

Abstract
The cycle of spliceosome assembly, intron excision, and spliceosome disassembly involves large-scale structural rearrangements of U6 snRNA that are functionally important. U6 enters the splicing pathway bound to the Prp24 protein, which chaperones annealing of U6 to U4 RNA to form a U4/U6 di-snRNP. During catalytic activation of the assembled spliceosome, U4 snRNP is released and U6 is paired to U2 snRNA. Here we show that point mutations in U4 and U6 that decrease U4/U6 base-pairing in vivo are lethal in combination. However, this synthetic phenotype is rescued by a mutation in U6 that alters a U6–Prp24 contact and stabilizes U2/U6. Remarkably, the resulting viable triple mutant strain lacks detectable U4/U6 base-pairing and U4/U6 di-snRNP. Instead, this strain accumulates free U4 snRNP, protein-free U6 RNA, and a novel complex containing U2/U6 di-snRNP. Further mutational analysis indicates that disruption of the U6–Prp24 interaction rather than stabilization of U2/U6 renders stable U4/U6 di-snRNP assembly nonessential. We propose that an essential function of U4/U6 pairing is to displace Prp24 from U6 RNA, and thus a destabilized U6–Prp24 complex renders stable U4/U6 pairing nonessential.
Keywords: pre-mRNA splicing, Prp24, snRNA, snRNP, RNA structure
INTRODUCTION
Pre-mRNA splicing is an essential and biochemically complex process that apparently occurs in all eukaryotes. It removes introns from pre-mRNA, yielding ligated exons that become mature mRNA. Pre-mRNA splicing is carried out in the nucleus by the spliceosome, which consists of five small nuclear RNAs (snRNAs)—U1, U2, U4, U5, and U6—and a large number of associated proteins. Spliceosome formation involves assembly of the five snRNAs and their bound proteins (small nuclear ribonucleoprotein particles or snRNPs) onto the pre-mRNA substrate (Wahl et al. 2009; Hoskins et al. 2011). At the core of the assembled spliceosome are conserved interactions between the snRNAs and the intron (Fig. 1).
FIGURE 1.
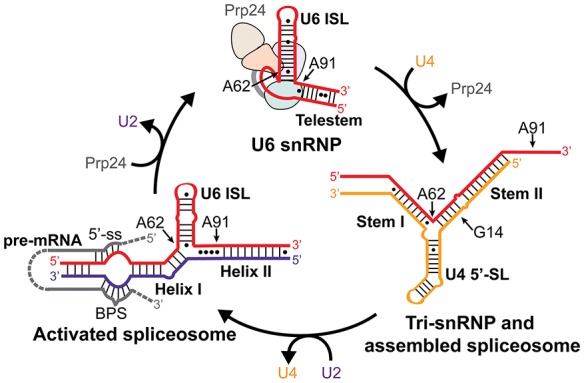
Changes in U6 snRNA conformation throughout the splicing cycle. The relevant region of U6 snRNA is diagrammed in red, U4 in yellow, U2 in purple, and pre-mRNA in gray. Important elements of each RNA structure and the corresponding spliceosomal complexes are labeled. Positions of bases mutated in this study are indicated. Positions of the 5′-splice site (5′-ss) and branch point sequence (BPS) are indicated on the pre-mRNA substrate.
During spliceosome assembly, U6 snRNA base-pairs with U4 snRNA to form the U4/U6 di-snRNP (Bringmann et al. 1984; Hashimoto and Steitz 1984; Brow and Guthrie 1988), a component of the U4/U6.U5 tri-snRNP. After assembly of all five snRNPs onto an intron spliceosome activation occurs, whereupon the U4/U6 complex is unwound (Cheng and Abelson 1987; Lamond et al. 1988) and U6 is paired with U2 snRNA (Madhani and Guthrie 1992; Sun and Manley 1995). The U2/U6 complex contains the U6 internal stem–loop (ISL) (Miura et al. 1983; Fortner et al. 1994) adjacent to U2/U6 Helix I (Madhani and Guthrie 1992), which juxtaposes the intron binding regions of U2 and U6 (Fig. 1; Parker et al. 1987; Sawa and Abelson 1992; Wassarman and Steitz 1992; Kandels-Lewis and Séraphin 1993; Lesser and Guthrie 1993). Thus, the U2/U6 structure scaffolds the first catalytic step of splicing: 5′ splice site cleavage via attack by the branchpoint adenosine. In yeast, U2/U6 forms a three-helix junction (Madhani and Guthrie 1992; Hilliker and Staley 2004; Mefford and Staley 2009; Burke et al. 2012), although evidence exists for formation of a competing four-helix junction in vitro (Sashital et al. 2004; Guo et al. 2009).
After the second catalytic step of splicing, the spliceosome is thought to disassemble into individual snRNPs, which are recycled for further rounds of splicing (Wahl et al. 2009). U6 recycling requires Prp24, an essential protein that binds free U6 RNA and catalyzes its base-pairing with U4 RNA (Shannon and Guthrie 1991; Ghetti et al. 1995; Raghunathan and Guthrie 1998b; Bell et al. 2002; Trede et al. 2007; Martin-Tumasz et al. 2011). Base-pairing in the U4/U6 complex mostly precludes U2/U6 base-pairing and vice versa, suggesting that U6 may be completely released from U2 RNA before pairing with U4 RNA (Fig. 1). However, U2/U6 Helix II can exist simultaneously with U4/U6 pairing (Hausner et al. 1990; Wassarman and Steitz 1992; Schneider et al. 2010) and appears to stabilize the human U4/U6 complex (Brow and Vidaver 1995), so it is possible that a U4/U6/U2 tri-snRNP is an intermediate in U6 recycling.
Several mutations that alter the relative stabilities of U6-containing complexes result in cold-sensitive growth in yeast. For example, substitution of U4–G14 with either a C or a U, which disrupts a G–C pair in U4/U6 Stem II (Fig. 1), is lethal at 22°C or below (Shannon and Guthrie 1991). A genome-wide selection for spontaneous suppressors of the cold-sensitive growth of a strain containing U4–G14C yielded four independent mutations in U6 RNA including U6–A91G, which is downstream from the U6 ISL and U4/U6 Stem II but adjacent to U2/U6 Helix II (Fig. 1; Shannon and Guthrie 1991). Likewise, substitution of U6–A62 with G, which converts an A–C mismatch at the base of the ISL to a stable G–C pair, results in a cold-sensitive growth defect that is suppressed by second-site mutations in U6 RNA and by a mutation in the Stem II region of U4 (Fortner et al. 1994; Vidaver et al. 1999). The cold-sensitivity of both U4–G14C and U6–A62G are suppressed by mutations in Prp24, consistent with its function in U4/U6 annealing (Shannon and Guthrie 1991; Vidaver et al. 1999; Montemayor et al. 2014).
Here we investigate the effect of three point mutations in U4 and U6 RNAs on the abundance and stability of U6-containing complexes. The combination of U6–A62G with U4–G14U is lethal, consistent with both mutations inhibiting formation of the U4/U6 complex. Strikingly, a second mutation in U6 RNA, U6–A91G, rescues the synthetic lethality of the U6–A62G/U4–G14U double mutant strain without restoring U4/U6 complex formation. The resulting triple mutant contains little or no detectable U4/U6 snRNP and instead accumulates free U4 snRNP, protein-free U6 RNA, and a novel complex containing U2/U6 snRNP. A substitution in the Prp24 residue that contacts U6–A91 partially rescues the U6–A62G/U4–G14U strain, suggesting that Prp24 release is an essential function of U4/U6 pairing. Further, the two U6 mutations increase the stability of U2/U6 in vitro, consistent with the observed accumulation of a U2/U6 complex in the triple mutant. Our data show that three point mutations in the RNA core of the spliceosome can profoundly alter the distribution of snRNP complexes in vivo without preventing the spliceosome from conducting its essential functions.
RESULTS
A genetic interaction network between two mutations in U6 RNA and one in U4 RNA
To better understand the molecular basis for the phenotypes of mutations in U4 and U6 RNAs, we examined the genetic interactions between three such mutations (Fig. 2A). Shannon and Guthrie (1991) showed that the U4–G14C and U4–G14U mutations, which disrupt a G–C base pair in U4/U6 Stem II, cause a strong cold-sensitive growth defect, with the G14C mutation being more severe. In our strain background, the U4–G14C mutation was slow-growing even at 30°C, so we used U4–G14U (Fig. 2B, row 2). Shannon and Guthrie isolated the U6–A91G mutation as a spontaneous suppressor of the cold-sensitivity of U4–G14C, and we show here that it also strongly suppresses the cold-sensitivity of U4–G14U (Fig. 2B, compare row 2 to row 3). However, because U6–A91 is outside of the base-paired region of U4/U6, its mechanism of suppression is not immediately clear.
FIGURE 2.
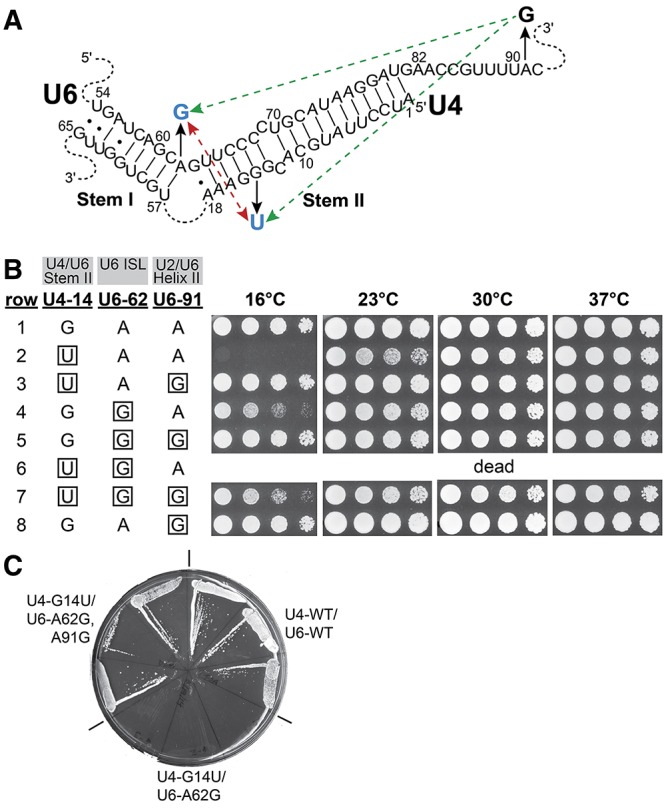
U6–A91G rescues a lethal double mutation in U4 and U6 RNAs. (A) Positions of the mutations in the U4/U6 base-paired region (see also Fig. 1). Substitutions are indicated by black arrows. Blue substitutions result in cold-sensitive growth. Green dashed arrows indicate suppression of cold-sensitivity and the double-headed, red dashed arrow indicates synthetic lethality. (B) Serial dilutions of isolates of yeast strain CJM000 that contain the indicated alleles of the U4 and U6 genes were grown on YEPD medium for 3 d (30°C and 37°C), 6 d (23°C), or 10 d (16°C). Mutant residues are boxed. The results are summarized in A. (C) The U4–G14U/U6–A62G double mutation is lethal, but is rescued by the U6–A91G mutation. Three colonies of yeast strain CJM000 transformed with each indicated combination of U4 and U6 alleles in pRS314-U4+U6 were streaked onto SC medium containing 0.75 mg/mL 5-fluoroorotic acid and incubated at 30°C to assess growth in the absence of pRS316-U4+U6. (WT) wild-type.
The U6–A62G mutation creates a stable G–C base pair from an A–C mismatch at the base of the U6 ISL (Fig. 1), and changes an A–U base pair in U4/U6 Stem I to a G–U pair (Fig. 2A). This mutation has a weaker cold-sensitive growth defect than U4–G14U (Fig. 2B, row 4), particularly when the U4 gene is on a plasmid and can therefore be amplified (Fortner et al. 1994), as is the case with the strain used here. Because both U4–G14U and U6–A62G result in a decrease in U4/U6 complex formation (Fortner et al. 1994; McManus et al. 2007), we hypothesized that U6–A91G might also suppress U6–A62G. An extensive but nonsaturating selection for suppressors of the cold-sensitivity of U6–A62G yielded mutations at 15 different positions in U6 RNA, but none in A91 (Fortner et al. 1994). We show here that the U6–A91G mutation is a suppressor of the cold-sensitivity of U6–A62G (Fig. 2B, row 5).
The fact that both U4–G14U and U6–A62G inhibit U4/U6 di-snRNP assembly and/or decrease U4/U6 stability in vivo (Shannon and Guthrie 1991; Vidaver et al. 1999; McManus et al. 2007) suggested that, in combination, the two mutations would have a stronger growth defect than either alone. Indeed, the U4–G14U/U6–A62G mutation is dead at 30°C (Fig. 2C). However, the U6–A91G mutation suppresses this synthetic lethality (Fig. 2C), although the triple mutant grows slower at low temperature than the U6–A91G mutant alone (Fig. 2B, cf. rows 7 and 8). Thus, the U6–A91G mutation can somehow compensate for a very severe defect in U4/U6 di-snRNP assembly and/or function.
Absence of stable U4/U6 pairing in the triple mutant strain
To determine if suppression of the lethal double mutant by U6–A91G is due to restoration of U4/U6 base-pairing in vivo, total RNA was extracted from wild-type and mutant strains under nondenaturing conditions and analyzed by solution hybridization (Fig. 3; Li and Brow 1993). As shown previously (Fortner et al. 1994; McManus et al. 2007), in wild-type cells 70%–85% of U4 RNA is paired with U6 RNA (lanes 1,2). Both U6–A62G (lanes 3,4) and U4–G14U (lanes 9,10) decrease the fraction of U4 RNA base-paired to U6 RNA even when cells are grown at permissive temperature (30°C), although only U6–A62G has a statistically significant effect in this study. Consistent with the absence of a growth defect, U6–A91G alone has no effect on the fraction of U4 paired with U6 (lanes 5,6). U6–A91G increases U4/U6 complex formation in the presence of U6–A62G (Fig. 3B, lanes 7,8), consistent with the observed suppression (Fig. 2). However, U4/U6 levels in this double mutant are still as low as in the cold-sensitive U4–G14U strain (Fig. 3B, lanes 11,12). Although U6–A91G strongly suppresses the cold-sensitivity of U4–G14U, it does not have a significant effect on the U4/U6 level in the U4–G14U strain. Thus, suppression by U6–A91G may function through another mechanism than restoration of U4/U6 assembly.
FIGURE 3.
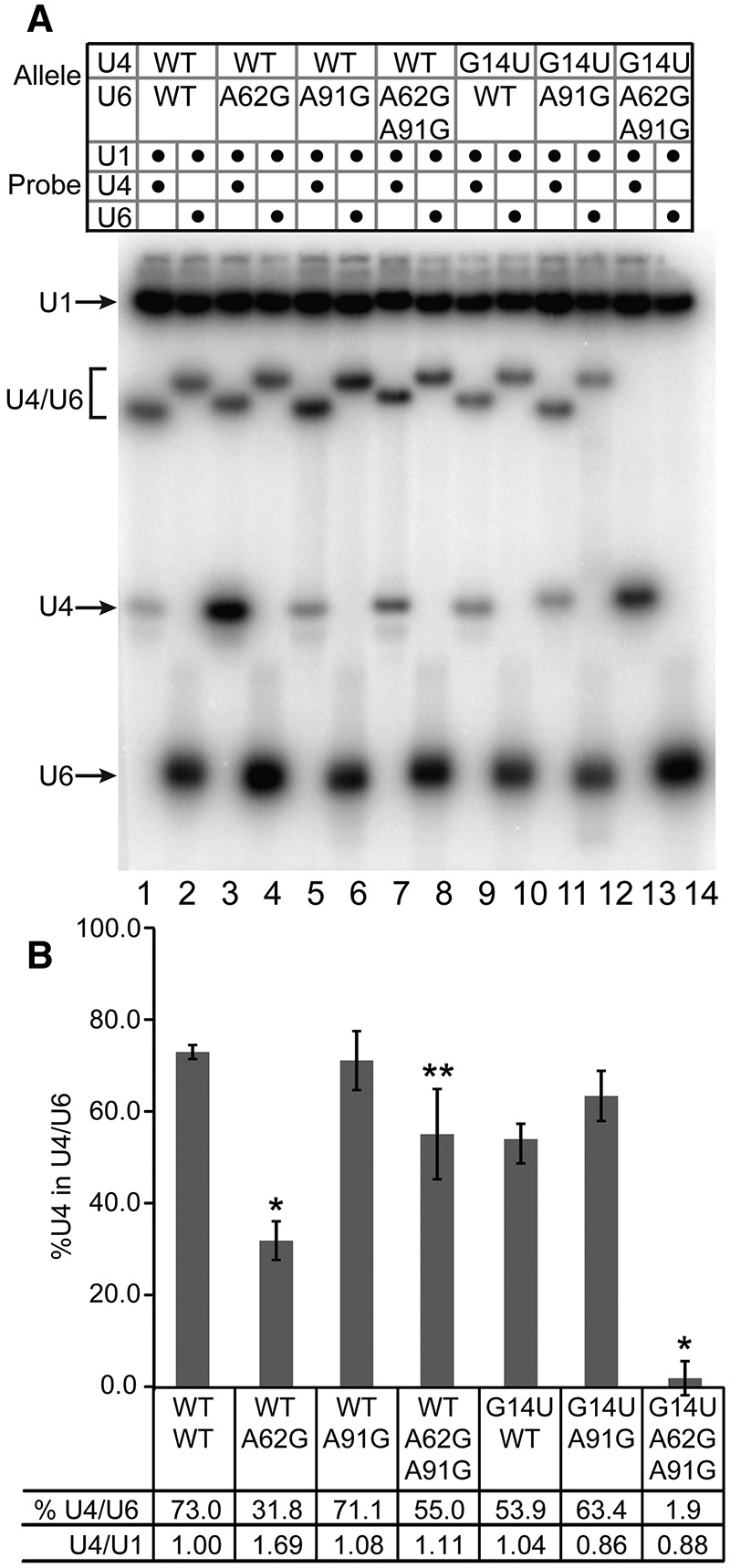
U6–A91G rescues the synthetic lethality of U4–G14U/U6–A62G without restoring U4/U6 pairing. (A) Solution hybridization of whole-cell RNA. RNA extracted under nondenaturing conditions from strain CJM000 bearing the U4 and U6 alleles listed (WT, wild-type) was hybridized to 32P-labeled DNA oligomers complementary to U1 and U4 or U6 (as indicated) at 37°C for 15 min, then analyzed by 9% nondenaturing polyacrylamide gel electrophoresis at 4°C. Note that the U4/U6 RNA complex has a different mobility when bound to a DNA oligomer complementary to U4 or U6. (B) Quantitative analysis of the fraction of U4 present as U4/U6 and total U4 levels normalized to U1 (average of two biological replicates; error bars indicate range). An asterisk indicates a P-value <0.05 in comparison with wild-type as determined by unpaired t-test. A double asterisk indicates a P-value of <0.05 in comparison with U6–A62G, U4-WT.
The strongest evidence that U6–A91G suppresses U6–A62G and U4–G14U by an alternative mechanism comes from RNA analysis of the triple mutant strain. Remarkably, the strain with all three mutations contains no detectable U4/U6 complex (Fig. 3, lanes 13,14), despite exhibiting near normal growth. This is the only viable strain of which we are aware that has been observed to exhibit <15% U4 RNA incorporation into U4/U6 complex by this or any other assay. However, any unstable U4/U6 complex present in the triple mutant strain could potentially dissociate during the solution hybridization procedure, which involves incubation with the labeled probe at 37°C, so we also performed Northern analysis of the RNA extracts, which were prepared at 4°C (Supplemental Fig. S1). Only 10% of the wild-type amount of U4/U6 complex is detected in the U4–G14U/U6–A62G,A91G triple mutant strain by Northern analysis. Since the small amount of U4/U6 detected by Northern analysis dissociates during the 37°C incubation required for solution hybridization, it must be highly unstable in the triple mutant. (Wild-type U4/U6 RNA complex has a melting temperature of ∼50°C.) Thus, suppression of the lethal double mutant by U6–A91G is not accompanied by restoration of stable U4/U6 pairing.
If stable U4/U6 pairing is no longer required in the triple mutant, then either U4 or Prp24 may be dispensable in this strain. However, U4 RNA and the U4/U6-annealing factor Prp24 are still required (Supplemental Results, Fig. S2), so either the unstable U4/U6 complex still has an essential function in this strain, or U4 RNA and Prp24 perform some other essential function.
Detection of a novel complex containing U2/U6 di-snRNP without U5 snRNP
The near absence of U4/U6 RNA complex in the triple mutant strain suggests either that the steady-state level of U4/U6 di-snRNP is very low, or that U4/U6 di-snRNP is abundant but lacks stable base-pairing between U4 and U6 RNAs. To distinguish between these possibilities, we prepared whole-cell extracts from wild-type and triple mutant strains using the liquid nitrogen/ball mill method and analyzed their steady-state snRNP composition using nondenaturing gel electrophoresis (Fig. 4). To sample any ATP-dependent changes in snRNP interactions, extracts were also incubated with or without 1 mM ATP for 30 min at 30°C (lanes 1–4) or 15 min at 23°C (lanes 5–8).
FIGURE 4.

Detection of a U2/U6 di-snRNP, U4 snRNP, and free U6 RNA in the triple mutant strain. Analysis of wild-type and triple mutant (starred lanes) whole-cell extracts with (dotted lanes) or without incubation with ATP by nondenaturing 4% polyacrylamide gel electrophoresis. RNA was transferred to a membrane, which was then serially probed with 32P-labeled DNA oligomer complementary to the RNA indicated above each panel. Bold labels indicate snRNPs and plain labels indicate snRNAs. Black boxes highlight stable U2/U6 RNA complexes present in the triple mutant strain. In lanes 11–12, extracts were deproteinized at low temperature by phenol/chloroform extraction, leaving only RNA. In lanes 13–14, DNA oligomer complementary to positions of 89–111 of U2 snRNA was added to promote RNase H cleavage of U2 (“cut U2”). U6* appears to be a truncated form of U6 RNA. Yeast U5 snRNA is produced as long (U5L) and short (U5S) species.
Consistent with the observed loss of U4/U6 pairing, cell extract from the triple mutant strain is essentially devoid of U4/U6 di-snRNP. Instead, the strain accumulates large amounts of free U4 snRNP (U4 probe, lanes 3,4) and free U6 snRNA (U6 probe, lanes 3,4). The large fraction of protein-free U6 snRNA in cell extract from the triple mutant strain suggests that one or both of the mutations in U6 disrupt binding of the U6 snRNP proteins, Prp24 and Lsm2-8. (The protein-free U6 RNA is not a consequence of U6 overexpression, since U6 RNA levels are similar in the wild-type and triple mutant strains.) Furthermore, an apparently truncated form of U6 RNA accumulates in the triple mutant (U6*), consistent with the absence of protection from nucleases by bound protein(s). Despite the absence of U4/U6 di-snRNP, the triple mutant strain contains U4/U6.U5 tri-snRNP, albeit substantially less than in the wild-type strain. U4 and U6 RNAs in the tri-snRNP must be either unstably paired or unpaired, since the deproteinized triple mutant extract contains no detectable U4/U6 complex (compare lanes 11,12, U4 and U6 probes). Thus, in the triple mutant strain the U4 snRNP and U6 snRNA may be incorporated into the tri-snRNP individually, via direct interactions with U5 snRNP. We will refer to this tri-snRNP with destabilized U4/U6 pairing as the U4.U5.U6 tri-snRNP.
A striking finding from this analysis is the identification of a novel complex that contains U2 and U6 snRNAs but not U5 snRNA, and that appears more abundant than the tri-snRNP or U2 snRNP in the triple mutant extract (U2 and U6 probes, lane 10). This complex is present in wild-type extract in lower amounts (U2 and U6 probes, lane 9), but is less stable as it dissociates upon incubation at 30°C (U2 and U6 probes, lane 1). Since it lacks the U5 snRNP, it cannot be the post-catalytic intron lariat spliceosome (ILS). Depletion of U2 snRNP in the triple mutant extract is consistent with elevated levels of the U2/U6-containing complex. This complex migrates faster than the U2 snRNP, possibly due to a greater net negative charge (from U6 RNA), more compact shape, and/or loss of U2 snRNP proteins. The U2/U6 RNA complex appears stable to deproteinization in the triple mutant extract, but not the wild-type extract (U6 probe, lanes 11,12). Because yeast U2 snRNA is 1175 nt long and yeast U6 is only 112 nt long, deproteinized U2 snRNA is not resolved from the U2/U6 snRNA complex in this gel (U2 probe, lanes 11,12).
To confirm that U6 RNA is paired with the 5′ end of U2 in the U2/U6-containing complex, as it is in the spliceosome (Fig. 1), we incubated the whole-cell extracts with a DNA oligomer complementary to positions 89–111 of U2 to direct cleavage of the U2 snRNA by endogenous RNase H. Since our U2 probe is complementary to residues 29–43, it will detect the short, 5′ fragment generated by cleavage. Such treatment of wild-type extract results in loss of the signal from U2 snRNP and appearance of a new band, the mobility of which is consistent with protein-free RNA of ∼100-nt length (U2 probe, lane 13). In the triple mutant extract, an additional, slower-migrating band is observed that also is detected by the U6 probe (U2 and U6 probes, lane 14), indicating that the 5′ end of U2 is base-paired with U6 in the U2/U6-containing complex. The low yield of the cut U2/U6 RNA complex relative to the free 5′-fragment of U2 may be due to dissociation during the incubation of whole-cell extract with the DNA oligomer. U4/U6.U5 tri-snRNP, U4/U6 di-snRNP, U6 snRNP, and U4 snRNP levels appear unaffected by addition of the U2 oligomer, as expected.
Given the homogeneous and high mobility of the U2/U6-containing complex (relative to the U2 snRNP), the absence of U5 snRNA, the presence of U2/U6 pairing, and the presence of the complex (in lower amounts) in wild-type extracts, we conclude that the complex is most likely U2/U6 di-snRNP. However, we cannot exclude the possibility that it also contains intron lariat and/or additional proteins. Accumulation of a U2/U6 di-snRNP in combination with U6 snRNP disruption suggests the possibility that Prp24 and/or Lsm2-8 are active participants in U6 RNA recycling from post-catalytic spliceosomes, but it is also possible that a U2/U6 snRNP forms de novo from U2 snRNP and protein-free U6 RNA.
The U6–A62G and U6–A91G mutations alter the structure and stability of U2/U6 in vitro
The unprecedented accumulation of U2/U6 observed in the triple mutant may be due to disruption of the U6 snRNP, stabilization of U2/U6, or both. To investigate the effects of these mutations on the structure and stability of the U2/U6 RNA interface, we used 2D NMR to determine the secondary structure of wild-type and mutant versions of an 83-nucleotide RNA construct that contains yeast U2/U6 intermolecular Helices I and II and the U6 ISL. For the wild-type construct, base-pairing is observed in the U6 ISL, Helix I, and Helix II (Fig. 5A; Supplemental Fig. S3). Chemical shifts are consistent with previous studies of related constructs (Sashital et al. 2004; Burke et al. 2012) and the observed secondary structure is consistent with a three-helix junction (Madhani and Guthrie 1992; Burke et al. 2012). Six broad uracil resonances that give strong NOE-cross peaks to one another are observed in the wobble pair region of the proton spectrum (∼11 ppm; Supplemental Fig. S3A,B) and are consistent with transient formation of U–U wobble pairs between U6-87 to 90 and U2-16 to 19 (Fig. 5A, gray dots). Pairing in this region is also consistent with previous crosslinking (Ryan et al. 2004) and structural (Burke et al. 2012) studies. The wild-type RNA has a melting temperature of 49.3°C ± 0.2°C in 100 mM potassium chloride and displays one predominantly cooperative folding/unfolding transition (Fig. 5B,D).
FIGURE 5.

U6–A91G structurally counteracts U6–A62G. (A,C,E) Secondary structures of the wild-type, U6–A62G, and U6–A62G/A91G RNAs, respectively, as determined by NMR spectroscopy. Residues are color-coded according to secondary structure as follows: Helix Ia and Ib (green), Helix II (magenta), and U6 ISL (blue). Mutated residues are shown in red boxes. Base pairs confirmed by the presence of an NOE cross-peak are shown as black lines (Watson–Crick) or circles (wobble). Base pairs inferred by chemical shift agreement are shown as dashed lines. Confirmed U–U wobble pairs are indicated with black circles, while inferred wobble pairs are indicated with gray dashed lines. See Supplemental Material for NMR spectra and chemical shift assignments. (B) Melting profiles of wild-type, U6–A62G, and U6–A62G/A91G U2/U6 RNAs. UV absorbance at 258 nm was normalized to the value at 10°C. Experiments conducted in triplicate were averaged and error bars denote standard deviation. (D,F) First derivatives of normalized hyperchromicity for the melting curves shown in B. Data were smoothed over three points.
The cold-sensitivity of the U6–A62G mutant is due at least in part to hyperstabilization of the U6 ISL, via substitution of an A–C mismatch with a stable G–C base pair at the base of the ISL (Fortner et al. 1994). In the U2/U6–A62G RNA the U6 ISL is extended by three base pairs, disrupting U2/U6 Helix Ib (Fig. 5C; Supplemental Fig. S4). We also observe formation of U2 Stem I (Sashital et al. 2004, 2007), indicating formation of a four-helix junction (Sashital et al. 2004; Cao and Chen 2006; Guo et al. 2009). Helix Ia and Helix II form in U2/U6–A62G, although fewer base-pairs are detected in Helix II than in wild-type (Fig. 5C). In contrast to wild-type, the A62G variant unfolds in two transitions (Fig. 5D). The new melting transition is centered at 64.6°C ± 0.2°C (Fig. 5D) and corresponds to the stabilized U6 ISL as determined by variable temperature 1D NMR (Supplemental Fig. S5B), consistent with previous studies of the U6–A62G ISL alone (Reiter et al. 2003).
Because U6–A91G suppresses the growth defect caused by U6–A62G, we hypothesized that inclusion of both mutations would restore a U2/U6 structure that resembles wild-type. Indeed, when both mutations are incorporated into U2/U6, base-pairing is detected in the U6 ISL, Helix II, Helix Ia, and Helix Ib (Fig. 5E; Supplemental Fig. S6). The combined effect nearly restores the cooperative folding of the wild-type RNA (compare A62G to A62G/A91G, Fig. 5D,F); however, the U6 ISL and Helix II are more stable in the U2/U6–A62G,A91G RNA than in wild-type (Fig. 5F; Supplemental Fig. S5C). Increased U2/U6 stability is consistent with accumulation of U2/U6 snRNP-containing complex in splicing extracts from the triple mutant strain.
The suppressor activity of U6–A91G does not require stabilization of U2/U6 Helix II
If stabilization of U2/U6 Helix II by U6–A91G is the mechanism of suppression of the cold-sensitivity of U4–G14U and U6–A62G, and lethality of the double mutant, then mutations in U2–C14 that strengthen its base pair with U6–A91 should also suppress these mutations. Conversely, mutations in U2 that disrupt the G–C base pair created by the U6–A91G mutation should restore the cold-sensitive or lethal phenotypes. To test these predictions, we determined the effects of changing U2–C14, to either G or U (Fig. 6A). In the presence of wild-type U4 and U6, these substitutions had no noticeable effect on growth at the four temperatures tested (Fig. 6B, rows 1–3). We next checked for suppression of U4–G14U-induced cold-sensitivity (Fig. 6B, row 4) by the U2 mutations. U2–C14U, which changes an A–C pair to a more stable A–U pair, does not suppress the cold-sensitivity (Fig. 6B, row 5). In fact, it appears to exacerbate the cold-sensitivity of U4–G14U, unlike U2–C14G, which creates an A–G mismatch and has little or no effect (Fig. 6B, row 6). Furthermore, mutation of U2–C14 to U or G has no effect on suppression of U4–G14U by U6–A91G (Fig. 6B, rows 7–9), despite disrupting the G–C base-pair formed by U6–A91G. Finally, neither U2 mutation alters rescue of the lethal U4–G14U/U6–A62G double mutant by U6–A91G, although both are expected to destabilize U2/U6 Helix II (Fig. 6B, rows 10–12). Thus, stabilization of U2/U6 Helix II is not required for the rescue of the U4–G14U/U6–A62G double mutant by U6–A91G, which must therefore be acting by some other mechanism.
FIGURE 6.
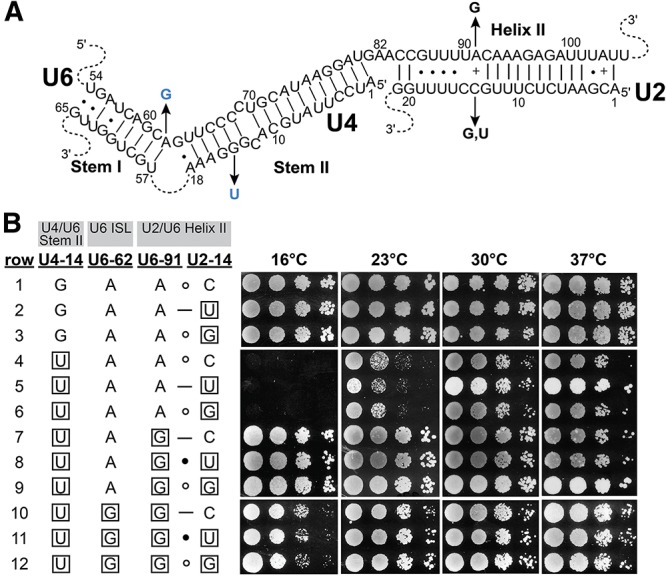
Suppression by U6–A91G does not correlate with stabilization of the U6-91/U2-14 base pair in Helix II. (A) Positions of the mutations in the U4/U6 and U2/U6 base-paired regions (see also Fig. 1). Black arrows indicate the substitutions. Blue substitutions confer a cold-sensitive growth defect. (B) Serial dilutions of isolates of yeast strain yJPS628 that contain the indicated alleles of the U4, U6, and U2 genes were grown on YEPD medium at the indicated temperatures. Mutant residues are boxed.
Substitution of the U6–A91-binding residue of Prp24 rescues U4–G14U/U6–A62G lethality
The mechanism by which the U6–A91G mutation suppresses U4–G14C was previously suggested to be disruption of binding of the U6 snRNP protein Prp24 (Shannon and Guthrie 1991; Jandrositz and Guthrie 1995). Such a mechanism would explain the accumulation of protein-free U6 RNA in cell extract from the triple mutant strain (Fig. 4). Our recent crystal structure of the core U6 snRNP reveals that the Prp24-S283 hydroxyl group hydrogen bonds with the U6–A91 base (Fig. 7A), and we isolated the Prp24-S283Y substitution as a spontaneous suppressor of U6–A62G (Montemayor et al. 2014). If U6–A91G rescues the lethal double mutant primarily by disrupting this contact with Prp24, then the Prp24-S283Y substitution should also rescue the lethality of U4–G14U/U6–A62G. Indeed, the U4–G14U/U6–A62G/Prp24-S283Y triple mutant strain is viable, but very cold-sensitive, growing somewhat slowly at 30°C and 37°C and not at all at 16°C and 23°C (Fig. 7B, row 6). Furthermore, Prp24-S283Y exhibits fairly strong suppression of both single mutants (Fig. 7B, rows 4 and 5). The fact that U6–A91G is a stronger suppressor than Prp24-S283Y may indicate that the former is more destabilizing to the U6 snRNP than the latter. Alternatively, the U6–A91G mutation may suppress the U4/U6 double mutation by more than one mechanism.
FIGURE 7.
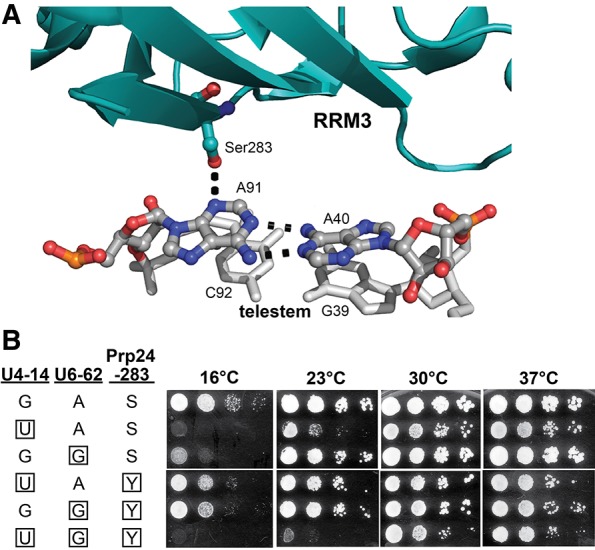
The Prp24-S283Y substitution rescues the lethal U4–G14U/U6-A62G mutation at higher temperatures. (A) Crystal structure (PDB: 4n0t) of the interface between Prp24 RRM3 (in cyan) and the “top” of the U6 RNA telestem, visualized with PyMOL (http://www.pymol.org/). The hydrogen bond between the hydroxyl group of serine 283 and N3 of A91 is shown as a dotted line, as are hydrogen bonds in the A40–A91 base pair. (B) Serial dilutions of isolates of yeast strain JEB100 that contain the indicated alleles of the U4, U6, and PRP24 genes were grown on YEPD medium at the indicated temperatures. Mutant residues are boxed. (S) serine; (Y) tyrosine.
DISCUSSION
Prp24 displacement from U6 may be an essential function of U4/U6 pairing
Here, we report the surprising finding that splicing can proceed without a stable U4/U6 di-snRNP as long as an additional single-residue substitution is present in U6 RNA or its binding protein Prp24. Under such conditions a U4.U5.U6 tri-snRNP forms, albeit less efficiently, possibly by independent binding of the U4 snRNP and U6 RNA to the U5 snRNP. Thus, rather than serving as a means of linking U6 RNA to the U5 snRNP, an essential function of U4/U6 pairing may be to displace Prp24 from U6 RNA, thereby exposing a binding site in U6 RNA for the U5 snRNP. It was shown previously that complete pairing of U4 with U6 is required for displacement of Prp24 from U6 RNA (Shannon and Guthrie 1991; Jandrositz and Guthrie 1995). In the presence of the U6–A91G mutation (or, presumably, the prp24-S283Y mutation at ≥30°C), the steady-state level of Prp24-free U6 RNA is high enough to allow an adequate level of U4–G14U.U5.U6–A62G tri-snRNP assembly.
U4 RNA still has an essential function in the presence of the U6–A62G/A91G double mutant (Supplemental Fig. S2). Perhaps binding of U4 snRNP to U5 snRNP is required to allosterically activate Prp8 or another U5 snRNP protein for U6 RNA binding, or U4 snRNP directly stabilizes U6 RNA binding to the U5 snRNP in the triple mutant strain, despite the apparent absence of free U4/U6 di-snRNP. We cannot exclude that transient, unstable pairing of U4 with U6 is required for association with the U5 snRNP. We attempted to observe transient formation of U4/U6 snRNP in triple mutant extracts by UV-induced psoralen crosslinking. Psoralen crosslinks human U4/U6 complex in Stem I and human U2/U6 complex in Helix II (Bringmann et al. 1984; Hausner et al. 1990; Wassarman and Steitz 1992). We did not observe a U4–U6 crosslink even in wild-type extracts, presumably because of sequence differences between yeast and human Stems I. However, we did observe a U6-containing crosslinked species consistent in mobility with U2/U6 complex, which was enriched in the triple mutant extract (data not shown).
Is Prp24 primarily a U6 RNA recycling factor?
Prp24 also is still essential in the triple mutant strain (Supplemental Fig. S2B,C), suggesting it has an essential function other than U4/U6 annealing. A possible essential function of Prp24 is recycling of U6 RNA from the U2/U6 complex. Thus, the observed accumulation of U2/U6 di-snRNP in the U4–G14U/U6–A62G,A91G triple mutant strain could be due to the combined effects of U2/U6 Helix II stabilization and weakening of Prp24-U6 binding. Using highly purified intron lariat spliceosome (ILS) and recombinant extrinsic factors, Fourmann et al. (2013) showed that the NTR complex (Prp43 and the Ntr1/Ntr2 heterodimer) and ATP are sufficient to disassemble the ILS, releasing free U6 RNA apparently unbound by any proteins. Similar results were obtained previously by Tsai et al. using immunoprecipitated ILS and affinity-purified NTR complex (Tsai et al. 2005). No Prp24 was added to these in vitro disassembly reactions. While these findings would seem to argue against an essential function for Prp24 in disruption of the U2/U6 complex, disassembly of purified ILS in vitro was slow (10–20 min) and the extreme dilution of the in vitro system would disfavor reassociation of released U6 with U2. In the crowded cell nucleus, Prp24 may be essential to shift the U2/U6 to U2+U6 equilibrium sufficiently to allow snRNP recycling to proceed at an adequate rate. Alternatively, or in addition, Prp24 may be essential to prevent nucleolytic degradation of released U6 RNA prior to its reincorporation into the spliceosome.
A possible U2/U6 di-snRNP
A U2/U6 di-snRNP has not previously been identified in yeast or humans, but our results suggest that it is present (in low amounts) even in wild-type yeast cell extracts. The stabilization afforded to the putative U2/U6 di-snRNP by the U6–A62G and A91G mutations, combined with its predicted wild-type-like three-helix junction, makes this mutant complex a potentially attractive target for future biochemical and structural characterization. We cannot currently discern if the U2/U6-containing complex we detect is an on-pathway intermediate of spliceosome disassembly or assembly.
The NMR-determined secondary structures of the wild-type and U6–A62G/A91G mutant U2/U6 constructs are consistent with recent studies demonstrating that the three-helix junction secondary structure is important for promoting a group II intron-like fold in the spliceosomal core (Fica et al. 2013, 2014). However, we do not observe the tertiary interactions described by Fica et al., presumably because they are stabilized by surrounding proteins and/or substrate binding. Stabilization of a correctly folded catalytic core in the context of the activated spliceosome may also explain why the U6–A62G mutant can grow normally at higher temperatures, despite the fact that we observe a predominately misfolded RNA species in vitro.
A pathway that accounts for our observations of the triple mutant strain is shown in Figure 8. After splicing, U6 RNA must be recycled from post-catalytic spliceosomes back into the tri-snRNP. The U6 double mutant (U6**, Fig. 8) stabilizes the U2/U6** snRNP and is expected to destabilize Prp24 binding (Shannon and Guthrie 1991; Jandrositz and Guthrie 1995), which results in a decreased forward equilibrium constant for recycling of U6 from post-catalytic spliceosomes and accounts for the observed accumulation of U2/U6** snRNP-containing complex. Our data suggest that Prp24 plays an essential role in the dissociation of U6 from U2/U6 snRNP. Since previous data indicate that Lsm2–8 proteins bind cooperatively with Prp24 (Rader and Guthrie 2002; Ryan and Abelson 2002), the decreased affinity of Prp24 for U6** also explains the observed accumulation of free U6** RNA in mutant cells (Figs. 4,8). Furthermore, since there is no detectable U4*/U6** di-snRNP in the triple mutant, we hypothesize that free U6** RNA can be incorporated directly into the tri-snRNP via protein-mediated interactions, perhaps involving the binding sites on Prp8 (Turner et al. 2006; Li et al. 2013). In support of this hypothesis, it was previously shown that overexpression of U6 renders Lsm8 protein dispensable for growth (Pannone et al. 2001).
FIGURE 8.
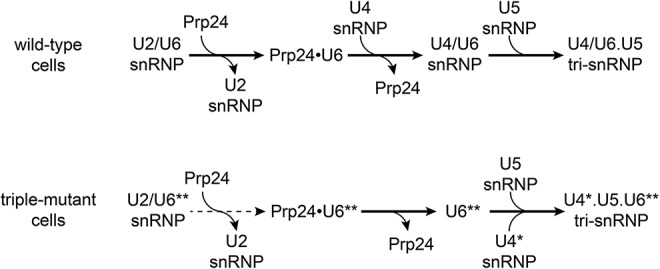
Proposed U6 recycling pathway from post-catalytic spliceosomes for wild-type (top) and U4–G41U/U6–A62G,A91G triple mutant (bottom) cells. Asterisks indicate mutations. Dashed line indicates a reduced forward equilibrium constant that results in an increased accumulation of U2/U6** snRNP. The Lsm2-8 proteins (not shown) likely act in concert with Prp24.
CONCLUSIONS
Our results highlight the remarkably fine balance between alternative conformational states in the spliceosomal RNAs, as well as the resilience of the spliceosome in accommodating large changes in snRNP stability and abundance. Finely balanced alternative conformations are presumably a necessary consequence of the mechanism of splice site recognition and proofreading, which entails an allosteric cascade of conformational changes prior to catalysis of intron excision. The resilience to changes in steady-state snRNP populations suggests that there is more than one way to assemble a functional spliceosome. By focusing on a small number of well-characterized mutations in the spliceosomal RNAs, and subjecting the variant RNAs to detailed genetic and biochemical analysis, we have uncovered the unanticipated result that spliceosome assembly can still proceed in the absence of stable U4/U6 pairing. Given that one proposed function of U4/U6 pairing is to negatively regulate the catalytic activity of U6 RNA until the spliceosome is properly assembled on correct splice sites (Brow and Guthrie 1989), it will be interesting to determine if the triple mutant is more error-prone in splicing than wild-type yeast that maintain highly stable, canonical U4/U6 pairing.
MATERIALS AND METHODS
Strain construction and in vivo growth studies
T4 DNA ligase and restriction enzymes were obtained from Promega or New England Biolabs. pRS314–U4+U6 was created by transferring the SpeI/EcoRI fragment of pRS316-U4wt-U6mini (McManus et al. 2007) to pRS314. pRS313–U4, pRS313-U4–G14U, pRS314–U6, and pRS314-U6–A62G have been described previously (Fortner et al. 1994; Kuhn and Brow 2000). All mutations were created using the Quikchange protocol (Stratagene) and confirmed by cycle sequencing with BigDye Terminator v3.1 (Life Technologies). Yeast transformations were done as described previously (Gietz et al. 1995).
Mutant U4 and U6 alleles were introduced into Saccharomyces cerevisiae strain CJM000 (Gietz et al. 1995; McManus et al. 2007) individually in plasmids pRS313 and pRS314, respectively, or together in pRS314-U4+U6. Transformants were streaked onto SC medium containing 0.75 mg/mL 5-fluoroorotic acid (5-FOA) to select for strains containing only the newly acquired alleles. Growth phenotypes were tested by spotting eightfold serial dilutions, starting with OD600 = 1 of each strain, onto solid YEPD medium and incubating at 16°C, 25°C, 30°C, and 37°C.
JEB100 U4/U6/PRP24 triple-disruption strain (MATa snr14Δ::trp1::ADE2 snr6Δ::LEU2 prp24-ΔORF::KANMX4 trp1 ura3 lys2 his3 ade2 [pRS316-U4/U6/PRP24]) was constructed from CJM000. The resident pRS316-U4+U6 plasmid of CJM000 was first replaced with pRS317-U4+U6 by transformation and selection on 5-FOA. pRS316-U4/U6/PRP24 was made by digesting pRS316-U4wt–U6mini with SacI and XbaI. The wild-type PRP24 gene was amplified from pRS313-PRP24 (Vidaver et al. 1999) by PCR with KOD polymerase (EMD Millipore) with the addition of XbaI and SacI restriction sites 100-bp upstream of and downstream from the protein-coding region, respectively. Digested PCR product and plasmid were then ligated. pRS316-U4/U6/PRP24 was confirmed by restriction digest and sequencing, and introduced into the pRS317-U4+U6-containing strain by plasmid shuffle to create strain JEB019. Growth of JEB019 on medium containing α-aminoadipate selected for loss of pRS317-U4+U6 to yield strain JEB020. The chromosomal PRP24 allele was knocked out via transformation with a PCR product containing the KANMX4 cassette flanked by ∼250 bp of sequence adjacent to the PRP24 open reading frame and selection on YEPD media containing 0.2 mg/mL of Geneticin (G418). The prp24-ΔORF::KANMX4 genotype was confirmed by transformation with individual plasmids containing wild-type U4 and U6 genes and confirming the absence of growth on medium containing 5-FOA, and by PCR amplification with PRP24 and KANMX4 primers. PRP24, U6, and U4 alleles were combined in JEB100 by transformation on pRS313, pRS314, and pRS317 plasmids, respectively, or using pRS313 and pRS314-U4+U6, followed by selection against the resident pRS316-U4/U6/PRP24 plasmid on 5-FOA. RRM deletion alleles of PRP24 were a kind gift from Sharon Kwan (Kwan 2005). pRS313-prp24-S283Y was described previously (Montemayor et al. 2014).
The U2/U4/U6 triple-disruption strain, yJPS628, was a kind gift from Jon Staley (University of Chicago) (Hilliker and Staley 2004). U2 alleles were introduced into this strain on pRS313-U2(SalI/NotI), a kind gift from Eric Steinmetz. This plasmid contains U2 (LSR1) sequences from 808 bp upstream of the transcription start site to 1658 bp downstream. U4 and U6 alleles were introduced on pRS314-U4+U6.
Whole-cell RNA extraction, solution hybridization, and Northern analysis
Whole-cell RNA was prepared under nondenaturing conditions as follows. Thirty milliliters YEPD cultures of each strain were incubated, shaking, at 30°C until reaching an OD600 of 0.6–0.8. Cells from enough culture to yield 10 OD600 units were pelleted at 4°C, washed in ice-cold RNA buffer (200 mM Tris, pH 7.5, 100 mM EDTA, and 500 mM NaCl), and resuspended in 300 μL RNA buffer. Two hundred microliters of acid-washed 0.5 mm glass beads and 300 μL of phenol/chloroform/isoamyl alcohol mixture (25:24:1) equilibrated to pH 6.6 (Ambion) were added and the tubes agitated for 2 min at 4°C in a Disruptor Genie (Scientific Industries). Nucleic acid in the aqueous phase was ethanol precipitated, washed with 70% ethanol, and resuspended in 20 μL Milli-Q (Millipore) H2O.
DNA oligomers used for solution hybridization and Northern analysis are as follows: U4-14B: 5′-AGGTATTCCAAAAATTCCC-3′ (Li and Brow 1993); U1-SH: 5′-CCGTATGTGTGTGTGACC-3′ and U6-SH: 5′-ATTGTTTCAAATTGACCAAAT-3′ (Kuhn et al. 1999); U2seq (RNase H digestion): 5′-GTGTATTGTAACAAATTAAAAGG-3′; U5B: 5′-AAGTTCCAAAAAATATGGCAAGC-3′; SRU2 (Northern analysis): 5′-CAGATACTACACTTG-3′ (McPheeters et al. 1989). Oligomers were 5′-end labeled with [γ-32P]ATP (PerkinElmer) using OptiKinase (USB).
Solution hybridization was performed as previously described (Li and Brow 1993). Total RNA from each strain was hybridized to 2–4 × 104 cpm of the desired DNA oligomer (U1-SH with U4-14B, or U6-SH) in 50 mM Tris [pH 7.5], 1 mM EDTA, 150 mM NaCl, for 15 min at 37°C. Following hybridization, the DNA/RNA hybrids were analyzed by nondenaturing 9% PAGE (29:1 acrylamide:bisacrylamide) in 50 mM TBE at 4°C. Gels were dried under vacuum and exposed to a phosphorimager screen, which was then imaged using a Typhoon FLA 9000 at 800 V.
Whole-cell RNA was also analyzed by Northern analysis. The RNA was separated by nondenaturing 6% PAGE (29:1 acrylamide:bisacrylamide) in 50 mM TBE at 4°C. The gel was soaked in 8.3 M urea, 0.1% SDS, 60 mM Tris [pH 6.8] for 10 min to denature RNA. RNA was transferred to a Hybond-N membrane at 10 V/cm for 12–16 h at 4°C in 12 mM Tris [pH 8.0], 6 mM NaOAc, 0.3 mM EDTA, and crosslinked to the membrane using a Stratagene Stratalinker at 1200 mJ for 90 sec. Membranes were prehybridized in 6× SSC, 0.2% SDS, 10× Denhardt's solution for 1.5–2 h at 65°C and hybridized to 32P-labeled U4-14B or U6-SH oligonucleotide (∼1 × 106 cpm/mL) in 6× SSC, 0.2% SDS, 5× Denhardt's solution for 16–18 h at 25°C. Following hybridization, membranes were washed in 6× SSC, 0.2% SDS twice for 10 min at 25°C and once for 10 min at 37°C and imaged as described for the solution hybridization.
Whole-cell extract preparation and snRNP analysis
Whole-cell extracts were prepared from wild-type or U6–A62G/A91G, U4–G14U strains using an adaptation of the liquid nitrogen method (Umen and Guthrie 1995). Two liters of each strain were grown until OD600 = 3–3.5 in YEPD. Cells were harvested by centrifugation and washed with AGK buffer (10 mM HEPES [pH 7.9], 1.5 mM MgCl2, 200 mM KCl, 10% glycerol). Cells were resuspended in 0.4 volumes AGK buffer with 1 mM PMSF and frozen in droplets in liquid nitrogen. The frozen droplets were ground to a fine powder for 5 × 3 min at 10 Hz in a Retsch MM400 Mixer Mill in 50 mL stainless steel jars with a 2.5-cm stainless steel ball, cooling the jars for 3 min in liquid nitrogen between cycles. The powder was thawed in a centrifuge tube rapidly at 25°C and centrifuged at 34,800g for 30 min at 4°C in a 70.1 Ti rotor. Samples were frozen in 100 µL aliquots in liquid nitrogen and stored at –80°C. Protein concentration as determined by Bradford assay using BSA as a standard was 15–25 mg/mL.
snRNPs were analyzed essentially as described (Raghunathan and Guthrie 1998a). Whole-cell extracts were incubated with or without 1 mM ATP in 2.5 mM MgCl2, 3% PEG 8000, 60 mM potassium phosphate [pH 7.0], 2 mM spermidine for 30 min at 30°C, 15 min at 23°C or kept on ice. For RNase H digestion, reactions were incubated under the 30°C conditions with 200 ng U2seq oligonucleotide and 1 mM ATP in a 10 µL final volume. Deproteinized RNA was isolated from cell extracts using phenol/chloroform as described above for whole cells. Samples were analyzed by nondenaturing 4% PAGE (80:1 acrylamide:bisacrylamide), 50 mM Tris base, 50 mM glycine, 2 mM MgCl2 (10 V/cm, 5 h) at 4°C. RNA was transferred to a Hybond-N membrane (Amersham) for 16–18 h at 2.3 V/cm (1500 mA) in 50 mM sodium phosphate [pH 6.5] at 4°C. RNA was crosslinked to the membrane, and membranes were probed as described above using the U4–14B, U6-SH, SRU2, and U5B DNA oligomers.
NMR sample preparation
DNA templates for all 83-nt constructs were prepared through phosphorylation and ligation of complementary, overlapping oligonucleotides (Integrated DNA Technologies) into pUC19 vector (New England Biolabs). A BsaI restriction site was included at the end of the template to allow for run-off transcription after digestion with BsaI enzyme (NEB). RNA was transcribed in vitro using purified His6-tagged T7 RNA polymerase (Milligan and Uhlenbeck 1989) in 40 mM TrisCl [pH 8.0], 1 mM spermidine, 0.01% Triton X-100, and 38 mM MgCl2. Nucleoside triphosphate concentrations were adjusted stoichiometrically to the RNA sequence with the lowest concentration at 5 mM. 13C–15N labeled samples were prepared in the same manner as the unlabeled RNA using 13C–15N labeled nucleotides (Cambridge Isotope Laboratories) and 5 mM GMP. RNA samples were purified using denaturing 6% polyacrylamide gel electrophoresis with 8 M urea. Impurities were removed by DEAE anion-exchange chromatography (Bio-Rad), using a low salt buffer (20 mM TrisCl [pH 7.6], 200 mM NaCl) to wash the sample and a high salt buffer (20 mM TrisCl [pH 7.6], 1.5 M NaCl) to elute the RNA. Each sample was heated to 90°C and cooled immediately on ice. Samples were dialyzed into 2 L of 20 mM sodium acetate [pH 6.0], for 24 h at 4°C and concentrated for NMR experiments using Millipore Amicon Ultra centrifugal filter devices (10-kDa molecular weight cutoff).
NMR spectroscopy
All spectra were obtained on Bruker Avance DMX 700 or 750 MHz spectrometers at the National Magnetic Resonance Facility at Madison. The spectrometers were equipped with cryogenic single z-axis gradient HCN probes. Exchangeable resonances were assigned using 1H–1H 2D NOESY spectra with a mixing time of 100 msec and 1H–15N TROSY-HSQC in 90% H2O, 10% D2O with 20 mM sodium acetate [pH 6.0] at 10°C. 1D 1H variable temperature NMR was performed in 10 mM potassium phosphate [pH 7.0]. RNA concentrations were in the range of 400–700 µM.
Temperature controlled UV spectrophotometry
UV spectrophotometry was performed using a Varian Cary 100-Bio. All RNAs were diluted to 0.6–0.8 µM in 10 mM potassium phosphate [pH 7.0], 100 mM potassium chloride. An identical cell containing the same buffer was used to monitor temperature. A matched reference cell containing buffer was used to collect background absorbance. Each sample was heated from 10°C to 95°C quickly (10°C/min) and cooled again without collecting data. The samples were heated and cooled slowly (1°C/min) eight times while monitoring the absorbance at 258 nm. Individual data sets were compared to check for RNA degradation and hysteresis, then averaged. Each experiment was conducted in triplicate.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
We thank Jon Staley and Eric Steinmetz for yeast strains and plasmids, Sharon Kwan for construction of the Prp24 RRM deletion alleles, Rebecca Breitlow, Dan Huettner, Mattie O'Sullivan, and Lynn Weaver for assistance with strain construction and spot tests, Christine Guthrie, Aaron Hoskins, Hiten Madhani, Megan Mayerle, and Stephen Tumasz for comments on drafts of the manuscript, and Dipali Sashital, Lawrence Clos II, and members of the Brow and Butcher laboratories for helpful discussions. This study made use of the National Magnetic Resonance Facility at Madison, which is supported by National Institutes of Health (NIH) grants P41RR02301 (BRTP/National Center for Research Resources) and P41GM103399 (National Institute of General Medical Sciences [NIGMS]). Additional equipment was purchased with funds from the University of Wisconsin, the NIH (RR02781, RR08438), the NSF (DMB-8415048, OIA-9977486, BIR-9214394), and the USDA. We thank all of the NMRFAM staff for technical support. J.E.B. was supported by NIH Predoctoral training grant T32 GM07215 (NIGMS) and the Wharton Predoctoral fellowship. This work was supported by NIH grant GM065166 (NIGMS) to D.A.B. and S.E.B.
Footnotes
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.048421.114.
REFERENCES
- Bell M, Schreiner S, Damianov A, Reddy R, Bindereif A 2002. p110, a novel human U6 snRNP protein and U4/U6 snRNP recycling factor. EMBO J 21: 2724–2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bringmann P, Appel B, Rinke J, Reuter R, Theissen H, Lührmann R 1984. Evidence for the existence of snRNAs U4 and U6 in a single ribonucleoprotein complex and for their association by intermolecular base pairing. EMBO J 3: 1357–1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brow DA, Guthrie C 1988. Spliceosomal RNA U6 is remarkably conserved from yeast to mammals. Nature 334: 213–218. [DOI] [PubMed] [Google Scholar]
- Brow DA, Guthrie C 1989. Splicing a spliceosomal RNA. Nature 337: 14–15. [DOI] [PubMed] [Google Scholar]
- Brow DA, Vidaver RM 1995. An element in human U6 RNA destabilizes the U4/U6 spliceosomal RNA complex. RNA 1: 122–131. [PMC free article] [PubMed] [Google Scholar]
- Burke JE, Sashital DG, Zuo X, Wang YX, Butcher SE 2012. Structure of the yeast U2/U6 snRNA complex. RNA 18: 673–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao S, Chen SJ 2006. Free energy landscapes of RNA/RNA complexes: with applications to snRNA complexes in spliceosomes. J Mol Biol 357: 292–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng SC, Abelson J 1987. Spliceosome assembly in yeast. Genes Dev 1: 1014–1027. [DOI] [PubMed] [Google Scholar]
- Fica SM, Tuttle N, Novak T, Li NS, Lu J, Koodathingal P, Dai Q, Staley JP, Piccirilli JA 2013. RNA catalyses nuclear pre-mRNA splicing. Nature 503: 229–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fica SM, Mefford MA, Piccirilli JA, Staley JP 2014. Evidence for a group II intron-like catalytic triplex in the spliceosome. Nat Struct Mol Biol 21: 464–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortner DM, Troy RG, Brow DA 1994. A stem/loop in U6 RNA defines a conformational switch required for pre-mRNA splicing. Genes Dev 8: 221–233. [DOI] [PubMed] [Google Scholar]
- Fourmann JB, Schmitzová J, Christian H, Urlaub H, Ficner R, Boon KL, Fabrizio P, Luhrmann R 2013. Dissection of the factor requirements for spliceosome disassembly and the elucidation of its dissociation products using a purified splicing system. Genes Dev 27: 413–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghetti A, Company M, Abelson J 1995. Specificity of Prp24 binding to RNA: a role for Prp24 in the dynamic interaction of U4 and U6 snRNAs. RNA 1: 132–145. [PMC free article] [PubMed] [Google Scholar]
- Gietz RD, Schiestl RH, Willems AR, Woods RA 1995. Studies on the transformation of intact yeast cells by the LiAc/SS-DNA/PEG procedure. Yeast 11: 355–360. [DOI] [PubMed] [Google Scholar]
- Guo Z, Karunatilaka KS, Rueda D 2009. Single-molecule analysis of protein-free U2-U6 snRNAs. Nat Struct Mol Biol 16: 1154–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto C, Steitz JA 1984. U4 and U6 RNAs coexist in a single small nuclear ribonucleoprotein particle. Nucleic Acids Res 12: 3283–3293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hausner TP, Giglio LM, Weiner AM 1990. Evidence for base-pairing between mammalian U2 and U6 small nuclear ribonucleoprotein particles. Genes Dev 4: 2146–2156. [DOI] [PubMed] [Google Scholar]
- Hilliker AK, Staley JP 2004. Multiple functions for the invariant AGC triad of U6 snRNA. RNA 10: 921–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoskins AA, Friedman LJ, Gallagher SS, Crawford DJ, Anderson EG, Wombacher R, Ramirez N, Cornish VW, Gelles J, Moore MJ 2011. Ordered and dynamic assembly of single spliceosomes. Science 331: 1289–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jandrositz A, Guthrie C 1995. Evidence for a Prp24 binding site in U6 snRNA and in a putative intermediate in the annealing of U6 and U4 snRNAs. EMBO J 14: 820–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandels-Lewis S, Séraphin B 1993. Involvement of U6 snRNA in 5′ splice site selection. Science 262: 2035–2039. [DOI] [PubMed] [Google Scholar]
- Kuhn AN, Brow DA 2000. Suppressors of a cold-sensitive mutation in yeast U4 RNA define five domains in the splicing factor Prp8 that influence spliceosome activation. Genetics 155: 1667–1682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn AN, Li Z, Brow DA 1999. Splicing factor Prp8 governs U4/U6 RNA unwinding during activation of the spliceosome. Mol Cell 3: 65–75. [DOI] [PubMed] [Google Scholar]
- Kwan SS 2005. “Protein-mediated U6 RNA conformational changes in the yeast spliceosome.” PhD thesis, University of Wisconsin, Madison, WI. [Google Scholar]
- Lamond AI, Konarska MM, Grabowski PJ, Sharp PA 1988. Spliceosome assembly involves the binding and release of U4 small nuclear ribonucleoprotein. Proc Natl Acad Sci 85: 411–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesser CF, Guthrie C 1993. Mutations in U6 snRNA that alter splice site specificity: implications for the active site. Science 262: 1982–1988. [DOI] [PubMed] [Google Scholar]
- Li Z, Brow DA 1993. A rapid assay for quantitative detection of specific RNAs. Nucleic Acids Res 21: 4645–4646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Zhang W, Xu T, Ramsey J, Zhang L, Hill R, Hansen KC, Hesselberth JR, Zhao R 2013. Comprehensive in vivo RNA-binding site analyses reveal a role of Prp8 in spliceosomal assembly. Nucleic Acids Res 41: 3805–3818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madhani HD, Guthrie C 1992. A novel base-pairing interaction between U2 and U6 snRNAs suggests a mechanism for the catalytic activation of the spliceosome. Cell 71: 803–817. [DOI] [PubMed] [Google Scholar]
- Martin-Tumasz S, Richie AC, Clos LJ 2nd, Brow DA, Butcher SE 2011. A novel occluded RNA recognition motif in Prp24 unwinds the U6 RNA internal stem loop. Nucleic Acids Res 39: 7837–7847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McManus CJ, Schwartz ML, Butcher SE, Brow DA 2007. A dynamic bulge in the U6 RNA internal stem–loop functions in spliceosome assembly and activation. RNA 13: 2252–2265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McPheeters DS, Fabrizio P, Abelson J 1989. In vitro reconstitution of functional yeast U2 snRNPs. Genes Dev 3: 2124–2136. [DOI] [PubMed] [Google Scholar]
- Mefford MA, Staley JP 2009. Evidence that U2/U6 helix I promotes both catalytic steps of pre-mRNA splicing and rearranges in between these steps. RNA 15: 1386–1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milligan JF, Uhlenbeck OC 1989. Synthesis of small RNAs using T7 RNA polymerase. Methods Enzymol 180: 51–62. [DOI] [PubMed] [Google Scholar]
- Miura K, Tsuda S, Harada F, Ueda T 1983. Chemical modification of cytosine residues of U6 snRNA with hydrogen sulfide (nucleosides and nucleotides. Part 49 [1]). Nucleic Acids Res 11: 5893–5901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montemayor EJ, Curran EC, Liao HH, Andrews KL, Treba CN, Butcher SE, Brow DA 2014. Core structure of the U6 small nuclear ribonucleoprotein at 1.7-Å resolution. Nat Struct Mol Biol 21: 544–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pannone BK, Kim SD, Noe DA, Wolin SL 2001. Multiple functional interactions between components of the Lsm2–Lsm8 complex, U6 snRNA, and the yeast La protein. Genetics 158: 187–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker R, Siliciano PG, Guthrie C 1987. Recognition of the TACTAAC box during mRNA splicing in yeast involves base pairing to the U2-like snRNA. Cell 49: 229–239. [DOI] [PubMed] [Google Scholar]
- Rader SD, Guthrie C 2002. A conserved Lsm-interaction motif in Prp24 required for efficient U4/U6 di-snRNP formation. RNA 8: 1378–1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raghunathan PL, Guthrie C 1998a. RNA unwinding in U4/U6 snRNPs requires ATP hydrolysis and the DEIH-box splicing factor Brr2. Curr Biol 8: 847–855. [DOI] [PubMed] [Google Scholar]
- Raghunathan PL, Guthrie C 1998b. A spliceosomal recycling factor that reanneals U4 and U6 small nuclear ribonucleoprotein particles. Science 279: 857–860. [DOI] [PubMed] [Google Scholar]
- Reiter NJ, Nikstad LJ, Allmann AM, Johnson RJ, Butcher SE 2003. Structure of the U6 RNA intramolecular stem–loop harboring an SP-phosphorothioate modification. RNA 9: 533–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan DE, Abelson J 2002. The conserved central domain of yeast U6 snRNA: importance of U2-U6 helix Ia in spliceosome assembly. RNA 8: 997–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan DE, Kim CH, Murray JB, Adams CJ, Stockley PG, Abelson J 2004. New tertiary constraints between the RNA components of active yeast spliceosomes: a photo-crosslinking study. RNA 10: 1251–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sashital DG, Cornilescu G, McManus CJ, Brow DA, Butcher SE 2004. U2-U6 RNA folding reveals a group II intron-like domain and a four-helix junction. Nat Struct Mol Biol 11: 1237–1242. [DOI] [PubMed] [Google Scholar]
- Sashital DG, Venditti V, Angers CG, Cornilescu G, Butcher SE 2007. Structure and thermodynamics of a conserved U2 snRNA domain from yeast and human. RNA 13: 328–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawa H, Abelson J 1992. Evidence for a base-pairing interaction between U6 small nuclear RNA and 5′ splice site during the splicing reaction in yeast. Proc Natl Acad Sci 89: 11269–11273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider M, Will CL, Anokhina M, Tazi J, Urlaub H, Lührmann R 2010. Exon definition complexes contain the tri-snRNP and can be directly converted into B-like precatalytic splicing complexes. Mol Cell 38: 223–235. [DOI] [PubMed] [Google Scholar]
- Shannon KW, Guthrie C 1991. Suppressors of a U4 snRNA mutation define a novel U6 snRNP protein with RNA-binding motifs. Genes Dev 5: 773–785. [DOI] [PubMed] [Google Scholar]
- Sun JS, Manley JL 1995. A novel U2-U6 snRNA structure is necessary for mammalian mRNA splicing. Genes Dev 9: 843–854. [DOI] [PubMed] [Google Scholar]
- Trede NS, Medenbach J, Damianov A, Hung LH, Weber GJ, Paw BH, Zhou Y, Hersey C, Zapata A, Keefe M, et al. 2007. Network of coregulated spliceosome components revealed by zebrafish mutant in recycling factor p110. Proc Natl Acad Sci 104: 6608–6613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai RT, Fu RH, Yeh FL, Tseng CK, Lin YC, Huang YH, Cheng SC 2005. Spliceosome disassembly catalyzed by Prp43 and its associated components Ntr1 and Ntr2. Genes Dev 19: 2991–3003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner IA, Norman CM, Churcher MJ, Newman AJ 2006. Dissection of Prp8 protein defines multiple interactions with crucial RNA sequences in the catalytic core of the spliceosome. RNA 12: 375–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umen JG, Guthrie C 1995. A novel role for a U5 snRNP protein in 3′ splice site selection. Genes Dev 9: 855–868. [DOI] [PubMed] [Google Scholar]
- Vidaver RM, Fortner DM, Loos-Austin LS, Brow DA 1999. Multiple functions of Saccharomyces cerevisiae splicing protein Prp24 in U6 RNA structural rearrangements. Genetics 153: 1205–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahl MC, Will CL, Luhrmann R 2009. The spliceosome: design principles of a dynamic RNP machine. Cell 136: 701–718. [DOI] [PubMed] [Google Scholar]
- Wassarman DA, Steitz JA 1992. Interactions of small nuclear RNA's with precursor messenger RNA during in vitro splicing. Science 257: 1918–1925. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.