

Abstract
Recent studies hint that endogenous dsRNA plays an unexpected role in cellular signaling. However, a complete understanding of endogenous dsRNA signaling is hindered by an incomplete annotation of dsRNA-producing genes. To identify dsRNAs expressed in Caenorhabditis elegans, we developed a bioinformatics pipeline that identifies dsRNA by detecting clustered RNA editing sites, which are strictly limited to long dsRNA substrates of Adenosine Deaminases that act on RNA (ADAR). We compared two alignment algorithms for mapping both unique and repetitive reads and detected as many as 664 editing-enriched regions (EERs) indicative of dsRNA loci. EERs are visually enriched on the distal arms of autosomes and are predicted to possess strong internal secondary structures as well as sequence complementarity with other EERs, indicative of both intramolecular and intermolecular duplexes. Most EERs were associated with protein-coding genes, with ∼1.7% of all C. elegans mRNAs containing an EER, located primarily in very long introns and in annotated, as well as unannotated, 3′ UTRs. In addition to numerous EERs associated with coding genes, we identified a population of prospective noncoding EERs that were distant from protein-coding genes and that had little or no coding potential. Finally, subsets of EERs are differentially expressed during development as well as during starvation and infection with bacterial or fungal pathogens. By combining RNA-seq with freely available bioinformatics tools, our workflow provides an easily accessible approach for the identification of dsRNAs, and more importantly, a catalog of the C. elegans dsRNAome.
Keywords: dsRNA, RNA editing, long introns, 3′ UTR, hyperediting
INTRODUCTION
Increasingly, double-stranded RNA (dsRNA) is being found to play important roles in cellular processes beyond its firmly established function as a potent activator of the host response to viral infection (Akira et al. 2006). For instance, in human cells, endogenous dsRNA formed by pairing of inverted Alu repeats is released from the nucleus during mitosis to activate the dsRNA- binding protein PKR, and repress translation (Kim et al. 2014). In a potentially related example, elevated levels of endogenous, repetitive Alu RNA, resulting from Dicer deficiency, leads to noncanonical activation of the NLRP3 inflammasome via MyD88 signaling (Tarallo et al. 2012; Kerur et al. 2013). Structured elements within mRNAs also serve as important regulatory elements, influencing translatability and stability. Double-stranded regions in 3′ UTRs of human mRNAs are bound by Staufen1, and subsequently turned over via Staufen-mediated mRNA decay (Gong and Maquat 2011; Gong et al. 2013). A Caenorhabditis elegans homolog of Staufen1 binds over 400 mRNAs, raising the possibility that Staufen-mediated decay is an evolutionarily conserved mRNA regulatory mechanism (LeGendre et al. 2013). Long 3′-UTR dsRNA formed by inverted Alus, and other repeats, affects the translatability of both human and C. elegans mRNAs (Hundley et al. 2008).
In most instances described above the identity of the dsRNA is known. However, in other instances the dsRNA responsible for the observed regulation has yet to be identified. In mouse models of diet-induced obesity, PKR is known to be activated, but the endogenous dsRNA ligand is unknown (Nakamura et al. 2010). Similarly, C. elegans strains deficient in the dsRNA processing factors DCR-1 (Dicer), RDE-4 and, RDE-1, display differential expression of genes associated with host defense, presumably due to accumulation of an undefined pool of cellular dsRNAs (Welker et al. 2007). The central role of these unidentified cellular dsRNAs in critical cellular processes underscores the need for new approaches for the identification of cellular dsRNAs.
Adenosine-to-inosine (A-to-I) RNA editing by ADAR enzymes requires a dsRNA substrate (Bass 2002; Nishikura 2010), and thus, identification of an editing site in an endogenous RNA indicates that the RNA is double-stranded in vivo. Early attempts to identify dsRNAs in C. elegans and H. sapiens took advantage of this by utilizing differential sensitivity of inosine-containing RNAs to digestion by RNase T1 (Morse and Bass 1997, 1999; Morse et al. 2002). While not high-throughput, these studies provided a first glimpse into the characteristics of cellular dsRNA, revealing multiple, extensive double-stranded regions in introns and 3′ UTRs of protein-coding genes, as well as a C. elegans long noncoding dsRNA (52G). 52G, which was later named rncs-1, has an interesting expression pattern, increasing and decreasing in response to the absence or presence of food, respectively (Hellwig and Bass 2008). A-to-I editing sites appear as A-to-G transitions in cDNA, and bioinformatics methods have been developed that identify RNA editing sites by the appearance of A-to-G transitions in high-throughput RNA sequencing data (Blow et al. 2004; Kim et al. 2004; Levanon et al. 2004). This approach has been used to identify editing sites in a variety of organisms, including humans, flies and mice, with astounding results. A recent survey of editing in humans revealed over 100 million editing sites covering the majority of coding genes (Bazak et al. 2014). These methods can be prone to false positives, as presumed editing sites can also derive from genomic SNVs (single-nucleotide variants) and sequencing errors. However, it was noted early on that identification of clustered RNA editing sites within the span of a few hundred bases, which are most commonly associated with long, mostly base paired, dsRNAs, improves the accuracy of editing site detection (Athanasiadis et al. 2004).
Our goal was to create a genome-wide annotation of the “dsRNAome” of C. elegans, rather than a list of editing sites. Thus, we applied a robust and effective bioinformatics approach for the identification of RNA editing clusters, a hallmark of long dsRNA, in high-throughput sequencing data sets. Using a combination of alignment algorithms to map both unique and repetitive reads, we detected editing-enriched regions (EERs) as markers of dsRNA-producing loci. C. elegans EERs are distributed throughout the genome with a particularly strong association with long introns and both annotated, and unannotated, 3′ UTRs. EERs are enriched for highly structured sequences and exhibit dynamic expression profiles during stress and development in C. elegans. Our findings detail an extensive in vivo survey of C. elegans dsRNAs, providing a valuable new resource for the analysis of dsRNA function in this organism.
RESULTS
Identification of C. elegans dsRNA using editing-enriched regions
Our goal was to obtain a comprehensive list of long dsRNAs expressed in C. elegans, so we isolated RNA from multiple strains and stages, immunoprecipitated in various ways to enrich for long dsRNA. Antibodies against DCR-1 (Tabara et al. 2002), the C. elegans Dicer homolog, and RDE-4 (Parker et al. 2006), a dsRNA-binding protein that facilitates DCR-1 processing, were used to coimmunoprecipitate dsRNA from embryo lysates. The dsRNA-specific J2 antibody, which binds RNA duplexes ≥40 base pairs (bps) (Schönborn et al. 1991; Lukács 1994; Bonin et al. 2000), was used to immunoprecipitate dsRNA from a mixed-stage lysate. Each immunoprecipitation was performed using wild-type (Bristol N2) C. elegans as well as a Dicer mutant strain (dcr-1(mg375)), which because of a mutation in the helicase domain of DCR-1, is deficient in processing endogenous siRNA, and consequently, predicted to accumulate long dsRNAs (Pavelec et al. 2009; Welker et al. 2010). The six barcoded samples were pooled and sequenced on an Illumina HiSeq 2000 sequencer, generating 627,627,876 raw 101 bp paired-end reads.
We developed a bioinformatics pipeline to identify hyperedited transcripts, or editing-enriched regions (EERs), in our high-throughput RNA sequencing data (Fig. 1); a summary of the total editing sites ≥1% edited within EERs, and the number of EERs identified, is shown in Table 1. After combining all A-to-G transition sites detected across our four data sets, and removing duplicates, we detected a total of 83,015 total sites, 34,749 of which are high-confidence editing sites located in EERs (Supplemental Table 1, Combined in EER tab). We used two sequence alignment algorithms to map reads to the C. elegans genome (UCSC October 2010 release ce10). The first was novoalign (Novocraft), which accurately aligns reads with higher error rates relative to other available aligners (Ruffalo et al. 2011; Borozan et al. 2013), and the second was a modified version of GNUMAP-bs (Genomic Next-generation Universal MAPper - bisulfite) customized to ignore A-to-G substitutions during alignment (Clement et al. 2010; Hong et al. 2013), thus allowing mapping of highly edited sequences. Using novoalign, 85.4% of reads aligned to the reference genome (51.7% mapped uniquely), and using GNUMAP, 48.9% aligned (45.9% mapped uniquely) (Table 1).
FIGURE 1.
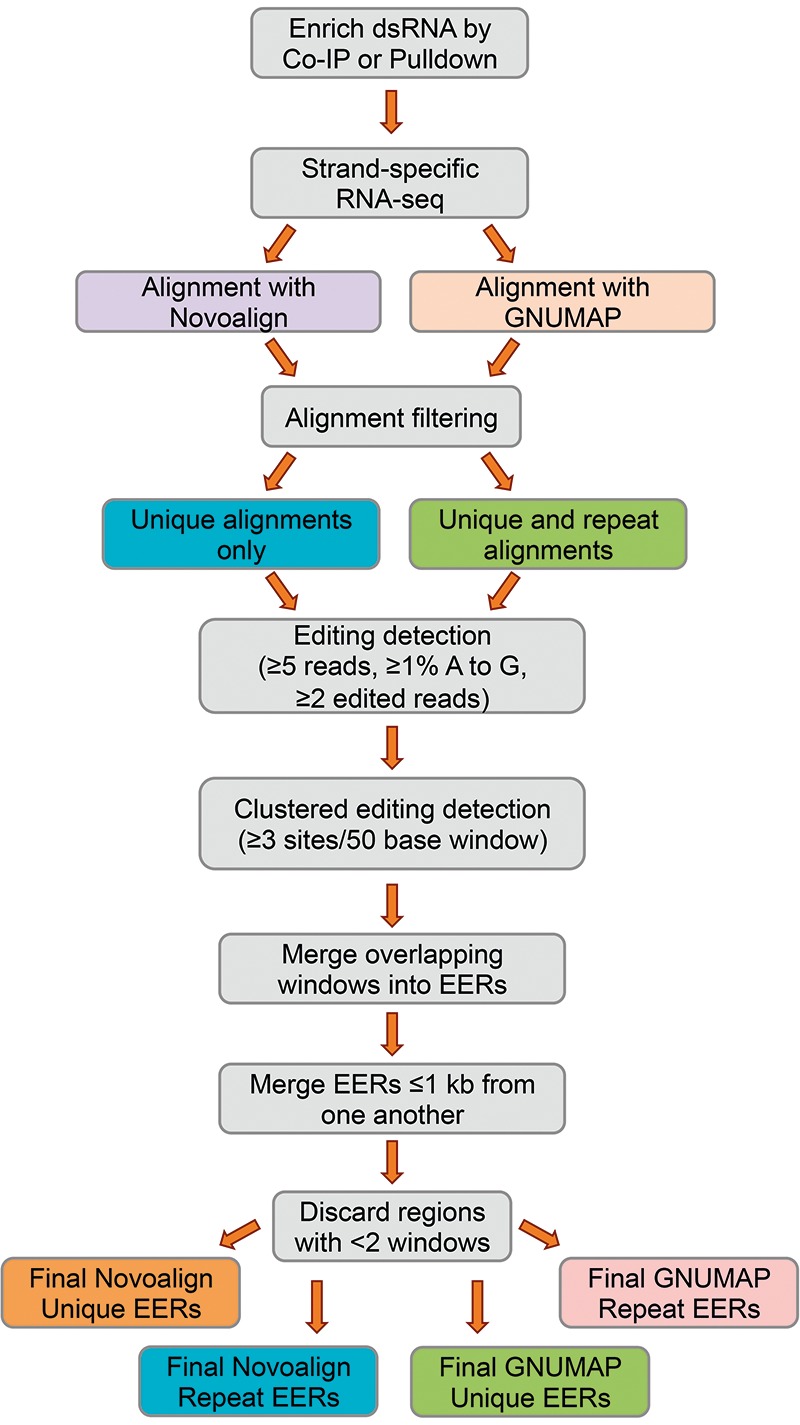
Clustered editing detection pipeline. See text and Materials and Methods for details. Gray, steps common among analyses.
TABLE 1.
Summary of alignments, editing, and EER analysis
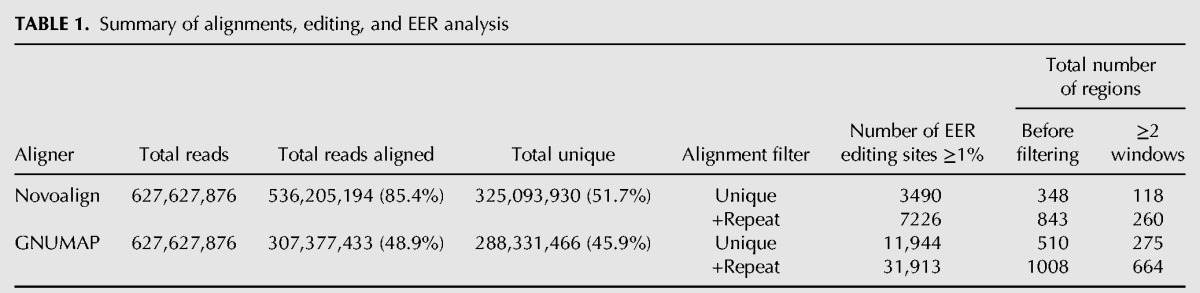
Studies in mammals indicate that the vast majority of RNA editing occurs in repetitive sequences, such as the Alu repeats of the primate lineage (Athanasiadis et al. 2004; Neeman et al. 2006). Thus, after filtering mapped reads based on the number of mismatches they had with the reference genome, we sorted alignments into two bins (see Materials and Methods): The first contained unique alignments only (i.e., a single mapped position, “Unique”), and the second combined both unique and repeat alignments (“+Repeat”). Filtered alignments were then scanned for evidence of A-to-G conversion. A strand-specific sliding window approach, using a 50-base window, was used to identify genomic regions with clusters of editing sites. Windows containing ≥3 editing sites, each with ≥1% editing were collected, and overlapping edited windows were combined into EERs. Candidate editing sites that were supported by only a single read were excluded. Non-overlapping EERs within 1 kilobase (kb) of another EER were merged into a single aggregate EER. Finally, to exclude short EERs from our final lists, which are likely to include more false positives, we required that EERs have a minimum of two overlapping editing-enriched windows. The final lists of EERs are detailed in Supplemental Table 2.
Having used two very different alignment algorithms to identify EERs, we were interested in comparing results from each aligner. We found that detection of RNA editing, and consequently EERs, was more sensitive when using GNUMAP alignments. Intersection of the genomic coordinates for EERs detected using novoalign or GNUMAP revealed that virtually all regions found using novoalign alignments were also found using GNUMAP, in addition to many GNUMAP-specific EERs (Fig. 2A). Moreover, in many instances a comparison of EERs detected in both novoalign and GNUMAP alignments showed that GNUMAP alignments revealed additional editing sites. For example, a common EER found within the 3′ UTR region of Y39H10A.6 (Fig. 2B), which forms a long and highly stable duplex (Fig. 2C), was more extensively edited in the GNUMAP analysis. Further, we detected 7226 editing sites ≥1% edited within all Novoalign +Repeat EERs, and in the same regions we detected 11,717 sites using GNUMAP +Repeat alignments. The increased sensitivity provided by GNUMAP is likely a consequence of its ability to ignore A-to-G substitutions during the alignment process, which allows for mapping of reads derived from extensively edited RNAs. As expected, both aligners detected more EERs when considering repeat alignments (Table 1).
FIGURE 2.

GNUMAP detects nearly all novoalign EERs in addition to detecting more editing sites. (A) Venn diagram showing overlap of GNUMAP and Novoalign Unique EERs. In one instance a GNUMAP Unique EER overlaps two Novoalign Unique EERs; thus, the true overlap is 94 (shown in parentheses). (B) Screen shot from Integrated Genome Browser (IGB) (http://bioviz.org/igb/) comparing editing levels and read coverage for a Novoalign +Repeat EER (red; NOVO_Repeat_MinusReg_10) and an overlapping GNUMAP +Repeat EER (green; GNUMAP_Repeat_Minus_1). Editing track, bar graph showing fraction of reads with A-to-G substitutions; coverage tracks, raw read counts. (C) Predicted secondary structure of GNUMAP_Repeat_Minus_1. Base pair color is indicative of the probability the base is paired (if shown as paired) or unpaired (if shown as unpaired). Structures were predicted using the ViennaRNA folding package (Lorenz et al. 2011).
EERs have properties of dsRNA edited by ADARs
The RNA editing activity of ADARs is specific for dsRNA, and thus, if an endogenous RNA is edited, it must exist as an intramolecular or intermolecular duplex in cells. To evaluate the propensity of the identified EERs to form dsRNA, we compared the predicted folding free energies of GNUMAP +Repeat EERs to a set of length-matched randomized sequences (Fig. 3A,B). We found that the EERs were far more stable than the random regions, with the predicted free-energy structures of the GNUMAP +Repeat EERs averaging −194.7 ± 537.1 (standard deviation) kcal/mol, while the random regions were less stable, averaging −94.1 ± 177.9 kcal/mol. Normalizing for length we found that the GNUMAP +Repeat EERs showed an average folding free energy per nucleotide (kcal/mol nucleotide) that was much lower than the corresponding random data set (−0.27 and −0.18 kcal/mol nucleotide, respectively). All data sets showed this trend (see ΔG/length column, Supplemental Table 2), consistent with the idea that the detected EERs were enriched for intramolecular duplexes. Representative structures from the GNUMAP +Repeat data set are shown in Figure 3C.
FIGURE 3.

EERs and the editing sites within them bear hallmarks of bona fide ADAR substrates. (A) GNUMAP +Repeat EERs are more stable than random length-matched controls. A comparison of folding free energy (kcal/mol) as a function of EER length is shown for GNUMAP +Repeat EERs (red diamonds) and three sets of randomized regions (black symbols). Folding free energies were calculated using UNAFold (Markham and Zuker 2008) (http://mfold.rna.albany.edu/?q=DINAMelt/software). EERs of all data sets showed a similar trend (Supplemental Table 2). (B) Same data as in A, but zoomed in on EERs ≤2000 nt long. (C) Representative secondary structures of three GNUMAP +Repeat EERs. Coloring as in Figure 2C. (D) The context of GNUMAP +Repeat EER editing sites is consistent with known ADAR site preferences. Editing site logo based on all editing sites found within GNUMAP +Repeat EERs that were ≥1% edited. The contribution of an editing site context to the logo was weighted according to the percentage of reads edited at the site (e.g., a CCAGG with an editing level of 15% would have a contribution of 0.15 to the logo, whereas a GUAGA site editing at 90% would have a contribution of 0.9). The logo was created using Seq2Logo (http://weblogo.threeplusone.com/create.cgi).
While the above analysis was effective at identifying stable intramolecular structures, it did not evaluate the possibility of intermolecular EER structures. In this case, editing should target both strands, and assuming each strand is present at a significant level, both strands should appear as separate EERs in our lists. We used the blat sequence alignment tool to query whether any GNUMAP +Repeat EERs were complementary to other EERs in the same list. We found that ∼27% of GNUMAP +Repeat EERs have >80% sequence complementarity with at least one other EER (Blat Stats sheet, Supplemental Table 3). Not surprisingly, many of the most highly complementary EER pairs appeared to be the result of overlapping transcription events, although we also found evidence for EER pairs in which the individual transcripts were located on different chromosomes (Supplemental Table 3). Considering the extensive complementarity we observed, it is very likely that intermolecular duplexes were also identified in our analysis.
ADARs prefer to target adenosines with certain 5′ and 3′ nearest neighbors. Thus, to gain further evidence that EERs are populated with true ADAR targets, we determined the sequence context of all sites detected by our GNUMAP +Repeat analysis workflow (Fig. 3D). Applying the same 1% editing threshold used for identifying EERs, we obtained a motif that recapitulates the 5′ nearest neighbor bias we expect based on studies of mammalian ADAR1 and ADAR2 (Lehmann and Bass 2000; Riedmann et al. 2008; Eggington et al. 2011). The neighboring 5′ base showed a preference for A > U > C > G, which bears a strong similarity to the previously reported preference of U > A > C > G for hADAR1 and hADAR2 (Fig. 3D; Lehmann and Bass 2000). Moreover, these preferences are consistent with the preferences found for C. elegans editing sites in a previous study by our laboratory (Morse and Bass 1999). In contrast to the 5′ nearest neighbor preference, the 3′ neighbor preference for A > G = U > C is considerably different than the reported preferences for both hADAR1 and hADAR2, which prefer a 3′ G or C. The disparity in preference could reflect intrinsic differences between C. elegans ADR-2, the only active adenosine deaminase in worms, and its human counterparts. Consistent with this idea, our 5′ and 3′ nearest neighbor preferences more closely resemble the preferences reported in a recent survey of 270 editing sites in C. elegans (Washburn et al. 2014). Regardless, considering the agreement between our editing site preferences and those of earlier reports, it is very likely that the editing sites comprising our EERs are generally bona fide ADAR editing events.
Most EERs are in close proximity to coding genes
As a first step in characterizing the identified EERs, we determined where EERs were located in the genome as well as their overlap with genomic annotations. Analyzing GNUMAP +Repeat EERs, our most abundant EER compilation, we found that EERs are distributed along all chromosomes, but with a visible enrichment on the ends of autosomes (Fig. 4A). We next intersected EER lists with a list of all C. elegans coding genes and sorted them into three bins, depending on whether they overlapped a protein-coding gene, a region ≤1000 nt from a coding gene, or a region >1000 nt from a coding gene (Fig. 4B). The majority of EERs overlapped a protein-coding gene, resulting in a significant fraction of coding genes having an associated EER. GNUMAP +Repeat EERs, for example, are associated with 1.7% of C. elegans annotated protein-coding genes. We further defined the EERs that overlapped coding genes and found that both novoalign and GNUMAP EERs predominantly overlapped noncoding elements of coding genes, such as introns and UTRs (Fig. 4C).
FIGURE 4.

EERs are most abundant on autosomes and primarily overlap noncoding regions of coding genes. (A) Chromosome map showing location of GNUMAP +Repeat EERs (white bars). Chromosome lengths are to scale. (B) Stacked bar graphs showing the location of all EERs with respect to protein-coding genes. Numbers of EERs from each category are shown inside bars in white. (C) Stacked bar graphs showing genome annotations of EERs that intersect with protein-coding genes. Overlap is defined by which annotation had the greatest overlap with the EER. (D) Annotations of EERs >1 kb from a coding gene. Intersection was defined as in C. Bases with no overlap were considered unannotated. EERs annotated as antisense intersected with coding genes on the opposite strand. (E) Annotation of EERs ≤1 kb from a coding gene. Annotations were determined as in C and D. (F) Distribution of EERs ≤1 kb from a coding gene between upstream of and downstream from a coding gene.
Considering the extensive overlap between EERs and coding genes, we were curious whether EERs associated with a particular class of genes. Gene Ontology (GO) analysis was performed using all genes having an overlapping GNUMAP +Repeat EER (Supplemental Table 4). The resulting GO terms were enriched for functions related to transcription initiation and nucleosome assembly and organization. One of the more strongly enriched GO categories was related to chromatin and nucleosome function and was populated predominantly by histone genes (GO:0000786∼nucleosome, GO Table sheet, Supplemental Table 4). Importantly, histone mRNAs have been observed to undergo RNA editing and are also overlapped by a population of ADAR-dependent small RNAs (Wu et al. 2011).
In each of our four analyses, EERs > 1 kb from any coding gene displayed the greatest overlap with ncRNAs, transposons, and antisense transcripts (Fig. 4, cf. D and E). However, the majority of EERs in the >1 kb category were completely unannotated (Fig. 4D). To determine whether these EERs were associated with unannotated coding genes we assessed their coding potential using Coding Potential Calculator application (http://cpc.cbi.pku.edu.cn/). The Coding Potential Calculator evaluates multiple sequence features, including homology and potential reading frames, to predict the coding potential of a transcript. We found that ∼93% of GNUMAP +Repeat EERs >1 kb from coding genes lacked predicted coding potential, with the majority of these regions having no overlapping annotation (Supplemental Table 5). These results indicated that these EERs are previously unannotated double-stranded ncRNAs. Several EERs in this category were located proximal to previously annotated lncRNAs (see column F in Supplemental Table 5). Here it should be noted that, while lncRNAs are not typically thought of as double-stranded, most of the EERs that are >1 kb from an annotated gene and lack coding potential would be called lncRNAs by conventional criteria (Ulitsky and Bartel 2013).
We anticipated that transposable elements, which have previously been shown to undergo RNA editing in C. elegans (Sijen and Plasterk 2003), would have significant overlap with EERs. However, we found that EERs defined using only unique alignments had little to no overlap with WormBase transposon annotations, regardless of which aligner was used (Fig. 4C–E). EERs >1 kb from an annotated gene had the greatest overlap (Fig. 4D), but in this case Novoalign Unique EERs had no transposon overlap, and just 5 out of 275 GNUMAP Unique EERs displayed overlap. Only when repetitive alignments were included in the analysis did we begin to see more overlap with annotated transposons. For EERs >1 kb from an annotated gene, we found that 10 out of 260 and 36 out of 664 Novoalign and GNUMAP +Repeat EERs, respectively, overlapped a transposon annotation. While transposon overlap is still small, this analysis emphasizes that meaningful information, particularly repetitive elements, is lost when repetitive reads with multiple alignments are discarded. Consistent with this idea, the majority of EERs in the repeat category were also supported by unique alignments. Of 664 GNUMAP +Repeat EERs, 532 (80.1%) had unique alignments covering ≥10% of their length; only 111 GNUMAP +Repeat EERs were comprised entirely of repetitive alignments (column O of GNUMAP +Repeat sheet, Supplemental Table 2). Thus, in our analysis the use of repetitive reads more often further substantiated the identification of an EER, rather than leading to artifactual EERs.
Many EERs are located in unannotated 3′ UTRs
Previous analyses of RNA editing in C. elegans, while not high-throughput, identified 3′ UTRs as sites of hyperediting (Morse and Bass 1999; Morse et al. 2002). While our analyses detected overlap between EERs and annotated 3′ UTRs (Fig. 4C), many genes do not have annotated UTRs. Thus, we considered the possibility that our analyses were underestimating the overlap of EERs with UTRs. Reasoning that unannotated UTRs would be in the category of EERs located <1 kb from an annotated gene, we parsed this category into those that were upstream of or downstream from an annotated gene (Fig. 4F). We found that the majority of EERs from each analysis were downstream from an annotated gene, as we would predict if these EERs were 3′ UTRs. In general EERs within 1 kb downstream from an annotated gene displayed continuous and above average read coverage extending from the upstream gene (Supplemental Fig. 1; see Y39H10A.6 in Fig. 2B for a specific example), suggesting these EERs were indeed part of the annotated mRNA transcript. In further support of this idea, several previously identified edited 3′ UTRs, not yet included in current annotations, were found in this category, including the 3′ UTRs of C35E7.6 and pop-1 (W10C8.2) (Morse et al. 2002). We also observed a significant number of EERs upstream of annotated coding genes, although fewer than those located downstream. The paucity of EERs in the 5′ UTR region may be a consequence of trans-splicing, which removes the encoded 5′ UTR sequences of many mRNAs in C. elegans (Blumenthal 2012). Nevertheless, our results suggest that hitherto unannotated UTR structures undergo editing, and that UTR editing is likely to be more extensive than previously known.
Introns that contain EERs are longer than typical C. elegans introns
A large fraction of EERs within coding genes were located within introns (Fig. 4C). We observed that many of the introns that contained EERs were quite long and wondered whether this was a general feature of EER-associated introns. We compared the length of EER-associated introns with all coding gene introns from the current ensemble genome build (Fig. 5). We found that for each data set the population of introns containing EERs was significantly longer than the average intron (Mann–Whitney U test, P < 0.0001). For example, introns with embedded GNUMAP +Repeat EERs were ∼eightfold longer than the average intron (2811 nt compared with 335 nt). Interestingly, the length of the EER did not correlate with the length of the intron, indicating that introns were not longer simply because of the added EER length (data not shown).
FIGURE 5.
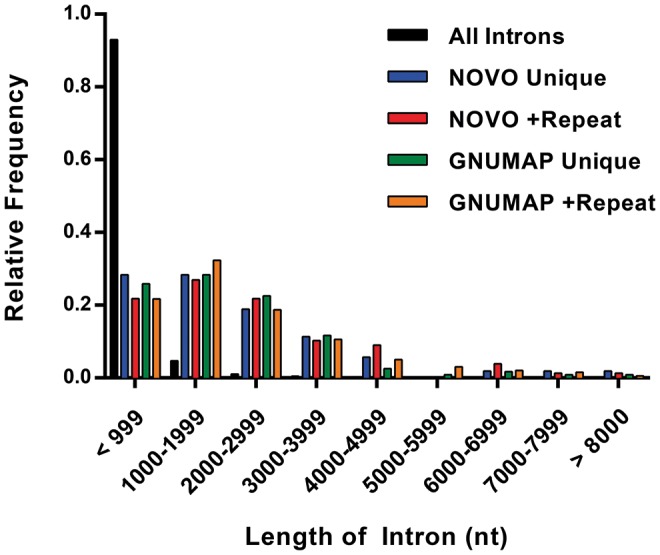
Introns containing EERs are longer than the average intron. Histogram comparing the length of introns with embedded EERs to all introns. Bin sizes were 1000 nt. Maximum length was set to 8000 nt for graphing purposes, but all intron lengths were used in calculating fractions. Duplicate introns (same genomic coordinates) were flattened into a single intron list prior to calculating fractions and plotting data.
Validation of EERs
We used RT-PCR and Sanger sequencing to validate the expression and editing of a representative set of EERs. This set was comprised of nine EERs that included potential ncRNAs and 3′ UTRs (Table 2). Of the nine EERs analyzed, we were able to successfully reverse transcribe six partial cDNAs, and in all six cases we verified that the RNAs were edited, as indicated by the appearance of A-to-G transitions in cDNAs (e.g., see Fig. 6C; Y71H2B.8 is GNUMAP_Repeat_Plus_241 in Table 2). Moreover, of the 180 sites within these 6 EERs that were ≥10% edited based on levels determined using GNUMAP +Repeat alignments, 160 (∼89%) were edited at ≥10% as measured using Sanger sequencing. Likewise, these same six EERs contained 61 editing sites according to Novoalign +Repeat alignments, and 58 of these (∼95%), many of which overlapped the same GNUMAP +Repeat sites, were also validated. These results indicated that alignments from either algorithm can be used to accurately identify EERs, but GNUMAP allows the detection of more editing sites, and consequently, more EERs. Indeed, visual inspection of GNUMAP and novoalign editing levels over EERs common to both data sets revealed that GNUMAP identified many more A-to-G mismatches (see example in Fig. 2B).
TABLE 2.
Validation of EERs

FIGURE 6.

An EER embedded in the 3′ UTR of Y71H2B.8. (A) Read coverage (blue) and editing levels (red) of GNUMAP_Repeat_Plus_241 (green) downstream from Y71H2B.8 (black). Y71H2B.8a represents the current Ensembl annotation, and Y71H2B.8b represents the conceptual gene structure based on our sequencing; exons, boxes; lines, introns. Data tracks were visualized using the Integrated Genome Browser (http://bioviz.org/igb/). (B) Secondary structure of the boxed sequence from A, encompassing the EER. Structure prediction was calculated using the Vienna RNAfold webserver (http://rna.tbi.univie.ac.at/) and visualized using VARNA (http://varna.lri.fr/). (C) Sanger sequencing trace of the region in B denoted by a red bracket. Edited sites detected by RNA-seq are marked with a red asterisk.
For some EERs, analyses using RT-PCR and Sanger sequencing also revealed information about gene structure. For example, we found that an EER >1 kb downstream from Y71H2B.8 is likely the 3′ UTR of an extended gene isoform (Fig. 6A). Y71H2B.8 encodes a nematode-specific protein that is predicted to function in lipid storage based on a reduced fat content RNAi phenotype (Ashrafi et al. 2003). RT-PCR using a primer pair encompassing the EER and final exon of the annotated Y71H2B.8, which we now call Y71H2B.8a, yielded a product significantly smaller than the expected length (cDNA alignment, Supplemental Fig. 2A). Sanger sequencing of the PCR product indicated the existence of a previously unannotated isoform, Y71H2B.8b, made by splicing from a previously unannotated 5′ splice site, upstream of the annotated stop codon, to a previously unannotated 3′ splice site, slightly upstream of the EER. Importantly, the Y71H2B.8b isoform we observe is also supported by C. elegans EST and RNA-seq data (Supplemental Fig. 2B). Conceptual translation of Y71H2B.8b revealed a stop codon just upstream of the EER, again suggesting that the EER is the 3′ UTR of the Y71H2B.8b isoform. Editing sites within this EER are located within a predicted duplex region (Fig. 6B,C).
Nonsynonymous codon changes resulting from editing are absent
In defining the C. elegans dsRNAome, we relied on the identification of clusters of editing sites (EERs), and all subsequent analyses and validation experiments indicated the majority of identified EERs were indeed expressed dsRNAs. Nonsynonymous editing of codons, which is known to have important functions in other organisms (Bass 2002; Nishikura 2010), although rare, has recently been observed in C. elegans transcripts (Washburn et al. 2014; Zhao et al. 2015). To determine whether any sites, both inside EERs and outside, resulted in nonsynonymous codon changes, we used the program Annovar to annotate all observed A-to-G changes in coding exons (Wang et al. 2010). We used the GNUMAP +Repeat alignments based on our earlier observations of its comprehensive nature. After removing known SNPs, a total of 1314 A-to-G substitutions were detected in exonic regions, and of these, 910 sites were predicted to cause nonsynonymous codon changes in 389 different genes (Supplemental Table 6). Only 66 of these nonsynonymous sites had at least 20 overlapping reads and were edited above 20%. A similar search of Novoalign +Repeat alignments for editing sites showed 55 A-to-G exonic substitutions, 28 of which were nonsynonymous changes, with 7 having ≥20 reads and ≥20% editing (Supplemental Table 7). Manual annotation indicated that none of these were likely to be true editing sites, since the A-to-G sites were consistently neighbored by variants not dependent on editing (non-A-to-G), or due to other alignment artifacts. It is important to note that these exonic sites are largely isolated, unlike our high-confidence sites identified in clusters. Thus, these relatively few false positives are not surprising. At present we have not been able to validate editing in a nonsynonymous codon, and while this was not the primary goal of this study, further analyses using less stringent coverage and editing cutoffs may yet yield new sites.
A subset of EERs overlaps endogenous siRNA loci
C. elegans have robust RNA interference (RNAi) machinery that plays critical roles in controlling expression of repetitive and exogenous genes, chromosome segregation, and viral defense (Grishok 2013). Some endogenous siRNAs are synthesized by an RNA-dependent RNA polymerase (RdRP) and are independent of Dicer and dsRNA. Others, however, require Dicer to produce a primary siRNA, which then facilitates recruitment of an RdRP to make secondary siRNAs that amplify the response. Given the requirement for dsRNA in primary siRNA production, we wondered whether there was overlap between EERs and annotated endo-siRNA loci. We intersected the GNUMAP +Repeat EERs with a compiled list of loci that produce endo-siRNA (1° and 2°) (Warf et al. 2012). We found that 207 out of 664 (31.2%) GNUMAP +Repeat EERs intersected with one or more annotated endo-siRNA loci, with the majority of these overlaps occurring on the same strand as the EER (Supplemental Table 8). Furthermore, 204 of these EERs overlapped coding genes; GO enrichment analysis for these genes produced multiple ontology terms related to growth, genital development, and both embryonic and post-embryonic development (GO Terms tab, Supplemental Table 8). EERs were distinct from the endo-siRNAs they overlapped, since the length of the reads mapped to the EERs largely exceeded the length of a siRNA. However, the EER could serve as the dsRNA precursor processed by Dicer to produce primary endo-siRNAs.
Subsets of EERs are differentially expressed during stress and development
Studies in higher organisms (Cabili et al. 2011; Ulitsky et al. 2011) and in C. elegans (Nam and Bartel 2012) show that lncRNAs display development-specific expression. To examine the expression patterns of EERs during embryogenesis we performed cluster analysis of GNUMAP +Repeat EERs, our most comprehensive EER list, across an embryogenesis time course available from the modEncode consortium (Gerstein et al. 2010). Expression values for EERs expressed in at least 70% of time points were clustered based on Pearson correlation of z-scores across the 720-min time course, starting from early embryos harvested from young adults. This subset of GNUMAP +Repeat EERs sorted into three primary expression clusters that we defined as early, mid, and late embryogenesis, with the largest number of EERs belonging to the midembryogenesis cluster (Fig. 7A, Supplemental Fig. 3).
FIGURE 7.

Subsets of EERs are differentially expressed during development and stress. (A) Clustering of GNUMAP +Repeat EER expression during early, mid, and late embryogenesis. Sorting was based on the z-score of each EER's expression across the 720 min time course (30-min intervals). (B) EERs are differentially expressed during Serratia marcescens infection. All GNUMAP +Repeat EERs displayed are differentially expressed greater than twofold between two biological replicates of E. coli, and three biological replicates of S. marcescens, both treated with pathogen for 24 h. (C) Same as B except that worms were treated with Harposporium. Expression data were obtained from the modEncode database (http://www.modencode.org/).
Recent studies reveal that cellular dsRNAs are capable of activating innate immune signaling pathways under various conditions (Nakamura et al. 2010; Kim et al. 2014). We were curious whether the expression of EERs could be regulated under conditions of stress, for example, during infection. We queried available modEncode RNA expression data from two infection models: the bacterial pathogen Serratia marcescens and the fungal pathogen Harposporium sp. A total of 22 GNUMAP +Repeat EERs were differentially expressed greater than twofold during infection with S. marcescens. Likewise, 16 EERs were differentially expressed during Harposporium sp. infection (Fig. 7B,C). Interestingly, rncs-1 is down-regulated by S. marcescens infection and up-regulated by Harposporium infection (GNUMAP_Repeat_Plus_20, see Supplemental Fig. 4). While EERs as a group are not significantly enriched in the infection data sets, EERs are found in diverse classes of RNA, only a subset of which may function in innate immunity and development.
The long noncoding dsRNA rncs-1 previously characterized by our laboratory is rapidly up-regulated during starvation, and down-regulated upon refeeding, suggesting it plays a role in the C. elegans response to nutrient stress (Hellwig and Bass 2008). However, as yet, a mutant phenotype has not been observed in strains containing a deletion of rncs-1. One possibility is that the role of rncs-1 is redundant with that of other cellular dsRNAs. To determine whether other dsRNAs identified in our analysis were regulated by changes in nutrient availability, we starved L4 larvae for 10 and 24 h, followed by refeeding for 24 h, and measured expression changes by Northern blot (Supplemental Fig. 5). We were able to detect northern signals for four EERs, and of these, two EERs (GNUMAP_Repeat_Plus_400; GNUMAP_Repeat_Minus_1, Table 2), were up-regulated during starvation and down-regulated by refeeding, similar to the pattern observed for rncs-1. Based on read coverage, both GNUMAP_Repeat_Plus_400 and GNUMAP_Repeat_Minus_1 are likely to be previously unannotated 3′ UTRs of coding genes (C17D12.7 and Y39H10A.6, respectively). Like rncs-1, both genes are expressed in the intestine (Spencer et al. 2011), providing further evidence for a shared role in the response to nutritional stress.
DISCUSSION
Previous studies have used bioinformatics approaches to identify ADAR editing sites (Athanasiadis et al. 2004; Cattenoz et al. 2013; Ramaswami et al. 2013; Bazak et al. 2014). The goal of our analysis was somewhat different, aimed at compiling a comprehensive catalog of very long dsRNAs expressed from the C. elegans genome. Using a combination of read alignment algorithms, we identified as many as 664 high-confidence EERs, and by several criteria showed that EERs consist of extended double-stranded structures. The vast majority of EERs are located proximal to, or embedded within, protein-coding genes, with most of these occurring in introns, and annotated, as well as unannotated, 3′ UTRs. Intriguingly, EERs are enriched at the distal arms of autosomal chromosomes, and subsets of EERs are dynamically expressed during infection, starvation, and development. Our data sets provide an invaluable resource for further studies aimed at understanding functions of genome-encoded, expressed dsRNA.
What approach is optimal for identifying dsRNAs?
In this study, we compared the EER detection performance of two aligners (novoalign and an editing-aware version of GNUMAP-bs), as well as a strict alignment filtering protocol that excludes repeat alignments (Unique) with a relaxed filter that allows repeats (+Repeat). Both aligners performed well at identifying EERs, but GNUMAP identified more editing sites, and thus, more EERs (Table 1; Supplemental Tables 1, 2). This is likely due to the ability of the editing-aware GNUMAP-bs version to ignore A-to-G mismatch penalties during alignment (Hong et al. 2013). We also found that the +Repeat filtering protocol produced a more inclusive set of EERs, based on nearly complete overlap with Unique EERs and more extensive overlap with genomic annotations known to encode edited transcripts (e.g., transposons). Thus, we conclude that using GNUMAP with the +Repeat filtering protocol provides the most comprehensive list of EERs.
We note that read coverage over EERs was low relative to surrounding unedited regions (e.g., see Fig. 2B). Low coverage could result from splicing that excises the EER. However, this is unlikely since, rather than the sharp drop that occurs at exon–intron boundaries, read coverage at EER boundaries showed a more gradual decrease (data not shown). Reverse transcriptase pausing at structured regions, long known to affect the efficiency of cDNA synthesis (Harrison et al. 1998), could also explain the reduced coverage. Our cDNA synthesis protocol was designed to ameliorate these effects (see Materials and Methods), and additionally, input RNA was fragmented prior to sequencing, which reduces the effect of secondary structure further. Nevertheless, the most straightforward explanation for reduced read coverage over an internal region of a transcript, in the absence of splicing, is interrupted cDNA synthesis.
EERs represent dsRNA structures
By several criteria we determined that EERs identified with both aligners were highly enriched for double-stranded structures. EERs had significantly higher predicted thermodynamic stability relative to randomized length-matched genomic regions (Fig. 3A,B). Moreover, in some cases EERs with limited intramolecular folding had significant sequence complementarity with other EERs (Supplemental Table 3), suggesting our approach is capable of identifying EERs with intermolecular structures. Long dsRNA is a prerequisite for the extensive and clustered ADAR editing that we detected with our pipeline. Indeed, in some instances we observed as many as 30 editing events in a single editing-enriched window of an EER (Supplemental Table 2, GNUMAP_Repeat_Minus_1, column I).
Our analysis builds substantially on the small set of previously identified long dsRNAs in C. elegans. Prior to this study, only 10 hyperedited substrates were known in C. elegans (Morse and Bass 1999; Morse et al. 2002), although studies have identified transcripts with multiple editing sites (Wu et al. 2011; Washburn et al. 2014; Zhao et al. 2015). Our final list of 664 GNUMAP +Repeat EERs extends this list considerably. Importantly, our pipeline identified 9 out of the 10 known hyperedited substrates, the lone instance where identification failed (M05B5.3 5′ UTR) being due to low coverage that did not meet our threshold. As further validation, Sanger sequencing confirmed 160 out of 180 GNUMAP +Repeat editing sites across six EERs. That such a large number of sites was confirmed using only 1% of our GNUMAP +Repeat EER list indicates that our pipeline is robust and accurate. Further, the strong agreement between the nearest neighbors of our sites (Fig. 3D) and those observed in C. elegans (Morse and Bass 1999; Washburn et al. 2014) and other organisms (Polson and Bass 1994; Lehmann and Bass 2000; Eggington et al. 2011) indicates that the majority of our sites are true editing sites.
Comparison of EER data sets with data sets of related studies
While other studies have identified dsRNAs in C. elegans, our analysis provides a largely unique set of dsRNAs that are likely to be biologically relevant. A previous study in C. elegans identified 9972 dsRNA regions by comparing RNA-Seq data sets of total RNA samples treated in vitro with dsRNA or ssRNA-specific nucleases (Li et al. 2012). While we find a modest overlap with the dsRNA regions reported in this study (18.3%; Supplemental Table 9), the majority of EERs are unique. This disagreement likely reflects differences in methodologies. Our approach identified clusters of editing sites, which are specific for long dsRNA and only occur in vivo. Moreover, our approach incorporated a coimmunoprecipitation step to enrich for highly structured RNAs prior to sequencing. The EERs we identified exist in vivo, which increases the likelihood they have important biological functions, and adding valuable information about the C. elegans dsRNAome.
A study in C. elegans by Wu et al. (2011) identified 454 dsRNA loci that serve as ADAR-dependent templates for small RNA biogenesis, termed ARLs (ADAR-modulated RNA loci). In wild-type worms, few small RNAs are produced over these regions while abundant small RNAs are observed in adr-1;adr-2 mutant animals, suggesting that ADAR competes with the RNAi machinery to engage these substrates, a recurring theme in multiple studies (Ekdahl et al. 2012; Vesely et al. 2012; Warf et al. 2012). The authors determined that a subset of ARLs (184) was edited, suggesting there might be overlap between our data sets. Indeed, 154 of 454 ARLs (∼34%) intersect a GNUMAP +Repeat EER, and 90 of these ARLs are also edited (∼49%). The high overlap between ARLs and EERs compared with the dsRNAs detected using nucleases (Li et al. 2012) likely reflects that ADAR is a shared effector in both studies. However, again, the majority of EERs do not overlap ARLs, suggesting that our study provides a largely novel analysis of dsRNA in C. elegans.
What are the biological roles of long dsRNA?
There are general features of the observed EERs, as well as specific features of individual EERs, that hint at biological functions. In general, EERs are clustered at the distal arms of autosomes, and there are fewer EERs on the X chromosome. EERs are also associated with long introns, which are similarly clustered at distal arms (Prachumwat et al. 2004). The distal arms of chromosomes in C. elegans are enriched for heterochromatin marks, such as H3K9 methylation, host chromosome pairing centers, and have elevated recombination rates relative to central, gene-rich chromosome regions (The C. elegans Sequencing Consortium 1998; Liu et al. 2011). The location of EERs and their potential association with heterochromatin and recombination hotspots suggests they may play a role in organization of chromatin domains and proper segregation of chromosomes. In support of this idea, small RNAs, which in some cases might derive from EERs, mediate homology-driven deposition of heterochromatin in C. elegans (Ashe et al. 2012; Buckley et al. 2012; Gu et al. 2012). In Drosophila, ADAR antagonizes heterochromatin deposition over transposable elements (Savva et al. 2013). ADAR antagonizes other dsRNA-mediated pathways, such as miRNA and siRNA biogenesis (Yang et al. 2005; Heale et al. 2009a,b; Vesely et al. 2012; Warf et al. 2012), and in a similar way, editing of C. elegans dsRNAs, or simply dsRNA binding, could regulate small RNA-mediated chromatin remodeling. Disrupting dsRNA-mediated chromatin remodeling would be predicted to have downstream effects on chromosome segregation, since heterochromatin is required in some cases for meiotic chromosome segregation (Dernburg et al. 1996; Karpen et al. 1996). Consistent with this, overexpression of rncs-1 results in X chromosome nondisjunction and an increase in male progeny (Hellwig and Bass 2008).
EERs might also function to control the specific mRNA they are associated with. For example, the majority of EERs are embedded within introns of protein-coding genes, and these may impact expression of the associated mRNA. In both humans and C. elegans, genes with longer introns are poorly expressed (Castillo-Davis et al. 2002). Furthermore, in C. elegans, longer introns correlate with a higher recombination rate (Prachumwat et al. 2004). Given the costs of having larger introns, it seems likely that embedded EERs are maintained because they provide a fitness benefit. Embedded EERs could serve as scaffolds for regulatory dsRNA-binding proteins, possibly to facilitate splicing or assembly of a ribonucleoprotein (RNP) complex prior to export to the cytoplasm. Alternatively, structures embedded within an intron could compete with spliceosome assembly to restrict pre-mRNA processing until certain conditions are met.
We observed that 3′ UTRs, many of which are unannotated, comprise a significant fraction of EERs. In some cases structured 3′ UTRs in mammals, Drosophila, and C. elegans, act as targeting sequences for the dsRNA-binding protein Staufen (St Johnston et al. 1992; Kim et al. 2005; LeGendre et al. 2013). In mammals, Staufen1 recruits RNA decay factors, triggering mRNA turnover via the Staufen-mediated decay (SMD) pathway (Kim et al. 2005, 2007; Gong and Maquat 2011). The conservation of Staufen1 in C. elegans, as well as many other SMD decay factors, suggests that turnover of mRNAs having dsRNA in their 3′ UTRs may also occur in C. elegans. Structured 3′ UTRs that are hyperedited by ADAR can be sequestered in nuclear paraspeckles (Zhang and Carmichael 2001) and, when not restricted to the nucleus, have reduced translatability (Hundley et al. 2008; Capshew et al. 2012). One or both of these mechanisms could act upon mRNAs with EERs in their 3′ UTRs. In fact, PKR is activated by certain cellular dsRNAs only during mitosis, when the nuclear envelope is not present (Kim et al. 2014), suggesting the presence of dsRNA in the cytoplasm might be dependent on cell cycle.
Finally, our analyses indicate that a subset of EERs is differentially expressed during stress and development (Fig. 7, Supplemental Figs. 3–5). Here there are parallels with dsRNA in other organisms. Human Alu repeat RNAs are differentially expressed in response to cellular stresses (Liu et al. 1995) and involved in human diseases such as macular degeneration (Kaneko et al. 2011; Tarallo et al. 2012). Recent studies in mouse show that the embryonic lethality of a homozygous Adar1−/− mutant is the result of aberrant dsRNA-induced activation of the innate immune response; animals can be rescued to live birth by deletion of Mavs, an intermediary interferon signaling protein (Mannion et al. 2014). The C. elegans dsRNA rncs-1 is up-regulated during starvation, and strains overexpressing rncs-1 produce more male progeny (Hellwig and Bass 2008), a typical response to stress (de Visser and Elena 2007). Unlike in the case of rncs-1, where response to starvation is regulated by the intestinal transcription factory elt-2, it is unclear how expression of EERs is regulated by stress. As many are embedded within coding genes, it is likely they are subject to the same control as their host gene. However, post-transcriptional regulation, such as cleavage by Dicer or sequestration into RNPs, may also play a role. Our study provides a useful data set to help address these questions in C. elegans, and using our pipeline, similar studies can be carried out in other organisms.
MATERIALS AND METHODS
Immunoprecipitation using DCR-1 and RDE-4 antibodies
Embryos were isolated from gravid adults of N2 (Bristol) and eri-4 (dcr-1(mg375)) strains by hypochlorite treatment (Emmons et al. 1979) and washed in M9 buffer. Embryos were homogenized in cold lysis buffer (25 mM HEPES–KOH pH 7.5, 10 mM KOAc, 2 mM Mg(OAc)2, 1 mM DTT, 1% Triton X-100, 1% protease inhibitor cocktail [Roche], 40 U/mL RNasin [Invitrogen]) using a stainless steel dounce. Lysates were centrifuged (14,000 rpm, 15 min) and KCl adjusted to 110 mM. Supernatants were incubated with anti-DCR-1 or anti-RDE-4 affinity-purified polyclonal antibodies (2 h, 4°C). The mixture was incubated with protein A/G agarose (Fisher Scientific) overnight at 4°C, and pellets washed three times with cold lysis buffer. RNA was extracted from beads with TRIzol (Invitrogen), treated with Turbo DNase (Invitrogen) and ethanol precipitated. RNA was resuspended in water and stored at −80°C.
RNA isolation and immunoprecipitation of dsRNA
Total RNA was extracted from mixed-stage N2 and eri-4 worms using TRIzol and treated with Turbo DNase. Total RNA (40 μg) was depleted of rRNA (RiboMinus kit, Invitrogen), and incubated with 20 μg of J2 monoclonal antibody (English and Scientific Consulting) in binding buffer (50 mM Tris pH 8, 150 mM NaCl, 1% NP-40 and 40 U/mL SUPERase-In [Invitrogen]) overnight at 4°C. J2-dsRNA complexes were purified on protein A/G agarose (Fisher Scientific) for 4 h at 4°C. Beads were washed five times with cold binding buffer and RNA recovered by TRIzol extraction and ethanol precipitation. RNA was resuspended in water and stored at −80°C.
Library preparation and high-throughput sequencing
RNAs extracted from J2, DCR-1 and RDE-4 immunoprecipitates were fragmented by incubating in fragmentation buffer (40 mM Tris–acetate pH 8.1, 30 mM Mg(OAc)2, 100 mM KOAc) for 5 min at 94°C. cDNA libraries were generated using Illumina small TruSeq RNA sample preparation kit according to manufacturer's recommended protocol, with the addition of 10% DMSO to the reverse transcriptase reaction. Library size distribution was evaluated on a Bioanalyzer DNA 1000. The library was subjected to 101-cycle paired-end sequencing on the Illumina HiSeq-2000 platform.
Sequence alignment using novoalign and GNUMAP
Paired-end sequencing reads were aligned to C. elegans genome WS220/ce10 using novoalign (V2.07.13, http://www.novocraft.com/products/novoalign/) with option “-r All” to retain repeats. Illumina universal adaptor sequences were stripped using the option “–c TGGAATTCTCGGGTGCCAAGG and GATCGTCGGACTGTAGAACTCTGAAC”. Alignments were filtered using the SamTranscriptomeParser application of the USEQ package (http://useq.sourceforge.net) using the following options: “-a 120” to remove reads with excessive mismatches (∼4/100 bp read), “-n 1” to keep only unique reads or “-n 1000000” to keep both unique and repeat reads, “-r” to reverse the strand of paired reads, and “-p” to merge paired reads.
The same paired-end data as above were also aligned to WS220/ce10 using an RNA editing-aware version of GNUMAP-bs (http://dna.cs.byu.edu/gnumap/) with the following GNUMAP arguments: “--lib_type unstrand --read_type rna --per_dist 150 --nt_conv a2i --top_k_hash 10 --map_quality sensitive”. GNUMAP alignments were also parsed using SamTranscriptomeParser as described above, except that the alignment score filter was set much higher (i.e., “-a 10000”). Consecutive low quality bases at the ends of reads were trimmed, and all alignments having more than 1 non-A>G mismatches were discarded (AlignmentEndTrimmmer, Useq). Alignments were then fed into the RNA detection pipeline outlined in Figure 1.
Detection of RNA editing and EERs
Pileup files containing genome position information were generated using the samtools mpileup application (http://www.htslib.org/) with SamTranscriptomeParser-filtered BAM files as input. Base alignment quality (BAQ) computation was disabled, and anomalous read pairs were used in generating pileup files. The USeq application RNAEditingPileupParser was used to parse pileup files for A-to-G transitions with ≥5 overlapping reads on the same strand. Sliding window scan utility (RNAEditingScanSeqs) scanned 50 bp windows, collecting windows with ≥ 3 editing sites with ≥1% editing. Overlapping editing-enriched windows were merged into EERs, and EERs within 1 kb were merged using the USeq application EnrichedRegionMaker. The 1 kb distance was chosen by evaluating effects on rncs-1, which is detected as two separate EERs with smaller distances.
Annotation of EERs and individual editing sites
A BED file of EERs was intersected with BED files for genome annotations (described below) using the Bedtools (http://bedtools.readthedocs.org/) application annotateBed (Quinlan and Hall 2010). Overlap is reported as percentage of EER bases overlapping an annotation. Bases that overlapped more than one annotation were counted in each category equally, and bases with no overlap were assumed to be intergenic. EERs >1 kb from a coding gene were identified by extending EER boundaries by 1 kb at both ends, and intersecting with coding genes using windowBed (Bedtools). In all intersections, regions were required to be on the same strand. EERs annotated as antisense intersected with coding genes on the opposite strand.
Variant calling to identify RNA editing sites was performed using Samtools essentially as described (http://samtools.sourceforge.net/mpileup.shtml). Variants with read coverage <5 were removed. Variant annotation was performed using Annovar (http://www.openbioinformatics.org/annovar/) against the WS220/ce10 genome build. We defined RNA editing sites as A-to-G variants on the forward strand or T>C variants on the reverse strand supported by nonreference bases from reads aligned to their respective strands. SNPs in dbSNP build 138 (https://www.ncbi.nlm.nih.gov/SNP/) were filtered from editing sites after liftover from ce6 to ce10 (http://genome.ucsc.edu/cgi-bin/hgLiftOver). Coordinates for editing sites detected using the USeq pipeline were converted from RNAEditingPileupParser .bar files to bed files.
Ensembl genome annotations (WS220/ce10) for coding exons, introns, pseudogenes, 3′ UTRs, and 5′ UTRs were obtained from the UCSC Genome Browser table browser in BED format. tRNAs, snoRNAs, rRNAs, snRNAs, miRNAs, and other ncRNAs were combined into a single ncRNA annotation BED file. Transposon annotations were obtained from Wormbase (http://www.wormbase.org/).
Validation of EERs by RT-PCR and northern blot
Total RNA was extracted from liquid cultures of N2 mixed-stage worms using TRIzol and treated with Turbo DNase (Ambion). Approximately 100 μg of RNA was enriched for polyadenylated RNA using Oligotex mRNA Midi column (Qiagen). cDNA was generated from poly(A)+ RNA using Superscript III (Invitrogen) and amplified by PCR. PCR products were excised and purified from 1% agarose gels for sequencing.
Poly(A) RNA was purified as above and resolved by formaldehyde agarose gel electrophoresis. RNA was transferred to Nytran SPC nylon membrane (GE Healthcare Life Sciences) and then UV cross-linked. Radiolabeled DNA probes were hybridized to the membrane at 42°C in Ultrahyb-Oligo hybridization buffer (Life Technologies), exposed on a PhosphorImager screen and scanned on a Typhoon Trio imager (GE Healthcare Life Sciences). Densitometry was carried out using Imagequant software (GE Healthcare Life Sciences). See Supplementary Table 10 for a list of oligos.
DATA DEPOSITION
RNA sequencing data were deposited in the NCBI Gene Expression Omnibus database (http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE61564.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful to Timothy Mosbruger for assistance with bioinformatics techniques and to members of the Bass Lab for valuable discussions and critical reviews of this manuscript. This work was supported by grants from the National Institutes of Health (NIH) to B.L.B. (National Institute on Aging: 8DP1AG044162; National Institute of General Medical Sciences: GM044073) and to W.E.J. (National Human Genome Research Institute: HG005692). J.M.W. was supported by an NIH Ruth L. Kirschstein NRSA award (NIGMS: F32GM106539). D.A.N. was supported by a National Cancer Institute (NCI) Cancer Center Support Grant (5P30CA042014).
Footnotes
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.048801.114.
Freely available online through the RNA Open Access option.
REFERENCES
- Akira S, Uematsu S, Takeuchi O 2006. Pathogen recognition and innate immunity. Cell 124: 783–801. [DOI] [PubMed] [Google Scholar]
- Ashe A, Sapetschnig A, Weick EM, Mitchell J, Bagijn MP, Cording AC, Doebley AL, Goldstein LD, Lehrbach NJ, Le Pen J, et al. 2012. piRNAs can trigger a multigenerational epigenetic memory in the germline of C. elegans. Cell 150: 88–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashrafi K, Chang FY, Watts JL, Fraser AG, Kamath RS, Ahringer J, Ruvkun G 2003. Genome-wide RNAi analysis of Caenorhabditis elegans fat regulatory genes. Nature 421: 268–272. [DOI] [PubMed] [Google Scholar]
- Athanasiadis A, Rich A, Maas S 2004. Widespread A-to-I RNA editing of Alu-containing mRNAs in the human transcriptome. PLoS Biol 2: e391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bass BL 2002. RNA editing by adenosine deaminases that act on RNA. Annu Rev Biochem 71: 817–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazak L, Haviv A, Barak M, Jacob-Hirsch J, Deng P, Zhang R, Isaacs FJ, Rechavi G, Li JB, Eisenberg E, et al. 2014. A-to-I RNA editing occurs at over a hundred million genomic sites, located in a majority of human genes. Genome Res 24: 365–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blow M, Futreal PA, Wooster R, Stratton MR 2004. A survey of RNA editing in human brain. Genome Res 14: 2379–2387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blumenthal T 2012. Trans-splicing and operons in C. elegans. WormBook: 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonin M, Oberstrass J, Lukacs N, Ewert K, Oesterschulze E, Kassing R, Nellen W 2000. Determination of preferential binding sites for anti-dsRNA antibodies on double-stranded RNA by scanning force microscopy. RNA 6: 563–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borozan I, Watt SN, Ferretti V 2013. Evaluation of alignment algorithms for discovery and identification of pathogens using RNA-Seq. PLoS One 8: e76935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckley BA, Burkhart KB, Gu SG, Spracklin G, Kershner A, Fritz H, Kimble J, Fire A, Kennedy S 2012. A nuclear Argonaute promotes multigenerational epigenetic inheritance and germline immortality. Nature 489: 447–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabili MN, Trapnell C, Goff L, Koziol M, Tazon-Vega B, Regev A, Rinn JL 2011. Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev 25: 1915–1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capshew CR, Dusenbury KL, Hundley HA 2012. Inverted Alu dsRNA structures do not affect localization but can alter translation efficiency of human mRNAs independent of RNA editing. Nucleic Acids Res 40: 8637–8645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castillo-Davis CI, Mekhedov SL, Hartl DL, Koonin EV, Kondrashov FA 2002. Selection for short introns in highly expressed genes. Nat Genet 31: 415–418. [DOI] [PubMed] [Google Scholar]
- Cattenoz PB, Taft RJ, Westhof E, Mattick JS 2013. Transcriptome-wide identification of A > I RNA editing sites by inosine specific cleavage. RNA 19: 257–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clement NL, Snell Q, Clement MJ, Hollenhorst PC, Purwar J, Graves BJ, Cairns BR, Johnson WE 2010. The GNUMAP algorithm: unbiased probabilistic mapping of oligonucleotides from next-generation sequencing. Bioinformatics 26: 38–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Visser JA, Elena SF 2007. The evolution of sex: empirical insights into the roles of epistasis and drift. Nat Rev Genet 8: 139–149. [DOI] [PubMed] [Google Scholar]
- Dernburg AF, Sedat JW, Hawley RS 1996. Direct evidence of a role for heterochromatin in meiotic chromosome segregation. Cell 86: 135–146. [DOI] [PubMed] [Google Scholar]
- Eggington JM, Greene T, Bass BL 2011. Predicting sites of ADAR editing in double-stranded RNA. Nat Commun 2: 319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekdahl Y, Farahani HS, Behm M, Lagergren J, Öhman M 2012. A-to-I editing of microRNAs in the mammalian brain increases during development. Genome Res 22: 1477–1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emmons SW, Klass MR, Hirsh D 1979. Analysis of the constancy of DNA sequences during development and evolution of the nematode Caenorhabditis elegans. Proc Natl Acad Sci 76: 1333–1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerstein MB, Lu ZJ, Van Nostrand EL, Cheng C, Arshinoff BI, Liu T, Yip KY, Robilotto R, Rechtsteiner A, Ikegami K, et al. 2010. Integrative analysis of the Caenorhabditis elegans genome by the modENCODE project. Science 330: 1775–1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong C, Maquat LE 2011. lncRNAs transactivate STAU1-mediated mRNA decay by duplexing with 3′ UTRs via Alu elements. Nature 470: 284–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong C, Tang Y, Maquat LE 2013. mRNA–mRNA duplexes that autoelicit Staufen1-mediated mRNA decay. Nat Struct Mol Biol 20: 1214–1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grishok A 2013. Biology and mechanisms of short RNAs in Caenorhabditis elegans. Adv Genet 83: 1–69. [DOI] [PubMed] [Google Scholar]
- Gu SG, Pak J, Guang S, Maniar JM, Kennedy S, Fire A 2012. Amplification of siRNA in Caenorhabditis elegans generates a transgenerational sequence-targeted histone H3 lysine 9 methylation footprint. Nat Genet 44: 157–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison GP, Mayo MS, Hunter E, Lever AM 1998. Pausing of reverse transcriptase on retroviral RNA templates is influenced by secondary structures both 5′ and 3′ of the catalytic site. Nucleic Acids Res 26: 3433–3442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heale BS, Keegan LP, McGurk L, Michlewski G, Brindle J, Stanton CM, Caceres JF, O'Connell MA 2009a. Editing independent effects of ADARs on the miRNA/siRNA pathways. EMBO J 28: 3145–3156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heale BS, Keegan LP, O'Connell MA 2009b. ADARs have effects beyond RNA editing. Cell Cycle 8: 4011–4012. [DOI] [PubMed] [Google Scholar]
- Hellwig S, Bass BL 2008. A starvation-induced noncoding RNA modulates expression of Dicer-regulated genes. Proc Natl Acad Sci 105: 12897–12902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong C, Clement NL, Clement S, Hammoud SS, Carrell DT, Cairns BR, Snell Q, Clement MJ, Johnson WE 2013. Probabilistic alignment leads to improved accuracy and read coverage for bisulfite sequencing data. BMC Bioinformatics 14: 337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hundley HA, Krauchuk AA, Bass BL 2008. C. elegans and H. sapiens mRNAs with edited 3′ UTRs are present on polysomes. RNA 14: 2050–2060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneko H, Dridi S, Tarallo V, Gelfand BD, Fowler BJ, Cho WG, Kleinman ME, Ponicsan SL, Hauswirth WW, Chiodo VA, et al. 2011. DICER1 deficit induces Alu RNA toxicity in age-related macular degeneration. Nature 471: 325–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karpen GH, Le MH, Le H 1996. Centric heterochromatin and the efficiency of achiasmate disjunction in Drosophila female meiosis. Science 273: 118–122. [DOI] [PubMed] [Google Scholar]
- Kerur N, Hirano Y, Tarallo V, Fowler BJ, Bastos-Carvalho A, Yasuma T, Yasuma R, Kim Y, Hinton DR, Kirschning CJ, et al. 2013. TLR-independent and P2X7-dependent signaling mediate Alu RNA-induced NLRP3 inflammasome activation in geographic atrophy. Invest Ophthalmol Vis Sci 54: 7395–7401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim DD, Kim TT, Walsh T, Kobayashi Y, Matise TC, Buyske S, Gabriel A 2004. Widespread RNA editing of embedded Alu elements in the human transcriptome. Genome Res 14: 1719–1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YK, Furic L, Desgroseillers L, Maquat LE 2005. Mammalian Staufen1 recruits Upf1 to specific mRNA 3′UTRs so as to elicit mRNA decay. Cell 120: 195–208. [DOI] [PubMed] [Google Scholar]
- Kim YK, Furic L, Parisien M, Major F, DesGroseillers L, Maquat LE 2007. Staufen1 regulates diverse classes of mammalian transcripts. EMBO J 26: 2670–2681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y, Lee JH, Park JE, Cho J, Yi H, Kim VN 2014. PKR is activated by cellular dsRNAs during mitosis and acts as a mitotic regulator. Genes Dev 28: 1310–1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeGendre JB, Campbell ZT, Kroll-Conner P, Anderson P, Kimble J, Wickens M 2013. RNA targets and specificity of Staufen, a double-stranded RNA-binding protein in Caenorhabditis elegans. J Biol Chem 288: 2532–2545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann KA, Bass BL 2000. Double-stranded RNA adenosine deaminases ADAR1 and ADAR2 have overlapping specificities. Biochemistry 39: 12875–12884. [DOI] [PubMed] [Google Scholar]
- Levanon EY, Eisenberg E, Yelin R, Nemzer S, Hallegger M, Shemesh R, Fligelman ZY, Shoshan A, Pollock SR, Sztybel D, et al. 2004. Systematic identification of abundant A-to-I editing sites in the human transcriptome. Nat Biotechnol 22: 1001–1005. [DOI] [PubMed] [Google Scholar]
- Li F, Zheng Q, Ryvkin P, Dragomir I, Desai Y, Aiyer S, Valladares O, Yang J, Bambina S, Sabin LR, et al. 2012. Global analysis of RNA secondary structure in two metazoans. Cell Rep 1: 69–82. [DOI] [PubMed] [Google Scholar]
- Liu WM, Chu WM, Choudary PV, Schmid CW 1995. Cell stress and translational inhibitors transiently increase the abundance of mammalian SINE transcripts. Nucleic Acids Res 23: 1758–1765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T, Rechtsteiner A, Egelhofer TA, Vielle A, Latorre I, Cheung MS, Ercan S, Ikegami K, Jensen M, Kolasinska-Zwierz P, et al. 2011. Broad chromosomal domains of histone modification patterns in C. elegans. Genome Res 21: 227–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenz R, Bernhart SH, Höner Zu Siederdissen C, Tafer H, Flamm C, Stadler PF, Hofacker IL 2011. ViennaRNA Package 2.0. Algorithms Mol Biol 6: 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukács N 1994. Detection of virus infection in plants and differentiation between coexisting viruses by monoclonal antibodies to double-stranded RNA. J Virol Methods 47: 255–272. [DOI] [PubMed] [Google Scholar]
- Mannion NM, Greenwood SM, Young R, Cox S, Brindle J, Read D, Nellåker C, Vesely C, Ponting CP, McLaughlin PJ, et al. 2014. The RNA-editing enzyme ADAR1 controls innate immune responses to RNA. Cell Rep 9: 1482–1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markham NR, Zuker M 2008. UNAFold: software for nucleic acid folding and hybridization. Methods Mol Biol 453: 3–31. [DOI] [PubMed] [Google Scholar]
- Morse DP, Bass BL 1997. Detection of inosine in messenger RNA by inosine-specific cleavage. Biochemistry 36: 8429–8434. [DOI] [PubMed] [Google Scholar]
- Morse DP, Bass BL 1999. Long RNA hairpins that contain inosine are present in Caenorhabditis elegans poly(A)+ RNA. Proc Natl Acad Sci 96: 6048–6053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morse DP, Aruscavage PJ, Bass BL 2002. RNA hairpins in noncoding regions of human brain and Caenorhabditis elegans mRNA are edited by adenosine deaminases that act on RNA. Proc Natl Acad Sci 99: 7906–7911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura T, Furuhashi M, Li P, Cao H, Tuncman G, Sonenberg N, Gorgun CZ, Hotamisligil GS 2010. Double-stranded RNA-dependent protein kinase links pathogen sensing with stress and metabolic homeostasis. Cell 140: 338–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nam JW, Bartel DP 2012. Long noncoding RNAs in C. elegans. Genome Res 22: 2529–2540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neeman Y, Levanon EY, Jantsch MF, Eisenberg E 2006. RNA editing level in the mouse is determined by the genomic repeat repertoire. RNA 12: 1802–1809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishikura K 2010. Functions and regulation of RNA editing by ADAR deaminases. Annu Rev Biochem 79: 321–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker GS, Eckert DM, Bass BL 2006. RDE-4 preferentially binds long dsRNA and its dimerization is necessary for cleavage of dsRNA to siRNA. RNA 12: 807–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavelec DM, Lachowiec J, Duchaine TF, Smith HE, Kennedy S 2009. Requirement for the ERI/DICER complex in endogenous RNA interference and sperm development in Caenorhabditis elegans. Genetics 183: 1283–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polson AG, Bass BL 1994. Preferential selection of adenosines for modification by double-stranded RNA adenosine deaminase. EMBO J 13: 5701–5711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prachumwat A, DeVincentis L, Palopoli MF 2004. Intron size correlates positively with recombination rate in Caenorhabditis elegans. Genetics 166: 1585–1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, Hall IM 2010. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26: 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramaswami G, Zhang R, Piskol R, Keegan LP, Deng P, O'Connell MA, Li JB 2013. Identifying RNA editing sites using RNA sequencing data alone. Nat Methods 10: 128–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riedmann EM, Schopoff S, Hartner JC, Jantsch MF 2008. Specificity of ADAR-mediated RNA editing in newly identified targets. RNA 14: 1110–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruffalo M, LaFramboise T, Koyutürk M 2011. Comparative analysis of algorithms for next-generation sequencing read alignment. Bioinformatics 27: 2790–2796. [DOI] [PubMed] [Google Scholar]
- Savva YA, Jepson JE, Chang YJ, Whitaker R, Jones BC, St Laurent G, Tackett MR, Kapranov P, Jiang N, Du G, et al. 2013. RNA editing regulates transposon-mediated heterochromatic gene silencing. Nat Commun 4: 2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schönborn J, Oberstrass J, Breyel E, Tittgen J, Schumacher J, Lukacs N 1991. Monoclonal antibodies to double-stranded RNA as probes of RNA structure in crude nucleic acid extracts. Nucleic Acids Res 19: 2993–3000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sijen T, Plasterk RH 2003. Transposon silencing in the Caenorhabditis elegans germ line by natural RNAi. Nature 426: 310–314. [DOI] [PubMed] [Google Scholar]
- Spencer WC, Zeller G, Watson JD, Henz SR, Watkins KL, McWhirter RD, Petersen S, Sreedharan VT, Widmer C, Jo J, et al. 2011. A spatial and temporal map of C. elegans gene expression. Genome Res 21: 325–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- St Johnston D, Brown NH, Gall JG, Jantsch M 1992. A conserved double-stranded RNA-binding domain. Proc Natl Acad Sci 89: 10979–10983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabara H, Yigit E, Siomi H, Mello CC 2002. The dsRNA binding protein RDE-4 interacts with RDE-1, DCR-1, and a DExH-box helicase to direct RNAi in C. elegans. Cell 109: 861–871. [DOI] [PubMed] [Google Scholar]
- Tarallo V, Hirano Y, Gelfand BD, Dridi S, Kerur N, Kim Y, Cho WG, Kaneko H, Fowler BJ, Bogdanovich S, et al. 2012. DICER1 loss and Alu RNA induce age-related macular degeneration via the NLRP3 inflammasome and MyD88. Cell 149: 847–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The C. elegans Sequencing Consortium. 1998. Genome sequence of the nematode C. elegans: a platform for investigating biology. Science 282: 2012–2018. [DOI] [PubMed] [Google Scholar]
- Ulitsky I, Bartel DP 2013. lincRNAs: genomics, evolution, and mechanisms. Cell 154: 26–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulitsky I, Shkumatava A, Jan CH, Sive H, Bartel DP 2011. Conserved function of lincRNAs in vertebrate embryonic development despite rapid sequence evolution. Cell 147: 1537–1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vesely C, Tauber S, Sedlazeck FJ, von Haeseler A, Jantsch MF 2012. Adenosine deaminases that act on RNA induce reproducible changes in abundance and sequence of embryonic miRNAs. Genome Res 22: 1468–1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K, Li M, Hakonarson H 2010. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 38: e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warf MB, Shepherd BA, Johnson WE, Bass BL 2012. Effects of ADARs on small RNA processing pathways in C. elegans. Genome Res 22: 1488–1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Washburn MC, Kakaradov B, Sundararaman B, Wheeler E, Hoon S, Yeo GW, Hundley HA 2014. The dsRBP and inactive editor ADR-1 utilizes dsRNA binding to regulate A-to-I RNA editing across the C. elegans transcriptome. Cell Rep 6: 599–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welker NC, Habig JW, Bass BL 2007. Genes misregulated in C. elegans deficient in Dicer, RDE-4, or RDE-1 are enriched for innate immunity genes. RNA 13: 1090–1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welker NC, Pavelec DM, Nix DA, Duchaine TF, Kennedy S, Bass BL 2010. Dicer's helicase domain is required for accumulation of some, but not all, C. elegans endogenous siRNAs. RNA 16: 893–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu D, Lamm AT, Fire AZ 2011. Competition between ADAR and RNAi pathways for an extensive class of RNA targets. Nat Struct Mol Biol 18: 1094–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W, Wang Q, Howell KL, Lee JT, Cho DS, Murray JM, Nishikura K 2005. ADAR1 RNA deaminase limits short interfering RNA efficacy in mammalian cells. J Biol Chem 280: 3946–3953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Carmichael GG 2001. The fate of dsRNA in the nucleus: a p54(nrb)-containing complex mediates the nuclear retention of promiscuously A-to-I edited RNAs. Cell 106: 465–475. [DOI] [PubMed] [Google Scholar]
- Zhao HQ, Zhang P, Gao H, He X, Dou Y, Huang AY, Liu XM, Ye AY, Dong MQ, Wei L 2015. Profiling the RNA editomes of wild-type C. elegans and ADAR mutants. Genome Res 25: 66–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.