

Abstract
The proteome of the amoebo-flagellate protozoan Naegleria gruberi is rich in candidate RNA repair enzymes, including 15 putative RNA ligases, one of which, NgrRnl, is a eukaryal homolog of Deinococcus radiodurans RNA ligase, DraRnl. Here we report that purified recombinant NgrRnl seals nicked 3′-OH/5′-PO4 duplexes in which the 3′-OH strand is RNA. It does so via the “classic” ligase pathway, entailing reaction with ATP to form a covalent NgrRnl–AMP intermediate, transfer of AMP to the nick 5′-PO4, and attack of the RNA 3′-OH on the adenylylated nick to form a 3′–5′ phosphodiester. Unlike members of the four known families of ATP-dependent RNA ligases, NgrRnl lacks a carboxy-terminal appendage to its nucleotidyltransferase domain. Instead, it contains a defining amino-terminal domain that we show is important for 3′-OH/5′-PO4 nick-sealing and ligase adenylylation, but dispensable for phosphodiester synthesis at a preadenylylated nick. We propose that NgrRnl, DraRnl, and their homologs from diverse bacteria, viruses, and unicellular eukarya comprise a new “Rnl5 family” of nick-sealing ligases with a signature domain organization.
Keywords: RNA repair, adenylyltransferase
INTRODUCTION
RNA ligases rectify RNA breaks, either as a means of reversing the effects of RNA damage or in the service of rearranging RNA primary structure. The classic ATP-dependent RNA ligases join 3′-OH and 5′-PO4 ends via three sequential nucleotidyl transfer steps, similar to those of DNA ligases. First, RNA ligase reacts with ATP to form a covalent ligase-(lysyl-N)–AMP intermediate plus pyrophosphate. Second, AMP is transferred from ligase-adenylate to the 5′-PO4 RNA end to form an RNA-adenylate intermediate (AppRNA). Third, ligase catalyzes attack by an RNA 3′-OH on the RNA-adenylate to seal the two ends via a 3′–5′ phosphodiester bond and release AMP.
The step 1 autoadenylylation reaction of RNA ligases is performed by a nucleotidyltransferase (NTase) domain that is conserved in ATP-dependent and NAD+-dependent DNA ligases and GTP-dependent RNA capping enzymes (Shuman and Lima 2004). The NTase domain includes six conserved peptide motifs (I, Ia, III, IIIa, IV, and V) that form the nucleotide-binding pocket. Motif I includes the lysine that becomes covalently attached to the NMP. It is proposed that RNA and DNA ligases and RNA capping enzymes evolved from an ancestral stand-alone NTase domain (Ho et al. 2004) via clade-specific acquisitions of flanking domain modules. For most RNA ligases, substrate specificity and dedication to a particular biological pathway of RNA sealing have evolved by fusions of the NTase domain to structurally diverse carboxy-terminal domain modules.
There are presently four structurally characterized RNA ligase families, exemplified by the following: bacteriophage T4 RNA ligase 1 (Rnl1 family) (El Omari et al. 2006); T4 RNA ligase 2 and Trypanosome REL1 (Rnl2 family) (Nandakumar et al. 2006); Pyrococccus abyssi RNA ligase (Rnl3 family) (Brooks et al. 2008); and Clostridium thermocellum RNA ligase (Rnl4 family) (Smith et al. 2012; Wang et al. 2012). The physiology and biochemistry of the Rnl families suggest a division of labor in RNA repair, whereby Rnl1 and Rnl4 ligases are tailored to seal single-strand breaks in the loop of RNA stem–loops (Amitsur et al. 1987; Wang et al. 2007; Nandakumar et al. 2008; Zhang et al. 2012), whereas Rnl2 ligases are designed to seal 3′-OH/5′-PO4 nicks in duplex RNAs and/or RNA:DNA hybrids (Blanc et al. 1999; Nandakumar et al. 2004; Nandakumar and Shuman 2004; Chauleau and Shuman 2013).
DraRnl, an ATP-dependent nick-sealing RNA ligase encoded by the bacterium Deinococcus radiodurans (Martins and Shuman 2004), does not belong to any of the established ligase families. The distinctive features of the DraRnl protein are that it consists of a carboxy-terminal NTase domain fused to a unique N-terminal module (Fig. 1A). DraRnl has been characterized biochemically (Martins and Shuman 2004; Raymond and Shuman 2007; Schmier and Shuman 2014) but its structure is tabula rasa.
FIGURE 1.
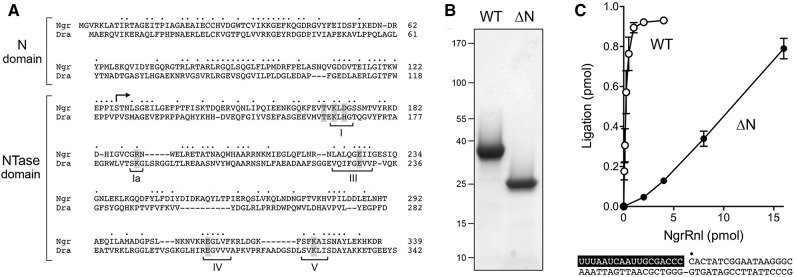
Recombinant NgrRnl and NgrRnl-▵N. (A) The amino acid sequence of NgrRnl (genbank accession XP_002669268.1) is aligned to that of DraRnl (accession NP_051627.1). Positions of amino acid side chain identity/similarity are denoted by dots above the NgrRnl sequence. Gaps in the alignment are indicated by dashes. The N domain and the nucleotidyltransferase (NTase) domain are indicated by brackets at left. NTase motifs I, Ia, III, IV, and V are indicated by brackets below the DraRnl sequence. Amino acids identified previously as essential for the nick-sealing activity of DraRnl are shaded gray. The translation start site of the truncated derivative NgrRnl-▵N at position 127 is indicated by the arrowhead above the NgrRnl sequence. (B) Aliquots (3 µg) of recombinant wild-type (WT) NgrRnl and NgrRnl-▵N were analyzed by SDS PAGE. The Coomassie blue-stained gel is shown. The positions and sizes (kDa) of marker polypeptide are indicated at left. (C) Nick-sealing reaction mixtures (10 µL) containing 50 mM Tris–acetate (pH 6.0), 5 mM MnCl2, 0.2 mM ATP, 5 mM DTT, 1 pmol (0.1 μM) of 32P-labeled nicked duplex (shown at the bottom, with the 32P label at the nick denoted by • and the RNAOH strand depicted in white on a black background), and WT NgrRnl or NgrRnl-▵N as specified were incubated for 30 min at 37°C. The products were analyzed by urea-PAGE. The extent of nick sealing, quantified by scanning the gel, is plotted as a function of input NgrRnl. Each datum is the average of three independent protein titration experiments ±SEM.
Homologs of DraRnl are encoded by 35 bacterial genera, representing 10 different phyla (Schmier and Shuman 2014). Bacteriophages that prey on Aeromonas, Caulobacter, Mycobacterium, and Sinorhizobium also encode homologs of DraRnl. An archaeal homolog is present in Methanobrevibacter ruminantium. We are especially intrigued by the presence of DraRnl homologs in many unicellular eukarya, including 13 genera of fungi, the amoebae Dictyostelium and Polysphondylium, and the amoebo-flagellate Naegleria.
Our aim in the present study was to biochemically characterize a eukaryal member of the DraRnl-like clade, specifically the homolog encoded by Naegleria gruberi. Naegleria are a genus of free-living fresh-water protozoa that have a complex life cycle whereby they are able to change rapidly from amoebal to flagellate morphology. A member of this taxon, N. fowleri (the “brain-eating amoeba”) is a virulent human pathogen, causing a devastating brain infection, primary amoebic meningoencephalitis. The 41 Mb genome of the avirulent species N. gruberi embraces 15,727 predicted protein-coding genes (Fritz-Laylin et al. 2010). To our inspection, the N. gruberi proteome is unusually rich in candidate RNA repair enzymes, including 13 putative ATP-dependent RNA ligases, seven of which belong to the Rnl1 family and five of which belong to the Rnl2 family. N. gruberi encodes a single homolog of DraRnl (Genbank XP_002669268.1), a 339-aa polypeptide that we will refer to henceforth as NgrRnl. Alignment of the NgrRnl and DraRnl polypeptides highlights conservation of both the clade-defining N domain and the carboxy-terminal NTase domain, to the extent of 82 positions of amino acid identity and 46 positions of side chain similarity (Fig. 1A). The NTase domain of NgrRnl includes readily identifiable counterparts of motifs I, Ia, III, VI, and V (Fig. 1A).
Here we ask (and answer) the following questions: (i) Is NgrRnl a bona fide RNA ligase? (Yes); (ii) What is its nucleic acid substrate preference? (Nicked duplex nucleic acid with a 3′-OH RNA strand); and (iii) Is the N-domain functionally relevant (Yes). Anent the latter issue, we have shown that the N-domain is important for 3′-OH/5′-PO4 nick sealing and ligase adenylylation, but is dispensable for nick sealing at a preadenylylated nick. Based on our findings, we propose that DraRnl, NgrRnl, and their many homologs comprise a new and widely distributed “Rnl5 family” of nick-sealing RNA ligases with a signature domain organization.
RESULTS
NgrRnl is a 3′-OH/5′-PO4 nick-sealing ligase
We produced recombinant full-length wild-type NgrRnl in E. coli as a His10Smt3·NgrRnl fusion and isolated it from a soluble bacterial extract by Ni-affinity chromatography. After cleaving the tag during overnight dialysis in the presence of the Smt3 protease Ulp1, we recovered tag-free NgrRnl in the flow-through fraction during a second Ni-affinity step. After subsequent steps of anion-exchange chromatography and gel filtration, during which NgrRnl eluted as a monomer, the preparation comprised a predominant ∼38 kDa polypeptide (Fig. 1B). Reaction of NgrRnl with a nicked duplex substrate, composed of an 18-mer 5′ 32P-labeled DNA strand and an unlabeled 18-mer 3′-OH RNA strand annealed to a 36-mer DNA strand (shown in Fig. 1C, with the nucleotides of the RNAOH strand lettered in white against a black background), in the presence of ATP and Mn2+ resulted in the formation of a 32P-labeled 36-mer RNApDNA strand that was resolved from the input 32P-labeled 18-mer DNA by urea-PAGE (e.g., see Fig. 2C). The extent of ligation was proportional to input NgrRnl and proceeded to >90% yield at saturating enzyme (Fig. 1C). In parallel, we produced and purified a truncated version, NgrRnl-▵N, that lacks the amino-terminal 126-aa segment (Fig. 1B). The specific activity of NgrRnl-▵N in nick sealing was 4% of the full-length wild-type NgrRnl (Fig. 1C).
FIGURE 2.

NgrRnl is a nick-sealing RNA ligase. (A) Aliquots (10 μg) of purified recombinant wild-type NgrRnl with motif I Lys170, or mutated versions in which Lys170 was replaced by Ala, Met, Gln, or His, were analyzed by SDS-PAGE. The Coomassie blue-stained gel is shown. The positions and sizes (kDa) of marker polypeptides are indicated on the left. (B) Nick-sealing activity requires Lys170. Reaction mixtures (10 μL) containing 50 mM Tris–acetate (pH 6.0), 5 mM MnCl2, 0.2 mM ATP, 5 mM DTT, 1 pmol (0.1 μM) of 32P-labeled nicked duplex (shown at the bottom, with the 32P label at the nick denoted by • and the RNAOH strand depicted in white on a black background), and either no NgrRnl (−) or 5 pmol of NgrRnl as specified were incubated for 30 min at 37°C. The products were resolved by urea-PAGE and visualized by autoradiography. (C) Requirement for RNA on the 3′-OH side of the nick. Reaction mixtures (10 μL) containing 50 mM Tris–acetate (pH 6.0), 5 mM MnCl2, 0.2 mM ATP, 5 mM DTT, 1 pmol (0.1 μM) of 32P-labeled nicked duplex as specified (the structures of the four substrates is illustrated at the bottom, with the 32P label at the nick denoted by • and RNA strands depicted in white on a black background), and either no enzyme (−) or 5 pmol NgrRnl (+) were incubated for 30 min at 37°C. (D) Reaction mixture containing 50 mM Tris–acetate (pH 6.0), 5 mM MnCl2, 0.2 mM ATP, 5 mM DTT, 1 pmol (0.1 µM) 32P-labeled nicked duplex containing 2, 3, or 12 ribonucleotides (in white lettering on a black background), and NgrRnl as specified were incubated for 30 min at 37°C.
NgrRnl NTase motif I 170KLDG embraces the predicted lysine nucleophile for adenylate transfer (Fig. 1A). We replaced Lys170 with alanine, methionine, glutamine, or histidine in the context of the full-length polypeptide (Fig. 2A). All of the mutants were inert in 3′-OH/5′-PO4 nick sealing (Fig. 2B).
A 3′-OH RNA strand is required for nick sealing
Ligase activity was tested with a series of singly nicked duplexes shown in Figure 2C, each 32P-labeled at the nick 5′-PO4, in which (i) the 3′-OH and 5′-PO4 strands were both RNA; (ii) the 3′-OH strand was RNA and the 5′-PO4 strand was DNA; (iii) the 3′-OH strand was DNA and the 5′-PO4 strand was RNA; or (iv) the 3′-OH and 5′-PO4 strands were both DNA. Whereas NgrRnl effected near-quantitative sealing of the two nicked duplexes in which the 3′-OH strand was RNA, it was unable to seal either of the two nicked duplexes in which the 3′-OH strand was DNA (Fig. 2C). The requirement by NgrRnl for a nicked duplex with a 3′-OH RNA strand, and its acceptance of either DNA or RNA in the 5′-PO4 strand, echoes the substrate specificities of DraRnl and T4 Rnl2 (Martins and Shuman 2004; Nandakumar et al. 2004).
We then compared variants of the nicked duplex substrate in which the 5′-PO4 and template strands were DNA and the 3′-OH strand consisted of 18 ribonucleotides, 15 deoxynucleotides and 3 ribonucleotides, or 16 deoxynucleotides and 2 ribonucleotides (Fig. 2D). Enzyme titration experiments showed that ligase specific activity was reduced sharply and progressively when the RNA content was limited to three or two nucleotides at the nick 3′-OH end (Fig. 2D). Whereas a similar trend was noted for DraRnl when RNA content was reduced (Martins and Shuman 2004), T4 Rnl2 remained fully active when the 3′-OH strand included only two ribonucleotides at the nick (Nandakumar and Shuman 2004).
All further characterization of the 3′-OH/5′-PO4 sealing reaction was performed with the nicked duplex substrate composed of a 3′-OH RNA strand, a 5′-PO4 DNA strand and a DNA template strand. Nick sealing was strictly dependent on a divalent cation cofactor (Fig. 3A, lane –). When various metals were tested at 5 mM concentration, manganese was the preferred cofactor. Magnesium also supported nick sealing, albeit with some accumulation of the AppDNA reaction intermediate, which migrated immediately above the 32P-labeled pDNA strand during urea-PAGE (Fig. 3A). Cobalt was less effective, while cadmium and nickel were feeble activators of ligation. Calcium permitted AppDNA formation, without subsequent ligation. Copper and zinc were inert. Divalent cation titrations (Fig. 3B) revealed that whereas nick sealing was optimal at 2.5 mM Mn2+ or 5 mM Mg2+, the adenylated nicked intermediate (RNAOH/AppDNA) was virtually the sole species formed at limiting metal concentrations, e.g., 0.31 mM Mn2+ and 0.62 mM Mg2+. Based on the results in Figure 3B, we routinely assayed nick sealing by NgrRnl in the presence of 5 mM manganese. Nick sealing was found to be optimal at pH 5.5 to 6.5 and declined sharply at pH ≥8.5 (Fig. 3C). The adenylylated nicked species accumulated as the sole reaction product at pH 4.5 (Fig. 3C). Omission of ATP did not efface nick-sealing activity (data not shown), reflecting the presence of preformed NgrRnl–AMP in the recombinant protein preparation. By plotting the extent of ATP-independent nick sealing as a function in input NgrRnl, we estimated that active NgrRnl–AMP comprised 50% of the enzyme preparation (data not shown).
FIGURE 3.
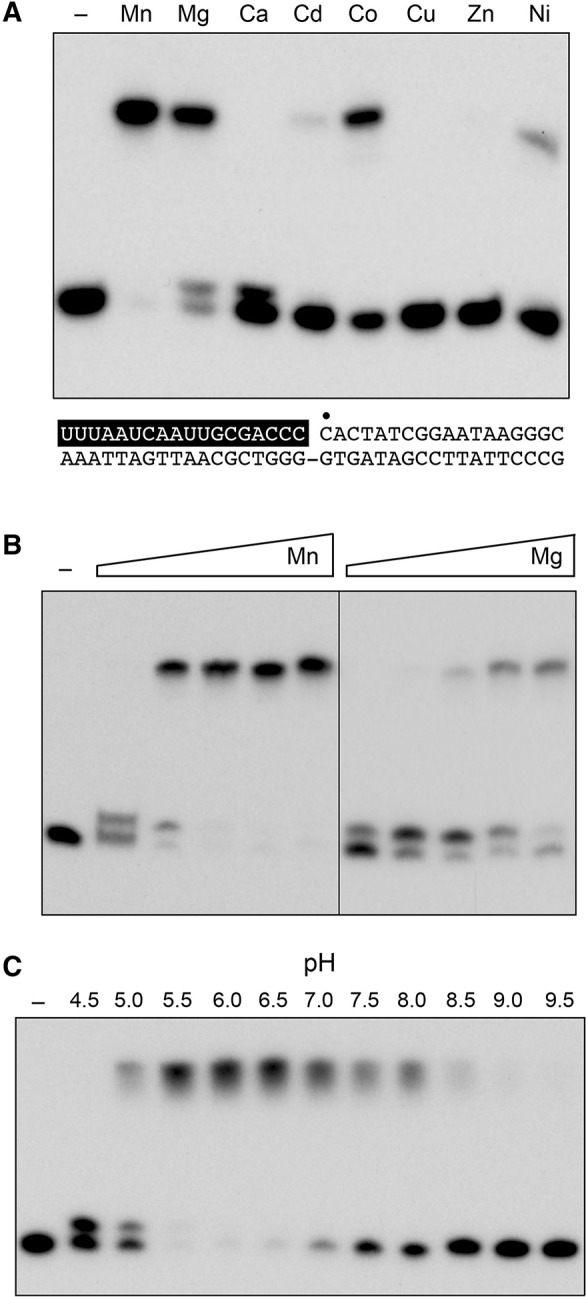
Optimal reaction conditions for nick sealing. (A) Divalent cation requirement. Reaction mixtures (10 μL) containing 50 mM Tris–acetate (pH 6.0), 0.2 mM ATP, 1 pmol (0.1 μM) of 32P-labeled nicked duplex (shown at the bottom, with the 32P label at the nick denoted by • and the RNAOH strand depicted in white on a black background), 1 pmol NgrRnl, and 5 mM of the indicated divalent cation (as the chloride salt) were incubated for 30 min at 37°C. Divalent cation was omitted from a control reaction in lane –. The products were resolved by urea-PAGE and visualized by autoradiography. (B) Divalent cation concentration dependence. Reaction mixtures (10 μL) containing 50 mM Tris–acetate (pH 6.0), 0.2 mM ATP, 1 pmol (0.1 μM) of 32P-labeled nicked duplex, 1 pmol NgrRnl, and increasing concentrations of MgCl2 or MnCl2 (0.31, 0.62, 1.25, 2.5, and 5 mM, proceeding from left to right) were incubated for 30 min at 37°C. Divalent cation was omitted from a control reaction in lane –. (C) pH profile. Reaction mixtures (10 μL) containing 50 mM Tris buffer (either Tris–acetate pH 4.5, 5.0, 5.5, 6.0, 6.5, and 7.0 or Tris–HCl pH 7.5, 8.0 8.5, 9.0, and 9.5), 5 mM MnCl2, 0.2 mM ATP, 1 pmol (0.1 μM) of 32P-labeled nicked duplex, and 1 pmol NgrRnl, were incubated for 30 min at 37°C. NgRnl was omitted from a control reaction in lane –.
Sealing of single-strand RNA
NgrRnl was comparatively feeble at ligating a 5′ 32P-labeled single-strand 18-mer pRNA substrate (the same labeled pRNA strand that was efficiently ligated when annealed to form a nicked duplex in Fig. 1). In the presence of ATP and Mn2+, a 10-fold molar excess of NgrRnl over pRNA converted 26% of the input 18-mer to AppRNA intermediate and 13% of the input 18-mer to an RNA circle (via intramolecular end joining). A trace amount of intermolecular sealing generated a dimer RNA product (Supplemental Fig. S1). No products were formed in the absence of a divalent cation. In the presence of Mg2+, NgrRnl converted 16% of the 18-mer pRNA to AppRNA, without subsequent end-joining (Supplemental Fig. S1). In contrast, virtually all of the input pRNA single-strand reacted with T4 Rnl1 in the presence of Mg2+. NgrRnl was unreactive with a 5′ 32P-labeled single-strand 18-mer pDNA substrate of identical nucleobase sequence in the presence of Mn2+ or Mg2+ (Supplemental Fig. S1).
Ligase adenylylation
Reaction of NgrRnl with [α-32P]ATP and 5 mM magnesium or manganese yielded a covalent enzyme-[32P]AMP adduct detectable by SDS-PAGE (Fig. 4A). Calcium, cobalt, copper, cadmium, nickel, and zinc were ineffective (Fig. 4A). NgrRnl–AMP formation was ablated by mutating Lys170 to Ala, Met, Gln, or His (Fig. 4B), consistent with motif I Lys170 being the active site nucleophile. Ligase adenylylation was optimal at pH 5.5–6.5 (Fig. 4C). The yield of NgrRnl–AMP was proportional to input NgrRnl (Fig. 4D). From the slope of the titration curve, we estimated that 50% of the NgrRnl protein was labeled in vitro with [32P]AMP. The extent of ligase adenylylation increased steadily with reaction time up to 20 min and plateaued thereafter (data not shown). The slowness of the ligase adenylylation step might account for the relatively low turnover number in nick sealing in the linear range of the enzyme titration experiments (e.g., ∼3 fmol of nicks sealed per fmol of NgrRnl) (Fig. 1C).
FIGURE 4.
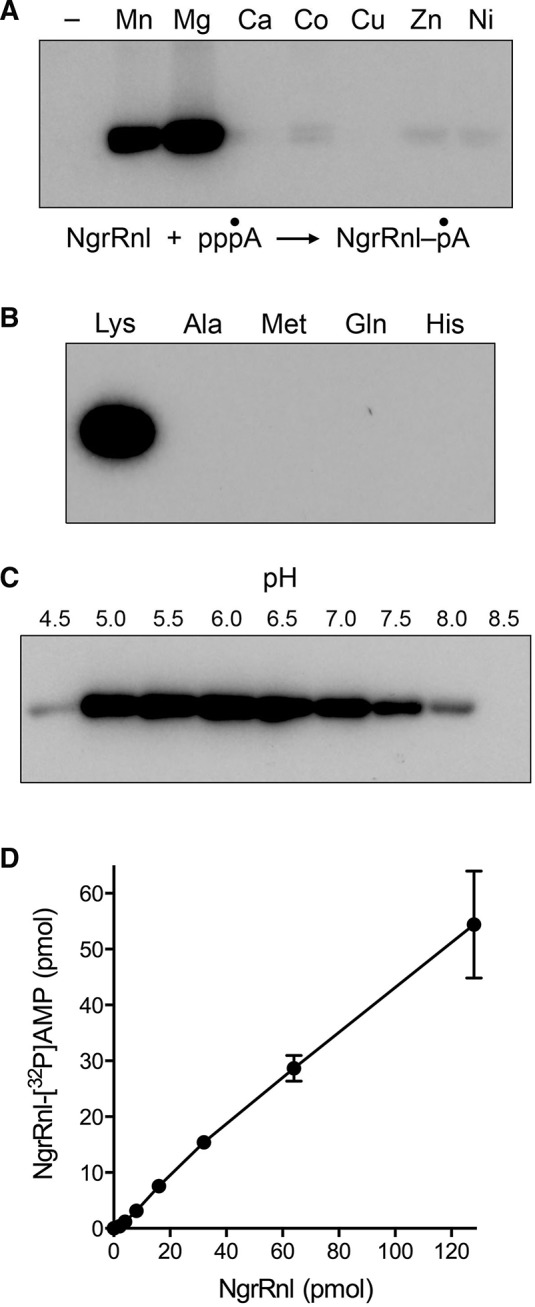
Ligase adenylylation. (A) Divalent cation requirement. Reaction mixtures (10 μL) containing 50 mM Tris–acetate (pH 6.0), 50 μM [α-32P]ATP, 100 pmol NgrRnl, and 5 mM of the indicated divalent cation (as the chloride salt) were incubated for 30 min at 37°C. Divalent cation was omitted from a control reaction in lane –. The products were resolved by SDS-PAGE and visualized by autoradiography. (B) Requirement for Lys170. Reaction mixtures (10 μL) containing 50 mM Tris–acetate (pH 6.0), 50 μM [α-32P]ATP, 5 mM MgCl2, and 100 pmol wild-type NgrRnl (with motif I Lys170), or mutated versions in which Lys170 was replaced by Ala, Met, Gln, or His, were incubated for 30 min at 37°C. (C) pH profile. Reaction mixtures (10 μL) containing 50 mM Tris buffer (either Tris–acetate pH 4.5, 5.0, 5.5, 6.0, 6.5, and 7.0 or Tris–HCl pH 7.5, 8.0 and 8.5), 50 μM [α-32P]ATP, and 5 mM MgCl2, and 100 pmol NgrRnl were incubated for 30 min at 37°C. (D) NgrRnl titration. Reaction mixtures (10 μL) containing 50 mM Tris–acetate (pH 6.0), 50 μM [α-32P]ATP, and 5 mM MgCl2, and NgrRnl as specified were incubated for 30 min at 37°C. The extent of covalent NgrRnl–[32P]AMP adduct formation is plotted as a function of input NgrRnl. Each datum is the average of three independent NgrRnl titration experiments ±SEM.
The NgrRnl-▵N mutant was inactive in covalent adenylylation in vitro under the reactions conditions that were optimal for full-length NgrRnl. Variations in reaction conditions showed that subtraction of the N domain caused an alkaline shift in the pH profile of the NgrRnl-▵N adenylylation reaction, which was optimal at pH 9.0 (Supplemental Fig. S2). An analogous alkaline shift in the adenylyltransferase pH profile was noted for T4 Rnl2 when its signature carboxy-terminal domain was deleted (Ho et al. 2004). Note that the extent of NgrRnl-▵N–[32P]AMP formation at its optimum of pH 9.0 was only 4% of the input enzyme, in contrast to 50% adenylylation of NgrRnl at its optimum of pH 6.0. We conclude that removing the N domain of NgrRnl cripples the composite nick-sealing reaction and, particularly, step 1 lysine adenylylation.
The N domain is dispensable for phosphodiester synthesis at a preadenylylated nick
The third step of the classic ligation reaction pathway (phosphodiester formation) can be studied in isolation by assaying the sealing of a preadenylylated nicked duplex. The preadenylylated substrate was composed of an unlabeled 18-mer RNA 3′-OH strand and a 5′ 32P-labeled 18-mer AppDNA strand annealed to a complementary 36-mer DNA template (Fig. 5C). The AppDNA strand was radiochemically pure and well resolved from the pDNA species by urea-PAGE (Fig. 5A). Reaction of the preadenylylated nicked duplex with NgrRnl-▵N and manganese in the absence of ATP resulted in near-quantitative joining of the RNAOH and AppDNA strands to form an RNApDNA product (Fig. 5A). (There was no detectable conversion of AppDNA to pDNA, signifying that the forward step 3 phosphodiester synthesis reaction was strongly favored over the reverse step 2 reaction on this substrate.) When we compared by enzyme titration the AppDNA nick-sealing activities of the full-length NgrRnl and the ▵N truncation, we were surprised to find that the specific activity of ▵N was 10-fold greater than that of wild-type NgrRnl (Fig. 5C). Taking into account that half of the wild-type NgrRnl preparation is NgrRnl–AMP, and that only the apoenzyme form of the ligase can bind and seal a preadenylylated substrate, we surmise that the ▵N protein is fivefold better than the full-length ligase in joining the RNAOH and AppDNA strands.
FIGURE 5.

The N domain is dispensable for sealing a preadenylylated nick. (A) Reaction mixtures (10 μL) containing 50 mM Tris–acetate (pH 6.0), 5 mM DTT, 5 mM MnCl2, 1 pmol (0.1 μM) of 32P-labeled preadenylylated nicked duplex substrates (depicted in C with the RNA strand lettered in white on a black background), and either no enzyme (lanes –) or 10 pmol of NgrRnl-▵N (lanes +) were incubated for 30 min at 37°C. The products were resolved by urea-PAGE (through a 40-cm gel) and visualized by autoradiography. (B) Reaction mixtures (10 µL) containing 50 mM Tris–acetate (pH 6.0), 5 mM MnCl2, 5 mM DTT, 1 pmol (0.1 μM) 32P-labeled preadenylylated nicked duplex with 3′-OH RNA strand, and 0.5, 1, 2, 4, or 8 pmol wild-type (WT) NgrRnl or NgrRnl-▵N were incubated for 30 min at 37°C. The extent of nick sealing, quantified by scanning the gel, is plotted as a function of input NgrRnl. Each datum is the average of three independent protein titration experiments ±SEM. (D) Divalent cation requirement. Reaction mixtures (10 μL) containing 50 mM Tris–acetate (pH 6.0), 1 pmol (0.1 μM) 32P-labeled preadenylylated nicked duplex with 3′-OH RNA strand, 1 pmol NgrRnl-▵N, and 5 mM of the indicated divalent cation (as the chloride salt) were incubated for 30 min at 37°C. Divalent cation was omitted from a control reaction in lane –. (E) pH profile. Reaction mixtures (10 μL) containing 50 mM Tris buffer (either Tris–acetate pH 4.5, 5.0, 5.5, 6.0, 6.5, 7.0 or Tris–HCl pH 7.5, 8.0 8.5, 9.0, 9.5), 5 mM MnCl2, 1 pmol (0.1 μM) 32P-labeled preadenylylated nicked duplex with 3′-OH RNA strand, and 1 pmol NgrRnl-▵N, were incubated for 30 min at 37°C. The products were resolved by urea-PAGE and visualized by autoradiography.
We considered the prospect that the inability of NgrRnl to seal a nick composed of 3′-OH and 5′-PO4 DNA strands (Fig. 2C) might reflect a need for a 3′-OH RNA strand exclusively during step 2 transfer of AMP from ligase-AMP to the nick 5′-PO4 to form AppDNA. If that were the case, then we might be able to bypass the 3′-OH RNA requirement by preadenylylating the 5′-PO4 of an all-DNA nick. To test this idea, we prepared a preadenylylated nicked duplex with a 3′-OH DNA strand (Fig. 5C). There was barely detectable joining of the DNAOH strand to the AppDNA strand by NgrRnl-▵N under the same conditions that allowed for full RNAOH to AppDNA ligation (Fig. 5A). We conclude that a 3′-OH RNA nick is critical for both step 2 and step 3 of the NgrRnl ligation pathway. We did detect a low extent of step 2 reversal, manifest by formation of radiolabeled pDNA, when ▵N was reacted with the all-DNA preadenylylated nick (Fig. 5A).
Sealing of the RNAOH and AppDNA strands by NgrRnl-▵N depended on a divalent cation, which could be either manganese or magnesium (Fig. 5D). Cobalt, zinc, and nickel were feeble cofactors for step 3 sealing; calcium and copper were inert (Fig. 5D). Phosphodiester synthesis was optimum at pH 5.5–6.5 and declined sharply at pH <5.0 or >8.0 (Fig. 5E).
DISCUSSION
The present study consolidates the existence and properties of a distinct family of RNA ligases, exemplified by eukaryal NgrRnl and bacterial DraRnl, that seal nicked 3′-OH/5′-PO4 duplexes in which the 3′-OH strand is RNA. We suggest that this family be designated henceforth as Rnl5. Although biochemically similar to the Rnl2 family ligases that also repair broken RNA nicks (e.g., T4 Rnl2 and kinetoplastid RELs), the Rnl5 clade has a unique domain organization. Specifically, it lacks a carboxy-terminal appendage to its NTase domain, and instead contains a defining N-terminal domain that we find is important for 3′-OH/5′-PO4 nick sealing and ligase adenylylation, but not for phosphodiester synthesis at a preadenylylated nick. The capacity of the isolated NTase domain of NgRnl to catalyze step 3 of the ligation pathway echoes analogous findings that T4 Rnl2 retains step 3 activity when its signature carboxy-terminal domain is deleted (Ho et al. 2004; Nandakumar and Shuman 2005).
Our observations that NgrRnl lacking the N domain is fivefold better than the full-length enzyme at phosphodiester synthesis on a preadenylylated substrate accords with the twofold improvement in RNAOH to AppDNA sealing by DraRnl when its N-domain was deleted (Raymond and Shuman 2007). We conclude that the signature Rnl5 family N-domain is an impediment to step 3 catalysis on an exogenous preadenylylated nick. This is analogous to the situation with T4 Rnl2, whereby deletion of its carboxy-terminal domain elicits a 10-fold gain of function in RNAOH/AppDNA sealing (Nandakumar and Shuman 2005).
The RNA specificity of the eukaryal Rnl5 enzyme NgrRnl is limited to the 3′-OH strand of the nicked duplex, implying that NgrRnl either strictly requires A-form helical conformation on the 3′-OH side of the nick; or requires a ribose 2′-OH at the nick for catalysis of steps 2 and 3 of the pathway. Our inability to bypass the RNAOH requirement by preadenylylating an all-DNA nick indicates that the 3′-OH RNA strand is required for step 3. Moreover, because this experiment was performed with the ▵N protein, we conclude that RNAOH specificity is inherent in the NTase domain of NgrRnl.
NgrRnl is the first eukaryal ligase of the Rnl5 family to be characterized biochemically. Rnl5-type ligases are also found in the proteomes of several other protozoa and of fungi. Although the physiological substrates that are repaired by Rnl5 enzymes are not known, their predilection for sealing nicks with RNAOH ends in vitro has prompted speculation that they (as well as Rnl2-family nick-sealing enzymes) could be involved either in the repair of guide-strand directed RNA damage (requiring the joining of RNAOH and pRNA ends) or of DNA damage in situations where single-strand gaps are filled in with a short RNA patch by a repair polymerase (entailing the joining of RNAOH and pDNA ends) (Schmier and Shuman 2014).
Our focus on the Rnl5-type enzyme from Naegleria is motivated in part by the remarkably rich “menu” of candidate RNA sealing enzymes in this organism's proteome, which dwarfs that of any other eukaryal model system. For example, the yeasts Saccharomyces cerevisiae and Kluyveromyces lactis and the plant Arabidopsis thaliana each have one ATP-dependent RNA ligase, thought to belong to the Rnl1 family (Wang and Shuman 2005; Nandakumar et al. 2008; Remus and Shuman 2013, 2014), that performs both tRNA splicing and nonspliceosomal mRNA splicing in the unfolded protein response (Sidrauski et al. 1996). A structurally and biochemically similar Rnl1-like tRNA ligase is found in the chordate Branchiostoma floridae (Englert et al. 2010). (The signature biochemical feature of the yeast, plant, and Branchiostoma tRNA ligases is their requirement for a terminal 2′-PO4 group on the 3′-OH RNA strand.) In contrast, N. gruberi encodes seven different Rnl1 family members (Genbank accessions: EFC42678.1, EFC38327.1, EFC38820.1, EFC38271.1, EFC38139.1, EFC37058.1, EFC41136.1) and five different Rnl2 family members (Genbank accessions: XP_002679917.1, XP_002679286.1, XP_002672691.1, XP_002676530.1, XP_002669886.1) in addition to the Rnl5 enzyme NgrRnl. As if 13 classic RNA ligases were not enough, N. gruberi also encodes two noncanonical RNA ligases of the RtcB family (Genbank accessions: EFC45815.1, EFC42397.1). RtcB is a GTP-dependent ligase that joins RNA 3′-PO4 and 5′-OH ends via covalent RtcB-(histinyl-N)–GMP and RNA3pp5G intermediates (Chakravarty et al. 2012). Mammals and the nematode Caenorhabditis elegans encode a single RtcB that is implicated in tRNA splicing and the unfolded protein response (Jurkin et al. 2014; Kosmaczewski et al. 2014). It is attractive to think that the plethora of RNA repair enzymes in Naegleria reflects functional specialization of the ligase families and paralogs for different types of RNA damage and/or distinct biological pathways, akin to the specialization among multiple DNA ligase paralogs in a given taxon for replication, excision repair, recombination, or nonhomologous end-joining. Taking speculation even further, it is worth considering that Naegleria RNA ligases might catalyze RNA rearrangements during the amoeba to flagellate developmental switch.
MATERIALS AND METHODS
Recombinant NgrRnl
A synthetic NgrRnl gene synthesized by GenScript USA Inc. was inserted between the BamHI and XhoI sites of pET28b-His10Smt3 to generate pET-His10Smt3-NgrRnl, which encodes the 339-aa NgRnl polypeptide fused to an amino-terminal His10Smt3 module. A truncated gene, encoding amino acids 127–339, was PCR-amplified and inserted between the BamHI and XhoI sites of pET28b-His10Smt3 to generate pET-His10Smt3-NgrRnl–▵N. Missense mutations of the Lys170 codon were introduced into the full-length NgrRnl gene by PCR amplification with mutagenic primers. The plasmid inserts were sequenced completely to exclude the acquisition of unwanted changes during PCR amplification and cloning. The plasmids were electroporated into E. coli BL21(DE3) cells. To produce and purify full-length NgrRnl, a culture (6 L) amplified from a single kanamycin-resistant transformant was grown at 37°C in Luria-Bertani medium containing 0.1 mg/mL kanamycin until the A600 reached 0.6. The culture was then adjusted to 1 mM isopropyl 1-thio-β-D-galactopyranoside, followed by incubation for 3 h at 37°C with constant shaking. Cells were harvested by centrifugation and stored at −80°C. All subsequent steps were performed at 4°C. The bacterial cell pellet was thawed and resuspended in 100 mL lysis buffer containing 50 mM Tris–HCl pH 8.0, 10% sucrose, 250 mM NaCl, 0.05% Triton X-100. Lysis was achieved by sonication for 10 min, after which insoluble material was removed by centrifugation in a Sorvall SS34 rotor for 60 min at 16,000 rpm. The soluble supernatant was applied to two 12-mL nickel-nitrilotetraacetic acid (NTA) agarose columns (Qiagen) that had been equilibrated with buffer A (50 mM Tris–HCl, pH 8.0, 10% glycerol, 250 mM NaCl). The columns were washed with buffer A and eluted stepwise with 20, 100, 200, and 1000 mM imidazole in buffer A. The polypeptide compositions of the eluate fractions were monitored by SDS-PAGE. The recombinant NgrRnl protein was recovered predominantly in the 200 mM imidazole eluates. Peak fractions were pooled, supplemented with Smt3-specific protease Ulp1 (at a NgrRnl:Ulp1 ratio of ∼500:1), and then dialyzed overnight against buffer A. The tag-less NgrRnl protein was separated from His10Smt3 by passage over a second nickel–agarose column equilibrated in buffer A. NgrRnl was recovered in the flow-through fraction. This material was dialyzed overnight against buffer B (50 mM Tris–HCl, pH 8.0, 10% glycerol, 20 mM NaCl). The dialysate was applied to a 5-ml DEAE Sephacel column that had been equilibrated with buffer B. NgrRnl was step eluted with 50 mM Tris–HCl, pH 8.0, 10% glycerol, 50 mM NaCl, concentrated by centrifugal ultrafiltration, then gel-filtered through a column of Superdex-200 equilibrated in 20 mM Tris pH 8.0, 100 mM NaCl. The peak NgrRnl fractions were pooled, concentrated by centrifugal ultrafiltration, and stored at −80°C. The NgrRnl K170A, K170M, K170Q, and K170H mutants were purified as described above from 3 L bacterial cultures. NgrRnl-▵N was purified from a 2 L bacterial culture. Protein concentrations were determined by using the BioRad dye binding reagent, with bovine serum albumin as the standard.
Ligase substrates
The substrates used were singly nicked duplexes composed of an 18-mer RNAOH strand and either a pDNA or pRNA 18-mer strand (labeled with 32P at the nick 5′-PO4) annealed to a 36-mer DNA template strand. RNA oligonucleotides were purchased from Dharmacon and deprotected according to the manufacturer's instructions. The 5′ 32P-labeled oligonucleotides were prepared by label transfer from [γ-32P]ATP catalyzed by T4 polynucleotide kinase, then purified free of ATP by electrophoresis through a nondenaturing 18% polyacrylamide gel. To form the nicked duplexes, the radiolabeled pDNA or pRNA strand, RNAOH strand, and DNA template strand were annealed at a 1:5:2 molar ratio in 200 mM NaCl, 10 mM Tris–HCl (pH 6.8), 1 mM EDTA by incubation for 10 min at 65°C, 15 min at 37°C, and then 30 min at 22°C.
Nick sealing
Reaction mixtures (10 µL) constituted as described in the figure legends were incubated for 30 min at 37°C and then quenched by adding 10 µL of 90% formamide, 50 mM EDTA. The mixtures were analyzed by electrophoresis through a 15-cm 20% polyacrylamide gel containing 7.5 M urea in 44.5 mM Tris–borate (pH 8.3), 1 mM EDTA. The radiolabeled nucleic acids were visualized by autoradiography. Where specified, the extent of sealing was quantified by scanning the gel with a Fuji BAS-2500 imagining apparatus.
Ligase adenylylation
Reaction mixtures (10 μL) constituted as described in the figure legends and including 50 µM [α-32P]ATP were incubated for 30 min at 37°C then quenched by adjustment to 2% SDS and 25 mM EDTA. The mixtures were analyzed by electrophoresis though a 10% polyacyrlamide gel containing 0.1% SDS. The ligase–[32P]adenylate adduct was visualized by autoradiography of the dried gel and quantified, where indicated, by scanning the gel with a Fuji BAS-2500 imaging apparatus.
5′-Adenylylated nicked duplex substrates
A preadenylylated 18-mer DNA strand (AppDNA) was prepared by RtcA-mediated AMP transfer to a 5′ 32P-labeled pDNA strand as described (Chakravarty and Shuman 2011). The AppDNA was separated from the pDNA by electrophoresis through a 40-cm 20% polyacrylamide gel containing 7 M urea. After locating the strands by autoradiography, a gel slice containing the AppDNA species was excised and the AppDNA was eluted into 300 μL of 0.5 M ammonium acetate (pH 8.0), 10 mM magnesium acetate (pH 8.0), 1 mM EDTA, 0.1% SDS during overnight incubation at 4°C. The AppDNA strand was ethanol-precipitated and resuspended in 50 µL of 10 mM Tris–HCl (pH 8.0), 1 mM EDTA. The preadenylylated nicked duplex substrates shown in the figures were formed by annealing the radiolabeled AppDNA strand, either an 18-mer RNAOH or DNAOH strand, and the complementary 36-mer DNA template strand at a 1:5:2 molar ratio in 200 mM NaCl, 10 mM Tris–HCl (pH 6.8), 1 mM EDTA.
Phosphodiester synthesis at a preadenylylated nick
Reaction mixtures (10 µL) containing 50 mM Tris–acetate (pH 6.0), 5 mM MnCl2, 5 mM DTT, 1 pmol (0.1 μM) 32P-labeled preadenylylated nicked duplex substrate and NgrRnl as specified were incubated for 30 min at 37°C, then quenched with formamide, EDTA. The mixtures were analyzed by urea-PAGE. The radiolabeled nucleic acids were visualized by autoradiography. Where specified, the extent of sealing was quantified by scanning the gel with a Fuji BAS-2500 imagining apparatus.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful to Brad Schmier for advice and design of the synthetic NgrRnl gene. This research was supported by National Institutes of Health (NIH) grant GM42498.
Footnotes
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.049197.114.
REFERENCES
- Amitsur M, Levitz R, Kaufman G 1987. Bacteriophage T4 anticodon nuclease, polynucleotide kinase and RNA ligase reprocess the host lysine tRNA. EMBO J 6: 2499–2503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanc V, Alfonso JD, Aphasizhev R, Simpson L 1999. The mitochondrial RNA ligase from Leishmania tarentolae can join RNA molecules bridged by a complementary RNA. J Biol Chem 274: 24289–24296. [DOI] [PubMed] [Google Scholar]
- Brooks MA, Meslet-Cladiére L, Graille M, Kuhn J, Blondeau K, Myllykallio H, Van Tilbeurgh H 2008. The structure of an archaeal homodimeric ligase which has RNA circularization activity. Protein Sci 17: 1336–1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakravarty AK, Shuman S 2011. RNA 3′-phosphate cyclase (RtcA) catalyzes ligase-like adenylylation of DNA and RNA 5′-monophosphate ends. J Biol Chem 286: 4117–4122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakravarty AK, Subbotin R, Chait BT, Shuman S 2012. RNA ligase RtcB splices 3′-phosphate and 5′-OH ends via covalent RtcB-(histidinyl)-GMP and polynucleotide-(3′)pp(5′)G intermediates. Proc Natl Acad Sci 109: 6072–6077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chauleau M, Shuman S 2013. Kinetic mechanism of nick sealing by T4 RNA ligase 2 and effects of 3′-OH base mispairs and damaged base lesions. RNA 19: 1840–1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Omari K, Ren J, Bird LE, Bona MK, Klarmann G, LeGrice SF, Stammers DK 2006. Molecular architecture and ligand recognition determinants for T4 RNA ligase. J Biol Chem 281: 1573–1579. [DOI] [PubMed] [Google Scholar]
- Englert M, Sheppard K, Gundllapalli S, Beier H, Söll D 2010. Branchiostoma floridae has separate healing and sealing enzymes for 5′-phosphate RNA ligation. Proc Natl Acad Sci 107: 16834–16839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritz-Laylin LK, Prochnik SE, Ginger ML, Dacks JB, Carpenter ML, Field MC, Kuo A, Paredez A, Chapman J, Pham J, et al. 2010. The genome of Naegleria gruberi illuminates early eukaryotic versatility. Cell 140: 631–642. [DOI] [PubMed] [Google Scholar]
- Ho CK, Wang LK, Lima CD, Shuman S 2004. Structure and mechanism of RNA ligase. Structure 12: 327–339. [DOI] [PubMed] [Google Scholar]
- Jurkin J, Henkel T, Nielsen AF, Minnich M, Popow J, Kaufmann T, Heindl K, Hoffmann T, Busslinger M, Martinez J 2014. The mammalian tRNA ligase complex mediates splicing of XBP1 mRNA and controls antibody secretion in plasma cells. EMBO J 33: 2922–2936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosmaczewski SG, Edwards TJ, Han SM, Eckwahl MJ, Meyer BI, Peach S, Hesselberth JR, Wolin SL, Hammarlund M 2014. The RtcB RNA ligase is an essential component of the metazoan unfolded protein response. EMBO Rep 15: 1278–1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martins A, Shuman S 2004. An RNA ligase from Deinococcus radiodurans. J Biol Chem 279: 50654–50661. [DOI] [PubMed] [Google Scholar]
- Nandakumar J, Shuman S 2004. How an RNA ligase discriminates RNA versus DNA damage. Mol Cell 16: 211–221. [DOI] [PubMed] [Google Scholar]
- Nandakumar J, Shuman S 2005. Dual mechanisms whereby a broken RNA end assists the catalysis of its repair by T4 RNA ligase 2. J Biol Chem 280: 23484–23489. [DOI] [PubMed] [Google Scholar]
- Nandakumar J, Ho CK, Lima CD, Shuman S 2004. RNA substrate specificity and structure-guided mutational analysis of bacteriophage T4 RNA ligase 2. J Biol Chem 279: 31337–31347. [DOI] [PubMed] [Google Scholar]
- Nandakumar J, Shuman S, Lima CD 2006. RNA ligase structures reveal the basis for RNA specificity and conformational changes that drive ligation forward. Cell 127: 71–84. [DOI] [PubMed] [Google Scholar]
- Nandakumar J, Schwer B, Schaffrath R, Shuman S 2008. RNA repair: an antidote to cytotoxic eukaryal RNA damage. Mol Cell 31: 278–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raymond A, Shuman S 2007. Deinococcus radiodurans RNA ligase exemplifies a novel ligase clade with a distinctive N-terminal module that is important for 5′-PO4 nick sealing and ligase adenylylation but dispensable for phosphodiester formation at an adenylylated nick. Nucleic Acids Res 35: 839–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remus BS, Shuman S 2013. A kinetic framework for tRNA ligase and enforcement of a 2′-phosphate requirement for ligation highlights the design logic of an RNA repair machine. RNA 19: 659–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remus BS, Shuman S 2014. Distinctive kinetics and substrate specificities of plant and fungal tRNA ligases. RNA 20: 462–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmier BJ, Shuman S 2014. Effects of 3′-OH and 5′-PO4 base mispairs and damaged base lesions on the fidelity of nick sealing by Deinococcus radiodurans RNA ligase. J Bacteriol 196: 1704–1712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shuman S, Lima CD 2004. The polynucleotide ligase and RNA capping enzyme superfamily of covalent nucleotidyltransferases. Curr Opin Struct Biol 14: 757–764. [DOI] [PubMed] [Google Scholar]
- Sidrauski C, Cox JS, Walter P 1996. tRNA ligase is required for regulated mRNA splicing in the unfolded protein response. Cell 87: 405–413. [DOI] [PubMed] [Google Scholar]
- Smith P, Wang LK, Nair PA, Shuman S 2012. The adenylyltransferase domain of bacterial Pnkp defines a unique RNA ligase family. Proc Natl Acad Sci 109: 2296–2301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang LK, Shuman S 2005. Structure–function analysis of yeast tRNA ligase. RNA 11: 966–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang LK, Nandakumar J, Schwer B, Shuman S 2007. The C-terminal domain of T4 RNA ligase 1 confers specificity for tRNA repair. RNA 13: 1235–1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P, Chan CM, Christensen D, Zhang C, Selvadurai K, Huang RH 2012. Molecular basis of bacterial protein Hen1 activating the ligase activity of bacterial protein Pnkp for RNA repair. Proc Natl Acad Sci 109: 13248–13253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Chan CM, Wang P, Huang RH 2012. Probing the substrate specificity of the bacterial Pnkp/Hen1 RNA repair system using synthetic RNAs. RNA 18: 335–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.