

Conspectus

Natural products chemistry has historically been the prime arena for the discovery of new chemical transformations and the fountain of insights into key biological processes. It remains a fervent incubator of progress in the fields of chemistry and biology and an exchange mediating the flow of ideas between these allied fields of science. It is with this ethos that our group has taken an interest in and pursued the synthesis of a complex family of natural products termed the dimeric epipolythiodiketopiperazine (ETP) alkaloids. We present here an Account of the highly complex target molecules to which we pegged our ambitions, our systematic and relentless efforts toward those goals, the chemistry we developed in their pursuit, and the insight we have gained for their translational potential as potent anticancer molecules.
The dimeric ETP alkaloids are fungal metabolites that feature a highly complex molecular architecture comprising a densely functionalized core structure with many stereogenic centers, six of which are fully substituted, and a pair of vicinal quaternary carbon stereocenters, decorated on polycyclic architectures in addition to the unique ETP motif that has been recognized as acid-, base-, and redox-sensitive. A cyclo-dipeptide consisting of an essential tryptophan residue and a highly variable ancillary amino acid lies at the core of these structures; investigation of the transformations that take this simplistic core to the complex alkaloids lies at the heart of our research program.
The dimeric epidithiodiketopiperazine alkaloids have largely resisted synthesis on account of their complexity since the 1970s when the founding members of this class, chaetocin A (Hauser D. et al. Helv. Chim. Acta 1970, 53, 1061.) and verticillin A (Katagiri K. et al. J. Antibiot. 1970, 23, 420.), were first isolated. This was despite their potent cytotoxic and bacteriostatic activities, which were well appreciated at the time of their discovery. In the past decade, an increasing number of studies have uncovered powerful new biological processes that these molecules can uniquely effect, such as the inhibition of histone methyltransferases by chaetocin A (Greiner D. et al. Nat. Chem. Biol. 2005, 1, 143.). In fact, the complete collection of hexahydropyrroloindoline alkaloids features a diverse range of potent biological properties including cytotoxic, antitumor, antileukemic, antiviral, antibiotic, and antinematodal activities (Jiang C.-S.; Guo Y.-W.. Mini-Rev. Med. Chem. 2011, 11, 728.). This mélange of activities is reflective of their structural diversity.
Under the precepts of retrobiosynthetic analysis, we have accomplished the syntheses of more than a dozen natural products, including members of the bionectin, calycanthaceous, chaetocin, gliocladin, naseseazine, and verticillin alkaloids. More importantly, these molecules have acted as venerable venues for the development of new strategies to address structural challenges including, but not limited to, C3–C3′ vicinal quaternary centers, heterodimeric linkages, C3–Csp2 linkages, diketopiperazine oxidation, stereoselective thiolation, homologue-specific polysulfidation, and C12-hydroxyl incorporation. Synthesis of these natural products has resulted in the structural confirmation, and sometimes revision such as the case of (+)-naseseazines A and B, as well as access to many plausible biogenetically relevant intermediates and new synthetic ETP derivatives. Furthermore, our studies have paved the way for the formulation of a comprehensive SAR profile and the identification of lead compounds with in vitro subnanomolar IC50’s against a broad range of cancer types.
Introduction
The dimeric epipolythiodiketopiperazine alkaloids are a fascinating collection of natural products derived from fungi across many genera.1,2 Their varied and potent biological activities have garnered much interest from biological and chemical circles since their first isolation many decades ago.3 Equally captivating, their molecular architecture features a plethora of chemical challenges that have until recently served as stalwart impediments to their synthesis (Figure 1). Establishing access to the complete spectrum of dimeric ETP alkaloids through the systematic development of selective, yet generalizable transformations was the primary objective of the research described henceforth.
Figure 1.
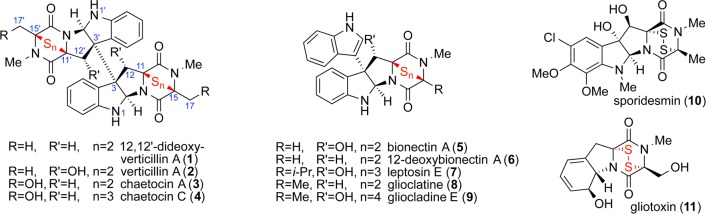
Representative structures of epipolythiodiketopiperazine alkaloids.
From the outset, we were inspired by the biogenesis of the dimeric ETPs, and we looked to nature to inform our synthetic design.4,5 Nature encodes valuable insights about a molecule’s chemical reactivity in the tools it utilizes as well as the trail of natural products it leaves behind. Employing what we termed retrobiosynthetic analysis,6 we attempted to deconstruct the complex target compounds into progressively simpler molecules utilizing only the predicted constituent transforms employed by nature. This analytical framework provides a more coarse-grained roadmap for bioinspired synthesis than a purely biomimetic approach,7 affording chemists sufficient flexibility to engage the full arsenal of chemical synthesis tools. We further attempted to fill in gaps between progenitor natural products by proposing reasonable structures for intermediates that have eluded isolation to date.
This analysis, at the inception of our program, was reconciled with the full complement of available data from the feeding studies of Kirby,8 Taylor,9 and Sammes,10 the genomic analysis of ETP gene clusters by Howlett,11 and the synthetic studies of Kishi,12 Schmidt,13 Rastetter,14 and others to formulate our specific hypothesis for ETP biosynthesis (Scheme 1) upon which our synthetic endeavors were founded.4,15
Scheme 1. Our Retrobiosynthetic Analysis of (+)-1 Highlighting Strategically Critical and Biogenetically Inspired Transforms.
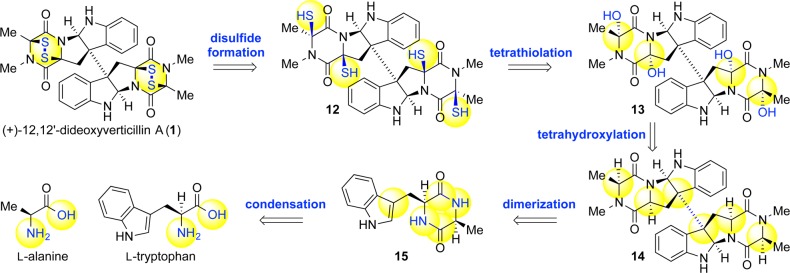
Mindful of the differing goals between nature and ourselves, as well as the distinctiveness of our respective chemical toolboxes, our synthetic approach to the dimeric ETPs was prudently and selectively inspired by our biosynthetic hypothesis.4 Fortunately, the generality and modularity of many of the natural transformations rendered much of nature’s processes suitable for adoption as a blueprint for our syntheses. Close adherence to the biosynthetic hypothesis enabled us to identify a collection of natural products of progressively increasing complexity each of which could be used as a platform for the development of novel solutions to successive challenges (Scheme 1).
C–C Bond Formation
The C3–C3′ linkage, replete with vicinal quaternary carbon stereocenters, is a defining feature of the large superfamily of dimeric hexahydropyrroloindoline alkaloids to which the dimeric ETP alkaloids belong; ubiquity of this motif was highly indicative of the antecedence of dimerization over most biosynthetic transformations including epipolythiodiketopiperazine synthesis. Accordingly, C3–C3′ bond formation was the first structural challenge we chose to address.16 We also identified the C2-symmetric calycanthaceous alkaloids (+)-chimonanthine (19), (+)-folicanthine (26), and (−)-calycanthine (27) as the optimal platforms for exploring new C–C bond forming strategies due to their minimalist structures. In formulating our synthetic plan, we took note of the biosynthetic hypothesis of Woodward17 and Robinson18 in which they posited that the C3–C3′ linkage arises from the oxidative dimerization of tryptamines (Scheme 2).
Scheme 2. (A) Nature’s Approach to Dimeric Cyclotryptamines and (B) Our Bio-inspired Total Synthetic Approach.
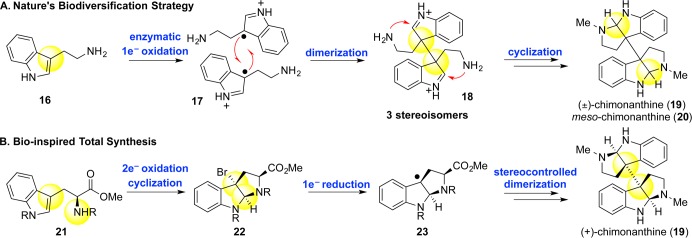
The prospect of harnessing nature’s approach to C3–C3′ bond construction through oxidative dimerization of tryptamine and tryptophan derivatives was appealing. Indeed, our initial foray into this field was marked by efforts to effect the inter- and intramolecular oxidative dimerization of indolic substrates.19 We, however, quickly appreciated the challenges of adopting a strategy whose ostensible aim was antithetical to our own. Nature uses oxidative dimerization as a biodiversification step, creating a medley of dimeric hexahydropyrroloindolines often without regard for features prized in total synthesis: regio-, chemo-, and stereoselectivity.
At this juncture, we hypothesized that the dimerization process could be parsed into its elementary components: single-electron oxidation and radical recombination. Rapid generation of molecular complexity arises from the latter, and that is the inspirational component. Single-electron oxidation, on the other hand, could be tactically accomplished through an alternative controlled process involving two-electron oxidation followed by single-electron reduction. We anticipated the strong preference for cis-fusions on [3.3.0]-bicyclic systems to allow the hexahydropyrroloindoline structures to maintain stereochemical integrity at the C3 position throughout the reduction and dimerization processes (Scheme 2). After significant experimentation, we discovered CoCl(PPh3)3 to be uniquely capable of reducing tricyclic bromides to tertiary benzylic free radicals and effecting dimerization efficiently (Scheme 3).16
Scheme 3. Implementation of Cobalt-Mediated Dimerization on Syntheses of Calycanthaceous Alkaloids.
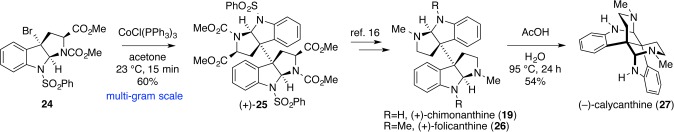
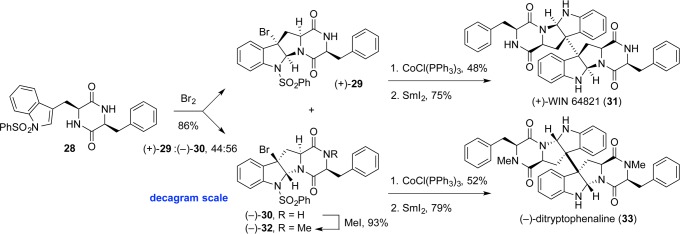
With a highly enabling method for the formation of C3–C3′ bonds in hand, dimeric diketopiperazines (+)-WIN 64821 (31) and (−)-ditryptophenaline (33) provided optimal platforms for exploring the synthesis of diketopiperazines, evaluating their stereoselectivities in halocyclization, managing their proclivity for epimerization, and verifying their compatibility with the dimerization conditions (Scheme 4).20 Indeed, we envisioned that two-electron oxidation of tryptophan through a bromocyclization reaction would enable synthesis of polycyclic tertiary bromides with absolute and relative stereocontrol. Total synthesis of these dimeric diketopiperazines moved us closer to the synthesis of more complex dimeric ETP alkaloids.
Scheme 4. Rapid Synthesis of Dimeric Diketopiperazine Alkaloids via Halocyclization and Cobalt-Promoted Dimerization.
Having observed the highly efficient and powerfully simplifying nature of the radical-mediated dimerization strategy in the construction of C3–C3′ vicinal quaternary centers, we wished to extend this transform to the full spectrum of dimeric cyclotryptamine and diketopiperazine alkaloids. The cobalt-mediated reaction, however, was not ideal for application beyond homodimeric structures, leaving the constitutional and stereochemical heterogeneity across the C3–C3′ junction as an unmet challenge. We instead embraced this opportunity to develop a new strategy for directed heterodimerization, specifically our diazene-based guided fragment coupling methodology.21
In order to take advantage of the power of radical recombination for mixed radical populations and do so selectively, a pair of radicals must be generated in close temporal and spatial proximity. Diazene fragmentation seemed a logical solution to this problem since photochemical decomposition of diazenes results in extrusion of dinitrogen with near-simultaneous formation of two tertiary benzylic radicals confined within a solvent cage;21 rapid radical–radical recombination would then afford a heterodimeric compound. This idea was reduced to practice by converting the C3-bromides of cobalt-dimerization precursors to the corresponding C3-amines. Stepwise union of two differentially functionalized C3-amines as a mixed sulfamide followed by oxidative extrusion of sulfur dioxide produced the key diazene. UV-irradiation successfully effected the first directed fusion of distinct cyclotryptamine fragments (Scheme 5).21
Scheme 5. Directed Fragment Assembly via Synthesis and Fragmentation of Dialkyl Diazenes.
A second-generation approach featuring rhodium-catalyzed intermolecular C–H amination demonstrated higher efficiency in providing the desired sulfamide intermediates (Scheme 6).22 Application of this streamlined process resulted in efficient, selective syntheses of (−)-calycanthidine (43), meso-chimonanthine (20), and (+)- and (−)-desmethyl-meso-chimonanthine.
Scheme 6. Heterodimeric Cyclotryptamine Synthesis via C–H Amination and Diazene-Mediated Coupling.

Cobalt-mediated dimerization enabled access to homodimeric C3sp3–C3′sp3 linkages, and diazenes provided complete control in C3sp3–C3′sp3 bond constructions even using dissimilar fragments.23 We wished to further extend the spectrum of dimerization modes to include C3sp3–Csp2 unions. The C3–C7′-linked (+)-naseseazines A and B24 presented an optimal venue for this new campaign.
Related natural products with C3-indolyl substituents ostensibly arise from the same oxidative radical-dimerization mechanism responsible for C3–C3′ bond formation but with displacement of a tryptophan-derived unit causing bond translocation. In contrast to prior radical-based strategies, a Friedel–Crafts-type reaction seemed particularly apt for a late-stage arylative dimerization approach to C3–C7′ bond construction.25,26
Tetracyclic bromide 48 could be rapidly accessed via our bromocyclization chemistry,20 and as hypothesized, a C3-carbocation could be generated by ionization of the tertiary benzylic bromide with silver salts in high-dielectric media. The carbocations proved to be highly reactive, and a range of π-nucleophiles could be added to the C3 position.25 Gratifyingly, diketopiperazine 46 proved to be a competent nucleophile providing regioisomeric C3–C6′ and C3–C7′ linked adducts in a 1:1.4 ratio favoring the desired C3–C7′ isomer (Scheme 7). Deprotection afforded (+)-naseseazines A and B, each in only six-steps from Boc-l-tryptophan, and enabled our stereochemical revision of these alkaloids.25
Scheme 7. Concise Total Synthesis of the Naseseazines.
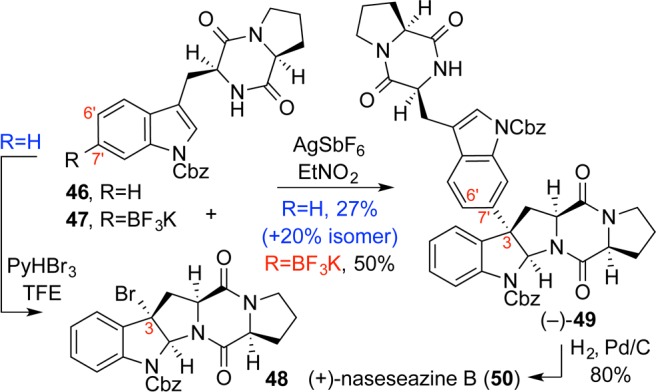
Striving to enhance the regioselectivity of the reaction, we sought a means for directing the Friedel–Crafts reaction toward the C7′ position of the tryptophan nucleophile. After evaluating numerous directing groups, the small, negatively charged trifluoroborate group27−29 proved supreme. Regioselectivity of the directed Friedel–Crafts-type reaction could be enhanced in the presence of crown ethers at low temperatures in non-Lewis-basic high-dielectric media such as nitroethane. Gratifyingly, C7′-trifluoroborate 47 was rapidly synthesized and united with the C3-bromide 48 to afford the desired product as a single regioisomer (Scheme 7). Our mild, highly regioselective Friedel–Crafts-based coupling strategy featured the formation of quaternary stereogenic centers and enabled the directed late-stage union of complex diketopiperazine structures.
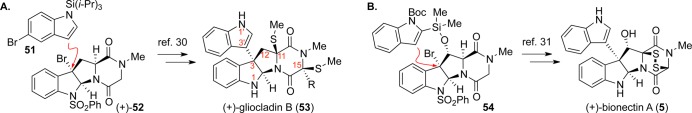
Our strategy for C3-arylation proved quite general as demonstrated by its application to the gliocladins (Scheme 8A).30 In an effort to continually expand the scope of our reactions, we attempted to apply the intermolecular arylation strategy to C12-hydroxylated ETP alkaloids like the bionectins. Unfortunately, the C12-alcohol negatively impacted the stability of the carbocation through adverse inductive effects as well as the approach of the nucleophile through adverse steric effects. Ultimately, we discovered that C3sp3–C3′sp2 bond formation could be accomplished by tethering a nucleophile to the C12-alcohol and performing an intramolecular Friedel–Crafts reaction (Scheme 8B).31
Scheme 8. Inter- and Intramolecular Friedel–Crafts C3-Arylation Strategies Applied to Syntheses of (+)-Gliocladin B (A) and (+)-Bionectin A (B).
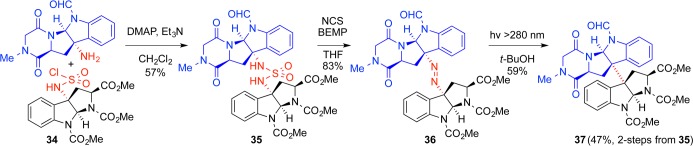
Oxidation
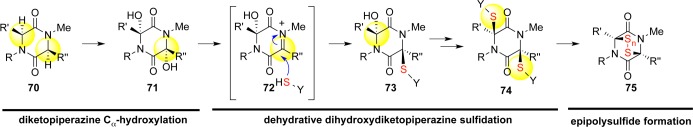
Having established a concise route to the carboskeleton of the dimeric ETP alkaloids, we hypothesized that oxidation of the core diketopiperazine would afford access to N-acyliminium ion intermediates and enable nucleophilic incorporation of hydrogen sulfide surrogates. Of the potential N-, Cα-, or Cβ-oxidation modes en route to these intermediates,4 our choice of Cα-oxidation was least conventional: β-elimination of β-hydroxylated amino acids by PLP-dependent serine/threonine ammonia lyases or kinase-dependent phosphoserine/threonine lyases is a well-documented posttranslational modification, and N-acyliminium ions could be derived by protonation of the resultant dehydroalanines. Alternatively, N-hydroxylated amides in diketopiperazine-containing natural products such as mycelianamide and astechrome provided support for the N-hydroxylation–elimination pathway.32 In contrast, direct evidence and precedence for the occurrence of Cα-hydroxylated amino acid residues as potential biosynthetic precursors for ETP alkaloids was notably lacking at the inception of our synthetic program targeting these alkaloids.4,15
Despite significant support for the β-hydroxylation/elimination strategy, this biosynthetic scheme was incongruent with the full spectrum of congeners in the ETP family. The cyclo-dipeptide that is central to the ETP motif comprises a highly variable amino acid in addition to the requisite tryptophan residue. The range of ancillary amino acids that could be incorporated into these natural products spans seven amino acids. A universal β-hydroxylation mechanism with the versatility to accommodate the diversity of side chains seemed highly unlikely if not impossible in the case of glycine.
Similarly, Ottenheijm’s proposal for the generation of N-acylimines via an N-hydroxylation scheme was appealing but unsuitable on account of the structural constraints imposed by the chronology of the biosynthetic process. Observation of unelaborated dimeric diketopiperazine structures such as (+)-WIN 64821 (31) and (−)-ditryptophenaline (33) made evident the precedence of dimerization over sulfuration. Quaternization of the C3 position leads to intramolecular cyclization, which gives rise to the hexahydropyrroloindoline core structure. This event saturates the valency of the N10 position and renders an N-hydroxylation event unlikely.
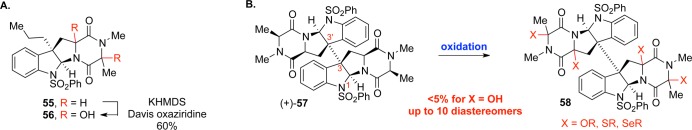
We adopted the hypothesis that chemical elaboration of the ETP core structure involves Cα-hydroxylation,4,15 and our initial explorations focused on Cα-hydroxylation via enolate chemistry. As predicted, double hydroxylation using potassium hexamethyldisilazane (KHMDS) and the Davis oxaziridine on model monomer 55 proceeded adequately to afford diol 56 in 60% yield as a mixture of three diastereomers (Scheme 9). While promising, we anticipated that translation of these reaction conditions to a dimeric system would be difficult. Doubling the number of hydroxylation events on the significantly more sterically congested dimer (+)-57 combined with the potential for generating 10 stereoisomers resulted in vanishingly low yields (<5%) even after significant optimization. Nitrogen-, oxygen-, sulfur-, and selenium-based oxidants were all insufficient in effecting a transformation plagued by low reactivity, selectivity, and stability.
Scheme 9. (A) Initial Oxidation Conditions and (B) Challenges in Translation to Dimeric Substrates.
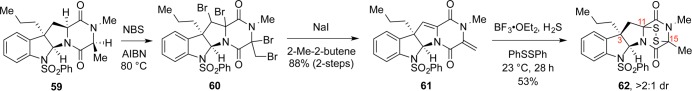
In light of these challenges, we revisited the monomeric model system and looked into the use of radical chemistry. Surprisingly, free-radical bromination of tetracycle 59 resulted in clean oxidation to a tetrabromide, which was reduced in situ to diene 61 (Scheme 10). The reaction was noteworthy for its mild conditions and overall efficiency, and although these conditions ultimately did not translate to the more sensitive dimers, they did offer the first example of late-stage conversion of a complex diketopiperazine to the corresponding ETP. Furthermore, careful analysis of each elementary step yielded key insights for the development of new transformations.
Scheme 10. First Successful Synthesis of a Tetracyclic ETP via Late-Stage Oxidation of the Corresponding Diketopiperazine.
The bromination event is initiated by abstraction of a captodatively stabilized Cα–H bond, whose bond dissociation energy (BDE) was estimated to be below that of H–Br and at the lower end of the typical range for amino acids. Recognizing the equivalence in BDEs between H–Br and formyl C–H bonds and cognizant of permanganate’s ability to oxidize aldehydes through a radical mechanism, we inferred permanganate’s unique ability to hydroxylate Cα–H bonds in diketopiperazines. Indeed, treatment of octacyclic dimer 57 with nBu4NMnO4 in pyridine afforded a diastereomeric mixture of tetraol 58 in 9% yield. The alcohols were dehydrated to provide an isomerically homogeneous population of a tetraene intermediate for facile analysis; this intermediate, however, was incompetent for subsequent thiolation due to the intransigence of the tryptophan-derived enamides toward protonation.
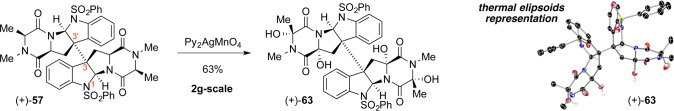
Improved purification conditions for the highly sensitive polar tetraol and optimization around the permanganate counterion dramatically improved the yield, while the move to an abasic solvent dispelled all issues related to diastereoisomerism. Treatment of dimer (+)-57 with Py2AgMnO4 in CH2Cl2 afforded tetraol (+)-63 as a single diastereomer in 63% yield (∼90% yield/hydroxylation) in a single step (Scheme 11). Analysis by X-ray diffraction of the tetrahydroxylation product revealed stereoretentive hydroxylation events, consistent with a radical abstraction–rebound mechanism. Radical-clock hydantoins bolstered this mechanism because the hydroxylation event occurs faster than the rate of cyclopropyl radical ring opening.4 A rare stereoinvertive hydroxylation observed during our synthesis of the bionectins offers further evidence in favor of a C–H abstraction mechanism over a direct C–H insertion mechanism, especially at more crowded C11 centers.31
Scheme 11. Our Biogenetically Inspired Tetrahydroxylation Reaction.
The tetraol proved highly sensitive, rapidly undergoing ring–chain tautomerization or elimination under basic or acidic conditions, respectively. Even simple dissolution in methanol leads to complex mixtures. The dearth of isolated natural products with hemiaminals may reflect its unique sensitivities rather than its absence as a legitimate intermediate in biological processes. Nevertheless, our diketopiperazine hydroxylation reaction has proven highly portable, successfully oxidizing the Cα–H bonds of serine and glycine residues in the chaetocins33 and gliocladins and bionectins,30,31 respectively, even in the presence of electron-rich indoles (Scheme 12).
Scheme 12. Representative Applications of Our Bioinspired Diketopiperazine Hydroxylation in Complex Settings.

Success of the permanganate-mediated tetrahydroxylation reaction led us to ponder the ramifications of such a transformation on our understanding of the biosynthesis of these compounds (Scheme 13). Our work, published in 2009, demonstrated the chemical viability of the Cα-hydroxylated species for the first time and its competency in further transformation to the epidisulfides of interest.4 In order for the Cα-hydroxylation event to be a bona fide biosynthetic transformation, however, an enzyme capable of executing the hydroxylation event must be encoded within the biosynthetic gene clusters responsible for the core ETP biosynthesis. Furthermore, we predicted that based on the mechanistic resemblance between our permanganate transformation and Fe(IV)-oxo-mediated oxidations, a cytochrome P450 may be responsible for the oxidation event. Consistent with this hypothesis, we discovered that cytochrome P450s with unassigned function were annotated within ETP gene clusters by Howlett and co-workers.11 In a full explication of our ETP biosynthetic hypothesis in 2009, we predicted the intermediacy of Cα-hydroxylated intermediates and identified the corresponding enzyme.4,15 We were gratified to learn in 2011 that the Hertweck group was able to confirm experimentally the accuracy of our detailed predictions made two years earlier.34
Scheme 13. Our Hypothesis for Biogenesis of Epipolythiodiketopiperazines via Cα-Hydroxylation and Dehydrative Sulfidation.
Thiolation
Robust, scalable access to N-acyliminium ion precursors paved the way for several iterations of our thiolation strategy, each improving on the last with respect to selectivity and generality. The first-generation synthesis of (+)-12,12′-dideoxyverticillin A (1) saw the implementation of our vision for a viable thiolation event in its purest form: Lewis acid-mediated ionization of the hemiaminals on tetraol 63 would generate N-acyliminium ions to which nucleophilic sulfur atoms could be added.4 The vision was carried out by condensation of hydrogen sulfide into a solution of tetraol 63 then addition of the highly oxophilic/weakly thiophilic Lewis acid hafnium trifluoromethanesulfonate in a pressure vessel. Removal of all volatiles followed by oxidative workup yielded the first synthetic samples of natural product (+)-1 in 2–15% yield (Scheme 14). While a vindication for our synthetic endeavor, this strategy left much to be desired.
Scheme 14. First-Generation Thiolation Strategy and Synthesis of (+)-12,12′-Dideoxyverticillin A (1).

Our second-generation strategy represented our effort to introduce selectivity and control into the thiolation process along with increased efficiency, reproducibility, and safety. With stereochemical control of chief concern, we envisioned application of the Woodward–Prévost cis-dihydroxylation principles by substituting dithioacetic acid in lieu of acetic acid. In practice, formation of an intractable mixture of products led us to decouple the thiolation and hydrolysis steps by employing a diprotic nucleophile (Scheme 15). A neutral double adduct would enable its isolation and hydrolysis in a subsequent step; potassium trithiocarbonate proved perfect for this application. Although this reagent had been used previously,13 its usage had been limited to SN2-type reactions in basic polar protic media, conditions incompatible with N-acyliminium ion generation. After careful design, we were able to balance the acid instability of the trithiocarbonate in nucleophilic solvents with its exclusive solubility in polar protic media. A solution of dipotassium trithiocarbonate and trifluoroacetic acid was added to tetraol (+)-63 in dichloromethane to afford the desired bisdithiepanethione (+)-83 in 56% yield. The bisdithiepanethione could then be unraveled under exceedingly mild conditions using ethanolamine to generate putative tetrathiol 84, which could undergo autoxidation or be subject to an oxidative workup to yield epidisulfide (+)-1 in 62% yield (Scheme 15).
Scheme 15. Second-Generation Thiolation Strategy for the Synthesis of (+)-1.

cis-Dithiolation notwithstanding, the transformation was noteworthy for an additional element of stereocontrol. Thiolation studies on monomeric systems featuring a n-propyl group at the C3 position resulted in a 2:1 endo/exo diastereoselection (Scheme 10), but trithiocarbonate addition across the dimeric diketopiperazines resulted in 5:1 endo/exo diastereofacial control per diketopiperazine owing to the enhanced steric pressures imposed by the larger C3 substituent.
In totality, this thiolation strategy was the first realization of our initial roadmap for stereochemical induction, which conceptually mirrored Seebach’s self-reproduction of chirality.35 All stereochemical information evident in the final product was encoded in the basic l-amino acid building blocks (Scheme 1). Their stereochemistry was relayed into the C2/C3 stereocenters, maintained through the C3–C3′ bond formation, stewarded through stereoablative processes at the C11/C15 stereocenters, and then reintroduced at the Cα-stereocenters whence the stereochemical information originated. To summarize, information was relayed from C15 out to C3 and back.
Our thiolation strategy was further refined as we expanded our work into other congeners within the dimeric ETP family. We wished to build upon the elements of control, regio, chemo, and stereo, that we had established in our prior work. Specifically, we hoped to introduce a level of precision and generality into the degree of sulfur concatenation in the synthesis of dimeric epipolythiodiketopiperazine alkaloids (Scheme 16). In order to achieve a controlled synthesis of sulfur homologues, we had to appreciate fully the nuances in the reactivity of the ETP alkaloids.
Scheme 16. Third-Generation Thiolation Strategy for Stereoselective Diketopiperazine Polysulfidation.

In our study of the chaetocins, we observed that the introduction of a heteroatom at C17 resulted in severe retardation of the C15-hemiaminal ionization event through inductive and stereoelectronic effects.33,36 We embraced this disparate reactivity and exploited it to introduce a thiol at C11 with complete regio- and stereoselectivity (Scheme 16). Accordingly, addition of trifluoroacetic acid to tetraol 65 in hydrogen sulfide-saturated nitromethane resulted in its clean conversion to a dithiol as a single diastereomer. Global acylation with isobutyryl chloride afforded dithioester (+)-86 while simultaneously protecting the C15-position from deleterious ring–chain tautomerization and activating it for future ionization.
The thiolation sequence was undertaken in the presence of the N1-benzenesulfonyl group to accommodate the enhanced acid sensitivity of the C2-aminals during sulfur incorporation on this substrate. This inevitable tactical maneuver was one we sought to avoid in previous routes because of the heightened redox lability of Cα-thiols at captodatively stabilized positions. Fortunately, realization that a photoinduced electron transfer reaction via exciplex formation could enable a targeted reduction led to the highly chemoselective unveiling of the aniline moieties. Chemoselective hydrazinolysis of the C11-thioisobutyrates at this juncture revealed the thiol functions to which one, two, or three sulfur atoms could be concatenated by appropriately sulfurated chloro(triphenylmethane)polysulfane reagents in prelude to the capstone event (Scheme 16).
Formation of the epipolysulfide motifs required a highly choreographed sequence of events carefully scripted to manage the instability of the rapidly disproportionating polysulfanes as well as the reactivity of the short-lived N-acyliminium ions. Application of Lewis acid boron trifluoride etherate to polysulfanes 89 led to the ionization of the preactivated isobutyrate esters to generate N-acyliminium ions at the C15/C15′ positions. These highly reactive species were immediately trapped by cyclization of the polysulfanes with concomitant loss of the triphenylmethyl cations. Highlighting the import of synchrony in the ionization, sulfuration, and detritylation events, successively slower rates of cyclization for tri- and tetrasulfanes onto their respective N-acyliminium ions resulted in competitive decomposition pathways involving the C2-aminal, as well as the inhibition of nucleophilic attack at C15 resulting from anchimeric engagement of the iminium ion by the proximal C17 acetates. The former could be obviated by temporary in situ N1-acylation while the latter could be attenuated through execution of the reaction in high-dielectric media where anchimeric assistance is rendered reversible. Global deacylation resulted in the successful syntheses of dimeric epidi-, epitri-, and epitetrathiodiketopiperazine alkaloids (+)-chaetocins A (3) and C (4) and (+)-12,12′-dideoxychetracin A (93), respectively, in a sulfur homologue-specific manner (Scheme 16). Crucially, this new method was faithful to our standards for chemo-, regio-, and stereoselectivity.
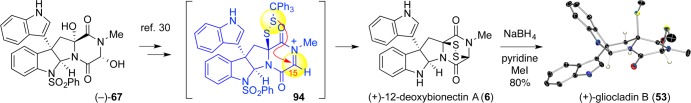
The generality of this thiolation scheme was validated on {(+)-gliocladin A/(+)-bionectin C} and (+)-gliocladin B.30 As noted previously, we discovered that the C11-hemiaminal of diol 67 could be selectively ionized over its glycine-derived counterpart (Scheme 17). Acid-mediated ionization and hydrogen sulfide addition was followed by global isobutyrylation. Chemoselective hydrazinolysis of the thioester then enabled sulfenylation to afford a triphenylmethyl disulfane. Finally, Lewis acid mediated ionization of the C15-alcohol initiated the intramolecular sulfur cyclization to afford the epidisulfide (+)-6.
Scheme 17. Intramolecular Sulfidation Chemistry Implemented in the Synthesis of (+)-Gliocladin B (53).
Our thiolation strategy continued to evolve with the changing structural landscape of our ever-diversifying portfolio of ETP targets. In drafting a synthetic route to the bionectins,31 we were faced with the prospect of being unable to effect sequential ionization of the C11/C15-alcohols; both would be similarly intransigent toward ionization, the former due to a vicinal heteroatom and the latter to the formation of a secondary carbocation. We hypothesized that C12-functionalization would enforce thiolation from the desired face, and prior observations from our gliocladin synthesis intimated the possibility of achieving diastereoselective thiolation at C15.30 Indeed, activation and ionization of the alcohols afforded N-acyliminium ions that could be functionalized by both thioacids and alkyl mercaptans of which only the latter afforded appreciable levels of diastereoselectivity (Scheme 18).
Scheme 18. Substrate-Directed Sulfidation Chemistry Applied to Synthesis of (+)-Bionectin A (5).
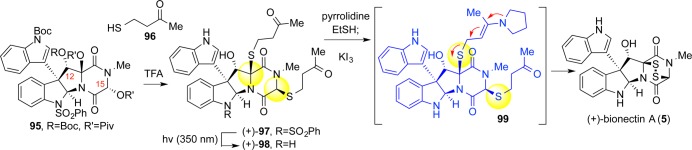
Common 2-cyanoethyl- and 2-trimethylsilylethylmercaptan reagents requiring incompatibly harsh basic dealkylation conditions notwithstanding, alkyl mercaptans serving as effective hydrogen sulfide surrogates were lacking. We developed alternative reagents 4-mercaptobutan-2-one (96, Scheme 18) and 3-mercaptopropiophenone as a result.31 These reagents capitalized on the reversibility of thiol additions to α,β-unsaturated carbonyl groups with thiol exchange or autoxidation of a dithiol driving the reaction forward. Furthermore, elimination could be facilitated by pyrrolidine-mediated enamine catalysis.
We proceeded to showcase the 4-mercaptobutan-2-one (96) reagent in our synthesis of the bionectins (Scheme 18). Acid-mediated hemiaminal-ionization on intermediate 95 in the presence of reagent 96 provided bisthioether (+)-97 in 80% yield and 3:1 dr. After N1-desulfonylation, the bisthioethers could be cleanly unraveled using pyrrolidine and ethanethiol in tetrahydrofuran. Finally, the putative dithiol was subject to oxidative workup to provide (+)-bionectin A (5).31
Four generations of thiolation strategies have now validated the chemical competency of a biosynthetic scheme involving the addition of thiol nucleophiles into hemiaminal-derived N-acyliminium ions.4 It was, however, eminently clear early in our synthetic endeavors that this was the most likely mode for ETP biosynthesis.33 In an exposition on our biosynthetic hypothesis (Scheme 19) in 2009,4,5,33 we took note of Kirby’s radiolabeled sulfur feeding studies,8 which showed high levels of sulfur incorporation from cysteine along with Howlett’s genomic analysis of ETP biosynthetic gene clusters and her annotation of an amino cyclopropane carboxylate synthase and a glutathione S-transferase of unknown functions.11 These clues, combined with our knowledge of chemical reactivity, allowed us to put forth a biosynthetic proposal in which glutathione S-transferase adds glutathione to the diketopiperazine motif (Scheme 19).5,33 Drawing a parallel to the leukotriene pathway, we suggested that sequential hydrolysis of the ancillary amino acids of glutathione would reveal a cysteine adduct. The amino cyclopropane carboxylate synthase of unassigned function may then act as a cysteine lyase responsible for C–S bond cleavage and formation of the dithiol in a PLP-dependent manner. It is exciting to note that our detailed original biosynthetic hypothesis4,5,33 has now been validated in its entirety through recent highly informative biosynthetic experiments by Hertweck34,37,38 and Doyle39 starting in 2011.
Scheme 19. Our Hypothesis for Biogenesis of Epipolythiodiketopiperazines via Divergent Sulfidation of a Diketopiperazine.

Biology
The chemistry described heretofore enabled the synthesis of a diverse collection of natural and unnatural products. We then undertook a structure–activity relationship (SAR) study using these compounds to identify sites relevant to their bioactivity (Figure 2).40 A panel of 60 compounds evaluated against up to five human cancer cell lines demonstrated that substitution at N1 or C17 does not significantly impact biological activity, increasing steric bulk at C3 is correlated with increasing bioactivity, and sulfuration at C11/C15 in a manner consistent with their conversion to β-epidisulfides is essential for cytotoxicity. Twenty-five derivatives have shown low nanomolar to subnanomolar IC50 values against a broad range of cancer types including U-937 (histiocytic lymphoma), HeLa (cervical carcinoma), H460 (lung carcinoma), 786-O (renal carcinoma), and MCF-7 (breast carcinoma) with no activity against human erythrocytes. Additionally, these ETPs induce caspase-dependent apoptotic cell death rather than necrosis. Further biological evaluation and expansion of our SAR study are ongoing and are adding to the promising translational potential of these alkaloids as potent anticancer agents.
Figure 2.
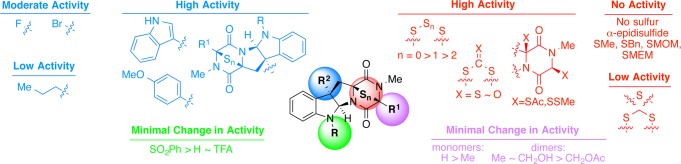
Overview of the results from our epipolythiodiketopiperazine structure–activity relationship study.
Conclusion
We embarked on our synthetic investigations into the dimeric epipolythiodiketopiperazine alkaloids by setting the most complex members of the family as fantastically lofty goals. Using nature as a guide and source of inspiration, we systematically tackled the various challenges that these natural products offered one-by-one: C3–C3′ vicinal quaternary centers, heterodimeric linkages, C3–Csp2 linkages, diketopiperazine oxidation, stereoselective thiolation, homologue-specific polysulfidation, and C12-hydroxyl incorporation, to name a few. In the course of these endeavors, we completed the syntheses of more than a dozen natural products thus far, many for the first time, enabling structure confirmation or reassignment as applicable. The transformations we developed enabled the synthesis of potential biosynthetic intermediates whose instabilities had precluded their isolation, allowing the formulation of a detailed biosynthetic hypothesis, and facilitated our assignment of enzymes with previously unassigned function. Our group’s original biosynthetic hypothesis4,5,15,33 for ETPs has since been nearly completely validated through detailed biosynthetic experiments by other research groups.34−39 Finally, the modularity and generality of our strategies enabled us to compile a collection of compounds for a comprehensive biological study rapidly. These studies resulted in the development of a complete SAR profile as well as the identification of several lead compounds with subnanomolar IC50’s against a broad range of cancers. The research we presented provides an example of exciting discoveries that are possible through the amalgamation of knowledge between allied fields of science and the continuing exciting interplay of chemistry and biology.
Biographies
Justin Kim graduated from Harvard University in 2007 with an A.B. in Chemistry and Physics and A.M. in Chemistry having conducted research under Prof. David Evans. A NDSEG fellow, he received his Ph.D. in chemistry in 2013 from Prof. Mo Movassaghi at MIT. He is currently a Miller Institute Basic Research in Science postdoctoral fellow at U. C. Berkeley pursuing studies in chemical biology with Prof. Carolyn Bertozzi.
Mohammad Movassaghi obtained his B.S. in chemistry from U. C. Berkeley in 1995 with Prof. Paul Bartlett as his research advisor. He completed his graduate studies under the direction of Prof. Andrew G. Myers as a Roche Predoctoral Fellow at Harvard University. After a Damon Runyon Cancer Research Foundation postdoctoral fellowship with Prof. Eric Jacobsen at Harvard University, he joined the faculty at MIT in 2003. His research program focuses on total synthesis of alkaloids in concert with the discovery and development of new reactions for organic synthesis.
This research program was supported by NIH-NIGMS (Grant GM089732). We thank Amgen for additional financial support.
The authors declare no competing financial interest.
Special Issue
Published as part of the Accounts of Chemical Research special issue “Synthesis, Design, and Molecular Function”.
Funding Statement
National Institutes of Health, United States
References
- Hauser D.; Weber H. P.; Sigg H. P. Isolierung und Strukturaufklärung von Chaetocin. Helv. Chim. Acta 1970, 53, 1061–1073. [DOI] [PubMed] [Google Scholar]
- Katagiri K.; Sato K.; Hayakawa S.; Matsushima T.; Minato H. Verticillin A, A New Antibiotic from Verticillium sp. J. Antibiot. 1970, 23, 420–422. [DOI] [PubMed] [Google Scholar]
- Jiang C.-S.; Guo Y.-W. Epipolythiodioxopiperazines from Fungi: Chemistry and Bioactivities. Mini-Rev. Med. Chem. 2011, 11, 728–745. [DOI] [PubMed] [Google Scholar]
- Kim J.; Ashenhurst J. A.; Movassaghi M. Total Synthesis of (+)-11,11′-Dideoxyverticillin A. Science 2009, 324, 238–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J.; Movassaghi M. Biogenetically Inspired Syntheses of Alkaloid Natural Products. Chem. Soc. Rev. 2009, 38, 3035–3050. [DOI] [PubMed] [Google Scholar]
- For representative discussions, see; a Movassaghi M.; Siegel D. S.; Han S. Total Synthesis of All (−)-Agelastatin Alkaloids. Chem. Sci. 2010, 1, 561–566. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Han S.; Movassaghi M. Concise Total Synthesis and Stereochemical Revision of all (−)-Trigonoliimines. J. Am. Chem. Soc. 2011, 133, 10768–10771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson W. S.; Semmelhack M. F.; Sultanbawa M. U. S.; Dolak L. A. A New Approach to Steroid Total Synthesis. A Nonenzymic Biogenetic-Like Olefinic Cyclization Involving the Stereospecific Formation of Five Asymmetric Centers. J. Am. Chem. Soc. 1968, 90, 2994–2996. [Google Scholar]
- Kirby G. W.; Robins D. J.. The Biosynthesis of Gliotoxin and Related Epipolythiodioxopiperazines. In The Biosynthesis of Mycotoxins; Steyn P. S., Ed.; Academic Press: New York, 1980; pp 301–326. [Google Scholar]
- Taylor A.The Toxicology of Sporidesmins and Other Epipolythiadioxopiperazines. In Microbial Toxins; Kadis S., Ciegler A., Ajl S. J., Eds.; Academic Press: New York, 1971; Vol. 7, pp 337–376. [Google Scholar]
- Sammes P. G.Naturally Occurring 2,5-Dioxopiperazines and Related Compounds. In Progress in the Chemistry of Organic Natural Products; Zechmeister L., Herz W., Grisebach H., Kirby G. W., Eds.; Springer: Vienna, Austria, 1975; Vol. 32, pp 51–118. [DOI] [PubMed] [Google Scholar]
- a Gardiner D. M.; Waring P.; Howlett B. J. The Epipolythiodioxopiperazine (ETP) Class of Fungal Toxins: Distribution, Mode of Action, Functions and Biosynthesis. Microbiology 2005, 151, 1021–1032. [DOI] [PubMed] [Google Scholar]; b Gardiner D. M.; Howlett B. J. Bioinformatic and Expression Analysis of the Putative Gliotoxin Biosynthetic Gene Cluster of Aspergillus fumigatus. FEMS Microbiol. Lett. 2005, 248, 241–248. [DOI] [PubMed] [Google Scholar]; c Fox E. M.; Howlett B. J. Biosynthetic Gene Clusters for Epipolythiodioxopiperazines in Filamentous Fungi. Mycol. Res. 2008, 112, 162–169. [DOI] [PubMed] [Google Scholar]
- Fukuyama T.; Nakatsuka S.-I.; Kishi Y. Total Synthesis of Gliotoxin, Dehydrogliotoxin and Hyalodendrin. Tetrahedron 1981, 37, 2045–2078. [Google Scholar]
- Poisel H.; Schmidt U. Syntheseversuche in der Reihe der 3.6-Epidithio-2.5-dioxo-piperazin-Antibiotika Gliotoxin, Sporidesmin, Aranotin und Chaetocin, II. Chem. Ber. 1971, 104, 1714–1721. [DOI] [PubMed] [Google Scholar]
- Williams R. M.; Rastetter W. H. Syntheses of the Fungal Metabolites (±)-Gliovictin and (±)-Hyalodendrin. J. Org. Chem. 1980, 45, 2625–2631. [Google Scholar]
- Miller S. J. Total Chemical Synthesis Peers into the Biosynthetic Black Box. Science 2009, 324, 186–187. [DOI] [PubMed] [Google Scholar]
- Movassaghi M.; Schmidt M. A. Concise Total Synthesis of (−)-Calycanthine, (+)-Chimonanthine, and (+)-Folicanthine. Angew. Chem., Int. Ed. 2007, 46, 3725–3728. [DOI] [PubMed] [Google Scholar]
- Woodward R. B.; Yang N. C.; Katz T. J.; Clark V. M.; Harley-Mason J.; Ingleby R. F.; Shepard N. Calycanthine: The Structure of the Alkaloid and Its Degradation Product, Calycanine. Proc. Chem. Soc., London 1960, 76–78. [Google Scholar]
- Robinson R.; Teuber H. J. Reactions with Nitrosodisulfonate. IV. Calycanthine and Calycanthidine. Chem. Ind. 1954, 783–784. [Google Scholar]
- Schmidt M. A.; Movassaghi M. New Strategies for the Synthesis of Hexahydropyrroloindole Alkaloids Inspired by Biosynthetic Hypotheses. Synlett 2008, 313–324. [Google Scholar]
- Movassaghi M.; Schmidt M. A.; Ashenhurst J. A. Concise Total Synthesis of (+)-WIN 64821 and (−)-Ditryptophenaline. Angew. Chem., Int. Ed. 2008, 47, 1485–1487. [DOI] [PubMed] [Google Scholar]
- Movassaghi M.; Ahmad O. K.; Lathrop S. P. Directed Heterodimerization: Stereocontrolled Assembly via Solvent-Caged Unsymmetrical Diazene Fragmentation. J. Am. Chem. Soc. 2011, 133, 13002–13005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lathrop S. P.; Movassaghi M. Application of Diazene-Directed Fragment Assembly to the Total Synthesis and Stereochemical Assignment of (+)-Desmethyl-meso-chimonanthine and Related Heterodimeric Alkaloids. Chem. Sci. 2014, 5, 333–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lathrop S. P.; Kim J.; Movassaghi M. Radical Mediated Dimerization and Oxidation Reactions for the Synthesis of Complex Alkaloids. Chimia 2012, 66, 389–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raju R.; Piggott A. M.; Conte M.; Aalbersberg W. G. L.; Feussner K.; Capon R. J. Naseseazines A and B: A New Dimeric Diketopiperazine Framework from a Marine-Derived Actinomycete, Streptomyces sp. Org. Lett. 2009, 11, 3862–3865. [DOI] [PubMed] [Google Scholar]
- Kim J.; Movassaghi M. Concise Total Synthesis and Stereochemical Revision of (+)-Naseseazines A and B: Regioselective Arylative Dimerization of Diketopiperazine Alkaloids. J. Am. Chem. Soc. 2011, 133, 14940–14943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For our biosynthetic hypothesis, see p S3 in the Supporting Information of ref (25).
- Lee S.; MacMillan D. W. C. Organocatalytic Vinyl and Friedel–Crafts Alkylations with Trifluoroborate Salts. J. Am. Chem. Soc. 2007, 129, 15438–15439. [DOI] [PubMed] [Google Scholar]
- Molander G. A.; Ellis N. Organotrifluoroborates: Protected Boronic Acids That Expand the Versatility of the Suzuki Coupling Reaction. Acc. Chem. Res. 2007, 40, 275–286. [DOI] [PubMed] [Google Scholar]
- Vedejs E.; Chapman R. W.; Fields S. C.; Lin S.; Schrimpf M. R. Conversion of Arylboronic Acids into Potassium Aryltrifluoroborates: Convenient Precursors of Arylboron Difluoride Lewis Acids. J. Org. Chem. 1995, 60, 3020–3027. [Google Scholar]
- Boyer N.; Movassaghi M. Concise Total Synthesis of (+)-Gliocladins B and C. Chem. Sci. 2012, 3, 1798–1803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coste A.; Kim J.; Adams T. C.; Movassaghi M. Concise Total Synthesis of (+)-Bionectins A and C. Chem. Sci. 2013, 4, 3191–3197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herscheid J. D. M.; Nivard R. J. F.; Tijhuis M. W.; Scholten H. P. H.; Ottenheijm H. C. J. α-Functionalized Amino Acid Derivatives. A Synthetic Approach of Possible Biogenetic Importance. J. Org. Chem. 1980, 45, 1880–1885. [Google Scholar]
- Kim J.; Movassaghi M. General Approach to Epipolythiodiketopiperazine Alkaloids: Total Synthesis of (+)-Chaetocins A and C and (+)-12,12′-Dideoxychetracin A. J. Am. Chem. Soc. 2010, 132, 14376–14378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Scharf D. H.; Remme N.; Habel A.; Chankhamjon P.; Scherlach K.; Heinekamp T.; Hortschansky P.; Brakhage A. A.; Hertweck C. A Dedicated Glutathione S-Transferase Mediates Carbon-Sulfur Bond Formation in Gliotoxin Biosynthesis. J. Am. Chem. Soc. 2011, 133, 12322–12325. [DOI] [PubMed] [Google Scholar]; b Scharf D. H.; Habel A.; Heinekamp T.; Brakhage A. A.; Hertweck C. Opposed Effects of Enzymatic Gliotoxin N- and S-Methylations. J. Am. Chem. Soc. 2014, 136, 11674–11679. [DOI] [PubMed] [Google Scholar]
- Seebach D.; Boes M.; Naef R.; Schweizer W. B. Alkylation of Amino Acids without Loss of the Optical Activity: Preparation of α-Substituted Proline Derivatives. A Case of Self-Reproduction of Chirality. J. Am. Chem. Soc. 1983, 105, 5390–5398. [Google Scholar]
- Iwasa E.; Hamashima Y.; Fujishiro S.; Higuchi E.; Ito A.; Yoshida M.; Sodeoka M. Total Synthesis of (+)-Chaetocin and Its Analogues: Their Histone Methyltransferase G9a Inhibitory Activity. J. Am. Chem. Soc. 2010, 132, 4078–4079. [DOI] [PubMed] [Google Scholar]
- Scharf D. H.; Chankhamjon P.; Scherlach K.; Heinekamp T.; Roth M.; Brakhage A. A.; Hertweck C. Epidithiol Formation by an Unprecedented Twin Carbon-Sulfur Lyase in the Gliotoxin Pathway. Angew. Chem., Int. Ed. 2012, 51, 10064–10068. [DOI] [PubMed] [Google Scholar]
- Scharf D. H.; Chankhamjon P.; Scherlach K.; Heinekamp T.; Willing K.; Brakhage A. A.; Hertweck C. Epidithiodiketopiperazine Biosynthesis: A Four-Enzyme Cascade Converts Glutathione Conjugates into Transannular Disulfide Bridges. Angew. Chem., Int. Ed. 2013, 52, 11092–11095. [DOI] [PubMed] [Google Scholar]
- Davis C.; Carberry S.; Schrettl M.; Singh I.; Stephens J. C.; Barry S. M.; Kavanagh K.; Challis G. L.; Brougham D.; Doyle S. The Role of Glutathione S-Transferase GliG in Gliotoxin Biosynthesis in Aspergillus fumigatus. Chem. Biol. 2011, 18, 542–552. [DOI] [PubMed] [Google Scholar]
- Boyer N.; Morrison K. C.; Kim J.; Hergenrother P. J.; Movassaghi M. Synthesis and Anticancer Activity of Epipolythiodiketopiperazine Alkaloids. Chem. Sci. 2013, 4, 1646–1657. [DOI] [PMC free article] [PubMed] [Google Scholar]