

Abstract
Alternative splicing is a key player in the creation of complex mammalian transcriptomes and its misregulation is associated with many human diseases. Multiple mRNA isoforms are generated from most human genes, a process mediated by the interplay of various RNA signature elements and trans-acting factors that guide spliceosomal assembly and intron removal. Here, we introduce a splicing predictor that evaluates hundreds of RNA features simultaneously to successfully differentiate between exons that are constitutively spliced, exons that undergo alternative 5′ or 3′ splice-site selection, and alternative cassette-type exons. Surprisingly, the splicing predictor did not feature strong discriminatory contributions from binding sites for known splicing regulators. Rather, the ability of an exon to be involved in one or multiple types of alternative splicing is dictated by its immediate sequence context, mainly driven by the identity of the exon's splice sites, the conservation around them, and its exon/intron architecture. Thus, the splicing behavior of human exons can be reliably predicted based on basic RNA sequence elements.
Keywords: alternative splicing, splicing predictor, bioinformatics, support vector machine
INTRODUCTION
Alternative splicing results in the production of multiple mRNA isoforms from a single pre-mRNA, thereby significantly enriching the proteomic diversity of higher eukaryotic organisms. It is carried out by the spliceosome, which catalyzes the removal of noncoding intronic sequences and concatenates remaining exons to generate the mature mRNA (Black 2003). Of the ∼25,000 genes encoded by the human genome (International Human Genome Sequencing Consortium 2004), >90% are believed to produce transcripts that are alternatively spliced (Pan et al. 2008; Wang et al. 2008). The most prevalent types of alternative splicing are the variable inclusion of an entire exon (cassette exon), the selection of alternative splice sites upstream of or downstream from the 3′ or the 5′ splice site, as well as intron retention (Wang et al. 2008). Defects in splicing lead to many human genetic diseases (Krawczak et al. 1992; Cartegni et al. 2002; Faustino and Cooper 2003) and splicing mutations in a number of genes involved in growth control have been implicated in multiple types of cancer (Carstens et al. 1997; Mercatante et al. 2001; Wang et al. 2003; Xu and Lee 2003; Bartel et al. 2004; Brinkman 2004). Insights into the basic mechanisms of pre-mRNA splicing and splice-site recognition are therefore fundamental to understanding regulated gene expression and human disease.
Regulation of the splicing process relies on the activity of multiple RNA signature elements, which include the strength of splice sites (Yeo and Burge 2004), the number of enhancers and silencers associated with the splicing unit (Black 2003), the exon/intron architecture (Fox-Walsh et al. 2005), RNA secondary structure (Eperon et al. 1988; Clouet d'Orval et al. 1991; Hiller et al. 2007; Shepard and Hertel 2008; McManus and Graveley 2011), and the process of transcription by RNA polymerase II (Kornblihtt 2005). Little is known about the extent to which these parameters influence each other to define the overall probability of exon recognition.
One goal in the splicing field is the creation of a “splicing code” that predicts splicing behavior based on RNA sequence alone (McManus and Graveley 2011). Several attempts of such a splicing code generation have been described. For example, Sorek et al. (2004) evaluated seven RNA features, which were combined to classify cassette exons with a true positive rate of 50% at a false positive rate of 1.8%. This initial classifier was refined by adding additional features and filtering for conserved exons (Dror et al. 2005) to improve prediction accuracy to a true positive rate of 50% for a false positive rate of 0.5%. The Blencowe and Frey laboratories reported on their efforts to increase the performance of the splicing code (Barash et al. 2010). Equipped with extensive microarray results that recorded the inclusion levels of >3500 cassette-type alternative exons from 27 diverse mouse tissues, an algorithm based on ∼200 features capable of predicting tissue-dependent changes in alternative splicing was introduced. This work was further improved using a Bayesian method on 1014 RNA features (Xiong et al. 2011; Barbosa-Morais et al. 2012).
While previous approaches impressively demonstrated that the simultaneous analysis of hundreds of splicing features supported cassette splicing predictions across different tissues, there is still significant room for improvement. First, the performance of the splicing code can be improved upon by adding new sequence features or by applying alternate classifiers (Xiong et al. 2011). Second, previous work only concentrated on differentiating between cassette-type exons and constitutive exons across various tissues or cell types, thus ignoring other major classes of alternative splicing, such as alternative 5′ or 3′ splice-site selection. To classify the alternative splicing behavior of internal exons we present the design and performance evaluation of a splicing predictor that differentiates between constitutive, cassette-type, and, for the first time, alternative 5′ and 3′ splice-site exons. Using support vector machines we identified RNA sequence features that characterize each category of alternative splicing.
RESULTS
Splice-site usage and exon inclusion levels
We analyzed the usage level of alternative 3′ and 5′ splice sites (referred to as “splice-site usage levels”) as well as the inclusion level of cassette exons (referred to as “exon inclusion levels”) using our data sets of alternative splicing events (Materials and Methods). As shown in Figure 1, the vast majority of alternative splicing events are biased toward high (≥80%) or low (≤20%) splice-site usage/exon inclusion levels, an observation consistent with inclusion levels derived from deep sequencing of individual cell lines (Shepard et al. 2011). This raises the question whether all alternative splicing events should be placed into the same training data set or whether it would be more advantageous to split exons into sets of high and low inclusion exon sets. Based on the bimodal distribution of alternative splicing events we filtered our training set of alternatively spliced exons into high inclusion exons or high usage splice sites (≥80% inclusion/usage) and low inclusion exons or low usage splice sites (≤20% inclusion/usage) to train support vector machines (SVMs).
FIGURE 1.
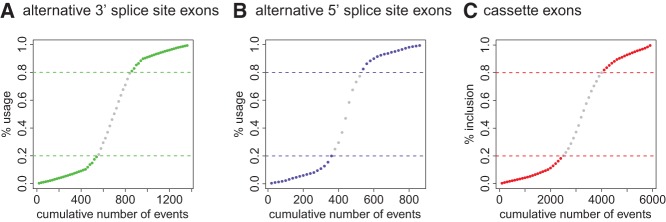
Splice-site usage (A) and (B) and exon inclusion (C) levels of exons in our data sets. The plots show the relationship between inclusion/usage levels and the cumulative number of events. Exons in each set were ordered based on their inclusion/usage level. Cassette exons were then grouped in sets of 100, exons with an alternative 3′ or 5′ splice-site were grouped in sets of 20 events and averaged.
Splicing code
To create a splicing code that differentiates between various types of alternative splicing, we applied the concept of a support vector machine (SVM), a widely used machine learning technique. A SVM is a binary classifier that uses input data for training and is then able to make predictions on new unclassified data based on the training data. Here, we applied it to distinguish between different classes of exons initially using 262 unique RNA features (Supplemental Table 1). To estimate the accuracy of the SVMs, we determined the area under the ROC curve (AUC), which plots the true positive rate (TRP) versus the false positive rate (FPR). When comparing constitutive exons and exons with a rarely used alternative 3′ or 5′ splice site, the splicing code was very efficient in differentiating between the splicing classes, generating ROC curves with an area under the curve (AUC) of 0.939 and 0.949, respectively (Table 1; Fig. 2A,B). In contrast, exons with frequently used alternative 3′ or 5′ splice sites are much harder to distinguish from constitutive exons (Table 1; Fig. 2A,B). The accuracy of classifying between exons with a moderately used alternative 3′ or 5′ splice site (i.e., exons with an alternative 3′ or 5′ splice-site usage up to 80%) and constitutive exons is also high (Fig. 3). We conclude that exons with a highly used alternative splice site behave very much like constitutive exons and the currently used RNA features do not allow efficient separation. However, the splicing code is very accurate in differentiating between constitutive exons and exons with moderately and rarely used alternative splice sites.
TABLE 1.
Performance of SVMs after a 10% cross-validation measured as the area under the ROC curve (AUC)
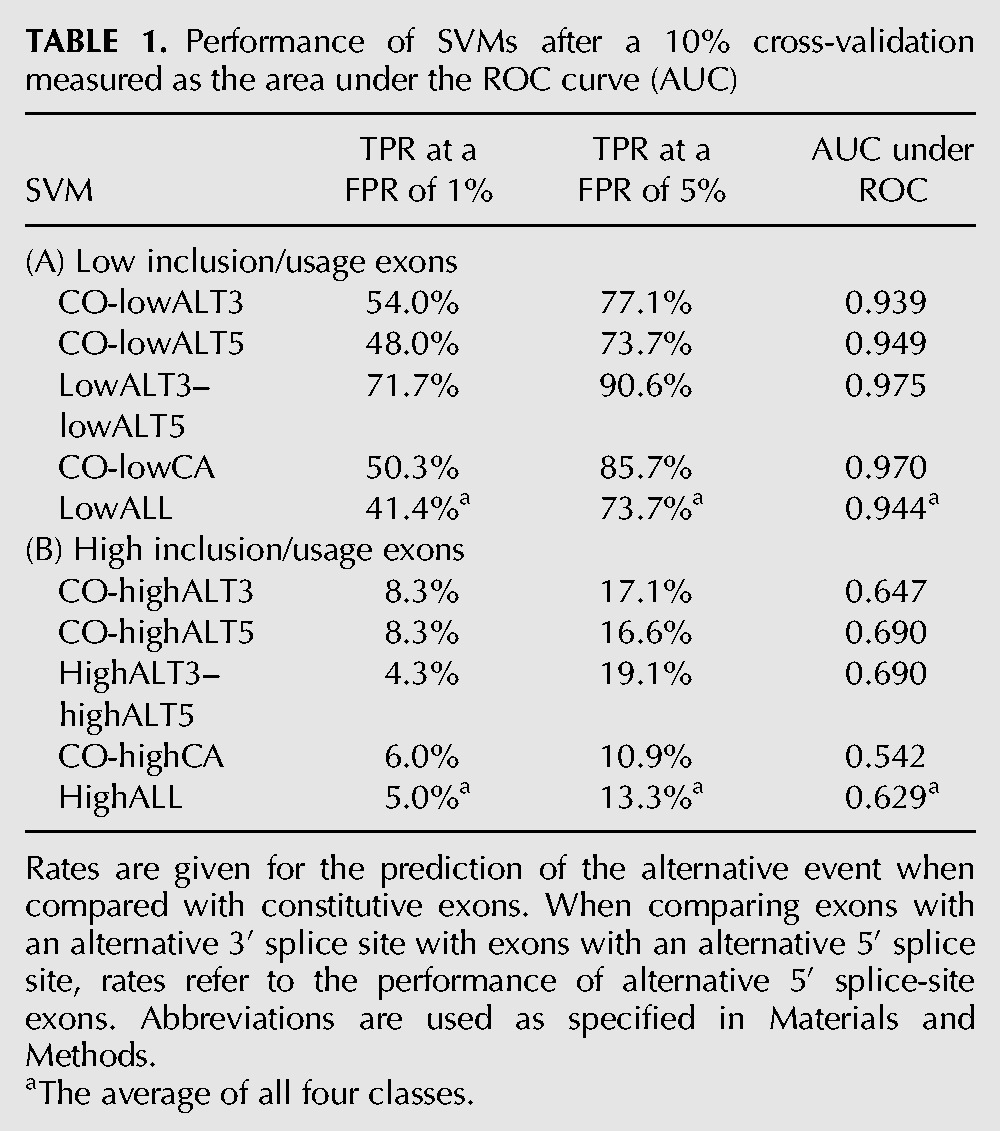
FIGURE 2.

ROC curves of SVMs comparing constitutive exons (CO) and (A) exons with an alternative 3′ splice site, (B) exons with an alternative 5′ splice site, (C) cassette exons, (D) rarely included cassette exons (lowCA), exons with a rarely used alternative 3′ splice site (lowALT3), and exons with a rarely used alternative 5′ splice site (lowALT5), and (E) frequently included cassette exons (highCA), exons with a frequently used alternative 3′ splice site (highALT3), and exons with a frequently used alternative 5′ splice site (highALT5). The true positive rate (TPR) is calculated as the number of true positives (TP) divided by the number of positive (P) samples in the test set. The false positive rate (FPR) is calculated as the number of false positives (FP) divided by the number of negative (N) samples in the test set.
FIGURE 3.

Accuracy of the predictions when comparing constitutive exons with alternatively spliced exons. Using the SVM that was trained on constitutive exons and (A) exons with a rarely used (up to 20%) alternative 3′ splice site (CO-lowALT3), (B) exons with a rarely used alternative 5′ splice site (CO-lowALT5), and (C) rarely included cassette exons, predictions were made for new test exons whose alternative splice sites were used with various different frequencies (x-axis). Accuracy is specified as AUC of the ROC curve (y-axis).
When comparing cassette and constitutive exons we observe a similar behavior. Rarely included exons are very well distinguishable from constitutively spliced exons leading to an AUC of 0.970 (Table 1; Fig. 2C). However, frequently included cassette exons are practically indistinguishable from constitutive exons (Table 1; Fig. 2C). Furthermore, the accuracy of classifying between constitutive exons and cassette exons with inclusion levels in the median range is still high for inclusion levels up to 60% (Fig. 3C).
Using a combination of several SVMs, we also trained a classifier (multiclassifier SVM) to differentiate between all four classes of exons: constitutive, exons with an alternative 3′ or 5′ splice site, and cassette exons. Internally, exons of each category are compared with exons of every other category, thus, six two-class SVMs are trained and their results are combined to make a final prediction. Depending on the exon class, we obtained excellent classifier performances of AUCs between 0.922 and 0.958 for rarely included exons and rarely used splice sites (Table 1; Fig. 2D). As was observed in the pairwise analysis, this performance drops significantly when comparing exons with highly used alternative splice sites and highly included exons (Table 1; Fig. 2E).
These results suggest that the splicing predictor might also be able to differentiate between different types of alternative splicing. To test this hypothesis we trained SVMs comparing cassette-type exons and exons with an alternative splice site. As was observed for the differentiation between constitutive and alternatively spliced exons (Fig. 2), the performance is excellent when comparing rarely included exons and exons with a rarely used alternative splice site. However, the predictive power drops significantly when comparing highly included exons and exons with a frequently used alternative splice site (Supplemental Fig. 1).
To further test the performance of the SVMs, we derived alternative splice-site usage and alternative exon inclusion levels from a HeLa cell mRNA high-throughput sequencing data set (Shepard et al. 2011) using MISO (Katz et al. 2010). For each splicing category (cassette exons and exons with an alternative 3′ or 5′ splice site) exons were chosen randomly and their observed alternative splicing phenotype was compared with SVM predictions. As illustrated in Figure 4, the SVM predictions correlate very well with the phenotype of alternative exons with low and intermediate inclusion or usage levels. We conclude that the SVMs display an excellent performance for predicting alternative splicing classes.
FIGURE 4.
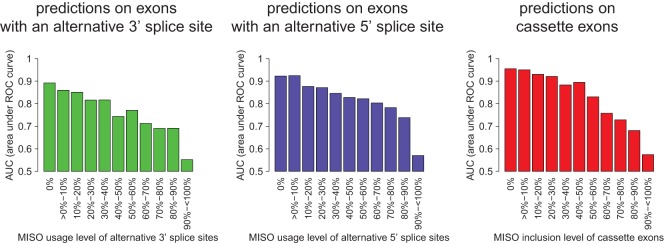
Experimental verification. Predictions were made using the SVMs CO-lowALT3 (for exons with an alternative 3′ splice site, drawn in green), CO-lowALT5 (for exons with an alternative 5′ splice site, drawn in blue), and CO-lowCA (for cassette exons, drawn in red). Inclusion/usage levels were determined based on RNA-seq data in HeLa cells (x-axes). The performance of the SVMs is shown on the y-axes as area under the ROC curve (AUC). Abbreviations for all SVMs are used as defined in Materials and Methods.
Information gain
In addition to using the exon sets to train SVMs, we extracted the features that showed the highest information gain with respect to the determination of the exon types. The information gain of each feature describes the “worth” of that feature when predicting the splicing type of an exon. We expect those features to be part of the basis that dictates the splicing behavior of an exon class. Thus, the RNA features with the highest information gain can be viewed as the most important molecular clues to classify exons as constitutive, as exons using alternative splice sites, or as alternatively included exons. When comparing constitutive exons with alternative 3′ splice-site exons we mainly detected high information gain for RNA features that describe the strength of the 3′ splice site (Fig. 5B, left panel). The combined strength of the 3′ and 5′ splice sites (a value measured by a Maximum Entropy Score, MES [Yeo and Burge 2004]) shows a high information gain as well, but presumably this RNA feature emerges due to the large effect of the 3′ splice site alone, which has an average 3′ MES of 1.68 for rarely used alternative 3′ splice sites compared with an average MES of 8.78 for constitutive 3′ splice sites. In contrast, frequently used alternative 3′ splice sites show an average MES of 7.40, a value much closer to this observed for constitutive 3′ splice sites. In addition, the conservation around the 3′ splice site has a significant information gain. While for constitutive exons the upstream intron is always intronic, these definitions are not so clear for exons with an alternative 3′ splice site. The area in between the alternative splice sites can be exonic and potentially protein coding or intronic and noncoding depending on whether the upstream or downstream splice site is used. Thus, for alternative 3′ splice-site exons the conservation of the upstream intronic region is much higher than the intronic conservation of constitutive exons. Based on similar arguments, the exonic region downstream from alternative 3′ splice sites is expected to display lower conservation scores than constitutive exons, as verified by our analysis (Fig. 5B). Thus, the fluctuating exonic character of alternative exon parts in addition to the presence of as of yet unidentified cis-acting RNA elements may reflect the phylogenetic differences between the constitutive and the alternative 3′ splice-site exon classes. However, none of the evaluated binding sites for known splicing regulators emerged as features with significant information gain.
FIGURE 5.

Most influential features and average conservation ±50 nt around the exon junctions after splitting the data sets into subsets. Color coding for different regions is depicted in A. Features that refer to a combination of several regions are given in gray. The surroundings of the 3′ and 5′ splices site were typically defined as ±50 nt around the exon/intron junctions. The left plots in B–D show the information gain of the dominant features when comparing (B) constitutive and exons with a rarely used alternative 3′ splice site, (C) constitutive and exons with a rarely used alternative 5′ splice site, (D) constitutive and rarely included cassette exons. Right plots in B–D show the average conservation (PhastCons score) ±50 nt around the exon junctions. The thick line in each box depicts the median, while the upper and lower ends of the box represent the 25% and 75% quantile, respectively. Smallest and largest observations are depicted by the upper and lower end of the whiskers. Constitutive exons (CO) are compared with (B) exons with a frequently or rarely used alternative 3′ splice site (highALT3 and lowALT3, respectively), (C) exons with a frequently or rarely used alternative 5′ splice site (highALT5 and lowALT5, respectively), and (D) frequently and rarely included cassette exons (highCA and lowCA, respectively).
In analogy to the alternative 3′ splice-site events, only features of and around the 5′ splice site show a high information gain when comparing constitutive exons with exons that have a rarely used alternative 5′ splice site (Fig. 5C, left panel). Especially the conservation around the 5′ splice site shows a strong influence on splicing categorization, while the conservation downstream from the 5′ splice site has a larger information gain than the upstream exonic conservation (Fig. 5C). In addition, a strong influence of the 5′ splice-site strength can be seen (an average MES of 8.28 for constitutive 5′ splice sites and an average MES of 0.15 for rarely used alternative 5′ splice sites, while frequently used alternative 5′ splice sites show an average MES of 6.72). The frequency and strength of possible alternative 5′ splice sites as well as the exon length complete the top 10 RNA features dictating alternative 5′ splice-site exon categorization. As was observed for alternative 3′ splice-site events, binding sites for splicing regulatory proteins did not emerge as RNA features with significant information gain.
When comparing constitutive exons with rarely included cassette exons, the phylogenetic conservation around the exon junctions shows by far the highest information gain and, thus, has the largest influence on the classifier between the two exon groups (Fig. 5D, left panel). Constitutive as well as frequently included cassette exons show a very high conservation within the exon and almost no conservation outside the exons (Fig. 5D, right panel, see median and quartiles around the median). In contrast, rarely included cassette exons show a rather low conservation both inside and outside the exon. This observation suggests that rarely included cassette exons are evolutionarily new or fast evolving because they are not fixed among the 46 vertebrates used to derive conservation values. Furthermore, rarely included cassette exons are more diverse in their conservation, as can be seen by the widespread quartiles around the median in Figure 5D (right panel). Besides phylogenetic conservation, the strength of the splice sites and the exon/intron architecture (defined in Materials and Methods) has a high impact on the prediction quality of the code. As previously reported (Stamm et al. 2000; Clark and Thanaraj 2002; Zheng et al. 2005), constitutive splice sites are stronger than alternative splice sites. While constitutive exons in our data sets showed an average combined MES of both splice sites of 17.06, rarely included cassette exons only showed a combined average MES of 13.71. Furthermore, introns downstream from constitutive exons have an average length of 3535 nt, whereas introns downstream from alternatively spliced exons are much larger with an average length of 11,201 nt. These observations support the notion that exons flanked by large introns are more likely to be involved in alternative splicing than exons flanked by short introns (Berget 1995; Fox-Walsh et al. 2005). Although splicing regulatory protein features are not represented in the top information gain list (Fig. 5), the binding sites for SRSF2 (SC35) and SRSF5 (Srp40) rank 12th and 13th in our feature compendium. However, their overall contribution in differentiating between constitutive and alternatively included exons is nearly negligible.
Surprisingly, the information gain analysis demonstrated that the SVMs did not feature strong discriminatory contributions from binding sites for known splicing regulators. With the exception of SRSF2 and SRSF5 features described above, none of the RNA features used in our classification scheme associated with the existence or strength of binding sites for splicing regulators showed significant information gain. To test whether this lack of discriminatory contributions from binding sites for known splicing regulators was the result of not including sufficient numbers of potential binding site sequences in our RNA feature compendium, we added additional 5mers, 6mers, and 7mers of known binding sites for splicing regulators as well as codon frequencies (Yeo et al. 2007; Barash et al. 2010) to increase the RNA feature compendium to 826 features. However, training our data sets with the extended RNA feature compendium did not improve the performance of our splicing code (Supplemental Fig. 2), nor did we observe a change in the identity of the RNA features that displayed the highest information gain (Supplemental Fig. 3).
Based on these observations, splice-site scores as well as the conservation around the exon junctions were identified as the RNA features with the highest information gain. To evaluate the performance gain derived from phylogenetic conservation, we re-trained our SVMs on the same training exons, but removed all conservation features. For all SVMs, the AUC decreased (Supplemental Fig. 4). Interestingly, the decrease in classification performance was less pronounced for alternative 5′ or 3′ splice-site usage. These results support the notion that the exon/intron architecture and the competing splice-site scores are the main determinants in dictating alternative 5′ or 3′ splice-site usage.
DISCUSSION
A splicing code was created to compare constitutive exons with exons exhibiting alternative 5′ or alternative 3′ splice site selection and alternative cassette-type exons. The splicing code permitted efficient classification of splicing events as long as overall splice-site selection levels (for alternative 5′ and 3′ splice-site exons) or inclusion levels (for alternative cassette exons) were incorporated. Based on the observation that the majority of exons undergo alternative splicing at either high (≥80%) or low (≤20%) frequencies, we carried out classification attempts in separate categories. By splitting alternative splicing events into groups, we were able to obtain excellent classification performances for exons with a rarely or moderately used alternative splice site as well as rarely and moderately included cassette exons (Fig. 3). However, for frequently occurring alternative splicing events, our splicing code was unable to make useful splicing categorizations. These observations demonstrate that none of the RNA features evaluated significantly differentiated the splicing behavior of alternative exons with high usage or high inclusion levels from constitutive exons. Furthermore, it suggests that the recognition of highly used splice sites and included exons is similar to the recognition of constitutive exons. This is indeed the case because highly included alternative exons are almost as efficient in discriminating against low inclusion exons as constitutive exons are (Supplemental Fig. 5).
Because our EST-derived data set originates from various tissues, it is unclear how the splice-site usage and exon inclusion rates of highly included exons are generated. For example, it is possible that the splice-site usage or exon inclusion rates are universally high in all tissues from which ESTs have been derived. Alternatively, exon splicing for those exons may be constitutive in most tissues, but infrequent in a small number of other tissues. Regardless, the overlap between constitutive exons and exons with highly used alternative splice sites as well as highly included exons raises the question of whether the definition of constitutive exons should remain as strict as it was defined here, i.e., no indication of alternative splicing at all, or whether it should be more relaxed permitting a small fraction of alternative splicing. We have recently argued that essentially all exons will engage in low level alternative splicing and its detection is limited by the sensitivity of the assays used to detect alternative splicing events (Fox-Walsh and Hertel 2009). Accordingly, a genome-wide investigation of alternative splicing using deep sequencing of unprecedented depth suggested widespread alternative splicing at low frequencies (Pickrell et al. 2010).
While the main purpose of the splicing code previously described (Barash et al. 2010, 2013) is to predict the directionality of change in exon inclusion between different tissues, when used to distinguish between constitutive and alternatively spliced cassette exons, its performance is comparable with the performance of our splicing code. However, our splicing code is the first to differentiate between constitutive exons, exons with an alternative 3′ or 5′ splice site, and cassette exons.
Mechanistic implication based on information gain analysis
When analyzing the information gain of single RNA features, phylogenetic conservation around the splice sites as well as the strength of the splice sites appeared to be the most significant discriminants in all classes of alternative splicing. Generally, constitutive exons show much higher conservation than rarely included cassette exons and conservation levels around alternative splice sites differ considerably from conservation values around constitutive splice sites. Even though phylogenetic conservation has a strong influence in the comparison of constitutively and alternatively spliced exons, conservation is not expected to be recognizable by the spliceosome. As suggested previously (Chen and Zheng 2008), the observed conservation features may reflect the existence of cis-acting RNA splicing elements that have not been defined yet. This interpretation leads to the expectation that increased conservation should be observed around alternative splicing events, presumably because regulatory elements are contained within. While this expectation is met when comparing flanking introns of cassette exons with constitutive exons (Fig. 5D, right panel), the more striking conservation difference detected in this splicing category was within the exon. Here, conservation was much lower for alternatively spliced cassette exons, an observation that does not support the association of unknown regulatory elements. Rather, this result indicates that a large proportion of the cassette exons with low inclusion rates are young or are changing fast on an evolutionary timescale.
After retraining our SVMs without conservation features we observed variable performance drops for SVMs trained on rarely included exons and/or exons with rarely used splice sites. As there is no significant difference between the conservation of constitutive exons and frequently included cassette exons and/or exons with frequently used alternative splice sites, the removal of those features had only a minor effect, if any, on the performance of the SVM. However, when comparing constitutive exons with rarely included cassette exons and/or exons with a rarely used alternative splice site, the performance of the SVMs decreased significantly. This observation supports the importance of the conservation features for the overall performance of our SVMs as well as those published previously (Barash et al. 2010; Shepard and Hertel 2010; Xiong et al. 2011).
We have previously demonstrated that the ability to form stable RNA secondary structures around splice sites correlates with an increased propensity to undergo alternative 5′ or 3′ splice-site selection (Shepard and Hertel 2008). While features characterizing the probability of splice sites to be involved in RNA secondary structures were incorporated into the splicing code, they did not emerge as one of the highest information gain features described in Figure 5. However, RNA secondary structure probability features were among the top 25% RNA features within the compendium tested when comparing constitutive exons and exons with an alternative 3′ splice site and among the top 28% RNA features when comparing constitutive and exons with an alternative 5′ splice site. As was argued before (Shepard and Hertel 2008), RNA secondary structures around splice sites are important in mediating alternative splice-site selection for a smaller fraction of alternative splicing events. Thus, the information across all alternative splicing events evaluated is limited.
Interestingly, our splicing code did not feature significant discriminatory contributions from binding sites for known splicing regulators. Although SRSF2 and SRSF5 binding sites did emerge as one of several weak contributors in differentiating between constitutive and cassette exons, none of the features associated with the existence or strength of binding sites for splicing regulators was among those identified with the highest information gain. Adding further features of known binding sites for splicing regulators as well as 2mer and 3mer frequencies within the exon and neighboring introns did not improve the performance of our splicing code. Furthermore, none of the extra binding site sequences emerged as features with high information gain. These observations suggest that the ability of an exon to be involved in one or multiple types of alternative splicing is dictated by its immediate sequence context, mainly driven by the identity of the exon's splice sites and exon/intron architecture. No set of the evaluated splicing regulatory proteins appears to generally promote one type of alternative splicing over another. Rather, the anticipated fluctuations in the expression of splicing regulators in different tissues may modulate the efficiency at which a particular exon, or an exon fraction, is recognized by the spliceosome. Thus, most splicing regulatory factors appear to work on a tissue-specific basis. As such, each exon has an intrinsic ability to participate in one or more types of alternative splicing that is based on the identity of the exon's splice sites and exon/intron architecture. Fluctuating inclusion rates for these exons are then achieved through regulated modifiers such as differential transcription kinetics or differential recruitment of splicing factors.
MATERIALS AND METHODS
To create a splicing code that differentiates between various types of alternative splicing, we applied the concept of a support vector machine (SVM), a widely used machine learning technique. A SVM is a binary classifier that uses input data for training and is then able to make predictions on new unclassified data based on the training data. Here, we applied it to distinguish between different classes of exons.
We used and trained the freely available SVM implementation in WEKA (version 3.6.2), a software package incorporating several machine learning techniques (Hall et al. 2009). Using WEKA, we also tested several other machine learning techniques like decision trees, a naïve Bayesian classifier as well as a Bayesian network on our data. Comparing their performance and predictive power, they were all outperformed by John Platt's sequential minimal optimization algorithm for training a support vector classifier (SMO, as implemented in WEKA) (Platt 1999). It normalizes training data automatically and uses a polynomial kernel.
Creating training sets
We created four sets of internal exons with known splicing behavior to train the SVMs: a set of constitutive exons, a set of exons having an alternative 3′ splice site, a set of exons with an alternative 5′ splice site, and a set of cassette exons, which can either be included or skipped. All sets are the results of very strict filtering to ensure that all exons display only one type of alternative splicing, i.e., exons with an alternative splice site will not be skipped and cassette exons do not have an alternative splice site. All splicing information was based on ∼4 million ESTs and known isoforms as well as the alternative events track (Alt Events) of the UCSC Genome Browser (GRCh37/hg19) (Meyer et al. 2013).
The set of constitutive exons includes internal human exons that are not involved in any type of alternative splicing and that are supported by at least 20 ESTs. Alternative exons in our sets are internal exons that show only one type of alternative splicing. All exons have a minimal length of 23 nt and a minimal length of neighboring introns of 78 nt. These numbers were set according to the length distributions of all internal exons as well as their neighboring introns, respectively, (see Supplemental Fig. 6). An exon length of 23 nt represents the 1% quantile of the exon length distribution, i.e., 99% of the internal human exons are at least 23 nt long. Equivalently, an intron length of 78 nt represents the 1% quantile of the intron length distribution.
Feature extraction
For all exons, 262 sequence features were evaluated (for a complete list, see Supplemental Table 1), covering all major parameters known to influence exon recognition.
Splice-site strength
The strength of the splice sites is measured by a maximum entropy score (MES) (Yeo and Burge 2004).
Exon/intron architecture
Features describing the exon/intron architecture include the length of the exon, the length of its neighboring introns, and its neighboring exons.
Local secondary structures
Secondary structures might influence splicing by either promoting the accessibility of certain sequence elements or by masking them and, thus, making them inaccessible. We evaluated the local secondary structure potential of both splice sites as well as those within ±70 nt around both exon junctions. For all areas, we calculated the probability of all 4mers to be unpaired, and thus accessible, using the Vienna RNA Package (Lorenz et al. 2011). Based on these values, we extracted average, minimal and maximal probabilities of a 4mer being unpaired in each area.
Binding sites for splicing regulators
To search for binding sites of known splicing regulators, we used the position-weighted matrices and thresholds as specified by ESEfinder (Cartegni et al. 2003) for the SR proteins SRSF1, SRSF2, SRSF5, and SRSF6. For each of them, we extracted the density, i.e., how many occurrences we find relative to the length of the exon, the average strength of all occurrences, and the maximal strength in each exon. Furthermore, we recorded the distance of the first binding site from the start of the exon as well as the distance of the last binding site to the end of the exon. In addition, we combined all these features with a local secondary structure measurement, i.e., we only accepted binding sites as real, if they were accessible. Accessibilities were calculated using the Vienna RNA Package (Lorenz et al. 2011). The accessibility threshold varies for each SR protein as previously described (Hiller et al. 2007). In addition, we scanned for a set of known binding sites for other regulators, including Nova binding sites (YCAY clusters) (Ule et al. 2006), Fox binding sites (Minovitsky et al. 2005), MBNL binding motifs (Ho et al. 2004), U-rich TIA1/TIAL1 motifs (Aznarez et al. 2008), UG-rich motifs (Faustino and Cooper 2005), splicing repressor sites that bind PTB (Ashiya and Grabowski 1997; Pérez et al. 1997; Oberstrass et al. 2005), and binding motifs of the Quaking protein (Galarneau and Richard 2005).
Extended binding motifs (only used in extended SVMs, Supplemental Fig. 2)
In addition to the aforementioned binding sites for splicing regulators, we extended our set of features by motif clusters identified in Yeo et al. (2007) and additional sequence features as described in Barash et al. (2010) as well as all possible 2mers and 3mers.
Alternative splice sites
To evaluate the potential of alternative splice-site selection around a recorded splice sites, we scanned for putative splice sites in the vicinity of the known splice sites. The number of found potential alternative splice sites as well as their strengths act as further features. To find a reasonable search radius around exon junctions, we evaluated the distance of all pairs of known alternative 3′ and 5′ splice sites in our data set to estimate a distribution of common distances of alternative 3′ and alternative 5′ splice sites. Using a 90% quantile limit of our distance distributions, potential alternative 3′ splice sites and alternative 5′ splice sites were searched within a ±200-nt window around both splice sites, assuring a remaining minimal exon length of 23 nt as well as a remaining minimal intron length of 78 nt. For each splice site, we extracted the total number of potential alternative splice sites found, the density (i.e., the total number of alternative splice sites found normalized by the length of the scanned area), the average strength of all sites and their maximal strength. Values were evaluated for several different MES thresholds.
Phylogenetic conservation
We extracted position-wise conservation of ±50 nt around the exon junctions and calculated the average conservation in each of these four regions (−50 nt of 3′ splice site, +50 nt of 3′ splice site, −50 nt of 5′ splice site, +50 nt of 5′ splice site). Conservation values are PhastCons scores (Siepel et al. 2005) based on a multiple alignment of 46 vertebrate genomes. PhastCons scores are calculated per position and represent probabilities of a nucleotide position to be part of a conserved sequence element. All conservation data were downloaded from the UCSC Genome Browser (Meyer et al. 2013). As previously shown, the conservation differs between constitutive and cassette exons and their flanking introns (Sorek and Ast 2003; Chen and Zheng 2008). Thus, we determined conservation features for the region of ±50 nt around the exon junctions.
For each exon, all evaluated features were concatenated into one vector. Their normalization was done automatically by the support vector package (SMO) used.
Splice-site usage and exon inclusion levels
In an attempt to improve the performance of the splicing code and to better understand differences between exon types, we analyzed the distribution of inclusion and usage levels of cassette exons and alternative splice sites, respectively. We extracted EST-based inclusion and usage levels of exons and splice sites in our training sets, respectively, that are supported by at least 10 ESTs. Exons in each category were sorted based on inclusion/usage levels, which are used as defined in Busch and Hertel (2013). The inclusion level equals the number of ESTs including the exon Cincl divided by the number of ESTs that either included Cincl or excluded Cexcl the exon, . Analogously, the usage level of a splice site equals the number of ESTs in which the exon occurs with the splice site of interest Csplice site of interest divided by the number of ESTs in which the exon occurs with any splice site Csplice site of interest+Cother splice sites,
As mentioned above, cassette exons in our data sets do not have alternative splice sites on either end.
SVM optimization
Based on frequently seen high and low inclusion levels, we carried out SVM optimization experiments using training exons in subgroups: exons showing inclusion levels of 80% or higher, exons included 20% or less, and exons showing intermediate inclusion levels between 20% and 80%. Similarly, alternative splice sites were split into subgroups based on their usage level: 80% or higher usage, 20% or lower usage, and a usage level between 20% and 80%. For most subsequent analyses, we focused on the two extreme groups of levels higher or equal to 80% and lower or equal to 20%.
We used 350 internal exons of each splicing subcategory to train the SVMs. As a quality control, a 10% crossvalidation available during the training process in WEKA was used. Here, 90% of the exons in our data sets were used to train the classifiers, which were then applied to the remaining 10% of the exons to test their performance. To estimate the classification accuracy of the SVMs, we determined the area under the ROC curve (AUC), which plots the true positive rate (TRP) vs. the false positive rate (FPR). The TPR is calculated as the number of true positives (TP) out of the number of positive (P) samples in the test set. The FPR is calculated as the number of false positives (FP) divided by the number of negative (N) samples in the test set. Additionally, we compared the TPR (recall) with the positive predictive value (PPV, precision). While the recall determines how many of the truly positive exons are predicted to be positive (e.g., how many of the cassette exons are predicted to be cassette exons), the precision describes how many of the positive predicted exons are real positives (e.g., how many of the exons predicted to be cassette exons are truly cassette exons). Supplemental Figure 7 illustrates the results of the precision/recall analysis for the data depicted in Figure 2A–C.
We created a general splicing code on mixed-tissue exon data sets based on ESTs. We trained SVMs distinguishing between the following data sets: (i) constitutive and exons with frequently used alternative 3′ splice sites (CO-highALT3), (ii) constitutive and exons with rarely used alternative 3′ splice sites (CO-lowALT3), (iii) constitutive and exons with frequently used alternative 5′ splice sites (CO-highALT5), (iv) constitutive and exons with rarely used alternative 5′ splice sites (CO-lowALT5), (v) exons with frequently used alternative 3′ splice sites and exons with frequently used alternative 5′ splice sites (highALT3–highALT5), (vi) exons with rarely used alternative 3′ splice sites and exons with rarely used alternative 5′ splice sites (lowALT3–lowALT5), (vii) constitutive and highly included cassette exons (CO-highCA), and (viii) constitutive and rarely included cassette exons (CO-lowCA). Even though a SVM typically classifies between two classes, we also applied a modified concept of a SVM to the comparison of all four exon types: (ix) constitutive, highly included cassette, exons with frequently used alternative 3′ splice sites and exons with frequently used alternative 5′ splice sites (highALL), as well as (x) constitutive, rarely included cassette, exons with rarely used alternative 3′ splice sites and exons with rarely used alternative 5′ splice sites (lowALL). Internally, exons of each category are compared with exons of every other category, thus, six two-class SVMs are trained and their results are combined to make a final prediction.
In addition to training the SVMs, we used WEKA (Hall et al. 2009) to evaluate the information gain of each feature with respect to the exon class.
SVM testing and verification
To test the performance of the SVMs derived, we used an RNA-seq data set of HeLa cells (Shepard et al. 2011). Exon inclusion and splice-site usage levels were calculated using MISO, a software that not only takes reads overlapping exon/exon junctions into account, but also reads that are within the exons neighboring the exon of question and mutual to both alternative isoforms (Katz et al. 2010). From each alternative splicing category (exons with an alternative 3′ or 5′ splice site or cassette exons) 3000 exons with at least 20 reads supporting either inclusion or exclusion were chosen randomly, with 250 from each subcategory out of 12 subcategories based on the inclusion/usage level of the exon or the splice site used. Subcategories of alternative splice-site usage or exon inclusion were: inclusion/usage level of 0% (exons or splice sites not used in HeLa cells), (0,10]%, (10,20]%, (20,30]%, (30,40]%, (40,50]%, (50,60]%, (60,70]%, (70,80]%, (80,90]%, (90,100)%, and 100% usage/inclusion. The last category was defined as being constitutive in HeLa cells. For all exons in each of these subsets, we made predictions using the SVMs previously trained on the comparison of constitutive exons and rarely included cassette exons (CO-lowCA) and constitutive exons and exons with a rarely used alternative 3′ or 5′ splice site (CO-lowALT3 or CO-lowALT5, respectively).
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by grants from the National Institutes of Health (NIH) (R01 GM62287 and R01 CA177651 to K.J.H.) and a fellowship within the Postdoc Programme of the German Academic Exchange Service, DAAD (A.B.).
Footnotes
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.048769.114.
REFERENCES
- Ashiya M, Grabowski PJ 1997. A neuron-specific splicing switch mediated by an array of pre-mRNA repressor sites: evidence of a regulatory role for the polypyrimidine tract binding protein and a brain-specific PTB counterpart. RNA 3: 996–1015. [PMC free article] [PubMed] [Google Scholar]
- Aznarez I, Barash Y, Shai O, He D, Zielenski J, Tsui LC, Parkinson J, Frey BJ, Rommens JM, Blencowe BJ 2008. A systematic analysis of intronic sequences downstream of 5′ splice sites reveals a widespread role for U-rich motifs and TIA1/TIAL1 proteins in alternative splicing regulation. Genome Res 18: 1247–1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barash Y, Calarco JA, Gao W, Pan Q, Wang X, Shai O, Blencowe BJ, Frey BJ 2010. Deciphering the splicing code. Nature 465: 53–59. [DOI] [PubMed] [Google Scholar]
- Barash Y, Vaquero-Garcia J, González-Vallinas J, Xiong HY, Gao W, Lee LJ, Frey BJ 2013. AVISPA: a web tool for the prediction and analysis of alternative splicing. Genome Biol 14: R114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbosa-Morais NL, Irimia M, Pan Q, Xiong HY, Gueroussov S, Lee LJ, Slobodeniuc V, Kutter C, Watt S, Colak R, et al. 2012. The evolutionary landscape of alternative splicing in vertebrate species. Science 338: 1587–1593. [DOI] [PubMed] [Google Scholar]
- Bartel F, Harris LC, Würl P, Taubert H 2004. MDM2 and its splice variant messenger RNAs: expression in tumors and down-regulation using antisense oligonucleotides. Mol Cancer Res 2: 29–35. [PubMed] [Google Scholar]
- Berget SM 1995. Exon recognition in vertebrate splicing. J Biol Chem 270: 2411–2414. [DOI] [PubMed] [Google Scholar]
- Black DL 2003. Mechanisms of alternative pre-messenger RNA splicing. Annu Rev Biochem 72: 291–336. [DOI] [PubMed] [Google Scholar]
- Brinkman BM 2004. Splice variants as cancer biomarkers. Clin Biochem 37: 584–594. [DOI] [PubMed] [Google Scholar]
- Busch A, Hertel KJ 2013. HEXEvent: a database of Human EXon splicing Events. Nucleic Acids Res 41: D118–D124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carstens RP, Eaton JV, Krigman HR, Walther PJ, Garcia-Blanco MA 1997. Alternative splicing of fibroblast growth factor receptor 2 (FGF-R2) in human prostate cancer. Oncogene 15: 3059–3065. [DOI] [PubMed] [Google Scholar]
- Cartegni L, Chew SL, Krainer AR 2002. Listening to silence and understanding nonsense: exonic mutations that affect splicing. Nat Rev Genet 3: 285–298. [DOI] [PubMed] [Google Scholar]
- Cartegni L, Wang J, Zhu Z, Zhang MQ, Krainer AR 2003. ESEfinder: a web resource to identify exonic splicing enhancers. Nucleic Acids Res 31: 3568–3571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Zheng S 2008. Identify alternative splicing events based on position-specific evolutionary conservation. PLoS One 3: e2806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark F, Thanaraj TA 2002. Categorization and characterization of transcript-confirmed constitutively and alternatively spliced introns and exons from human. Hum Mol Genet 11: 451–464. [DOI] [PubMed] [Google Scholar]
- Clouet d'Orval B, d'Aubenton Carafa Y, Sirand-Pugnet P, Gallego M, Brody E, Marie J 1991. RNA secondary structure repression of a muscle-specific exon in HeLa cell nuclear extracts. Science 252: 1823–1828. [DOI] [PubMed] [Google Scholar]
- Dror G, Sorek R, Shamir R 2005. Accurate identification of alternatively spliced exons using support vector machine. Bioinformatics 21: 897–901. [DOI] [PubMed] [Google Scholar]
- Eperon LP, Graham IR, Griffiths AD, Eperon IC 1988. Effects of RNA secondary structure on alternative splicing of pre-mRNA: Is folding limited to a region behind the transcribing RNA polymerase? Cell 54: 393–401. [DOI] [PubMed] [Google Scholar]
- Faustino NA, Cooper TA 2003. Pre-mRNA splicing and human disease. Genes Dev 17: 419–437. [DOI] [PubMed] [Google Scholar]
- Faustino NA, Cooper TA 2005. Identification of putative new splicing targets for ETR-3 using sequences identified by systematic evolution of ligands by exponential enrichment. Mol Cell Biol 25: 879–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox-Walsh KL, Hertel KJ 2009. Splice-site pairing is an intrinsically high fidelity process. Proc Natl Acad Sci 106: 1766–1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox-Walsh KL, Dou Y, Lam BJ, Hung SP, Baldi PF, Hertel KJ 2005. The architecture of pre-mRNAs affects mechanisms of splice-site pairing. Proc Natl Acad Sci 102: 16176–16181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galarneau A, Richard S 2005. Target RNA motif and target mRNAs of the Quaking STAR protein. Nat Struct Mol Biol 12: 691–698. [DOI] [PubMed] [Google Scholar]
- Hall M, Frank E, Holmes G, Pfahringer B, Reutemann P, Witten IH 2009. The WEKA data mining software: an update. ACM SIGKDD Explor Newsl 11: 10–18. [Google Scholar]
- Hiller M, Zhang Z, Backofen R, Stamm S 2007. Pre-mRNA secondary structures influence exon recognition. PLoS Genet 3: e204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho TH, Charlet BN, Poulos MG, Singh G, Swanson MS, Cooper TA 2004. Muscleblind proteins regulate alternative splicing. EMBO J 23: 3103–3112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- International Human Genome Sequencing Consortium. 2004. Finishing the euchromatic sequence of the human genome. Nature 431: 931–945. [DOI] [PubMed] [Google Scholar]
- Katz Y, Wang ET, Airoldi EM, Burge CB 2010. Analysis and design of RNA sequencing experiments for identifying isoform regulation. Nat Methods 7: 1009–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornblihtt AR 2005. Promoter usage and alternative splicing. Curr Opin Cell Biol 17: 262–268. [DOI] [PubMed] [Google Scholar]
- Krawczak M, Reiss J, Cooper DN 1992. The mutational spectrum of single base-pair substitutions in mRNA splice junctions of human genes: causes and consequences. Hum Genet 90: 41–54. [DOI] [PubMed] [Google Scholar]
- Lorenz R, Bernhart SH, Höner Zu Siederdissen C, Tafer H, Flamm C, Stadler PF, Hofacker IL 2011. ViennaRNA Package 2.0. Algorithms Mol Biol 6: 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McManus CJ, Graveley BR 2011. RNA structure and the mechanisms of alternative splicing. Curr Opin Genet Dev 21: 373–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercatante DR, Bortner CD, Cidlowski JA, Kole R 2001. Modification of alternative splicing of Bcl-x pre-mRNA in prostate and breast cancer cells. analysis of apoptosis and cell death. J Biol Chem 276: 16411–16417. [DOI] [PubMed] [Google Scholar]
- Meyer LR, Zweig AS, Hinrichs AS, Karolchik D, Kuhn RM, Wong M, Sloan CA, Rosenbloom KR, Roe G, Rhead B, et al. 2013. The UCSC Genome Browser database: extensions and updates 2013. Nucleic Acids Res 41: D64–D69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minovitsky S, Gee SL, Schokrpur S, Dubchak I, Conboy JG 2005. The splicing regulatory element, UGCAUG, is phylogenetically and spatially conserved in introns that flank tissue-specific alternative exons. Nucleic Acids Res 33: 714–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberstrass FC, Auweter SD, Erat M, Hargous Y, Henning A, Wenter P, Reymond L, Amir-Ahmady B, Pitsch S, Black DL, et al. 2005. Structure of PTB bound to RNA: specific binding and implications for splicing regulation. Science 309: 2054–2057. [DOI] [PubMed] [Google Scholar]
- Pan Q, Shai O, Lee LJ, Frey BJ, Blencowe BJ 2008. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat Genet 40: 1413–1415. [DOI] [PubMed] [Google Scholar]
- Pérez I, Lin CH, McAfee JG, Patton JG 1997. Mutation of PTB binding sites causes misregulation of alternative 3′ splice site selection in vivo. RNA 3: 764–778. [PMC free article] [PubMed] [Google Scholar]
- Pickrell JK, Pai AA, Gilad Y, Pritchard JK 2010. Noisy splicing drives mRNA isoform diversity in human cells. PLoS Genet 6: e1001236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platt JC 1999. Fast training of support vector machines using sequential minimal optimization. MIT Press, Cambridge, MA. [Google Scholar]
- Shepard PJ, Hertel KJ 2008. Conserved RNA secondary structures promote alternative splicing. RNA 14: 1463–1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepard PJ, Hertel KJ 2010. Embracing the complexity of pre-mRNA splicing. Cell Res 20: 866–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepard PJ, Choi EA, Busch A, Hertel KJ 2011. Efficient internal exon recognition depends on near equal contributions from the 3′ and 5′ splice sites. Nucleic Acids Res 39: 8928–8937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siepel A, Bejerano G, Pedersen JS, Hinrichs AS, Hou M, Rosenbloom K, Clawson H, Spieth J, Hillier LW, Richards S, et al. 2005. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res 15: 1034–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorek R, Ast G 2003. Intronic sequences flanking alternatively spliced exons are conserved between human and mouse. Genome Res 13: 1631–1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorek R, Shemesh R, Cohen Y, Basechess O, Ast G, Shamir R 2004. A non-EST-based method for exon-skipping prediction. Genome Res 14: 1617–1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamm S, Zhu J, Nakai K, Stoilov P, Stoss O, Zhang MQ 2000. An alternative-exon database and its statistical analysis. DNA Cell Biol 19: 739–756. [DOI] [PubMed] [Google Scholar]
- Ule J, Stefani G, Mele A, Ruggiu M, Wang X, Taneri B, Gaasterland T, Blencowe BJ, Darnell RB 2006. An RNA map predicting Nova-dependent splicing regulation. Nature 444: 580–586. [DOI] [PubMed] [Google Scholar]
- Wang Z, Lo HS, Yang H, Gere S, Hu Y, Buetow KH, Lee MP 2003. Computational analysis and experimental validation of tumor-associated alternative RNA splicing in human cancer. Cancer Res 63: 655–657. [PubMed] [Google Scholar]
- Wang ET, Sandberg R, Luo S, Khrebtukova I, Zhang L, Mayr C, Kingsmore SF, Schroth GP, Burge CB 2008. Alternative isoform regulation in human tissue transcriptomes. Nature 456: 470–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong HY, Barash Y, Frey BJ 2011. Bayesian prediction of tissue-regulated splicing using RNA sequence and cellular context. Bioinformatics 27: 2554–2562. [DOI] [PubMed] [Google Scholar]
- Xu Q, Lee C 2003. Discovery of novel splice forms and functional analysis of cancer-specific alternative splicing in human expressed sequences. Nucleic Acids Res 31: 5635–5643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeo G, Burge CB 2004. Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J Comput Biol 11: 377–394. [DOI] [PubMed] [Google Scholar]
- Yeo GW, Van Nostrand EL, Liang TY 2007. Discovery and analysis of evolutionarily conserved intronic splicing regulatory elements. PLoS Genet 3: e85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng CL, Fu XD, Gribskov M 2005. Characteristics and regulatory elements defining constitutive splicing and different modes of alternative splicing in human and mouse. RNA 11: 1777–1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.