

Abstract
The small noncoding RNAs (sncRNAs) are considered as post-transcriptional key regulators of male germ cell development. In addition to microRNAs (miRNAs) and PIWI-interacting RNAs (piRNAs), other sncRNAs generated from small nucleolar RNAs (snoRNAs), tRNAs, or rRNAs processing may also play important regulatory roles in spermatogenesis. By next-generation sequencing (NGS), we characterized the sncRNA populations detected at three milestone stages in male germ differentiation: primordial germ cells (PGCs), pubertal spermatogonia cells, and mature spermatozoa. To assess their potential transmission through the spermatozoa during fertilization, the sncRNAs of mouse oocytes and zygotes were also analyzed. Both, microRNAs and snoRNA-derived small RNAs are abundantly expressed in PGCs but transiently replaced by piRNAs in spermatozoa and endo-siRNAs in oocytes and zygotes. Exhaustive analysis of miRNA sequence variants also shows an increment of noncanonical microRNA forms along male germ cell differentiation. RNAs-derived from tRNAs and rRNAs interacting with PIWI proteins are not generated by the ping-pong pathway and could be a source of primary piRNAs. Moreover, our results strongly suggest that the small RNAs-derived from tRNAs and rRNAs are interacting with PIWI proteins, and specifically with MILI. Finally, computational analysis revealed their potential involvement in post-transcriptional regulation of mRNA transcripts suggesting functional convergence among different small RNA classes in germ cells and zygotes.
Keywords: sncRNAs, germ cell development, miRNAs, piRNAs, snoRNAs, next-generation sequencing
INTRODUCTION
Noncoding RNAs (small and long) play key roles as negative post-transcriptional gene regulators in cell differentiation and development (Gomes et al. 2013). Small noncoding RNAs (sncRNAs) comprise different types: microRNAs (miRNA); repeat-associated RNAs (rasRNAs) including PIWI-interacting RNAs (piRNAs), and endogenous small interfering RNAs (endo-siRNAs). Other classes of small noncoding RNAs are those derived from tRNAs, rRNAs, and small nucleolar RNAs (snoRNAs) that could share functions with miRNAs but their roles and biogenesis have not been well established. All these different types of RNAs differ in their biogenesis as well as their functional activity. The miRNAs act through RNA-induced silencing complex as negative regulators of gene expression, and are present in all cells. Targeting occurs through partial complementarity between the miRNA and the target mRNA (Huang et al. 2010; Leonardo et al. 2012). Although the functional mechanisms of action of rasRNAs (principally endo-siRNAs and piRNAs) and other sncRNAs are less clear, interactions with complementary RNAs and/or chromatin-modifying complexes have been described (Castel and Martienssen 2013; Cook and Blelloch 2013; Gomes et al. 2013).
Germ cell specification, differentiation, and development involve complex regulatory networks at both transcriptional and post-transcriptional levels, including epigenetic mechanisms and the participation of small noncoding RNAs. In mammals, Primordial Germ Cells (PGCs) are derived from pluripotent proximal epiblast cells of the embryo (McLaren 2003). In males, these cells differentiate into gonocytes (also named pro-spermatogonia) entering in mitotic arrest at 13.5 d post-coitum (dpc) in the mouse. Gonocytes resume mitotic proliferation after birth to become spermatogonial cells which undergo spermatogenic differentiation and give rise to meiotic spermatocytes, haploid spermatids, and finally the most differentiated mammalian germ cell: the spermatozoa (Nagano and Yeh 2013). In contrast, female PGCs enter meiosis during the same developmental period that male gonocytes enter mitotic arrest. The oocyte at birth is arrested at meiotic prophase I and resumes meiosis to metaphase II at ovulation in response to luteinizing hormone. As consequence of fertilization the oocyte completes the meiotic process generating the zygote that starts a critical reprogramming of gene expression (Rivera and Ross 2013; Teperek and Miyamoto 2013).
In contrast to somatic cells, all types of sncRNAs are present in germ cells making them good candidate regulators of germline development and fertilization. The presence and functions of sncRNAs in mammalian spermatogenesis have been analyzed in different studies (Papaioannou and Nef 2010; McIver et al. 2012; Modzelewski et al. 2012; Yadav and Kotaja 2014). In particular, the miRNAs have been defined as robust regulators of germ cell development (Kedde and Agami 2008). The piRNAs were discovered in developing male germ cells but their analysis has been mainly restricted to their presence in meiocytes (Aravin et al. 2006; Girard et al. 2006; Grivna et al. 2006a; Grivna et al. 2006b; Lau et al. 2006). On the other hand, endo-siRNAs were initially associated to oocytes (Tang 2010; Banisch et al. 2012) and their functional presence in male germ cells has not been assessed.
The involvement of other sncRNAs during germ cell differentiation remains still unknown. In this sense, small RNAs derived from ribosomal RNAs (rRNAs) and small RNAs derived from transfer RNAs (tRNAs) have emerged as new small molecules with important regulatory roles during cell differentiation processes (Deato and Tjian 2008) and cancer (Martens-Uzunova et al. 2013). In addition, small RNAs derived from small nucleolar RNAs (snoRNAs) have been reported to have miRNA-like functions (Falaleeva and Stamm 2013). The snoRNAs are involved in preribosomal processing reactions and guide methylation and pseudouridylation of rRNAs, spliceosomal small nuclear RNAs, and other cellular RNAs (Watkins and Bohnsack 2012) in a process which involves RNA:RNA complementarity events similar to observed for miRNAs, piRNAs or endo-siRNAs. Classically, the snoRNAs have been grouped into C/D Box, H/ACA Box, and Cajal Body classes, depending on their functional activity and their sequence motifs (for review, see Watkins and Bohnsack 2012).
The comparative analysis of small noncoding RNAs in early spermatogenic stages and terminally differentiated male germ cells, represented by the spermatozoa, could allow the progress in defining the role of these regulatory RNAs in male germ cell differentiation. Moreover, similar analysis in oocytes before and after fertilization will contribute to our understanding of the potential role of these RNAs in early post-zygotic development during embryogenesis. Therefore, in the present study, we characterize different populations of small ncRNAs by next-generation sequencing (NGS) comparing the early male gametogenic cells: PGCs at 13.5 dpc, spermatogonia cells at 8.5 d post-natal (dpn) and the final differentiated spermatozoa cells. The data were compared with those obtained from sncRNA populations detected in oocytes and zygotes to decrypt the potential sncRNA contribution from the spermatozoa to the zygote. Moreover, we delve into the miRNA roles during early germ cell development analyzing the mRNA transcriptome profiles of mouse PGCs and spermatogonia cells.
Globally, the landscape of sncRNAs showed the preponderance of miRNAs in PGCs and spermatogonia cells with respect to the other types of sncRNAs. In contrast, rasRNAs were the most abundant type of sncRNAs in both the terminal differentiated gametes and in zygotes. Finally, we further characterized the group of snoRNAs-derived small RNAs that could act as microRNA-like molecules during germ cell sex differentiation, development, and fertilization.
RESULTS
Libraries from small RNAs (18–32 nt) were prepared from total RNA isolated from mouse male PGCs, spermatogonia cells, spermatozoa, oocytes, and zygotes (see Material and Methods) and sequenced using next-generation sequencing (NGS) technology. After adapter trimming and removal of low quality sequences, the analysis of small RNA libraries returned >30 million of reads in each sample corresponding to an average of 1.6 million of different sequences (Table 1).
TABLE 1.
Global analysis of small RNA population
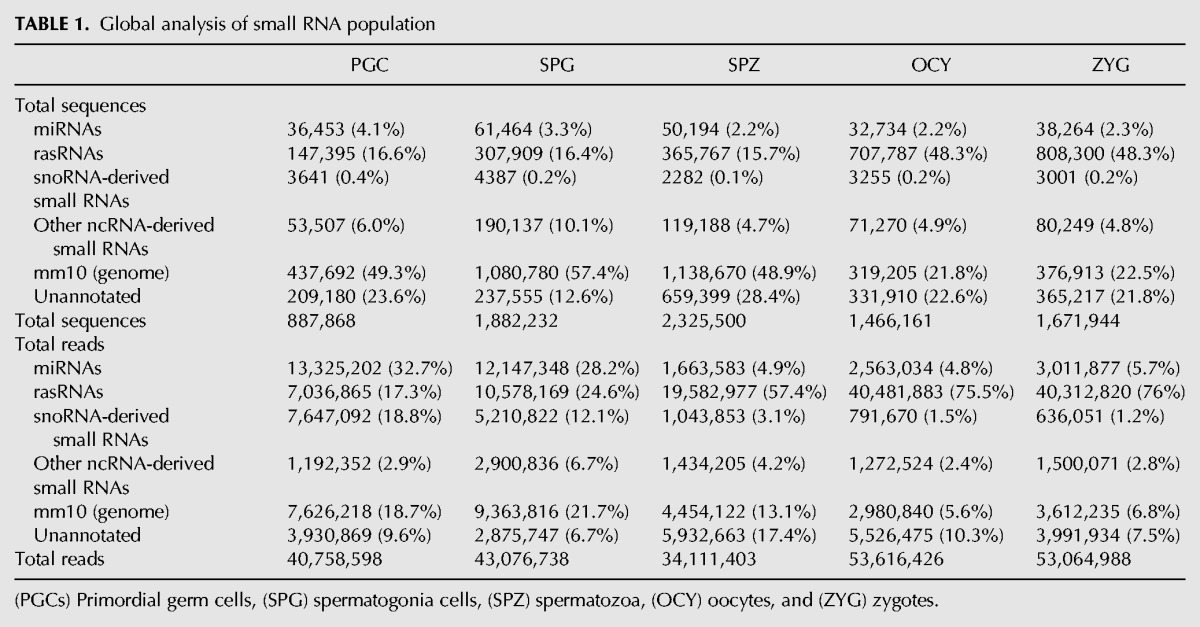
By means of bioinformatics analysis we classified all these RNA sequences according to their reference identities retrieved from public databases and following different biogenesis pathways (Supplemental Fig. 1). The RNA sequences were grouped into four major categories: miRNAs, rasRNAs, snoRNAs-derived, and other ncRNA-derived small RNAs (including heterogeneous small RNAs reported in the Rfam database). Those sequences not included in these categories but aligned with any region of the mouse genome were grouped under the “mm10 genome” (version 10 of the mouse genome) category. Finally, the small RNA remaining sequences with no identity in any annotated genome region were classified as “unannotated” (Table 1; Supplemental Fig. 1).
The comparative analysis in the different cell types showed that miRNAs are the major group in PGC and spermatogonia cells, while for both gametes and zygotes, the principal read accumulation corresponds to rasRNAs (Table 1). This suggested that the roles of the rasRNAs are mainly associated with final steps in the gamete differentiation and maintained in the zygote. However, miRNAs could have a predominant role associated with the pluripotency of PGCs. In fact, although the proportion of miRNAs was also high in spermatogonia cells, we observed a decrease in miRNA reads with respect to PGC. This suggests a correlation between abundance of miRNA and cell self-renewal processes and pluripotency (Leonardo et al. 2012; Choi et al. 2013). Curiously, the level of expression of snoRNAs-derived small RNAs decreased from PGCs to spermatozoa differentiating cells (Table 1).
Analysis of the miRNA population
The number of miRNAs detected in each sample varied from 964 different miRNAs detected in spermatogonia cells to 720 detected in oocytes. Hierarchical clustering analysis of miRNA expression showed, as expected, similar profiles of miRNAs in PGC and spermatogonia cells on the one hand and in oocytes and zygotes on the other (Fig. 1A). Although the proportion of sequences classified as miRNAs was relatively similar in all samples, the total reads corresponding to miRNAs were more than fivefold higher in PGC and spermatogonia cells compared with the other cell types (Table 1). This initial paradox about similar numbers of miRNA detected but significant differences in the accumulation, i.e., in their expression levels, suggested that different regulatory mechanisms operated in the miRNA biogenesis in each sample. Analyzing the global characteristics of miRNA sequences for each cell type we detected high variability in the sequence length that indicates laxity during miRNA precursor processing. To test this hypothesis of lax precursor processing, we classified the miRNA sequences as canonical and noncanonical forms. An example of sequence variants is shown in the Figure 2A. The proportion of noncanonical forms was increased according to the progress of differentiation process. In spermatozoa (84%) the rate of noncanonical forms was double that of PGC (42%), and was also higher than in both oocytes (68%) and zygotes (67%) (Fig. 2B). This suggested more efficient processing of miRNA precursors in early differentiating male germ cells. The increased proportion of sequences classified as “noncanonical processing” in both gametes and the zygote could represent presence in these cells of functional hairpin loops as was recently reported (Okamura et al. 2013). However, mature forms without sequence changes seem to be positively selected in the five cellular types studied, because the number of reads was higher than other miRNA variant forms (Fig. 2B). On the contrary, the low rate of sequences that showed one polymorphism in the first nucleotide indicates a negative selection of miRNAs with changes in this position (Fig. 2B). This nucleotide is considered critical during strand miRNA selection, enhancing the assembly of Argonaute proteins and the miRNAs to the RISC complex (Seitz et al. 2011).
FIGURE 1.

miRNA profiling of male germ cells, oocytes, and zygotes. (A) Heat map with unsupervised hierarchical clustering of miRNA detected in the five cell types studied: primordial germ cells (PGCs), spermatogonia cells (SPG), spermatozoa (SPZ), oocytes (OCY), and zygotes (ZYG). The color scale shown in the map illustrates the relative expression levels (in logarithmic scale) of each miRNA across all samples: Red represents an expression level above the mean; green represents expression lower than the mean. Black represents median expression. (B) Cluster of miRNAs differentially expressed in spermatozoa (SPZ). (C) Cluster of miRNAs differentially expressed in PGC and spermatogonia cells (SPG). (D) Cluster of miRNAs down-regulated in spermatozoa (SPZ). (E) Cluster of miRNAs highly expressed in all cell types. (F) Cluster of miRNAs differentially expressed in spermatozoa (SPZ) and zygotes (ZYG).
FIGURE 2.
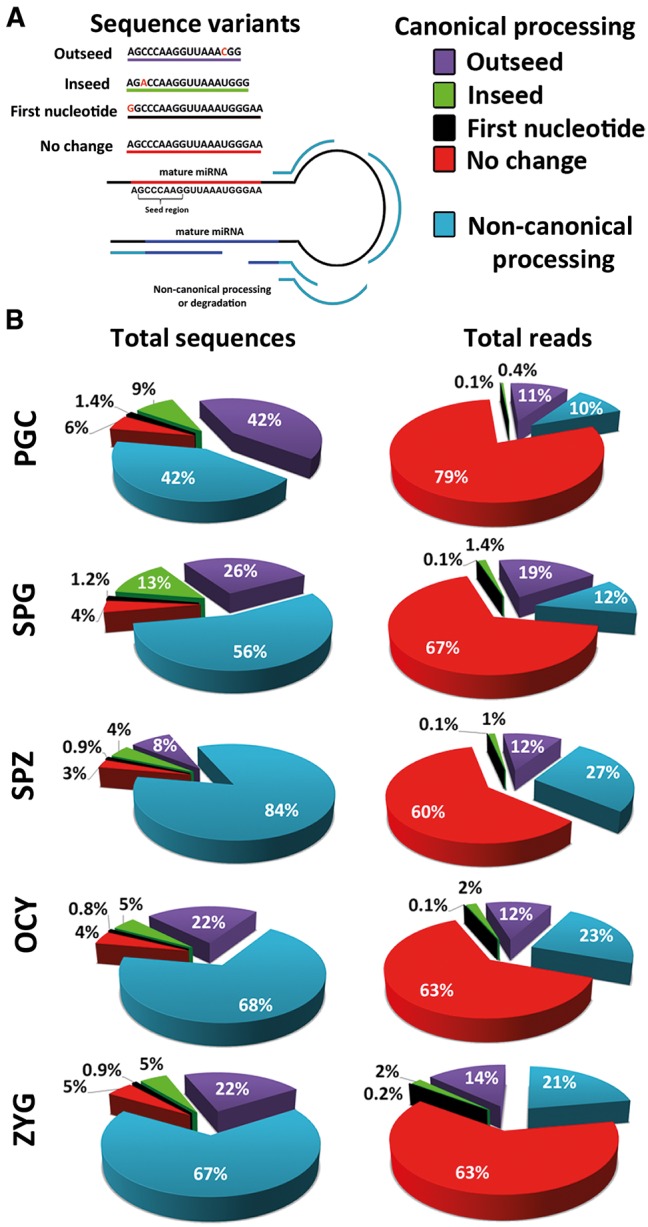
Analysis of miRNA sequence features. (A) The image shows the categories used to classify all the sequences mapped into miRNA precursor molecules. (B) The circular diagrams show the percentage of sequences and reads associated with each sequence feature category.
Of the total of 1316 miRNAs detected in all cell types studied, 37% (478 miRNAs) were detected in all cell samples (Supplemental Fig. 2). Spermatozoa contained the highest number of specific miRNAs (140 miRNAs), whereas PGC and spermatogonia cells showed the highest number of shared miRNAs (745 miRNAs) (Supplemental Fig. 2). Moreover, common miRNAs in PGCs and SPGs showed similar profiles of expression level (Fig. 1C). In PGCs and spermatogonia cells, we detected 867 and 964 distinct miRNAs, respectively. However, >75% of total reads (quartile Q3) corresponded to 20 miRNAs in PGCs and to 27 in spermatogonia cells (Supplemental Table 1). To assess the potential functions of highly expressed miRNAs that we detected in PGCs and spermatogonia cells, we searched their validated targets by bioinformatics approaches. Most of validated targets of such highly expressed miRNAs are mainly involved in MAPK signaling pathway, Jak-STAT signaling pathway and TGF-β signaling pathway (Supplemental Fig. 3). All highly expressed miRNAs detected in PGCs were also detected in spermatogonia cells. So, in order to assess the different regulatory pathways where miRNAs of PGC and spermatogonia cells were involved, we searched the specific miRNAs detected in each cell type. We detected a small set of miRNAs that were specifically expressed in PGCs or spermatogonia cells. For example, as specifically expressed in PGCs we found the miR-302 family members (miR-302a-3p/-5p; miR-302d-3p; miR-302c-3p), whose validated target gene is cyclin-dependent kinase inhibitor 1A (Cdkn1a). Also specifically expressed in PGCs, we detected the miR-7662-5p and the miR-421-5p. However, these last two miRNAs have not validated targets. Despite their specificity, all these miRNAs were poorly expressed (below of 100 reads). Similarly, the most representative of unique miRNAs in spermatogonia cells were miR-3102-5p.2-5p and miR-1188-5p. Neither of them has validated target genes and showed a level of expression corresponding to below 50 reads.
Despite detecting 745 common miRNAs in PGC and spermatogonia cells, both cell types presented differences in their miRNAomes. To assess the functional consequences of such differences, we compared the miRNA data obtained by NGS with mRNA profiles in PGCs and spermatogonia cells that we obtained from microarray analysis of the global transcriptome. For comparative purposes, total RNA from both whole adult testis, as source of tissue expressing all male germ cells, and 13.5 dpc embryos, that were depleted of their gonads before to RNA isolation (“gonad-less” embryos), as source of somatic-line; were also analyzed and used for normalization. At the level of global transcriptome, the 14,569 genes detected were grouped in clusters relating to their expression profiles of gene expression, to define some signatures of mRNA expression in PGCs and spermatogonia cells (Supplemental Fig. 4). By Gene Ontology tools we selected genes involved in biological processes concerning to germ cell differentiation. Differential expression patterns of these genes in PGCs and spermatogonia cells were observed and reported (Fig. 3). Regulation of gene expression during differentiation involves upstream effectors and post-transcriptional regulators. To assess the contribution of miRNAs down-regulating their validated mRNA targets we combined the data obtained by NGS and transcriptome arrays. We identified a group of miRNAs that showed inversed correlation profiles with their respective targets in PGCs or spermatogonia cells. For example, Pten and Stat3 increased their expression levels in spermatogonia cells respect to the PGCs. Pten is a validated target of miR-21a-5p, miR-17-5p, miR-20a-5p, miR-18a-5p, and miR-19b-3p. Similar occurs with Stat3 and miR-93-5p, miR-20a-5p, and miR-17-5p (Fig. 4). As expected, the accumulation rate of Pten and Stat3 transcripts was reverse to the relative expression of their potential regulatory miRNAs, suggesting important roles of these miRNAs in Pten and Stat3 regulation in the differentiation of PGCs to spermatogonia cells (Fig. 4).
FIGURE 3.

Gene expression profiling of genes involved in germ cell development and differentiation. Bar charts show the log2 expression ratios obtained by microarray analysis.
FIGURE 4.

Comparative analysis of miRNA expression levels in relation with fold change of their validated mRNA targets in PGCs and spermatogonia cells (SPG).
Members of miRNA clusters have been reported as coregulated in differentiation processes. However, only two members of the cluster miR-183/96/182: miR-182-5p and miR-183-5p; accumulated >35% of total miRNA reads detected in PGCs. In spermatogonia cells only 3.5% of total miRNA reads corresponded to miR-182-5p. This suggests a potential role of specific members of this cluster in germ cell differentiation showing down-regulation in the transition from PGCs to spermatogonia cells (Supplemental Table 1).
Similarly, the members of the family miR-290-295 that promote pluripotency are also located into a genome cluster (Lichner et al. 2011; Kaspi et al. 2013). Corroborating previous studies carried out by real-time PCR (Hayashi et al. 2008) we detected by deep sequencing that the members of this family showed their highest expression levels in PGCs (Fig. 1D). The data stressed the role of this miRNA family in cell pluripotency. In addition, RT-qPCR analysis of miR-290-295 family served us to validate the data obtained by NGS results (Supplemental Fig. 5).
In spermatozoa, mRNAs were barely detectable. However, these cells are highly rich in miRNAs. Analyzing the miRNA population of spermatozoa, we found that the vast majority of miRNAs accumulated in spermatozoa were also detected in spermatogonia cells but showing different expression levels (Supplemental Table 1). For example, one of them, the miR-143-3p, that has been reported as involved in cell proliferation and differentiation (Huang et al. 2012; Fan et al. 2013), was the highest accumulated in spermatozoa being also the most abundant in spermatogonia cells. This miRNA was detected in all cell types, but especially in spermatogonia cells and spermatozoa (Supplemental Table 1). The transient increase of miR-143-3p expression from PGC to spermatogonia cells and spermatozoa suggested that this miRNA could be involved in negative control of cell proliferation driving the germ cell differentiation needed when meiosis is initiated. This occurs soon after 8.5 dpn in most of the mouse spermatogonia and agrees with its role as tumor suppressor (Noguchi et al. 2011; Pramanik et al. 2011). In stark contrast, miR-143-3p and miR-34c-5p showed similar expression level in spermatozoa. However, miR-34c-5p was far less abundant in SPG (0.09%), suggesting specific roles in late stages of spermatogenesis. Although miR-34c family has been considered as spermatozoa-specific (Liu et al. 2012) we were able to detect the most abundant strand: miR-34c-5p in all cell types analyzed (Fig. 1D). The other strand of miR-34c, miR-34c-3p along with other members of this family including miR-34b-3p, miR-34b-5p, were also overexpressed in spermatozoa with respect to the rest of samples (Fig. 1B).
The miRNAs detected as common in spermatozoa and ZYG but not present in oocytes are the perfect candidates to be transferred from spermatozoa to the zygotes. A total of 48 miRNAs showing this characteristic was detected (Supplemental Fig. 2), but their expression levels were low in both samples (<100 reads). In fact, the most expressed of these 48 miRNAs was the mmu-miR-125b-2-3p accumulating 65 reads in spermatozoa and 37 reads in zygotes. Similarly, a smaller group of miRNAs were expressed more highly in spermatozoa and zygotes than in the other cell types. These included two miRNAs families such as miR-3473 and miR-221 (Fig. 1F). Although we cannot underestimate the potential relevance of these particular miRNAs in early development, the global analysis of miRNAs in spermatozoa, oocytes, and zygotes revealed that miRNAs detected in zygotes are mainly from maternal origin as reflects the similar expression profiles of miRNAs in oocytes and zygotes (Supplemental Table 1).
rasRNAs involved in gametogenesis
Between 17% and 76% of total small RNA reads that we detected by NGS mapped to highly repeated genome elements and were classified under the repeat-associated small RNAs (rasRNAs) category (Table 1). In male germ cells, the relative expression rates of rasRNAs increased with the differentiation from PGC to spermatozoa. In spermatozoa, these rasRNAs reached >50% of the total reads detected (Table 1), and were even higher in oocytes and zygotes, suggesting important roles for rasRNAs during the last stages of female gametogenesis and early embryogenesis. As shown in Figure 5, the rasRNAs detected mapped to three types of transposable elements, LTRs, LINEs, SINE, and two ncRNAs, tRNAs, and rRNAs. Moreover, and mainly in male germ cells, a restricted group of small RNAs, mapped to genome regions enriched in non-TE derived piRNAs (Fig. 5).
FIGURE 5.

Analysis of rasRNA population. The circular diagrams show the read percentage of rasRNAs associated with different genomic elements. (A–C) Pie charts show the results of rasRNA mapping reads in male germ cells. (D,E) Pie charts show the results of rasRNA mapping reads in oocytes (OCY) and zygotes (ZYG), respectively.
In general, germline rasRNAs have been included into two major categories: piRNAs and endo-siRNAs (Yang et al. 2010; García-López et al. 2014) sharing functions as safeguards of genome integrity but differing in their biogenesis pathways. First, we classified as piRNAs the small sequences detected by NGS identical to those previously reported in the RNA database (RNAdb) (Supplemental Fig. 1). The RNAdb contains small RNA sequences classified as piRNAs on the basis of their inmunoprecipitation with PIWI proteins (Aravin et al. 2006; Girard et al. 2006; Lau et al. 2006). This initial approach revealed that PIWI proteins could interact with a miscellaneous group of small RNAs as transposon-derived small RNAs, tRNA-derived small RNAs, or rRNA-derived small RNAs (Supplemental Fig. 6). The sequences that could not be classified as either miRNAs or piRNAs using the aforementioned criteria were classified using instead biogenesis criteria. The endo-siRNAs arise from dsRNA that occur as result of convergent transcription of gene and pseudogene, as well as from transposons and other transcripts which are sense and antisense in different loci of the genomes (Röther and Meister 2011). Hence, the rasRNA sequences that mapped in the genome sequences with these characteristics were classified as endo-siRNAs. Conversely, and according to published criteria (Beyret et al. 2012), the rasRNAs with sense transcription that showed a small number of mapping hits (<10 hits) on the genome were classified as piRNAs (Table 2). According to this classification, we observed clear differences between the types of rasRNAs detected in male germ cells and those detected in oocytes or zygotes (Table 2; Supplemental Fig. 6). Whereas in male germ cells the predominant rasRNA group consisted of piRNAs, in oocytes and zygotes these were predominantly of the endo-siRNAs, accumulating the 64% and >76% of total reads, respectively.
TABLE 2.
Analysis of rasRNA population
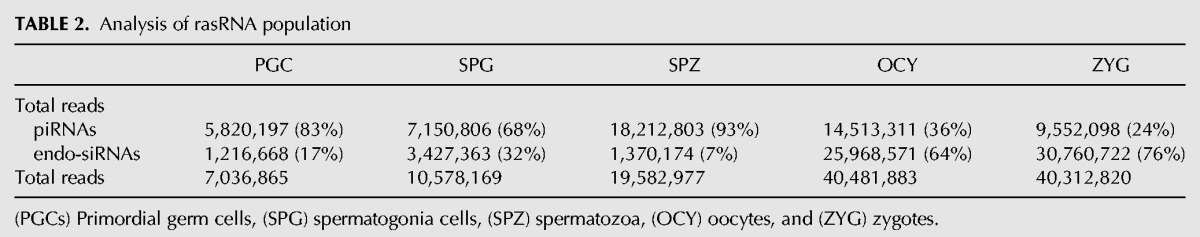
Based on mapping results we identified a large group of piRNAs that could arise from tRNAs and rRNAs processing. In male germ cells, the small sequences derived from tRNAs or rRNAs represented more than half of total rasRNA sequences (Fig. 5). In contrast, their relative expression rates decreased in oocytes (29% of total rasRNA reads) and zygotes (14% of total rasRNA reads) (Fig. 5). Curiously, between 54% and 92% of the rRNA-derived small RNAs detected, were previously included in RNAdb as piRNAs (Fig. 5). This indicates that small RNAs derived from rRNA processing interact with PIWI proteins. The rRNA-derived piRNAs were highly accumulated in spermatozoa as we also previously reported (García-López et al. 2014). Although some authors support that PIWI proteins and piRNAs are depleted in late spermatogenesis stages (Zhao et al. 2013), others have hypothesized that spermatozoa piRNAs play important roles during fertilization and embryonic genome activation (Krawetz et al. 2011). Here we report that piRNAs exist in spermatozoa and, rather than being eliminated, their levels actually increase through spermatogenesis.
We also detected tRNA-derived small RNAs, out of which one-third has been previously included in RNAdb, indicating their binding to PIWI proteins. However, the different PIWI-association rates detected in rRNA-derived with respect to tRNA-derived small RNAs (Fig. 5), suggest that the tRNA-derived could participate in other silencing pathways.
In the piRNA biogenesis a complex mechanism known as the ping-pong cycle is involved mediated by the overlapping of the first 10 nt of sense and target antisense RNAs (Brennecke et al. 2007; Gunawardane et al. 2007; O'Donnell and Boeke 2007). It was observed a singular signature in the sequences that entered into the ping-pong cycle including a bias for 5′ uracil in the first nucleotide position (5U) (primary piRNAs) or adenine at the tenth position (10A) (secondary piRNAs) (Brennecke et al. 2007; Gunawardane et al. 2007; Grimson et al. 2008). To assess the involvement of rRNA-derived and tRNA-derived small RNAs in the piRNA pathway, we first analyzed the sequence features of both small RNA populations, related to the ping-pong cycle. In all cell types, the highly expressed sequences corresponding to tRNA-derived sequences fit into “primary” piRNA group or “other” (different to 5U, 10A, or 5U10A) piRNA group (Fig. 6A). Consequently, the sequences showing signatures proper to secondary piRNAs and those showing both motifs, 5′ uracil at first and adenine at tenth positions (5U10A), were underrepresented. These results indicate that tRNA processing could serve as a source of primary piRNAs during gametogenesis and fertilized oocytes. In contrast, the analysis of rRNA-derived sequences showed that sequences without ping-pong signatures represented >80% of normalized reads detected in male germ cells (Fig. 6A). These results strongly diverged from data obtained in oocytes and zygotes where the expression levels of rRNA-derived sequences including an adenine at the tenth position (secondary piRNAs) were significantly increased (Fig. 6A).
FIGURE 6.

Analysis of tRNA- and rRNA-derived small RNAs. (A) The bar charts show the analysis of sequence features observed in tRNA- and rRNA-derived small RNAs. (B) The pie charts show the results of ping-pong partner quest using the small sequences derived from tRNAs. (C) The pie charts show the results of ping-pong partner quest using the small sequences derived from rRNAs.
To analyze whether these different sequence features could be related to functional activities of piRNAs through ping-pong mechanisms, we tested the complementary binding of tRNA- and rRNA-derived with transposons or other antisense transcripts (see Materials and Methods). The results confirmed that both tRNA- and rRNA-derived small RNAs, present complementarity not only with small sequences derived from TEs (Fig. 6B,C), but also with piRNAs arisen from non-TE transcripts. However, most of the tRNA- and rRNA-derived sequences lack ping-pong signatures and complementary sequences necessary for ping-pong cycle, suggesting that these piRNAs are not participating in such amplification cycle.
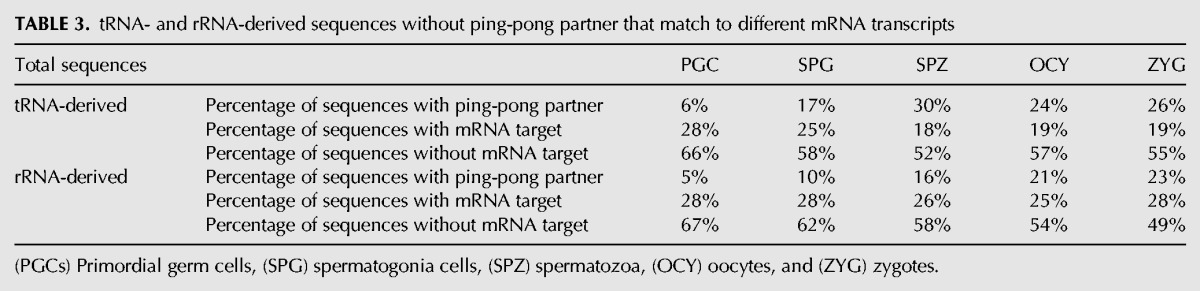
To assess whether tRNA- or rRNA-derived RNAs could also be acting as potential regulators of mRNAs transcripts, the sequences detected as having no ping-pong partner were aligned in a reverse complementary manner with those sequences contained in the Mus_musculus.GRCm38.75.cdna database (Table 3). From this analysis, between 18% and 28% of tRNA- and rRNA-derived sequences could potentially bind to mRNA sequences (Table 3). One example of potential mRNA transcript regulation by tRNA-derived piRNAs was found in the PIR59423 and ornithine decarboxylase antizyme 1 (Oaz1) mRNA. In testis, the mRNA (Oaz-t) encoding OAZ-t protein, was specifically expressed in haploid germ cells and involved in the formation of the junction between the head and the sperm tail (Tokuhiro et al. 2009). In contrast, the mRNA level of somatic OAZ1 decreased markedly at the late stages of haploid germ cell differentiation (Tosaka et al. 2000). OAZ1 differ from OAZ-t in a functional region that binds to ornithine decarboxylase (ODC). The PIR59423 derives from tRNA-Gln-CAG processing and was classified as primary piRNA (5′-TCCCTGGTCTAGTGGTTAG). We detected that PIR59423 increased their expression levels from PGC and SPG to SPZ 1.7- and 1.5-fold, respectively. The binding region of PIR59423 is just in the differential sequence region between Oaz1 and Oaz-t. If PIR59423 acts on the regulation of Oaz1, Oaz-t could be skipped for the negative regulation mediated by this piRNA. This suggests a double role of piRNAs derived from tRNAs and rRNAs that could act as safeguards of genome integrity preventing TE insertion, and alternatively acting as gene expression regulators.
TABLE 3.
tRNA- and rRNA-derived sequences without ping-pong partner that match to different mRNA transcripts
Among all small RNAs identified in this study, the most abundant sequence found in male germline cells was CGACTCTTAGCGGTGGATCACTC. This sequence has been previously annotated as PIR64428 after its recovery through inmunoprecipitation assays with PIWI proteins (Aravin et al. 2006; Girard et al. 2006; Lau et al. 2006). We mapped this sequence to the Rs5.8s1 RNA, which is a product of ribosomal RNA (rRNA) processing. Neither a ping-pong partner nor mRNA target was detected for PIR64428. Initially, the function of this piRNA is a clue. However, in oocytes and in zygotes the PIR64428 expression levels were significantly lower than in male germ cells, suggesting some enhanced roles during male germline differentiation and especially during spermiogenesis. The relative expression level of the PIR64428 was also confirmed by RT-qPCR along with other differentially expressed rasRNAs in the different cell types (Supplemental Fig. 7).
Analysis of snoRNAs-derived small RNAs and other ncRNA-derived small RNAs
According to our sequencing data analysis, between 5% and 10% of the sequences mapped to ncRNAs were neither miRNAs nor rasRNAs (Table 1). In this category, a miscellaneous group of sequences mapped to snoRNAs or long noncoding RNAs (lncRNAs) (Supplemental Fig. 8). We subclassified these sequences into two different classes: snoRNAs-derived and other ncRNA-derived small RNAs (Table 1). Interestingly, those sequences mapping to small nucleolar RNAs (snoRNAs) showed higher expression levels in PGCs and spermatogonia cells. We performed a systematic analysis of small RNAs mapped to snoRNAs and found evidence for snoRNA processing into small RNAs in male germ cells, oocytes, and zygotes. As described previously (Taft et al. 2009; Brameier et al. 2011; Falaleeva and Stamm 2013), we found snoRNAs-derived from C/D Box and H/ACA Box RNAs, but also from the third type of snoRNAs known as Cajal bodies. The small RNAs derived from C/D box snoRNAs were highly expressed in all cell samples analyzed (Supplemental Fig. 9). The distinct length distribution attributed to the three classes of snoRNAs-derived (Fig. 7), was in agreement with the distribution previously described (Taft et al. 2009). Such three snoRNAs-derived populations showed three distinctive length-peaks at 22–24 nt, 25 nt, and 31 nt, suggesting the existence of highly regulated machinery of biogenesis acting on snoRNAs to originate this derivative small RNAs.
FIGURE 7.

Analysis of snoRNA-derived population. (A) The bar diagrams show the read length distribution of sequences derived from different types of snoRNAs. The right panel shows three examples of C/D snoRNAs-derived small RNAs found in all samples analyzed. (B) SNORD45. (C) SNORD21. (D) SNORD60. Predicted secondary structures of the snoRNAs SNORD21, SNORD45, and SNORD60 were calculated with RNAfold web server program. Secondary structure sequences are given in dot-bracket annotation with base-pairings represented by two complementary parentheses “(” and “)” and nonpairing bases by dots “.”. The sequences of highest reads from snoRNAs SNORD21, SNORD45, and SNORD60 are indicated with red arrows. Box D elements are underlined in the sequences and potential base-pairing interactions between C/D Box snoRNAs and rRNA are shown in SNORD21 and SNORD60 based on previous studies (Hertel et al. 2008).
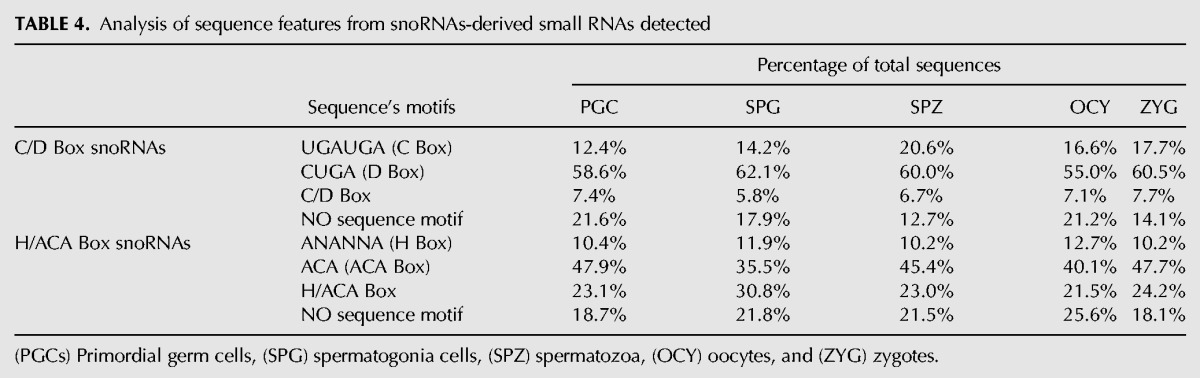
Recently, it has been demonstrated that several small RNAs derived from snoRNAs can act as gene silencers. Analyzing some sequences that we identified in this group of snoRNAs-derived we observed miRNA-like processing signatures (Fig. 7B–D) similar to those previously described by other authors (Ender et al. 2008; Brameier et al. 2011). In fact, small RNAs derived from human SNORD45 and SNORD21 showed miRNA activity (Brameier et al. 2011). All of the small RNAs derived from them conserved the D Box motif (CUGA) into their sequences (Fig. 7B–D). The D Box motif serves as a guide for rRNA methylation, which occurs 4–5 nt upstream of it. In general, >50% of C/D-box-derived small RNAs contains the D Box motif into their sequences (Table 4) suggesting that many small RNAs derived from snoRNAs could be generated from rRNA overlapping regions. The targets of snoRNAs-derived small RNAs are not clearly validated and future studies must be performed to elucidate their role as mRNA silencers and post-transcriptional regulators.
TABLE 4.
Analysis of sequence features from snoRNAs-derived small RNAs detected
Finally, many of the small RNAs detected mapped into untranslated genome regions. All these small RNAs (>45% total reads as it is shown in Table 1) were classified under mm10-genome category. Due to the high complexity and extreme variability existing in this small RNA population, we will address these species in future studies.
DISCUSSION
In recent years different types of endogenous small interference RNAs have emerged as key regulators of gene expression (Morris and Mattick 2014). Initially, the different small RNA classes were studied separately as post-transcriptional regulators of gene expression arising from different biogenesis mechanisms. However, after delving into the knowledge of miRNAs, snoRNAs, piRNAs, and endo-siRNAs, the frontiers of their established regulatory activities tend to blur. The current study provides some examples of both diversity and potential functional convergence of sncRNAs expressed during early spermatogenesis, the fully differentiated gamete, and the zygote. We compared the sncRNAs identified in male germ cells with those found in female gametes and zygotes to improve the understanding of their regulatory roles.
Regardless of being detected in all samples studied, globally the miRNA population decreased their expression levels from PGCs to gametes (Table 1). It has been previously reported that miRNA populations increase during early embryogenesis from oocytes to blastocysts (Ohnishi et al. 2010; García-López and del Mazo 2012), which suggests a strong correlation between miRNA and pluripotency. In fact, in zygotes the miRNA expression rates increased soon after fertilization respect to male and female gametes (Table 1). PGCs and the progressive differentiation to spermatogonia cells showed the highest expression levels of miRNAs but also the most diverse miRNA population with 867 and 964 different miRNAs detected, respectively. By comparing the mRNA array data and NGS data, we observed the complex regulatory networks between miRNAs and their mRNA targets (Fig. 4). The expression profiles found in PGCs and spermatogonia cells of certain transcripts could be explained by its possible interaction with highly expressed miRNAs, as exemplified by Pten, Stat3, Snai1, or Cdkn1a, which are important regulators of cell cycle and germ cell differentiation (Fig. 3). The transcripts of these genes are validated targets of several miRNAs and their expression profiles were inverse to the miRNA profiles detected. Our analysis reflects a cooperative activity of some miRNAs (Fig. 4). However, in other cases, the expression profile fluctuations were poorly explained through miRNA levels, as exemplified by Runx1, Wnt3a, or Mef2c (Fig. 4), reflecting the complex regulatory networks that operate during gene expression regulation in the germline development.
Some miRNAs were found to be preferentially associated with germ cell differentiation. Interestingly, miR-182-5p and miR-183-5p are the two highest expressed miRNAs in PGCs. In contrast, their expression levels decreased in spermatogonia cells and spermatozoa. The members of the cluster mir-183/96/182 are considered oncomirs involved in cell proliferation and migration in different types of cancer (Sarver et al. 2010). Their presence in testicular cells has been associated with testicular tumors (Novotny et al. 2012). Surprisingly, expression of miR-182 in oocytes and zygotes represented >10% of all detected miRNAs, suggesting that the elements of this cluster, and more specifically miR-182 and miR-183, may participate in the control of cell proliferation and differentiation.
Inverse expression pattern to miRNAs was observed for rasRNAs. The rasRNA group encompasses small RNAs derived from repeat genome elements as transposons and classically has been divided into piRNAs and endo-siRNAs. Coincident with expression of PIWI proteins (MIWI, MIWI2, and MILI) different piRNA populations have been reported. Initially, piRNAs were divided in prepachytene and pachytene groups. Prepachytene piRNAs were principally detected in male gonocytes and associated with MIWI2, whereas pachytene piRNAs are expressed during meiosis and associate with MIWI. Here, we expand the group of piRNAs potentially associated with MILI proteins (Supplemental Fig. 10) that have recently been implicated in mRNA targeting (Vourekas and Mourelatos 2014). Such piRNAs were principally mapped to rRNAs and tRNAs (Supplemental Fig. 6B), suggesting that MILI-associated piRNAs may arise as secondary products of tRNA and rRNA processing machineries. This could imply cross-talk between translational and interfering pathways (Sobala and Hutvagner 2011). The alternative roles of some piRNAs as potential regulators of mRNAs have been recently reported (García-López et al. 2014) and experimentally confirmed in species such as Aplysia (Rajasethupathy et al. 2012) and silkworm (Kiuchi et al. 2014). Some new examples of this convergent role and expanded function among diverse piRNAs are described here.
In oocytes and zygotes, the piRNA population is replaced by endo-siRNAs principally derived from LTR and LINE transposons (Supplemental Fig. 6A). The reason for this replacement is not fully understood, but it has been reported that differential expression of transposable elements triggers sequential reprogramming on the embryonic genome (Peaston et al. 2004; Bui et al. 2009). First, by NGS and later confirmed by RT-qPCR, we detected high expression levels of mmur001272954 small RNA, which was classified as endo-siRNA. Their expression levels were elevated especially in oocytes and zygotes (Supplemental Fig. 7). This endo-siRNA arises from a long terminal repeat element (LTR) of ERVL-MaLR family. In humans and rodents it has been demonstrated that LTR sequences can act as promoters regions for multiple coding genes (Buzdin et al. 2006; Romanish et al. 2007) some of them related to embryogenesis (Chang-Yeh et al. 1991). Moreover, recent work suggests the involvement of long-non-coding RNAs (lncRNAs) derived from LTRs of ERV family in embryo stem cell as key regulators of totipotence (Lu et al. 2014) that could explain the abundance of rasRNAs derived from LTR transposons in oocytes and zygotes (Fig. 5).
As miRNAs, the snoRNAs have been defined as ancient mobile genetic elements that lost the capacity to copy themselves to other genomic location (Weber 2006). Growing evidence supports the hypothesis that snoRNAs molecules can serve as a source of small RNAs with miRNA-like functions. Here, we show a detailed characterization of miRNA and snoRNA populations and observe strong similarities between them. Besides similar sequence length and processing signatures, both types of small RNAs show similar expression patterns through male gametogenesis stages. However, whether snoRNAs combine their chemical modification activities with small RNA interference functions is a clue. More than 50% of C/D box snoRNAs-derived small RNAs conserve into their sequences the D Box motif (Table 4). This sequence motif is critical for snoRNAs:rRNA assembly. In fact, methylation of rRNAs begins 4 nt upstream of this region. In consequence, the small RNAs, which contains D box motif in their 3′ ends, have also the cognate sites of ribose methylation in rRNA. Furthermore, the involvement of DICER or DGCR8 in snoRNAs processing is additional evidence that both small RNA classes can act through the same pathway, as it has been demonstrated with the decay of snoRNAs-derived small RNAs in Dicer1 Δ/Δ and Dgcr8 Δ/Δ mouse ES cells (Taft et al. 2009).
Accumulating evidence suggest that sncRNAs play key regulatory roles in cell differentiation and development (Tang 2010; Meikar et al. 2013). The presence and functional activity of some sncRNAs as piRNAs or endo-siRNAs were restricted to germline, but recent studies have also identified them in different somatic cell types (Akkouche et al. 2013; Peng and Lin 2013; Juliano et al. 2014), opening a new fascinating and unexpected dimension of their regulatory roles. By NGS we have analyzed and cataloged sncRNAs and highlighted differences among male early germ cells, gametes and the zygote. In parallel, we also quantify mRNA levels by transcriptome arrays in PGCs and spermatogonia cells and endos-siRNAs and piRNAS by RT-qPCR. Here, we show a reprogramming of diverse sncRNAs along the germline differentiation. However, our data illustrate functional convergence between miRNAs and other types of sncRNAs. In that sense, piRNAs and snoRNAs may act as transcript regulators. Furthermore, tRNA- and rRNA-derived sncRNAs may act as a subtype of piRNAs. Further studies in other cell context or developmental processes will provide insight into the regulatory pathways mediated by the sncRNA networks.
MATERIALS AND METHODS
Animals
All procedures relating to the care and handling of mice used in the present study, were carried out in the CIB-CSIC bioterium under specific pathogen-free (SPF), temperature (22°C ± 1°C) and humidity-controlled (50%–55%) conditions. All animals were housed on a 12 h light/dark cycles with ad libitum access to food and water. Animal care and handling was carried out in accordance with the regulations of the Bioethics Committee of the Consejo Superior de Investigaciones Científicas (CSIC) that approved the study, and adhering to the European Commission guidelines.
PGCs and SPG isolation
To purify fetal PGCs and post-natal spermatogonia cells, a cell suspension was prepared from 13.5 dpc male gonads and 8.5 dpn testes, respectively. The PGCs were purified from the cell suspension by immunopurification using anti-CD15 (anti-SSEA-1 in mouse) magnetic beads in MS columns (Miltenyi Biotech) following the manufacturer's instructions. The purity of the PGCs (93%–96%) was validated by alkaline phosphatase staining with the naphtol AS-MX/ FAST-RED (Sigma-Aldrich). The spermatogonia cells were purified from the cell suspension by performing a short cell culture discrimination procedure (3–4 h) that promotes the sedimentation and adhesion of somatic cells. Accordingly, the spermatogonia cells suspended in the supernatant can be readily recovered. The purity of the spermatogonia cells was validated by the presence of Stra8 (germline-specific gene) and the absence of Fn1 (soma-specific gene). Total RNAs was extracted by TRIzol (Invitrogen) and then evaluated quantitatively and qualitatively and stored at −80°C until use.
Spermatozoa collection
Cauda epididymis and vasa deferentia from mature CD-1 males were collected in 500 µL of M2 medium after which adipose tissue and blood vessels were removed. Cleaned structures were placed in a new 200 µL drop of M2 medium covered with mineral oil. Epididymal fluid was squeezed out and sperm was suspended in M2 medium. Concentrations were determined with a Neubauer hemocytometer. The sperm suspension was layered in a 15 mL conical tube on a discontinuous 90%/45% Percoll gradient in a 1:1:1 ratio (cell sample: 45% Percoll: 90% Percoll) and centrifuged at 700g during 20 min. The pellet was washed in PBS and centrifuged at 350g during 5 min. In order to entirely remove contaminant cells, a hiposmotic shock was induced by resuspension of spermatozoa in H2O-DEPC. Finally, after new centrifugation of the cell suspension the pellet was resuspended and homogenized in 100 µL of TRIzol Reagent (Invitrogen) and frozen at −80°C until use.
Oocyte and zygote collection
Fully grown oocytes and zygotes were collected from the oviducts of mice as described previously (Hogan et al. 1994). Briefly, superovulation was induced in 4–5-wk-old C57BL6 female mice by intraperitoneal injection of 5 IU pregnant mare serum gonadotropin (PMSG), followed 48 h later by 5 IU of human chorionic gonadotropin (hCG). After hCG administration, female mice were mated with DBA/6J males. Fully grown oocytes and zygotes were treated with hyaluronidase (300 µg/mL, Sigma H3884) in order to remove cumulus cells by passage through several drops of M2 medium (Sigma M7167). To carry out the high-throughput sequencing a total of 15,210 fully grown oocytes and 15,416 zygotes were collected. In all cases, the zona pellucida was removed by incubation in 30 μL of prewarmed acidic Tyrode solution under mineral oil during approximately 15 sec, followed by careful washing in three drops of M2 medium under a stereomicroscope. Samples were stored in TRIzol Reagent (Invitrogen) and frozen at −80°C until use.
Microarray analysis and data normalization
Total RNA from the testes was extracted using TRIzol Reagent (Invitrogen) according to the manufacturer's suggested protocol. RNA concentrations were quantified measuring absorbance (A260/280 ratio) on a NanoDrop Spectrophotometer ND-1000 (NanoDrop). RNA integrity, considering the particular profile of spermatozoa, was assessed using an Agilent 2100 Bioanalyzer (Agilent Technologies). RNA from PGCs and spermatogonia cells samples were reverse transcribed and labeled for microarray analysis using standard techniques as previously describe (López-Casas et al. 2012). Briefly, 1 µg from total RNA was amplified using the Amino Allyl MessageAmp aRNA kit (Ambion) obtained between 15 and 60 μg of amino-allyl amplified RNA (aRNA). For each sample, 2.5 μg of aRNA was labeled with Cy3 or Cy5 and it was purified with the Amino Allyl MessageAmp aRNA kit (Ambion). Cy3 and Cy5 incorporation was measured in NanoDrop Spectrophotometer ND-1000 (NanoDrop). Labeled targets were resuspended in hybridization buffer and applied to glass microarrays. Hybridizations were performed overnight at 42°C. Washed and dried slides were imaged in an Axon GenePix 4000A scanner.
Raw data were normalized using limma package (Wettenhall and Smyth 2004) and R Bioconductor open software (Gentleman et al. 2004). The background was subtracted from each individual hybridization and they were Lowess normalized (Smyth and Speed 2003). For each condition, a technical replicate was performed by dye-swapping (two hybridizations) in order to minimize a possible bias in the labeling process. Gene expression values are shown as log2FC (fold change). Clustering analysis was performed to define the expression profiles of genes involved in germ cell differentiation; we applied the intensity, fold change, and pair-wise P values as additional filters. Genes were grouped by k-means algorithm into clusters that behave similarly in the different samples (Fig. 3A). After this initial analysis, genes whose normalized expression values were twofold induced in PGCs or spermatogonia cells respect to gonad-less embryos were filtered to search their biological functions using GO terms from STRING 9.0 Software (Snel et al. 2000). The genes involved in germ cell differentiation processes were further analyzed to found subsets of differential expressed genes in PGCs and spermatogonia cells (Fig. 3B).
RNA purification and sequencing
After total RNA isolation from PGC, spermatogonia cells, spermatozoa, oocytes, and zygotes using TRIzol Reagent (Invitrogen), RNA concentrations were quantified using the NanoDrop Spectrophotometer ND-1000 (NanoDrop). The integrity of total RNA isolated was assessed using Agilent 2100 Bioanalyzer (Agilent Technologies). The small RNA libraries from total RNA were constructed as previously described (García-López et al. 2014). Briefly, 3′ and 5′ adapters were ligated to each RNA molecule in order to accomplish the cDNA synthesis. In order to avoid any bias, the cDNA was PCR amplified using common primers and after amplification the small RNA fraction was purified. Such small RNA fraction was sequenced by high-throughput deep sequencing using an Illumina Hiseq2000 sequencer according to Illumina protocols (available on the Illumina website at www.illumina.com/support).
Data analysis
Sequencing using the Illumina platform yielded sequences with a length range of 18–32 nt. Small sequences were classified following the bioinformatic pipeline described in Supplemental Figure 1. In order to compare the sequence representation of different samples, we applied the DeSeq tool of the R/Bioconductor software package (Anders and Huber 2010; Anders et al. 2013). Bowtie aligner was used to align the sequences to the miRBase 20, RNAdb, and Deepbase, allowing up to three mismatches (bowtie -v 3 -l 10 --tryhard -a --strata --best --al), whereas alignments to the Rfam were performed allowing for no mismatches (bowtie -v 0 -l 10 --tryhard -a --strata --best --al). The permissive alignment (up to three mismatches) was due to the potential editing of double-stranded small RNA precursor molecules (Heale et al. 2009; Mallela and Nishikura 2012).
miRNA sequence analysis
By means of bioinformatic approaches we used the information reported in bowtie alignments along with the annotation data contained in miRBase (release 20) (Kozomara and Griffiths-Jones 2011). The classification was performed according to structural criteria that depend on the location of the sequences relative to the canonical miRNA regions within the precursor sequence of every miRNA gene defined in miRBase (Kozomara and Griffiths-Jones 2011).
Search of validated miRNA targets
To avoid false negative cases we did not use target prediction algorithms and directly used experimentally validated miRNA–target interactions which are collected in miRTarBase (Hsu et al. 2011). Only the miRNA-target interactions validated through reporter assay experiments, qPCR, or Western blot were used in the present work.
rasRNA analysis
We classified the sequences obtained as piRNA or endo-siRNA according to the following criteria: (a) The sequences that reported >84% of identity with the sequences contained in the RNAdb were classified as piRNAs. The interaction between the sequences contained in the RNAdb and PIWI proteins has previously demonstrated (Aravin et al. 2006; Girard et al. 2006; Lau et al. 2006). (b) The rest of the sequences were aligned against small RNA sequences contained in the Deepbase (Yang et al. 2010). In DeepBase were deposited small RNA sequences that are derived from the processing of repeat genome elements as transposon, pseudogenes, tRNAs, or rRNAs. Both piRNAs and endo-siRNAs can be derived from these types of repeated elements so we divided the sequences that have been aligned to the Deepbase into two major groups according to biogenesis criteria that we detailed in results. The piRNAs classified after Deepbase alignment were added to the piRNAs classified after RNAdb alignment forming a single group (Supplemental Fig. 1).
Ping-pong analysis
Ping-pong signatures were searched in tRNA- and rRNA-derived sequences as previously has been reported (García-López et al. 2014). Briefly, we analyzed by bioinformatics approaches, the proportion of tRNA- or rRNA-derived showing complementarity between the first 10 nt with the sequences of the other rasRNAs detected by NGS. Then, the sequences without ping-pong partner were aligned in a reverse complementary manner with the sequences contained in Mus_musculus.GRCm38.75.cdna database, using Bowtie aligner program (bowtie --nofw -k 10 -v 0 -p 8 --quiet --best --strata).
RT-qPCR amplification of rasRNA molecules
Commercial stem–loop RT-primers and TaqMan (Applied Biosystems) qPCR primers were used to amplify the selected rasRNAs, PIR64428, PIR63462, PIR75142, and mmur001272954. All custom primers used were designed in collaboration with Applied Biosystems. Due to the difficulty to find a consensus housekeeping gene for all cell types analyzed we decided to normalize the RT-qPCR data using two different small RNAs: let-7a-1-3p, miR-1-3p, and the U6 gene. The miRNAs let-7a-1-3p and miR-1-3p showed similar normalized reads in all cell types after the NGS data analysis, so we considered them useful for RT-qPCR normalization.
We used RNA extracted from PGC, spermatogonia cells, spermatozoa, oocytes, and zygotes to generate cDNAs by reverse transcription. All reactions were performed at 16°C for 30 min, 42°C for 30 min, 85°C for 5 min, and then held at 4°C. The reactions were performed in a total volume of 15 μL containing the following components: NP-40 0.1%, 1× RT buffer, 0.25 mM of each dNTP, 50 U of MultiScribe Reverse Transcriptase (Applied Biosystems), 38 U of RNase inhibitor, and the specific primer. To reverse transcribe the mature miRNA molecules used as reference genes, stem–loop commercial RT primers were used according to the manufacturer's recommendations (Applied Biosystems).
Quantitative PCR reactions were performed using specific TaqMan real-time PCR primers and precursor primers in an iQ5 thermocycler Detection System (Bio-Rad). The 20 μL PCR reaction volume contained: 4.5 μL RT product, 1× TaqMan Universal PCR master mix, 1 μL of primers, and the probe mix of the TaqMan MicroRNA Assay kit. Reactions were incubated at 95°C for 10 min, followed by 45 cycles of 95°C for 15 sec and 60°C for 10 min. The expression levels of rasRNAs that we obtained by RT-qPCR, were determined using the 2−ΔΔCt method (Schmittgen et al. 2004) and let-7a-1-3p, miR-1-3p, and U6 gene were used as reference genes.
DATA DEPOSITION
Public data base accessions are provided for all raw data sets and all data obtained as result of analysis here mentioned have been deposited in the NCBI Gene Expression Omnibus (GEO) (http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE59255.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
We thank M. Quesada and M. Moreno for technical assistance, and Dr. P. Cohen for critical review of the manuscript. M.A.B.-E. was supported by Postdoctoral Grant from CONACYT Mexico (165418). This work was supported by grants from MEDDTL, France (11-MRES-PNRPE-9-CVS-072); CSIC (PIE-201020E016) and MINECO (BFU2013-42164-R), Spain.
Footnotes
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.048215.114.
REFERENCES
- Akkouche A, Grentzinger T, Fablet M, Armenise C, Burlet N, Braman V, Chambeyron S, Vieira C 2013. Maternally deposited germline piRNAs silence the tirant retrotransposon in somatic cells. EMBO Rep 14: 458–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Huber W 2010. Differential expression analysis for sequence count data. Genome Biol 11: R106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, McCarthy DJ, Chen Y, Okoniewski M, Smyth GK, Huber W, Robinson MD 2013. Count-based differential expression analysis of RNA sequencing data using R and Bioconductor. Nat Protoc 8: 1765–1786. [DOI] [PubMed] [Google Scholar]
- Aravin A, Gaidatzis D, Pfeffer S, Lagos-Quintana M, Landgraf P, Iovino N, Morris P, Brownstein MJ, Kuramochi-Miyagawa S, Nakano T, et al. 2006. A novel class of small RNAs bind to MILI protein in mouse testes. Nature 442: 203–207. [DOI] [PubMed] [Google Scholar]
- Banisch TU, Goudarzi M, Raz E 2012. Small RNAs in germ cell development. Curr Top Dev Biol 99: 79–113. [DOI] [PubMed] [Google Scholar]
- Beyret E, Liu N, Lin H 2012. piRNA biogenesis during adult spermatogenesis in mice is independent of the ping-pong mechanism. Cell Res 22: 1429–1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brameier M, Herwig A, Reinhardt R, Walter L, Gruber J 2011. Human box C/D snoRNAs with miRNA like functions: expanding the range of regulatory RNAs. Nucleic Acids Res 39: 675–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennecke J, Aravin AA, Stark A, Dus M, Kellis M, Sachidanandam R, Hannon GJ 2007. Discrete small RNA-generating loci as master regulators of transposon activity in Drosophila. Cell 128: 1089–1103. [DOI] [PubMed] [Google Scholar]
- Bui LC, Evsikov AV, Khan DR, Archilla C, Peynot N, Hénaut A, Le Bourhis D, Vignon X, Renard JP, Duranthon V 2009. Retrotransposon expression as a defining event of genome reprogramming in fertilized and cloned bovine embryos. Reproduction 138: 289–299. [DOI] [PubMed] [Google Scholar]
- Buzdin A, Kovalskaya-Alexandrova E, Gogvadze E, Sverdlov E 2006. At least 50% of human-specific HERV-K (HML-2) long terminal repeats serve in vivo as active promoters for host nonrepetitive DNA transcription. J Virol 80: 10752–10762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castel SE, Martienssen RA 2013. RNA interference in the nucleus: roles for small RNAs in transcription, epigenetics and beyond. Nat Rev Genet 14: 100–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang-Yeh A, Mold DE, Huang RC 1991. Identification of a novel murine IAP-promoted placenta-expressed gene. Nucleic Acids Res 19: 3667–3672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi E, Choi E, Hwang KC 2013. MicroRNAs as novel regulators of stem cell fate. World J Stem Cells 5: 172–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook MS, Blelloch R 2013. Small RNAs in germline development. Curr Top Dev Biol 102: 159–205. [DOI] [PubMed] [Google Scholar]
- Deato MD, Tjian R 2008. An unexpected role of TAFs and TRFs in skeletal muscle differentiation: switching core promoter complexes. Cold Spring Harb Symp Quant Biol 73: 217–225. [DOI] [PubMed] [Google Scholar]
- Ender C, Krek A, Friedlander MR, Beitzinger M, Weinmann L, Chen W, Pfeffer S, Rajewsky N, Meister G 2008. A human snoRNA with microRNA-like functions. Mol Cell 32: 519–528. [DOI] [PubMed] [Google Scholar]
- Falaleeva M, Stamm S 2013. Processing of snoRNAs as a new source of regulatory non-coding RNAs: snoRNA fragments form a new class of functional RNAs. Bioessays 35: 46–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan X, Chen X, Deng W, Zhong G, Cai Q, Lin T 2013. Up-regulated microRNA-143 in cancer stem cells differentiation promotes prostate cancer cells metastasis by modulating FNDC3B expression. BMC Cancer 13: 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-López J, del Mazo J 2012. Expression dynamics of microRNA biogenesis during preimplantation mouse development. Biochim Biophys Acta 1819: 847–854. [DOI] [PubMed] [Google Scholar]
- García-López J, Hourcade Jde D, Alonso L, Cardenas DB, del Mazo J 2014. Global characterization and target identification of piRNAs and endo-siRNAs in mouse gametes and zygotes. Biochim Biophys Acta 1839: 463–475. [DOI] [PubMed] [Google Scholar]
- Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, et al. 2004. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol 5: R80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girard A, Sachidanandam R, Hannon GJ, Carmell MA 2006. A germline-specific class of small RNAs binds mammalian Piwi proteins. Nature 442: 199–202. [DOI] [PubMed] [Google Scholar]
- Gomes AQ, Nolasco S, Soares H 2013. Non-coding RNAs: multi-tasking molecules in the cell. Int J Mol Sci 14: 16010–16039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimson A, Srivastava M, Fahey B, Woodcroft BJ, Chiang HR, King N, Degnan BM, Rokhsar DS, Bartel DP 2008. Early origins and evolution of microRNAs and Piwi-interacting RNAs in animals. Nature 455: 1193–1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grivna ST, Beyret E, Wang Z, Lin H 2006a. A novel class of small RNAs in mouse spermatogenic cells. Genes Dev 20: 1709–1714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grivna ST, Pyhtila B, Lin H 2006b. MIWI associates with translational machinery and PIWI-interacting RNAs (piRNAs) in regulating spermatogenesis. Proc Natl Acad Sci 103: 13415–13420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunawardane LS, Saito K, Nishida KM, Miyoshi K, Kawamura Y, Nagami T, Siomi H, Siomi MC 2007. A slicer-mediated mechanism for repeat-associated siRNA 5′ end formation in Drosophila. Science 315: 1587–1590. [DOI] [PubMed] [Google Scholar]
- Hayashi K, Chuva de Sousa Lopes SM, Kaneda M, Tang F, Hajkova P, Lao K, O'Carroll D, Das PP, Tarakhovsky A, Miska EA, et al. 2008. MicroRNA biogenesis is required for mouse primordial germ cell development and spermatogenesis. PLoS One 3: e1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heale BS, Keegan LP, McGurk L, Michlewski G, Brindle J, Stanton CM, Caceres JF, O'Connell MA 2009. Editing independent effects of ADARs on the miRNA/siRNA pathways. EMBO J 28: 3145–3156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hertel J, Hofacker IL, Stadler PF 2008. SnoReport: computational identification of snoRNAs with unknown targets. Bioinformatics 24: 158–164. [DOI] [PubMed] [Google Scholar]
- Hogan B, Constantini F, Lacy E 1994. Manipulating the mouse embryo: a laboratory manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- Hsu SD, Lin FM, Wu WY, Liang C, Huang WC, Chan WL, Tsai WT, Chen GZ, Lee CJ, Chiu CM, et al. 2011. miRTarBase: a database curates experimentally validated microRNA–target interactions. Nucleic Acids Res 39: D163–D169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Shen XJ, Zou Q, Zhao QL 2010. Biological functions of microRNAs. Bioorg Khim 36: 747–752. [DOI] [PubMed] [Google Scholar]
- Huang S, Guo W, Tang Y, Ren D, Zou X, Peng X 2012. miR-143 and miR-145 inhibit stem cell characteristics of PC-3 prostate cancer cells. Oncol Rep 28: 1831–1837. [DOI] [PubMed] [Google Scholar]
- Juliano CE, Reich A, Liu N, Götzfried J, Zhong M, Uman S, Reenan RA, Wessel GM, Steele RE, Lin H 2014. PIWI proteins and PIWI-interacting RNAs function in Hydra somatic stem cells. Proc Natl Acad Sci 111: 337–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaspi H, Chapnik E, Levy M, Beck G, Hornstein E, Soen Y 2013. Brief report: miR-290–295 regulate embryonic stem cell differentiation propensities by repressing Pax6. Stem Cells 31: 2266–2272. [DOI] [PubMed] [Google Scholar]
- Kedde M, Agami R 2008. Interplay between microRNAs and RNA-binding proteins determines developmental processes. Cell Cycle 7: 899–903. [DOI] [PubMed] [Google Scholar]
- Kiuchi T, Koga H, Kawamoto M, Shoji K, Sakai H, Arai Y, Ishihara G, Kawaoka S, Sugano S, Shimada T, et al. 2014. A single female-specific piRNA is the primary determiner of sex in the silkworm. Nature 509: 633–636. [DOI] [PubMed] [Google Scholar]
- Kozomara A, Griffiths-Jones S 2011. miRBase: integrating microRNA annotation and deep-sequencing data. Nucleic Acids Res 39: D152–D157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krawetz SA, Kruger A, Lalancette C, Tagett R, Anton E, Draghici S, Diamond MP 2011. A survey of small RNAs in human sperm. Hum Reprod 26: 3401–3412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau NC, Seto AG, Kim J, Kuramochi-Miyagawa S, Nakano T, Bartel DP, Kingston RE 2006. Characterization of the piRNA complex from rat testes. Science 313: 363–367. [DOI] [PubMed] [Google Scholar]
- Leonardo TR, Schultheisz HL, Loring JF, Laurent LC 2012. The functions of microRNAs in pluripotency and reprogramming. Nat Cell Biol 14: 1114–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichner Z, Páll E, Kerekes A, Pállinger E, Maraghechi P, Bosze Z, Gócza E 2011. The miR-290-295 cluster promotes pluripotency maintenance by regulating cell cycle phase distribution in mouse embryonic stem cells. Differentiation 81: 11–24. [DOI] [PubMed] [Google Scholar]
- Liu WM, Pang RT, Chiu PC, Wong BP, Lao K, Lee KF, Yeung WS 2012. Sperm-borne microRNA-34c is required for the first cleavage division in mouse. Proc Natl Acad Sci 109: 490–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López-Casas PP, Mizrak SC, López-Fernández LA, Paz M, de Rooij DG, del Mazo J 2012. The effects of different endocrine disruptors defining compound-specific alterations of gene expression profiles in the developing testis. Reprod Toxicol 33: 106–115. [DOI] [PubMed] [Google Scholar]
- Lu X, Sachs F, Ramsay L, Jacques PÉ, Göke J, Bourque G, Ng HH 2014. The retrovirus HERVH is a long noncoding RNA required for human embryonic stem cell identity. Nat Struct Mol Biol 21: 423–425. [DOI] [PubMed] [Google Scholar]
- Mallela A, Nishikura K 2012. A-to-I editing of protein coding and noncoding RNAs. Crit Rev Biochem Mol Biol 47: 493–501. [DOI] [PubMed] [Google Scholar]
- Martens-Uzunova ES, Olvedy M, Jenster G 2013. Beyond microRNA—novel RNAs derived from small non-coding RNA and their implication in cancer. Cancer Lett 340: 201–211. [DOI] [PubMed] [Google Scholar]
- McIver SC, Roman SD, Nixon B, McLaughlin EA 2012. miRNA and mammalian male germ cells. Hum Reprod Update 18: 44–59. [DOI] [PubMed] [Google Scholar]
- McLaren A 2003. Primordial germ cells in the mouse. Dev Biol 262: 1–15. [DOI] [PubMed] [Google Scholar]
- Meikar O, Da Ros M, Kotaja N 2013. Epigenetic regulation of male germ cell differentiation. Subcell Biochem 61: 119–138. [DOI] [PubMed] [Google Scholar]
- Modzelewski AJ, Holmes RJ, Hilz S, Grimson A, Cohen PE 2012. AGO4 regulates entry into meiosis and influences silencing of sex chromosomes in the male mouse germline. Dev Cell 23: 251–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris KV, Mattick JS 2014. The rise of regulatory RNA. Nat Rev Genet 15: 423–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagano MC, Yeh JR 2013. The identity and fate decision control of spermatogonial stem cells: Where is the point of no return? Curr Top Dev Biol 102: 61–95. [DOI] [PubMed] [Google Scholar]
- Noguchi S, Mori T, Hoshino Y, Maruo K, Yamada N, Kitade Y, Naoe T, Akao Y 2011. MicroRNA-143 functions as a tumor suppressor in human bladder cancer T24 cells. Cancer Lett 307: 211–220. [DOI] [PubMed] [Google Scholar]
- Novotny GW, Belling KC, Bramsen JB, Nielsen JE, Bork-Jensen J, Almstrup K, Sonne SB, Kjems J, Rajpert-De Meyts E, Leffers H 2012. MicroRNA expression profiling of carcinoma in situ cells of the testis. Endocr Relat Cancer 19: 365–379. [DOI] [PubMed] [Google Scholar]
- O'Donnell KA, Boeke JD 2007. Mighty Piwis defend the germline against genome intruders. Cell 129: 37–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohnishi Y, Totoki Y, Toyoda A, Watanabe T, Yamamoto Y, Tokunaga K, Sakaki Y, Sasaki H, Hohjoh H 2010. Small RNA class transition from siRNA/piRNA to miRNA during pre-implantation mouse development. Nucleic Acids Res 38: 5141–5151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamura K, Ladewig E, Zhou L, Lai EC 2013. Functional small RNAs are generated from select miRNA hairpin loops in flies and mammals. Genes Dev 27: 778–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papaioannou MD, Nef S 2010. microRNAs in the testis: building up male fertility. J Androl 31: 26–33. [DOI] [PubMed] [Google Scholar]
- Peaston AE, Evsikov AV, Graber JH, de Vries WN, Holbrook AE, Solter D, Knowles BB 2004. Retrotransposons regulate host genes in mouse oocytes and preimplantation embryos. Dev Cell 7: 597–606. [DOI] [PubMed] [Google Scholar]
- Peng JC, Lin H 2013. Beyond transposons: the epigenetic and somatic functions of the Piwi-piRNA mechanism. Curr Opin Cell Biol 25: 190–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pramanik D, Campbell NR, Karikari C, Chivukula R, Kent OA, Mendell JT, Maitra A 2011. Restitution of tumor suppressor microRNAs using a systemic nanovector inhibits pancreatic cancer growth in mice. Mol Cancer Ther 10: 1470–1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajasethupathy P, Antonov I, Sheridan R, Frey S, Sander C, Tuschl T, Kandel ER 2012. A role for neuronal piRNAs in the epigenetic control of memory-related synaptic plasticity. Cell 149: 693–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera RM, Ross JW 2013. Epigenetics in fertilization and preimplantation embryo development. Prog Biophys Mol Biol 113: 423–432. [DOI] [PubMed] [Google Scholar]
- Romanish MT, Lock WM, van de Lagemaat LN, Dunn CA, Mager DL 2007. Repeated recruitment of LTR retrotransposons as promoters by the anti-apoptotic locus NAIP during mammalian evolution. PLoS Genet 3: e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Röther S, Meister G 2011. Small RNAs derived from longer non-coding RNAs. Biochimie 93: 1905–1915. [DOI] [PubMed] [Google Scholar]
- Sarver AL, Li L, Subramanian S 2010. MicroRNA miR-183 functions as an oncogene by targeting the transcription factor EGR1 and promoting tumor cell migration. Cancer Res 70: 9570–9580. [DOI] [PubMed] [Google Scholar]
- Schmittgen TD, Jiang J, Liu Q, Yang L 2004. A high-throughput method to monitor the expression of microRNA precursors. Nucleic Acids Res 32: e43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seitz H, Tushir JS, Zamore PD 2011. A 5′-uridine amplifies miRNA/miRNA* asymmetry in Drosophila by promoting RNA-induced silencing complex formation. Silence 2: 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smyth GK, Speed T 2003. Normalization of cDNA microarray data. Methods 31: 265–273. [DOI] [PubMed] [Google Scholar]
- Snel B, Lehmann G, Bork P, Huynen MA 2000. STRING: a web-server to retrieve and display the repeatedly occurring neighbourhood of a gene. Nucleic Acids Res 28: 3442–3444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobala A, Hutvagner G 2011. Transfer RNA-derived fragments: origins, processing, and functions. Wiley Interdiscip Rev RNA 2: 853–862. [DOI] [PubMed] [Google Scholar]
- Taft RJ, Glazov EA, Lassmann T, Hayashizaki Y, Carninci P, Mattick JS 2009. Small RNAs derived from snoRNAs. RNA 15: 1233–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang F 2010. Small RNAs in mammalian germline: tiny for immortal. Differentiation 79: 141–146. [DOI] [PubMed] [Google Scholar]
- Teperek M, Miyamoto K 2013. Nuclear reprogramming of sperm and somatic nuclei in eggs and oocytes. Reprod Med Biol 12: 133–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokuhiro K, Isotani A, Yokota S, Yano Y, Oshio S, Hirose M, Wada M, Fujita K, Ogawa Y, Okabe M, et al. 2009. OAZ-t/OAZ3 is essential for rigid connection of sperm tails to heads in mouse. PLoS Genet 5: e1000712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tosaka Y, Tanaka H, Yano Y, Masai K, Nozaki M, Yomogida K, Otani S, Nojima H, Nishimune Y 2000. Identification and characterization of testis specific ornithine decarboxylase antizyme (OAZ-t) gene: expression in haploid germ cells and polyamine-induced frameshifting. Genes Cells 5: 265–276. [DOI] [PubMed] [Google Scholar]
- Vourekas A, Mourelatos Z 2014. HITS-CLIP (CLIP-Seq) for mouse Piwi proteins. Methods Mol Biol 1093: 73–95. [DOI] [PubMed] [Google Scholar]
- Watkins NJ, Bohnsack MT 2012. The box C/D and H/ACA snoRNPs: key players in the modification, processing and the dynamic folding of ribosomal RNA. Wiley Interdiscip Rev RNA 3: 397–414. [DOI] [PubMed] [Google Scholar]
- Weber MJ 2006. Mammalian small nucleolar RNAs are mobile genetic elements. PLoS Genet 2: e205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wettenhall JM, Smyth GK 2004. limmaGUI: a graphical user interface for linear modeling of microarray data. Bioinformatics 20: 3705–3706. [DOI] [PubMed] [Google Scholar]
- Yadav RP, Kotaja N 2014. Small RNAs in spermatogenesis. Mol Cell Endocrinol 382: 498–508. [DOI] [PubMed] [Google Scholar]
- Yang JH, Shao P, Zhou H, Chen YQ, Qu LH 2010. deepBase: a database for deeply annotating and mining deep sequencing data. Nucleic Acids Res 38: D123–D130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao S, Gou LT, Zhang M, Zu LD, Hua MM, Hua Y, Shi HJ, Li Y, Li J, Li D, et al. 2013. piRNA-triggered MIWI ubiquitination and removal by APC/C in late spermatogenesis. Dev Cell 24: 13–25. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.