIBSVM (Chang and Lin 2011). We obtained 0.79 in sensitivity and 0.98 in specificity in the cross-validation, while miRBoost, with the boosting technique, gives 0.84 in sensitivity and 0.97 in specificity. In the prediction of new sequences, SVM reaches 0.78 in sensitivity and 0.97 in specificity, while miRBoost produces 0.88 in sensitivity and 0.91 in specificity. In both cases, g-mean from miRBoost (0.90) is always higher than that from SVM (0.88 and 0.87).
As expected, despite the effort to correct the imbalance in training data sets via optimized parameters, the standard SVM technique tends to classify the sequences into the larger negative data set. The result of this is that the number of predicted positive pre-miRNAs is lower than when using the boosting technique for the training on imbalanced data. The boosting technique therefore allows prediction of more positive pre-miRNAs, but more importantly gives a better compromise between sensitivity and specificity. This suggests that beside our defined pre-miRNA features and feature selection process, which contribute to the high performance in discrimination between pre-miRNAs and non-miRNAs, the performance of miRBoost is also improved with the boosting method.
miRBoost performance in comparison with the state-of-the-art methods
Here we show that miRBoost is a fast and accurate machine learning-based computational approach for classifying pre-miRNAs. Our method shows over 0.95 accuracy in the cross-validation test, and over 0.88 sensitivity and 0.91 specificity in predicting the novel sequences. Compared with existing methods in the literature, miRBoost gives the best compromise between sensitivity and specificity, with a g-mean of 0.90, in addition to between prediction efficiency and execution time. Furthermore, it is the only method dealing with imbalanced data that has a fast execution time. miRBoost takes ∼2.5 min, while the other methods (microPred, MiPred, HeteroMirPred) take several hours.
The two most rapid methods, triplet-SVM and MiReNA, take less than a minute. Nevertheless, while the results obtained in the cross-validation of these two tools are relatively low, their performances in prediction of new sequences are much worse than that of miRBoost. The rapidity of triplet-SVM and MiReNA is principally due to the use of a filter, which quickly rejects the sequences that do not satisfy some constraints and thus rapidly classifies those sequences as negative samples. In miRBoost, most of the running time is for quantifying the sequence features, i.e., computing numeric feature values, which are necessary as the input to SVM classifiers. We try to fold every given sequence into a hairpin structure with miRNAFold (Tempel and Tahi 2012), then subsequently generate feature values for the classification (see Pre-miRNA feature selection section), instead of using foremost a filter as in triplet-SVM and MIReNA. Though the use of such a filter can significantly reduce the execution time, we prefer to keep it as pre-miRNA features to see how our classification method is able to deal with different discriminative levels of the features.
Prospectus
The performance of miRBoost could be improved with an enhancement on feature selection using boosting (Redpath and Lebart 2005; Chen et al. 2010). With regard to the efficiency of boosting, a study on the diversity of weakened SVM component classifiers might provide an insight into the concept of diversity and into the correlation between boosting and diversity (Li et al. 2005). Furthermore, other types of SVM that manage imbalanced data better (Akbani et al. 2004) might also benefit from boosting in dealing with the imbalance issue.
One of our future prospects is to integrate miRBoost to our algorithm miRNAFold, which was previously developed for identifying pre-miRNAs in genomes. miRNAFold is very fast in comparison with the other tools in the literature (it takes <30 sec to analyze a sequence of 1 Mb). It also shows high sensitivity (>90%), but its specificity is unfortunately low. An efficient integration of miRBoost to miRNAFold should therefore allow fast and selective identification of pre-miRNAs in whole genomes.
Software availability
Our software miRBoost is provided on the web server http://EvryRNA.ibisc.univ-evry.fr with 14 human and 18 cross-species specific features determined through our feature selection process on the whole data sets of human and cross-species (see Data sets), respectively. The model trained on the whole cross-species data is also available for prediction of new sequences.
MATERIALS AND METHODS
Data sets
We use different sets of positive (pre-miRNA) and negative (non-miRNA) data to perform the cross-validation on human and cross-species genomes and to realize the feature selection process.
Positive data set for cross-validation
The genomes of eukaryotes containing at least 100 miRNAs in the miRBase database (version 18) (Kozomara and Griffiths-Jones 2011) are studied. We take from these genomes pre-miRNAs of <400 nt. As it is known that miRBase contains a number of mis-annotated miRNAs, we first remove the sequences reported as mis-annotated in the later versions (19 and 20). The remaining pre-miRNAs are filtered by ncRNAclassifier (Tempel et al. 2012) to discard the ones that are mis-annotated because corresponding to transposable elements. The obtained sequences are then considered as positive data. They include 1279 sequences for human and 3082 sequences for cross-species. To avoid overfitting, we remove the sequences that have an identity of >97% with the other ones using EMBOSS skipredundant (Rice et al. 2000). Finally, we obtain 863 pre-miRNAs for human and 1677 pre-miRNAs for cross-species.
Negative data set for cross-validation
From selected genomes, we randomly choose the exonic regions from protein-coding genes at NCBI (http://www.ncbi.nlm.nih.gov/genome). We also take the noncoding RNAs that are not miRNA, including tRNA, siRNA, snRNA and snoRNA, from fRNAdb (Kin et al. 2007), NONCODE (Liu et al. 2005), and snoRNA-LBME-db (Lestrade and Weber 2005) database. All of them contain <400 nt. We use miRNAFold (Tempel and Tahi 2012) to predict a hairpin-like structure in each selected sequence (see Fig. 3). miRNAFold first identifies the longest exact stem from the given sequence. The identified stem is then extended into the longest nonexact stem (i.e., succession of exact stems separated by symmetric internal loops). The hairpin secondary structure corresponding to this nonexact stem is finally predicted. Various constraints are applied to the structure prediction. The hairpin-like structure should have a folding free energy ▵G0 < −25.0 kcal/mol, while its hairpin is formed with at least one exact stem of >5 nt. Moreover, at least 90% of the features introduced in miRNAFold must be satisfied. We reduce the sequence redundancy to 97% using EMBOSS skipredundant, giving 7123 human and 7916 cross-species sequences from exonic regions that are not pre-miRNAs, and 299 human and 350 cross-species noncoding RNA sequences.
FIGURE 3.
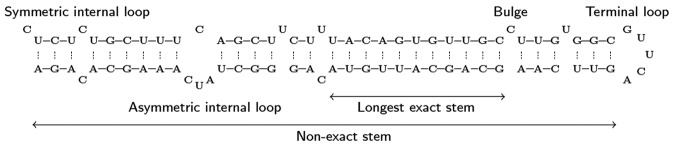
Example of a pre-miRNA hairpin structure (hsa-mir-107).
The two sets of coding and noncoding sequences are then combined to constitute the negative data set for cross-validation. We have then in total 7422 and 8266 sequences of human and cross-species, respectively.
Novel pre-miRNAs for sensitivity validation
Furthermore, we evaluate the sensitivity of our algorithm, i.e., its ability to identify pre-miRNAs, through a prediction on novel cross-species pre-miRNA sequences. These pre-miRNAs are taken from newly added sequences in the versions 19 and 20 of miRBase database. We also use ncRNAclassifier to eliminate false pre-miRNAs, after removing the misannotated ones that are reported in miRBase. Their redundancy is then reduced to 97%, giving 690 sequences.
Non pre-miRNAs for specificity validation
We also measure the specificity of miRBoost, i.e., its ability to reject non-miRNA sequences, with a prediction on other non-miRNAs. This set is constructed similarly to the negative data set for cross-validation described above, and is reduced to 97% of redundancy to the latter. It contains 7916 exonic region sequences and 330 noncoding RNAs.
Algorithm
Boosting is a machine learning method in which weak component classifiers are subsequently added to an ensemble in such a way that they emphasize the samples misclassified by the existing classifiers in the ensemble, where a weak classifier is one that performs slightly better than a random guess. AdaBoost (or Adaptive Boosting), the most popular boosting method formulated by (Freund and Schapire 1997), iteratively learns weak classifiers with a weight distribution on the training samples, then adds them to a final strong classifier. The AdaBoost algorithm is presented as follows:
Algorithm AdaBoost
Input: a set of training samples with labels ; a maximum number of iterations T.
Initialize: the weights of training samples:
For t = 1 to T do
Build a weak classifier ht on with .
-
Calculate the training error of ht on :
If , stop.
- Set the weight of component classifier ht:
- Update the weights of training samples:
where Ct is a normalization constant, and .
Output: .
The core of AdaBoost procedure is to construct appropriate weak component classifiers. Each weak classifier is adaptively built with a favor to the samples misclassified by previous classifiers via weights associated to training samples.
For a weak classifier, the condition that its training error is lower than 1/2 is widely used in the literature (Rangel et al. 2005; Li et al. 2008). SVM, as a relatively strong classifier, does not seem to be suitable for the principle of boosting and may lead to performance degradation (Wickramaratna et al. 2001). However, the use of a weakened SVM model is still efficient, as shown in (Rangel et al. 2005; Li et al. 2008; Ting and Zhu 2009; Wang and Japkowicz 2010). The boosting on SVM classifiers can perform as well as SVM and shows a better generalization performance than that of SVM.
Different methods for building weak classifiers have been implemented. Li et al. (2008) proposed weak RBFSVM (Radial Basis Function) classifiers using large Gaussian widths . They set a large initial value to and gradually decreased it to obtain moderately accurate SVM component classifiers. Ting and Zhu (2009) subdivided the feature space into nonoverlapping regions and trained the local SVM component classifiers on those. Wang and Japkowicz (2010) reduced the diversity of SVM component classifiers by updating weights on training samples. Nonetheless, these methods do not well clarify or quantify the weakness, i.e., control the training error, of component classifiers. Rangel et al. (2005) weakened the component classifiers by not using the whole data set but only its subsets for training in such a way that the training errors of constructed -SVM classifiers were bounded above by 1/2.
In this work, we propose a new definition of classifier weakness, which allows controlling the weakness of component classifiers by implying a lower bound of , for some small , and an upper bound of on their training error. The SVM classifiers are also weakened by training on subsets of the whole data set. We select the training subsets in such a way that the training errors on the whole data set are bounded between and . We implement the weighted C-SVM instance from LIBSVM (Chang and Lin 2011), which allows penalizing the imbalance among training samples via their different weights. The boosting with such weakened SVM classifiers can improve the computation time of the training algorithm, as the training is realized on a smaller data set and requires a smaller number of support vectors.
Let be a set of labeled samples, where , and be a weight distribution over , , where . To build a weak SVM classifier over , we discard that consists of the samples of weights bounded above by from the original data set , and train an SVM classifier on the training subset :
where J has a minimum cardinality, and .
Our algorithm for building a weak SVM classifier is then presented as follows:
Algorithm WeakSVM
Input: set of training samples with labels ; weight distribution ; parameter .
Select such that and J has a minimum cardinality.
- Train a RBFSVM component classifier ht on with the weight distribution
Output: ht
We clarify here the choice of for the weakened SVM classifier built. Let and be the training errors of ht over and , respectively:
where is the indicator function. We hereby deduce . As the training error of ht over is required to be lower than , we have , and thus
| (1) |
Moreover, given that ht classifies the samples of independently with probability , the expected value of is . The weakness of ht on requires the lower bound on : , which implies
| (2) |
For a tiny , as ht is still a strong classifier on , we can deduce, from Equations (1) and (2), , which implies .
Hence, we may leave δ as a parameter and choose for some δ in [1/4, 1/2]. As δ is involved in the lower bound of the training error, the choice of δ plays an important role in determining the weakness of SVM component classifiers, and thus for the performance of miRBoost. When δ is small (close to 0.25), the training errors get close to each other, and thus the diversity among component classifiers is reduced. Contrarily, when δ is large (close to 0.5), the weakness is not always guaranteed. We found a critical value of for both data sets (human and cross-species), at which miRBoost performs its Pareto optimality regarding different measures, i.e., the optimal state that no measure could be made better off without making any other measure worse off (see Fig. 1 in Supplemental Data 1). This may suggest appropriate bounds for the diversity between component classifiers, which is correlated with the efficiency of the boosting technique.
Pre-miRNA feature selection
A given sequence is identified as either a pre-miRNA or a non-miRNA based on its features. It is thus important to select an appropriate set of features of pre-miRNAs for classification. In this work, we did an exhaustive study to get all (or at least most) pre-miRNA characteristics representing intrinsic properties on sequence and structure used by different teams in their published works on pre-miRNA prediction.
We first calculate 62 features that we newly introduced, among which 26 features are used in miRNAFold (Tempel and Tahi 2012). These features describe the intrinsic properties of the pre-miRNA hairpin: size, energy, nucleotide composition of exact and nonexact stems in total and in average, size and number of bulges and loops, and hairpin asymmetry.
We also extract 125 features from the literature, which are used in several pre-miRNA prediction algorithms, including microPred (Batuwita and Palade 2009), MiPred (Jiang et al. 2007), miR-abela (Sewer et al. 2005), miRank (Xu et al. 2008), and triplet-SVM (Xue et al. 2005). Among them, 32 structural features are taken from triplet-SVM (Xue et al. 2005). They represent the structure-sequence information of every three adjacent nucleotides, described by the middle nucleotide among the three and the pairing status of all the three, e.g., U.((. In addition, 63 features are taken from microPred (Batuwita and Palade 2009), 14 features from miR-abela (Sewer et al. 2005), and 16 features from miRank (Xu et al. 2008) and miPred (Jiang et al. 2007). These features correspond to the primary sequence of pre-miRNAs such as ratio of dinucleotides (Batuwita and Palade 2009), the secondary structure of pre-miRNAs such as the number of base pairs (Sewer et al. 2005), and the average size of internal loops (Batuwita and Palade 2009).
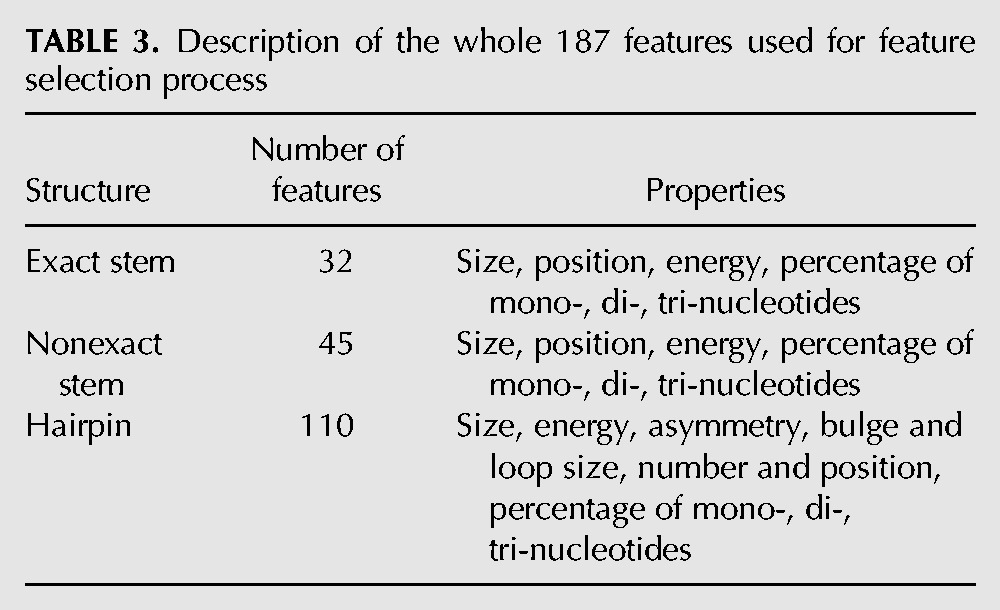
Thus, we study in total 187 features, which give information on structure and sequence of pre-miRNAs. They can be gathered into three groups describing size, position, composition, asymmetry, and energy of the exact stem, nonexact stem and hairpin (see Table 3; Supplemental Data 2).
TABLE 3.
Description of the whole 187 features used for feature selection process
For each data set, to select the consistent and nonredundant features from those 187 ones, we exploit feature selection techniques proposed by the WEKA workbench (Hall et al. 2009): Best First, Linear Forward Selection, Greedy Stepwise, Scatter Search, and Subset Size Forward Selection. These search-based techniques (Devijver and Kittler 1982; Jain and Zongker 1997) find highly scored low-dimensional projections of the given data, and thus select the features giving the largest projections in lower dimensional spaces. From the output of those five methods, we choose the features discovered by at least two of them.
Existing software
A number of computational tools were introduced for pre-miRNA classification. As mentioned in the Results, we used several of them for comparison with miRBoost: CSHMM (Agarwal et al. 2010), triplet-SVM (Xue et al. 2005), microPred (Batuwita and Palade 2009), HeteroMirPred (Lertampaiporn et al. 2013), MiPred (Jiang et al. 2007), mirExplorer (Guan et al. 2011), and MIReNA (Mathelier and Carbone 2010), which are described as follows.
CSHMM used a context-sensitive hidden Markov model (HMM) to represent pre-miRNA structures and identify pre-miRNAs in genomes. CSHMM extended the idea of HMM by introducing a memory, in the form of a stack or a queue, between certain states in the model.
The proposed CSHMM structure had two context sensitive states that were linked to the same pairwise-emission state through a stack in order to separate states for the stem and symmetric bulge generation and to keep information about what was emitted earlier. The known human pre-miRNA sequences were used to assess the transition and emission probabilities for CSHMM.
triplet-SVM consisted of an SVM classifier applied on the features of local contiguous structure-sequence information to distinguish real and pseudo pre-miRNAs. The SVM model trained on human miRNA data showed the ability to predict pre-miRNAs from other species across animals, plants, and viruses with high accuracy. It suggested that their 32 features of triplet elements reflected discriminative and conserved characteristics of pre-miRNAs, which were consistent across all species.
microPred applied filter methods to select discriminative features (they filtered 48 initially proposed features to 21) and utilized an SVM classifier, which was able to deal with the imbalance problem via techniques of random over/under-sampling, synthetic minority over-sampling technique (SMOTE), different error costs, and z-SVM. SMOTE was an over-sampling technique that created new synthetic samples in the neighborhood of the existing minority (positive) samples. In this technique, they randomly selected a positive class sample, and determined its k-nearest neighbors. A set of synthetic data points was then generated such that each one was located between the original data point and one of its nearest neighbors. In z-SVM method, firstly, an SVM model was developed using the imbalanced training data set. Then, the decision boundary of the resulted model was modified to remove the bias of the classifier toward the majority (negative) class. This was done by multiplying the coefficients of the minority (positive) support vectors by a particular value referred to as z (z > 1). The value of z, which gave the best classification for the training data set, was selected as the optimal z value. The prediction model of microPred was built on human data and then validated on other animal and viral data.
HeteroMirPred aggregated the prediction of different heterogeneous algorithms, including SVM, k-nearest neighbors, and random forest in order to create a high level of diversity and to reduce bias of each individual classifier. The authors proposed a modified version of SMOTE to solve the imbalanced data problem. The feature selection was realized via filter methods of ReliefF, Information Gain, and Correlation-based feature selection from 125 features on sequence, secondary structure, base pairs, triplet elements, and structural robustness. This cooperative combination was validated across various organisms, from animals, plants to viruses.
MiPred used random forests to separate the real pre-miRNAs from the pseudo ones. Random forest is an ensemble classifier that consists of several decision trees (Breiman 2001). It could be considered as a combination of a further development of the bagging technique and a random feature selection technique. For bagging, each tree was trained on a bootstrap sample of the training data, and predictions were made by a majority vote of trees. For feature selection, a hybrid feature that incorporated 34 features on the local contiguous structure-sequence composition, the minimum free energy of the secondary structure and the P-value of randomization test was used. The model was only trained on human pre-miRNA sequences and tested on data from other species.
mirExplorer proposed a visual pre-miRNA prediction using a set of transition probability and miRNA biogenesis features. Adaptive Boosting was applied to boost several weak classifiers built from each of those features whose discriminative power had been analyzed by F-score. SMOTE and under-sampling methods were also used to resolve the imbalanced data issue. The method could distinguish real pre-miRNAs from pseudo pre-miRNAs in a genome from a wide range of species, including animals, plants, and viruses.
MIReNA explored a multidimensional space defined by three combinatorial criteria (unfolding property of a miRNA in its precursor, size relation of a miRNA and its complementary sequence, percentage of unmatched nucleotides) and two physical (adjusted minimum folding free energy and minimum free energy index) criteria to identify pre-miRNAs. The thresholds for these criteria were defined using knowledge from known miRNAs in miRBase. The discriminative ability of MIReNA was validated on five species H. sapiens, Arabidopsis thaliana, Caenorhabditis elegans, Oryza sativa, and Rattus norvegicus.
Other existing classification methods, such as miR-abela (Sewer et al. 2005), mir-KDE (Chang et al. 2008), miPred (Ng and Mishra 2007), miRPara (Wu et al. 2011), and yasMiR (Pasaila et al. 2011), were not tested due to their unavailability or configuration problems for executing their programs. Especially, we failed to validate HuntMi (Gudys et al. 2013), which has recently appeared as an efficient tool, implementing ROC-select and applied random forest to get the best balance between sensitivity and specificity. Twenty gigabytes of memory on our machine was not enough to build its models on our data. Moreover, it took a lot of time for the feature selection process, which was inspired from microPred. Besides, we could neither get the source code of miRenSVM (Ding et al. 2010), which used bagging technique on SVM to deal with imbalanced data, nor receive the classification results via the web server of miRD (Zhang et al. 2011), which used a boosting method to combine two different SVM models.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
We thank Daniel Gautheret and Mark Ibberson for their reading of the paper and their helpful comments. This work was supported by the Council of Essonne Region (Pôle System@tic, OpenGPU project).
Footnotes
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.043612.113.
Freely available online through the RNA Open Access option.
REFERENCES
- Agarwal S, Vaz C, Bhattacharya A, Srinivasan A 2010. Prediction of novel precursor miRNAs using a context-sensitive hidden Markov model (CSHMM). BMC Bioinformatics 11Suppl 1:S29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akbani R, Kwek S, Japkowicz N 2004. Applying support vector machines to imbalanced datasets. In Mach Learn volume 3201 of Lect Notes Comput Sci (ed. Boulicaut JF, et al. ), pp. 39–50. [Google Scholar]
- Batuwita R, Palade V 2009. microPred: effective classification of pre-miRNAs for human miRNA gene prediction. Bioinformatics 25: 989–995. [DOI] [PubMed] [Google Scholar]
- Bauer E, Kohavi R 1999. An empirical comparison of voting classification algorithms: bagging, boosting, and variants. Mach Learn 36: 105–139. [Google Scholar]
- Brameier M, Wiuf C 2007. Ab initio identification of human microRNAs based on structure motifs. BMC Bioinformatics 8: 478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breiman L 1996. Bagging predictors. Mach Learn 24: 123–140. [Google Scholar]
- Breiman L 2001. Random forests. Mach Learn 45: 5–32. [Google Scholar]
- Chang CC, Lin CJ 2011. LIBSVM: a library for support vector machines. ACM Trans Intell Syst Technol 2: 1–27. [Google Scholar]
- Chang D, Wang CC, Chen JW 2008. Using a kernel density estimation based classifier to predict species-specific microRNA precursors. BMC Bioinformatics 9Suppl 12:S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chawla NV, Bowyer KW, Hall LO, Kegelmeyer WP 2002. SMOTE: synthetic minority over-sampling technique. J Artif Intell Res 16: 321–357. [Google Scholar]
- Chen S, Wang J, Liu Y, Xu C, Lu H 2010. Fast feature selection and training for AdaBoost- based concept detection with large scale datasets. In Proceedings of the international conference on multimedia, pp. 1179–1182, New York, NY. [Google Scholar]
- Dash M, Liu H 1997. Feature selection for classification. Intell Data Anal 1: 131–156. [Google Scholar]
- de Pontual L, Yao E, Callier P, Faivre L, Drouin V, Cariou S, Van Haeringen A, Genevieve D, Goldenberg A, Oufadem M, et al. 2011. Germline deletion of the miR-17∼92 cluster causes skeletal and growth defects in humans. Nat Genet 43: 1026–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devijver PA, Kittler J 1982. Pattern recognition: a statistical approach. Prentice Hall, Englewood Cliffs, NJ. [Google Scholar]
- Ding J, Zhou S, Guan J 2010. MiRenSVM: towards better prediction of microRNA pre-cursors using an ensemble SVM classifier with multi-loop features. BMC Bioinformatics 11Suppl 11:S11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freund Y, Schapire RE 1997. A decision-theoretic generalization of on-line learning and an application to boosting. J Comput Syst Sci 55:119–139. [Google Scholar]
- Guan DG, Liao JY, Qu ZH, Zhang Y, Qu LH 2011. mirExplorer: detecting microRNAs from genome and next generation sequencing data using the AdaBoost method with transition probability matrix and combined features. RNA Biol 8: 922–934. [DOI] [PubMed] [Google Scholar]
- Gudys A, Szczesniak M, Sikora M, Makalowska I 2013. HuntMi: an efficient and taxon-specific approach in pre-miRNA identification. BMC Bioinformatics 14: 83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall M, Frank E, Holmes G, Pfahringer B, Reutemann P, Witten IH 2009. The WEKA data mining software: an update. SIGKDD Explor Newsl 11: 10–18. [Google Scholar]
- He L, Thomson JM, Hemann MT, Hernando-Monge E, Mu D, Goodson S, Powers S, Cordon-Cardo C, Lowe SW, Hannon GJ, et al. 2005. A microRNA polycistron as a potential human oncogene. Nature 435: 828–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hertel J, Stadler PF 2006. Hairpins in a Haystack: recognizing microRNA precursors in comparative genomics data. Bioinformatics 22: e197–e202. [DOI] [PubMed] [Google Scholar]
- Huang TH, Fan B, Rothschild M, Hu ZL, Li K, Zhao SH 2007. MiRFinder: an improved approach and software implementation for genome-wide fast microRNA precursor scans. BMC Bioinformatics 8: 341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain A, Zongker D 1997. Feature selection: evaluation, application, and small sample performance. IEEE Trans Pattern Anal Mach Intell 19: 153–158. [Google Scholar]
- Japkowicz N 2000. The class imbalance problem: significance and strategies. In Proceedings of the international conference artificial intelligence, pp. 111–117, Las Vegas, NV. [Google Scholar]
- Jiang P, Wu H, Wang W, Ma W, Sun X, Lu Z 2007. MiPred: classification of real and pseudo microRNA precursors using random forest prediction model with combined features. Nucleic Acids Res 35: W339–W344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Q, Wang Y, Hao Y, Juan L, Teng M, Zhang X, Li M, Wang G, Liu Y 2009. miR2Disease: a manually curated database for microRNA deregulation in human disease. Nucleic Acids Res 37Suppl 1:D98–D104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kin T, Yamada K, Terai G, Okida H, Yoshinari Y, Ono Y, Kojima A, Kimura Y, Komori T, Asai K 2007. fRNAdb: a platform for mining/annotating functional RNA candidates from non-coding RNA sequences. Nucleic Acids Res 35: D145–D148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozomara A, Griffiths-Jones S 2011. miRBase: integrating microRNA annotation and deep-sequencing data. Nucleic Acids Res 39Suppl 1:D152–D157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagos-Quintana M, Rauhut R, Lendeckel W, Tuschl T 2001. Identification of novel genes coding for small expressed RNAs. Science 294: 853–858. [DOI] [PubMed] [Google Scholar]
- Lai E, Tomancak P, Williams R, Rubin G 2003. Computational identification of Drosophila microRNA genes. Genome Biol 4: R42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legendre M, Lambert A, Gautheret D 2004. Profile-based detection of microRNA precursors in animal genomes. Bioinformatics 21: 841–845. [DOI] [PubMed] [Google Scholar]
- Lertampaiporn S, Thammarongtham C, Nukoolkit C, Kaewkamnerdpong B, Ruengjitchatchawalya M 2013. Heterogeneous ensemble approach with discriminative features and modified-SMOTE bagging for pre-miRNA classification. Nucleic Acids Res 41: e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lestrade L, Weber MJ 2005. snoRNA-LBME-db, a comprehensive database of human H/ACA and C/D box snoRNAs. Nucleic Acids Res 34Suppl 1:D158–D162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis DD, Yang Y, Rose TG, Li F 2004. RCV1: a new benchmark collection for text categorization research. J Mach Learn Res 5: 361–397. [Google Scholar]
- Li Y, Shawe-Taylor J 2003. The SVM with uneven margins and Chinese document categorization. In Proceedings of the 17th Pacific Asia conference on language, information and computation, pp. 216–227, Singapore. [Google Scholar]
- Li X, Wang L, Sung E 2005. A study of AdaBoost with SVM based weak learners. In Proceedings of the IEEE international joint conference on neural networks, Vol. 1, pp. 196–201, Montreal, Quebec, Canada. [Google Scholar]
- Li X, Wang L, Sung E 2008. AdaBoost with SVM-based component classifiers. Eng Appl Artif Intell 21: 785–795. [Google Scholar]
- Lim LP, Lau NC, Weinstein EG, Abdelhakim A, Yekta S, Rhoades MW, Burge CB, Bartel DP 2003. The microRNAs of Caenorhabditis elegans. Genes Dev 17: 991–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling CX, Li C 1998. Data mining for direct marketing: problems and solutions. In Proceedings of the fourth international conference on knowledge discovery and data mining, pp. 73–79, New York, NY. [Google Scholar]
- Liu C, Bai B, Skogerbø G, Cai L, Deng W, Zhang Y, Bu D, Zhao Y, Chen R 2005. NONCODE: an integrated knowledge database of non-coding RNAs. Nucleic Acids Res 33(Database issue): D112–D115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maes OC, Chertkow HM, Wang E, Schipper HM 2009. MicroRNA: implications for Alzheimer disease and other human CNS disorders. Curr Genomics 10: 154–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathelier A, Carbone A 2010. MIReNA: finding microRNAs with high accuracy and no learning at genome scale and from deep sequencing data. Bioinformatics 26: 2226–2234. [DOI] [PubMed] [Google Scholar]
- Mencía A, Modamio-Høybjør S, Redshaw N, Morin M, Mayo-Merino F, Olavarrieta L, Aguirre LA, del Castillo I, Steel KP, Dalmay T, et al. 2009. Mutations in the seed region of human miR-96 are responsible for nonsyndromic progressive hearing loss. Nat Genet 41: 609–613. [DOI] [PubMed] [Google Scholar]
- Morik K, Brockhausen P, Joachims T 1999. Combining statistical learning with a knowledge-based approach—a case study in intensive care monitoring. In Proceedings of the 16th international conference on machine learning, pp. 268–277, Bled, Slovenia. [Google Scholar]
- Mraz M, Pospisilova S, Malinova K, Slapak I, Mayer J 2009. MicroRNAs in chronic lymphocytic leukemia pathogenesis and disease subtypes. Leuk Lymphoma 50: 506–509. [DOI] [PubMed] [Google Scholar]
- Nam JW, Shin KR, Han J, Lee Y, Kim VN, Zhang BT 2005. Human microRNA prediction through a probabilistic co-learning model of sequence and structure. Nucleic Acids Res 33: 3570–3581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng KL, Mishra SK 2007. De novo SVM classification of precursor microRNAs from genomic pseudo hairpins using global and intrinsic folding measures. Bioinformatics 23: 1321–1330. [DOI] [PubMed] [Google Scholar]
- Opitz D, Maclin R 1999. Popular ensemble methods: an empirical study. J Artif Intell Res 11: 169–198. [Google Scholar]
- Pasaila D, Sucila A, Mohorianu I, Pantiru S, Ciortuz L 2011. MiRNA recognition with the yasMiR system: the Quest for further improvements. In Software Tool Algorithms Biol Syst volume 696 of Adv Exp Med Biol (ed. Arabnia HR, Tran QN), pp. 17–25. [DOI] [PubMed] [Google Scholar]
- Rangel P, Lozano F, Garcia E 2005. Boosting of support vector machines with application to editing. In Proceedings of the fourth international conference on machine learning and applications, pp. 374–382, Los Angeles, CA. [Google Scholar]
- Redpath D, Lebart K 2005. Boosting Feature Selection. In Pattern Recognit Data Mining volume 3686 of Lect Notes Comput Sci (ed. Singh S, et al. ), pp. 305–314. [Google Scholar]
- Rice P, Longden I, Bleasby A 2000. EMBOSS: the European molecular biology open software suite. Trends Genet 16: 276–277. [DOI] [PubMed] [Google Scholar]
- Schapire RE 1990. The strength of weak learnability. Mach Learn 5: 197–227. [Google Scholar]
- Sewer A, Paul N, Landgraf P, Aravin A, Pfeffer S, Brownstein M, Tuschl T, van Nimwegen E, Zavolan M 2005. Identification of clustered microRNAs using an ab initio prediction method. BMC Bioinformatics 6: 267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stepanowsky P, Kim J, Ohno-Machado L 2012. A robust feature selection method for novel pre-microRNA identification using a combination of nucleotide-structure triplets. In Proceedings of the second IEEE conference on healthcare informatics, Imaging and systems biology, p. 61, La Jolla, CA. [Google Scholar]
- Tempel S, Tahi F 2012. A fast ab-initio method for predicting miRNA precursors in genomes. Nucleic Acids Res 40: e80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tempel S, Pollet N, Tahi F 2012. ncRNAclassifier: a tool for detection and classification of transposable element sequences in RNA hairpins. BMC Bioinformatics 13: 246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terai G, Komori T, Asai K, Kin T 2007. miRRim: a novel system to find conserved miRNAs with high sensitivity and specificity. RNA 13: 2081–2090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thum T, Galuppo P, Wolf C, Fiedler J, Kneitz S, van Laake LW, Doevendans PA, Mummery CL, Borlak J, Haverich A, et al. 2007. MicroRNAs in the human heart: a clue to fetal gene reprogramming in heart failure. Circulation 116: 258–267. [DOI] [PubMed] [Google Scholar]
- Ting K, Zhu L 2009. Boosting support vector machines successfully. In Mult Classifier Syst volume 5519 of Lect Notes Comput Sci (ed. Benediktsson J, et al. ), pp. 509–518. [Google Scholar]
- Tyagi S, Vaz C, Gupta V, Bhatia R, Maheshwari S, Srinivasan A, Bhattacharya A 2008. CID-miRNA: a web server for prediction of novel miRNA precursors in human genome. Biochem Biophys Res Commun 372: 831–834. [DOI] [PubMed] [Google Scholar]
- van Rooij E 2011. The art of microRNA research. Circ Res 108: 219–234. [DOI] [PubMed] [Google Scholar]
- Vapnik VN 1998. Statistical learning theory. Wiley, New York. [DOI] [PubMed] [Google Scholar]
- Wang B, Japkowicz N 2010. Boosting support vector machines for imbalanced data sets. Knowl Info Syst 25: 1–20. [Google Scholar]
- Wang X, Zhang J, Li F, Gu J, He T, Zhang X, Li Y 2005. MicroRNA identification based on sequence and structure alignment. Bioinformatics 21: 3610–3614. [DOI] [PubMed] [Google Scholar]
- Wickramaratna J, Holden S, Buxton B 2001. Performance degradation in boosting. In Mult Classifier Syst volume 2096 of Lect Notes Comput Sci (ed. Kittler J, Roli F), pp. 11–21. [Google Scholar]
- Wu G, Chang EY 2003. Class-boundary alignment for imbalanced dataset learning. In Proceedings of the workshop learning from imbalanced datasets, pp. 49–56, Washington, DC. [Google Scholar]
- Wu Y, Wei B, Liu H, Li T, Rayner S 2011. MiRPara: a SVM-based software tool for prediction of most probable microRNA coding regions in genome scale sequences. BMC Bioinformatics 12: 107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Zhou X, Zhang W 2008. MicroRNA prediction with a novel ranking algorithm based on random walks. Bioinformatics 24: 50–i58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue C, Li F, He T, Liu GP, Li Y, Zhang X 2005. Classification of real and pseudo microRNA precursors using local structure-sequence features and support vector machine. BMC Bioinformatics 6: 310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yousef M, Nebozhyn M, Shatkay H, Kanterakis S, Showe LC, Showe MK 2006. Combining multi-species genomic data for microRNA identification using a Naïve Bayes classifier. Bioinformatics 22: 1325–1334. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Yang Y, Zhang H, Jiang X, Xu B, Xue Y, Cao Y, Zhai Q, Zhai Y, Xu M, et al. 2011. Prediction of novel pre-microRNAs with high accuracy through boosting and SVM. Bioinformatics 27: 1436–1437. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.